

Reduced nutrient intake has been shown to increase life span. In this study, O'Rourke et al. show that transcriptional up-regulation of a lipase is part of a physiological response to fasting that leads to an enrichment of ω-6 polyunsaturated fatty acids. This in turn activates autophagy, which leads to starvation resistance and increases C. elegans life span. Their results suggest that a similar activation program may exist in humans after dietary supplementation with ω-3/6 polyunsaturated fatty acids.
Keywords: C. elegans, fasting, lipid, autophagy, polyunsaturated fatty acids (PUFA), longevity
Abstract
Adaptation to nutrient scarcity depends on the activation of metabolic programs to efficiently use internal reserves of energy. Activation of these programs in abundant food regimens can extend life span. However, the common molecular and metabolic changes that promote adaptation to nutritional stress and extend life span are mostly unknown. Here we present a response to fasting, enrichment of ω-6 polyunsaturated fatty acids (PUFAs), which promotes starvation resistance and extends Caenorhabditis elegans life span. Upon fasting, C. elegans induces the expression of a lipase, which in turn leads to an enrichment of ω-6 PUFAs. Supplementing C. elegans culture media with these ω-6 PUFAs increases their resistance to starvation and extends their life span in conditions of food abundance. Supplementation of C. elegans or human epithelial cells with these ω-6 PUFAs activates autophagy, a cell recycling mechanism that promotes starvation survival and slows aging. Inactivation of C. elegans autophagy components reverses the increase in life span conferred by supplementing the C. elegans diet with these fasting-enriched ω-6 PUFAs. We propose that the salubrious effects of dietary supplementation with ω-3/6 PUFAs (fish oils) that have emerged from epidemiological studies in humans may be due to a similar activation of autophagic programs.
Nutrient deprivation triggers homeostatic mechanisms that allow organisms to survive until food becomes available again. Animals bearing mutations that make them resistant to starvation are frequently long-lived under normal feeding regimens (Pan et al. 2007; Lee and Ashrafi 2008; Rogers et al. 2011), suggesting the existence of common molecular mechanisms underlying the physiological responses to nutrient deprivation and the regulation of life span. Although some of the components that sense and transduce nutrient availability into adaptive physiological responses—such as the AMP-sensing AMP kinase, NAD-responsive sirtuins, or multiple input target of rapamycin (TOR) protein kinase—are implicated in aging (Vellai et al. 2003; Apfeld et al. 2004; Jia et al. 2004; Berdichevsky et al. 2006), the common metabolic changes underlying resistance to nutritional stress and extended life span are mostly unknown. Moreover, some compounds have been proposed to extend life span through mimicking the effects of dietary restriction; however, their proposed mechanisms of action have been questioned (Bass et al. 2007; Miller and Birnbaum 2010; Miller et al. 2011).
To uncover genes and metabolites that both promote resistance to nutritional deprivation and have an impact on aging, we searched in data sets from multiple species of animals for conserved metabolic genes that are transcriptionally responsive to fasting. We then asked which of those nutrient-responsive genes were reported to affect Caenorhabditis elegans life span. Lip3, a predicted lipase strongly up-regulated in glucose-deprived Drosophila (Zinke et al. 2002; Bass et al. 2007), has a C. elegans homolog, lipl-4, which is required to extend the life span of germline-deficient worms (Wang et al. 2008). The physiological role of lipl-4 and the nature of the link between LIPL-4 lipase activity and longevity are not known. Here we show that lipl-4 transcriptional up-regulation is part of a physiological response to fasting and that constitutive fasting levels of expression of lipl-4 in ad libitum-fed animals, which mimic the transcriptional up-regulation observed in fasted worms, cause an enrichment of ω-3/6 polyunsaturated fatty acids (PUFAs). Enrichment of ω-3/6 PUFAs has been observed in fasted C. elegans (Van Gilst et al. 2005b), but the biological relevance of this response has not been established. Here we show that these fasting-enriched ω-6 PUFAs activate autophagy, a cell recycling mechanism that promotes starvation survival. In addition to being a major response to caloric restriction, autophagy can slow aging and the onset of age-related disorders (Cuervo 2008; Rubinsztein et al. 2011). We show that dietary supplementation with these fasting-enriched ω-6 PUFAs increases C. elegans life span in well-fed animals by activating autophagy. We also show that the ability to respond to ω-6 PUFAs by activating autophagy is conserved in mammalian cells. Hence, our results reveal that ω-6 PUFAs act as a link between lipolysis and life span in C. elegans and suggest that these ω-6 PUFAs activate responses normally occurring in calorically restricted animals across metazoans.
Results
ω-6 PUFAs enriched upon fasting increase C. elegans resistance to starvation
The C. elegans lipase gene lipl-4 is induced in germline-deficient mutants and is necessary for the increased longevity of these animals (Wang et al. 2008). A Drosophila homolog, Lip3, is the most up-regulated gene in glucose-deprived Drosophila (Zinke et al. 2002). We tested whether lipl-4 is responsive to nutritional stress in C. elegans and found that it is transcriptionally up-regulated up to sevenfold in fertile adults fasted for 2, 6, or 12 h relative to ad libitum-fed age-matched animals (Fig. 1A). However, despite its strong response to fasting and its predicted role as a triglyceride lipase, lipl-4 deficiency [lipl-4(tm4417)] does not affect lipid levels in ad libitum-fed or fasted worms (Supplemental Fig. S1A). One likely explanation for the lack of a lipid phenotype in the lipl-4 knockout mutant is redundancy: lipl-4 is one member of a family of eight C. elegans paralogous genes (lipl-1 to lipl-8). We therefore used an unregulated gene expression approach to study the metabolic function of lipl-4. We constructed transgenic lines in which the expression of lipl-4 is driven by the strong, constitutively active promoter ges-1. In this way, we uncoupled LIPL-4 expression from its normal transcriptional regulation. Animals carrying the ges-1 promoter∷lipl-4 gene fusion express lipl-4 only in the pharynx and gut (Supplemental Fig. S1B) at 12 times higher levels than ad libitum-fed nontransgenic sibling animals (Fig. 1B). In ad libitum-fed conditions, these ges-1 promoter∷lipl-4 gene fusion-expressing animals show levels of expression of lipl-4 mRNA that are physiological for fasting worms; for simplicity, we refer to these animals expressing fasting levels of lipl-4 constitutively as LIPL-4Ox (lipl-4 overexpression). To uncover the potential contribution of lipl-4 to energy homeostasis, we tested the ability of LIPL-4Ox animals to resist nutritional stress. LIPL-4Ox animals are resistant to starvation (Fig. 1C; Supplemental Fig. S2), which suggests that lipl-4 induction is part of a response that improves C. elegans' ability to resist nutritional deprivation. To study the mechanism by which lipl-4 promotes resistance to starvation, we performed a more detailed lipid profiling of LIPL-4Ox worms. Fatty acid methyl-ester (FAME) analysis of lipids isolated from the lipl-4-overexpressing strain (Watts and Browse 2002; O'Rourke et al. 2009) revealed an enrichment of arachidonic acid (AA) and eicosapentanoic acid (EPA), 20-carbon ω-6 and ω-3 fatty acids, respectively (Fig. 1D). Fasted wild-type animals are enriched in AA, EPA, and di-homo-γ-linoleic acid (DGLA) (Fig. 1D; Van Gilst et al. 2005b), but the function of this physiological response has not been established. Because ω-3/6 PUFAs are enriched in fasted and LIPL-4Ox animals and because constitutive high expression of LIPL-4 induces starvation resistance, we hypothesized that ω-3/6 PUFAs were triggering a response that promotes resistance to nutritional deprivation. We therefore tested the impact of supplementing fasted worms with AA, DGLA, and EPA at concentrations comparable with those observed in the LIPL-4-overexpressing strains (Supplemental Fig. S3). AA and DGLA supplementation enhances the ability of wild-type animals to resist starvation (Fig. 1E), suggesting that these ω-6 PUFAs promote C. elegans survival during starvation. In contrast, supplementation with EPA, a similarly absorbable (Supplemental Fig. S3) 20-carbon ω-3 fatty acid, does not improve C. elegans resistance to starvation (Fig. 1E), suggesting that AA and its precursor, DGLA, are not simply used as a carbon source.
Figure 1.

LIPL-4 overexpression mimics a physiological response that increases C. elegans resistance to starvation. (A) lipl-4 is transcriptionally up-regulated in fasting animals. lipl-4 mRNA abundance in fasting wild-type young adults relative to ad libitum feeding animals of the same age at different times from food withdrawal is depicted as mean ± SEM of two independent experiments. Statistical differences are depicted (*). (B) LIPL-4Ox animals express lipl-4 at twice the levels observed in fasted wild-type worms. lipl-4 mRNA levels in well-fed ges-1P∷LIPL-4∷SL2∷GFP young adults compared with control (nontransgenic siblings) is presented from three independent experiments, and all values are corrected for the efficiency of the primer pairs and normalized to ama-1 as internal control as well as to transcript levels in same age nontransgenic sibling animals. All quantitative RT–PCR (qRT–PCR) reactions were performed at least in triplicate. Median ± SEM of ΔΔCt is reported (Pfaffl 2001). (C) LIPL-4 overexpression promotes resistance to starvation. LIPL-4Ox and control (nontransgenic sibling [NTS]) hatchlings were sorted (COPAS Biosort) and independently grown on OP50 bacteria, then harvested as young L4 larvae and rotated at 20°C in 1 μM Sytox orange in S-basal. Every other day, aliquots were sorted to calculate the percentage of red (dead) worms. The average percentage of LIPL-4Ox animals alive on the same day that ∼50% of control worms were alive from three independent experiments ± SEM is depicted. See also representative L4 survival curve in Supplemental Figure S2. (D) Fasting and LIPL-4Ox lead to an enrichment of ω-3/6 PUFAs. Levels of AA and EPA relative to total FAMEs (Watts and Browse 2002; Van Gilst et al. 2005b) are depicted as the mean percentage ± SEM for wild-type well-fed and 8-h fasted and well-fed young adults (similar results were obtained when compared with nontransgenic siblings) (data not shown). (E) AA and DGLA, but not EPA, promote resistance to starvation. Ten micromolar AA, 20 μM EPA, 10 μM DGLA (concentrations of supplemented fatty acids that lead to levels of incorporation comparable with those observed in LIPL-4Ox animals) (see Supplemental Fig. S3), or an equal volume of 50% ethanol was added, and the percentage of animals alive was determined and presented as in C but using Sytox green to stain dead worms.
ω-6 PUFAs activate autophagy in well-fed C. elegans
To investigate the subcellular localization of LIPL-4, we constructed a protein fusion to TagRFP. This LIPL-4∷TagRFP protein fusion is observed in cytoplasmic bodies resembling autophagic granules (Supplemental Fig. S4A). We tested whether LIPL-4 colocalized with active sites of autophagy by simultaneously looking at the localization of LIPL-4∷TagRFP and LGG-1∷GFP (Melendez et al. 2008). The coexpression of LIPL-4∷TagRFP and the autophagic marker LGG-1 shows that the LIPL-4 red fluorescent bodies do not colocalize with the LGG-1 green autophagic punctae (Supplemental Fig. S4A, insets in the top panel), suggesting that, at least in the conditions tested, LIPL-4 does not reside in active autophagic bodies. However, this experiment revealed that there are more abundant LGG-1 punctae in LIPL-4-overexpressing animals compared with nontransgenic siblings, suggesting that high expression of LIPL-4 activates autophagy (Fig. 2A; Supplemental Figs. S4A,B [for bleed-through and autofluorescence controls], S5). Our finding that constitutively increased lipl-4 expression activates autophagy is in agreement with the previous observation that overexpression of lipl-4 from its own promoter also increases the number of autophagic structures (Lapierre et al. 2011). Hence, lipl-4 is induced upon fasting, a nutritional state that activates autophagy, and high expression of lipl-4 triggers some of the physiological changes of fasting, such as enrichment of ω-6 PUFAs and activation of autophagy. In addition, we observed that the supplementation with fasting-enriched ω-6 PUFAs increases C. elegans resistance to starvation (Fig. 1E). As autophagy is a major survival mechanism when food is scarce, we hypothesized that the fasting-enriched ω-6 PUFAs alone might activate autophagy. We found that supplementation with AA and DGLA, but not with EPA, was sufficient to activate autophagy in ad libitum-fed C. elegans (Fig. 2B). AA or DGLA supplementation did not affect C. elegans food consumption (Supplemental Fig. S6), suggesting that ω-6 PUFAs induce cellular responses characteristic of the response to caloric restriction but do not actually induce caloric restriction.
Figure 2.

ω-6 PUFAs activate autophagy in C. elegans. (A) LIPL-4Ox activates autophagy. LGG-1∷GFP signal in L3 animals overexpressing LIPL-4 from a constitutively highly expressed intestinal promoter versus control animals is shown (control corresponds to siblings not carrying LIPL-4∷TagRFP but carrying LGG-1∷GFP); exposure time = 100 msec. Quantification of LGG-1∷GFP punctae per anterior intestinal cell from at least three independent experiments is depicted as mean ± SEM using the same exposure time for LIPL-4∷TagRFP and control worms. At longer exposure times (500 msec), more LGG-1∷GFP punctae than at 100 msec can be observed in control animals; for comparison at 500 msec, see Supplemental Figure S5. For bleed-through and autofluorescence controls, see Supplemental Figure S4. (B) AA and DGLA activate autophagy in C. elegans. LGG-1∷GFP punctae per anterior intestinal cell in supplemented (50 μM) or mock-treated (50% ethanol) L3 animals are shown; data from three independent experiments are depicted as mean ± SEM. (C) fat-6, fat-7, or fat-1 inactivation is insufficient to activate autophagy in C. elegans. LGG-1∷RFP worms were egg-laid in fat-1, fat-6, fat-7, or vector control RNAi plates (qPCR analysis showed that the levels of expression of fat-1, fat-6, and fat-7 after RNAi treatment were 0.2 ± 0.1, 0.3 ± 0.05, 0.3 ± 0.1 of vector control treated animals, respectively). The pattern and intensity of the LGG-1∷RFP signal was scored in young adult worms. Representative control and fat RNAi-treated animals are shown. No difference was observed in the intensity or pattern of the expression of LGG-1∷RFP in treated animals when compared with empty vector-treated worms.
In C. elegans, fat-6 and fat-7 are required for the synthesis of long chain unsaturated fatty acids, and fat-1 is required for the conversion of ω-6 PUFAs to ω-3 PUFAs. Thus, impaired fat-6 and fat-7 function leads to reduced ω-6 PUFA levels, and fat-1 loss of function leads to reduced ω-3 PUFA levels (Watts and Browse 2002; Van Gilst et al. 2005a). To address whether differences in the ratios of ω-6 to ω-3 PUFAs would determine the levels of autophagy, we tested whether the fat genes affected the basal levels of autophagy in well-fed animals. RNAi inactivation of fat-6, fat-7, or fat-1 did not affect the intensity or the distribution of LGG-1∷RFP in well-fed animals (Fig. 2C), suggesting that the effect of ω-6 fatty acids on autophagy is achieved only when their levels relative to fatty acids other than ω-3 fatty acids are above a certain threshold, a condition that cannot be mimicked by inactivating the enzymes responsible for the synthesis of ω-6 fatty acids or their conversion to ω-3 fatty acids. The absence of an autophagy phenotype in the fat-1 RNAi-treated animals, at least in the conditions tested, suggests that it is not the ratio ω-6 to ω-3 that determines the levels of autophagy in worms but does not rule out that ω-3 PUFAs or some of their derivatives could also have an effect on autophagy levels.
ω-6 PUFAs activate autophagy in mammalian cells
Dietary supplementation with ω-3/6 PUFAs can improve several human conditions that intriguingly overlap with the pathologies associated with autophagy malfunction (Cao et al. 2000; Suresh and Das 2001; Das 2008; Deckelbaum and Calder 2010; Kuballa and Xavier 2010). We tested whether human cells would also respond to ω-6 PUFAs by activating autophagy. Supplementation with AA and its precursor, DGLA, but not with EPA, leads to an enrichment of the autophagic marker phosphatydilethanolamine-LC3 (LC3II) in HeLa cells growing in rich medium (Fig. 3A), which suggests that human cells also activate autophagy upon exposure to ω-6 PUFAs even if nutrients are available. As LC3II enrichment can be due to either the activation of autophagy or the blockage of the final steps of the autophagic process (Klionsky et al. 2008), which would result in the accumulation of the lipidated form of LC3, we looked at the accumulation of LC3II in the presence of lysosomal inhibitors and directly tested the degradative capacity of the autophagolysosome. Direct fluorescence microscopy to monitor the distribution of GFP-hLC3B shows an increase in the number of large GFP-hLC3B punctae after 30 h of treatment with 100 μM AA or DGLA (Fig. 3B, top row and table). Treatment with inhibitors of lysosomal enzymes (6 h in 10 μg/mL pepstatin and 5 μg/mL E64d) causes further accumulation of GFP-hLC3B (Fig. 3B, second row and table), suggesting that activation of autophagy and not blockage of lysosomal function is the most likely explanation for the increase in GFP-hLC3B punctae in the presence of AA or DGLA. To rule out the possibility that the increase in the levels of punctate GFP-hLC3B would result from the aggregation of the overexpressed reporter protein, we looked at the response of a form of GFP-hLC3B that cannot be lipidated due to a Gly120-to-Ala substitution. Unlike the wild-type version of hLC3B, the mutated form of hLC3B does not produce punctate structures in AA- or DGLA-supplemented cells (Supplemental Fig. S7A). Immunoblot analysis of p62/SQSTM1 levels, a protein degraded in the autophagolysosome that serves as a proxy for the autophagic status of the cells (Mizushima and Yoshimori 2007), shows that AA or DGLA treatment does not inhibit autophagolysosome-driven protein degradation (Supplemental Fig. S7B). As p62/SQSTM1 is also degraded by the proteasome and its transcriptional levels are affected by a number of treatments (Ishii et al. 2000; Kuusisto et al. 2001), we further assessed autophagic activity by monitoring the degradation of a fluorophore-conjugated form of BSA (DQ-BSA). DQ-BSA accumulates in the lysosome and remains quenched unless it is proteolytically cleaved by lysosomal proteases. This fluorescent probe allows monitoring of both autophagosome formation (GFP-hLC3B) and, in parallel, the functionality of the autophagolysosomal compartment (red DQ-peptide signal) (Klionsky et al. 2008; Vazquez and Colombo 2009). Active breakdown of DQ-BSA, even in the presence of AA and DGLA, and the overlap between GFP-hLC3B and the red DQ-peptides observed after AA or DGLA treatment show that the enriched GFP-hLC3B punctae are active sites of autophagic breakdown (Fig. 3B, bottom panels). Finally, inactivation of the essential autophagy gene ATG161L impairs the accumulation of GFP-hLC3B punctae (Fig. 3C), further supporting the hypothesis that AA- and DGLA-triggered enrichment of punctate GFP-hLC3B is due to an increase in the autophagic flux in the treated cells and not the inhibition of the post-sequestration steps of the autophagic process. The combination of data from C. elegans and mammalian cells in culture strongly suggests that ω-6 PUFAs can activate autophagy across metazoans.
Figure 3.

ω-6 PUFAs activate autophagy in mammalian cells. (A) AA and DGLA supplementation leads to an increase in LC3II in HeLa cells. Fifty percent confluent cells were treated with 100 μM AA, DGLA, or EPA or mock-treated with an equal volume of 50% ethanol for 24 h. Representative Western blot analysis of endogenous LC3 in supplemented or mock-treated cells is shown, and quantification of LC3II (autophagy-specific form) signal intensity relative to actin from three independent experiments is depicted as mean ± SEM. (B) AA and DGLA activate autophagy in HeLa cells. The top row depicts HeLa cells stably expressing GFP-hLC3B grown in rich culture medium supplemented with 100 μM AA or DGLA or equal volume of ethanol and imaged after 30 h of total treatment. Quantification of GFP-hLC3B punctae is presented in the table below. The second row shows the increase in GFP-hLC3B punctae observed when the lysosomal inhibitors E64d (5 μg/mL) and pepstatin (10 μg/mL) were added to cells pretreated for 24 h with carrier or fatty acids and incubated for another 6 h with the inhibitors. Quantification of GFP-hLC3B punctae is presented in the table below. The third row shows lysosomal breakdown capacity in the presence of fatty acids as revealed by the cleavage and consequent dequenching of 10 μg/mL red DQ-BSA added after a 24-h treatment with fatty acids and incorporated and cleaved for 6 h. The fourth row shows that the GFP-hLC3B and red DQ-peptide signals overlap in the AA- and DGLA-treated cells (merges between the top and third row images are shown). For all experiments, images were taken after 30 h of total treatment. Images are representative from at least three independent experiments. Bars, 20 μm. The table shows GFP-hLC3B punctae quantification and statistical analyses for three independent experiments. Proportion of cells showing more than five GFP-hLC3B punctae ≥2 μm in the presence or absence of the lysosomal inhibitors is shown as percentages of the total number of cells (n) analyzed per treatment. The number of dots >2 μm per cell was quantified from at least 10 different fields using SPOT software. P-values relative to mock-treated cells are presented at the bottom of the columns. Protein aggregation control is shown in Supplemental Figure S7A, and p62/SQSTM1 degradation control is shown in Supplemental Figure S7B. (C) AA- and DGLA-triggered activation of autophagy in mouse embryonic fibroblasts (MEFs) is ATG16L1-dependent. The top row shows wild-type MEFs stably expressing GFP-hLC3B grown in rich culture medium supplemented with 100 μM AA or DGLA or equal volume of ethanol and imaged after 24 h of total treatment. The bottom row shows ATG16L1−/− MEFs stably expressing GFP-hLC3B grown in rich culture medium supplemented with 100 μM AA or DGLA or equal volume of ethanol and imaged after 24 h of total treatment.
ω-6 PUFAs increase C. elegans life span through the activation of autophagy in well-fed conditions
lipl-4 up-regulation is required to extend the life span of germline-deficient C. elegans (Wang et al. 2008), and autophagy is required for life span extension under caloric restriction (Hansen et al. 2008). Because LIPL-4 overexpression causes increased levels of autophagy, we tested whether autophagy mediates the life span extension triggered by overexpression of LIPL-4. Inactivation of the essential autophagy genes bec-1 (the ortholog of beclin-1, which is required for autophagosome assembly), lgg-1 (the ortholog of Atg-8/MAPLC3, which is incorporated into preautophagosomal membranes), and atg-16.2 (the ortholog of ATG16p/ATG16L1, which localizes to the preautophagosome and is required for autophagy), suppress the life span extension induced by LIPL-4 overexpression (Fig. 4A; Supplemental Table S1A). Using LIPL-4Ox, transgenic lines in which lipl-4 is constitutively expressed at levels normally observed in fasted worms (ges-1 promoter), we showed that the LIPL-4 overexpression-triggered increase in life span is partially suppressed by inactivation of pha-4, a FoxA transcription factor that binds to the promoters of some autophagy genes (Fig. 4B; Supplemental Table S1B; Zhong et al. 2010; Niu et al. 2011). In contrast, the inactivation of the FoxO transcription factor daf-16, which controls gene expression upon fasting but not autophagy genes in particular, does not suppress the LIPL-4 overexpression longevity phenotype (Fig. 4B; Supplemental Table S1B), further supporting the model that LIPL-4 overexpression extends C. elegans life span through the activation of autophagy. A similar autophagy-dependent extension of life span by overexpression of LIPL-4 from its own promoter has been observed (Lapierre et al. 2011).
Figure 4.
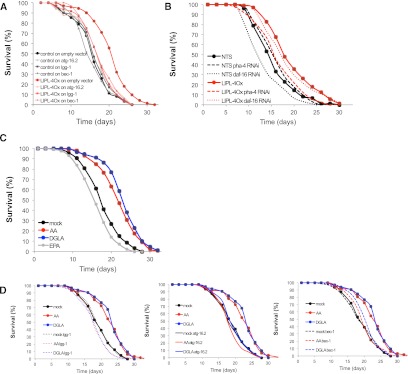
ω-6 PUFAs extend life span through the activation of autophagy. (A) Autophagy contributes to LIPL-4Ox longevity. Representative survival curve of LIPL-4Ox or control (nontransgenic siblings) incubated at 20°C in OP50 plates and transferred as L4 larvae to vector control or RNAi against core autophagy genes is presented (mean life spans: control = 16.1, LIPL-4Ox = 19.0; P = 3.21 × 10−5). Mean life span and statistical significance of the differences compared with control of an independent experiment are presented in Supplemental Table S1A. (B) The autophagy transcriptional regulator PHA-4 (FoxA), but not the starvation regulator DAF-16 (FoxO), contributes to the LIPL-4Ox-extended life span phenotype. Representative survival curve of LIPL-4Ox (ges-1 promoter) or nontransgenic siblings (NTS) grown at 20°C in OP50 plates and transferred as L4 larvae to vector control, pha-4, or daf-16 RNAi is presented. Mean life span and statistical significance of the differences compared with control are presented in Supplemental Table S1B. (C) AA and DGLA extend C. elegans life span. Survival curves of worms grown in OP50 plates and transferred as L4 larvae to plates supplemented with ω-3/6 PUFAs or carrier are presented. Worms were incubated at 20°C and transferred every other day to fresh plates containing 10 μM AA, 10 μM DGLA, 20 μM EPA, or an equal volume of carrier until cessation of reproduction. Worms were then transferred every 7 d to maintain the strength of the treatments. Statistical analysis of an independent experiment is presented in Supplemental Table S2A. (D) AA and DGLA extend C. elegans life span through activation of autophagy. Survival curves of worms grown in OP50 plates, transferred as L4 larvae to RNAi plates supplemented with ω-3/6 PUFAs or carrier, and/or treated with RNAi against autophagy genes or vector control are presented. Worms were incubated at 20°C and transferred every other day to fresh control L4440 or RNAi plates containing 10 μM AA, 10 μM DGLA, 20 μM EPA, or an equal volume of carrier until cessation of reproduction. Worms were then transferred every 7 d to maintain the strength of the treatments. Statistical analyses of independent experiments are presented in Supplemental Table S2B.
We showed that LIPL-4 overexpression leads to an enrichment of AA and DGLA and that these ω-6 PUFAs activate autophagy. We tested whether supplementation with these fasting-enriched ω-6 PUFAs would phenocopy the increase in life span induced by activation of autophagy (Cuervo 2008; Rubinsztein et al. 2011). Chronic long-term dietary supplementation with AA and DGLA, but not EPA, extends C. elegans life span in conditions of food abundance (Fig. 4C,D; Supplemental Table S2A). Furthermore, this life span extension is bec-1-, atg-16.2-, and lgg-1-dependent (Fig. 4D; Supplemental Table S2B). These results show not only that dietary supplementation with ω-6 PUFAs activates a conserved cellular response normally triggered by fasting, but also that long-term administration of ω-6 PUFAs can render the beneficial effects of low-caloric intake even in ad libitum feeding conditions, and suggest that these lipid metabolites would act as signals of nutrient scarcity.
Although the LIPL-4Ox construct is expressed only in the digestive tract (ges-1 or vha-6 promoters), increased autophagic signal is also observed in the hypodermal seam cells (Figs. 2A, 5C). This observation suggests that ω-6 PUFAs might be secreted and able to improve cellular clearance distal from their site of production. Out of the nine predicted lipid-binding proteins (LBPs) in C. elegans, four (lbp-1, lbp-3, lbp-5, and lbp-7) are transcriptionally up-regulated upon food withdrawal (Fig. 5A), and LIPL-4Ox induces lbp-3, lbp-5, and lbp-8 transcription (Fig. 5B). We tested the effect of inactivating those LBPs that are transcriptionally activated by fasting and LIPL-4 overexpression—namely, lbp-3 and lbp-5—on the activation of autophagy induced by LIPL-4 overexpression. lbp-3 and lbp-5 reduction of function by RNAi hinders the hypodermal seam cell activation of autophagy (Fig. 5C) and partially suppresses the LIPL-4Ox longevity phenotype (Fig. 5D; statistical analysis of independent experiment in Supplemental Table S3), suggesting that intestinal activation of autophagy is insufficient to fully extend C. elegans life span and that, instead, systemic activation of autophagy is necessary.
Figure 5.
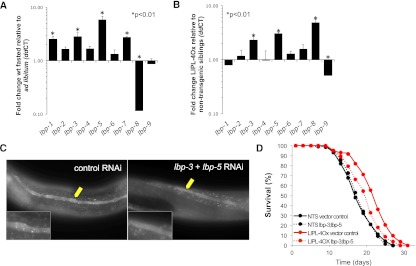
Lipolysis products activate autophagy in C. elegans distal from their production site with the contribution of LBPs. (A) C. elegans genes encoding the lipid transporter LBPs are transcriptionally up-regulated upon fasting. lbp-1 to lbp-9 mRNA levels of expression in 6-h-fasted wild-type young adults relative to ad libitum-fed animals of the same developmental age are depicted as mean ± SEM of three independent experiments. (B) LIPL-4Ox is sufficient to transcriptionally activate the fasting-responsive lipid transporters lbp-3, lbp-5, and lbp-8. lbp-1 to lbp-9 mRNA levels of expression in LIPL-4Ox relative to control (nontransgenic siblings) incubated at 20°C in OP50 plates are presented as mean ± SEM of three independent experiments. (C) LBP-3 and LBP-5 facilitate activation of autophagy distal from the LIPL-4 site of expression. Representative images of LGG-1∷GFP signal in intestinal and hypodermal seam cells (yellow arrow) of L3–L4 animals overexpressing LIPL-4 only in the digestive tract (vha-6 promoter), treated or not from the L4 stage of the first generation with RNAi against lbp-3 and lbp-5 (50% each), are presented. (D) LBP-3 and LBP-5 contribute to the LIPL-4 overexpression extended life span phenotype. Survival curve of LIPL-4Ox or control (nontransgenic siblings) incubated at 20°C in OP50 plates and transferred as L4 larvae to control or lbp-3/lbp-5 RNAi plates is presented. Worms were incubated at 20°C and transferred every other day to fresh control L4440 or RNAi plates until cessation of reproduction. Worms were then transferred every 7 d to maintain the strength of the treatments. Detailed statistical analysis of an independent experiment is presented in Supplemental Table S3.
Discussion
The link between lipid metabolism and longevity remains obscure. In C. elegans, animals with decreased fat stores (the caloric restriction mutant model eat-2) (Lakowski and Hekimi 1998) or with increased adiposity (the insulin receptor mutant daf-2) (Kimura et al. 1997) can be long-lived. Even in mammals, the connection between adiposity and life span is not clear. Some murine genetic models with extended life span show increased adiposity (Coschigano et al. 2000; Flurkey et al. 2001; Berryman et al. 2004), and food restriction makes genetically obese mice live longer than wild-type animals even though these mice accumulate twice as much fat as wild-type mice fed ad libitum (Harrison et al. 1984), suggesting a complex link between lipid metabolism and life span regulation. Autophagy, a major cellular clearance mechanism that delays aging (Melendez et al. 2003; Cuervo 2008; Hansen et al. 2008; Rubinsztein et al. 2011), also controls lipid metabolism through a specialized form of autophagy named lipophagy, whose genetic or chemical inactivation leads to an increase in cytosolic fat stores (Singh et al. 2009) in mammals. Germline-deficient C. elegans activate the expression of the lipase gene lipl-4 (Wang et al. 2008) and have increased levels of autophagy (Lapierre et al. 2011). The coordinated and interdependent activation of LIPL-4 and autophagy in germline-deficient worms (glp-1) led to the hypothesis that enhanced LIPL-4-driven lipophagy could explain the glp-1 extended life span phenotype (Lapierre et al. 2011). However, the following observations make this hypothesis unlikely: (1) Despite having increased LIPL-4 lipase activity and higher levels of autophagy (Lapierre et al. 2011), glp-1 animals accumulate more cytosolic fats than wild-type animals (O'Rourke et al. 2009). (2) lipl-4-deficient worms show normal cytosolic fat levels in well-fed and fasting conditions (Supplemental Fig. S1A). (3) LIPL-4 does not localize to the sites of active autophagy (Supplemental Fig. S4A). (4) LIPL-4 is a neutral lipase (Lapierre et al. 2011), and the lipases breaking down fats through lipophagy are expected to act in the autophagolysosome and consequently be active at low pHs (Czaja and Cuervo 2009). The alternative hypothesis in which a lipase breakdown product might activate autophagy was also proposed (Lapierre et al. 2011). Here we show evidence supporting this latter model; specifically, we show that lipl-4 induction leads to an enrichment of ω-6 PUFAs and that ω-6 PUFAs or some of their derivatives act as metabolic signals that activate autophagy and extend C. elegans life span.
Lipids continue to emerge as a class of molecules that affect behavior, growth, and organismal survival, not only because of their energetic value, but also because of their structural and signaling functions (Vrablik and Watts 2012). In C. elegans, ω-3/6 PUFAs are required for normal development and neural function (Watts et al. 2003; Kahn-Kirby et al. 2004). Additionally, eicosapentaenoylethanolamide (EPEA) and arachidonoylethanolamide (AEA) promote C. elegans growth, as reduced levels of these N-acylethanolamines (NAEs) are necessary to enter and maintain the dauer state, a starvation-resistant diapause stage (Lucanic et al. 2011). Here we showed that, opposite to their NAE derivatives (Supplemental Table S4), EPA and AA are enriched in fasted worms and that AA and its precursor, DGLA, promote resistance to starvation. Also different from their NAE derivatives, AA and its precursor, DGLA, extend C. elegans life span in an abundant food regime. NAEs were proposed to act as high nutrient availability signals, and consequently, they promote reproduction and the correlated program of somatic aging. We propose that AA and DGLA or some derivatives of these lipid metabolites act as signals of reduced food availability and consequently trigger a fasting program that extends life span. It would be interesting to determine whether AA and/or DGLA derive from triglycerides or phospholipids. Alternatively, these ω-6 PUFAs might be breakdown products of cardiolipin, an AA-rich lipid whose levels accompany the drop in mitochondrial content observed in fasting C. elegans (EJ O'Rourke, unpubl.). The fasting-enriched ω-3/6 PUFAs are precursors and products of the fasting-depleted NAEs. It is fascinating to find that such closely related lipid molecules have opposite effects on organismal survival. We propose that the relative proportion of these lipids or some of their derivatives would signal nutrient abundance or scarcity and in this way would help to coordinate organismal responses that maximize the chances of survival. It would also be of value to investigate whether glp-1 and daf-2 mutant animals, genetic models of extended life span where lipl-4 is constitutively induced, also have elevated levels of AA and DGLA. NAEs were proposed to function by activating a cannabinoid signaling pathway through yet unknown nematode cannabinoid-like receptors and possibly regulating nutrient intake and energy balance as previously shown in mammals (Lucanic et al. 2011). Here we showed that the mechanism by which ω-6 PUFAs promote survival is by activating autophagy. Autophagy provides nutrients when external sources of energy are not available and mediates the turnover of damaged organelles and long-lived proteins. Therefore, the activation of autophagy that we documented could explain the prosurvival effects of the ω-6 PUFAs AA and DGLA in conditions of food deprivation and food abundance.
LIPL-4 overexpression is sufficient to not only enrich ω-6 PUFAs and activate autophagy, but also transcriptionally activate nutrient-responsive genes (specifically, genes encoding the fatty acid-binding proteins [FABPs] lbp-3 and lbp-5), revealing a direct link between the activity of LIPL-4 and the regulation of the transcriptional programs activated in C. elegans upon nutritional stress. Lipid metabolites may modulate the activity of fatty acid-regulated transcription factors. AA, DGLA, some derivatives of these lipids, or LIPL-4 products yet to be found could directly control the function of transcription factors such as PPARα/CeNHR-49, SREBP-1/CeSBP-1, or other NHRs. Additionally, we showed data supporting the hypothesis that the LBPs LBP-3 and LBP-5 contribute to the activation of autophagy distally from the tissue where LIPL-4 is expressed. LBP-3 and LBP-5 are required for the full extension of life span observed in LIPL-4-overexpressing worms, suggesting that systemic activation of autophagy is necessary to fully extend C. elegans life span. LBP-3 and LBP-5 are unique FABPs. Unlike most FABPs, LBP-3 has a signal sequence that drives the secretion of the protein into the interstitial space of C. elegans (Plenefisch et al. 2000). LBP-5, on the other hand, has been characterized as an intracellular lipid transporter that binds AA (Xu et al. 2011). We propose that LIPL-4 activation leads to an enrichment of ω-6 PUFAs, directly or indirectly acting as metabolic signals that communicate a state of alarm to distal tissues and therefore help coordinate molecular and cellular responses that enhance organismal survival in the absence and presence of food.
In humans, dietary supplementation with ω-3/6 PUFAs (fish oils) can reduce the incidence and improve the condition of patients suffering from inflammatory, neurodegenerative, and neoplastic pathologies (Deckelbaum and Calder 2010; Kuballa and Xavier 2010). The reduction of the pools of proinflammatory molecules, such as AA, is the most accepted mechanism of action for ω3/6 PUFAs (Fritsche 2008). However, type 2 diabetes is associated with low levels of AA (Suhre et al. 2010), and the addition of exogenous AA ameliorates diabetes (Suresh and Das 2001). Additionally, AA supplementation reduces the risk of cardiovascular disease (Harris et al. 2009) and neurodegeneration (Das 2008), suggesting that the beneficial effects of ω-3/6 fatty acids are not only or necessarily mediated by reducing AA levels. On the contrary, these observations seem to indicate that ω-6 long chain fatty acid supplementation can have beneficial effects despite being precursors of proinflammatory molecules. Additionally, modulation of the endocannabinoid system has been shown to mediate some of the beneficial effects of long chain PUFA dietary supplementation, but these studies had been restricted to analyzing the impact of ω-3 PUFAs (Batetta et al. 2009). Our data show that supplementation with ω-6 PUFAs activates autophagy in human epithelial cells. As in worms, fasted mammals show enrichment in AA in serum, liver, and/or extrahepatic tissues (de Alaniz et al. 1980; Garg et al. 1992; Zhou and Nilsson 1999). Moreover, unlike wild-type animals, mice deficient for PPARα, a transcription factor that coordinates essential metabolic responses to fasting (Kersten et al. 1999; Leone et al. 1999), are unable to enrich AA upon fasting (Lee et al. 2004). This result suggests that enrichment of AA or some of its derivatives is part of an essential response to food deprivation that is conserved across metazoans. The mechanism by which cells sense or uptake ω-6 PUFAs and how these lipid metabolites or some derivatives actually activate autophagy remains to be elucidated. ω-6 PUFA-triggered activation of autophagy seems to be additive to treatment of HeLa cells with rapamycin (EJ O'Rourke, unpubl.), suggesting that ω-6 fatty acids would activate autophagy through a mammalian TOR (mTOR)-independent mechanism. Several stimuli have been shown to induce autophagy independent of mTOR, all of which reduce intracellular inositol levels (Ravikumar et al. 2010). It would be interesting to test whether PI3 kinase inhibitors affect the ω-6 PUFA-triggered activation of autophagy.
The malfunctioning of autophagy has been linked to a wide range of pathological conditions, including neurodegenerative diseases, chronic inflammation, infection, and cancer (Kuballa and Xavier 2010). Several of these pathologies are ameliorated by dietary supplementation with ω-3/6 PUFAs. The overlap between the beneficial effects of activating autophagy and the salubrious effects of ω-3/6 PUFA consumption, together with the data presented here, suggests that ω-3/6 PUFAs could counteract the deterioration of the autophagic system normally occurring during aging. Our data suggest that ω3/6 PUFAs could be used, and/or analogs could be developed, to activate or even inhibit autophagy, both of which would be of great help in the prevention and treatment of multiple pathologies.
Materials and methods
C. elegans strains
lipl-4(tm4417) was backcrossed four times. LIPL-4∷TagRFP corresponds to Ex[vha-6P∷lipl-4 ORF∷TagRFP] (three independent lines). LIPL-4Ox corresponds to Ex[ges-1P∷lipl-4ORF∷SL2∷GFP] (two independent lines). These animals with constitutive fasting levels of expression of lipl-4 (several independent lines and two different constructs) are developmentally delayed, so animals were synchronized based on vulva and/or gonadal development and not chronological age. LGG-1∷GFP from DA2123 (Kang et al. 2007) was crossed to LIPL-4:TagRFP.
Transcriptional analyses
Young adults (adult vulva with less than five eggs) were harvested and washed in a 35-μm nylon mesh. For fasting experiments, animals were seeded back onto empty NGM plates (fasting) or back onto OP50 plates (well-fed control). After the fasting period, treated and controls were harvested, washed in a 35-μm nylon mesh, and frozen in liquid nitrogen. Total RNA was isolated using TriReagent (Molecular Research Center, Inc.), and RT–PCR was performed as previously described (Troemel et al. 2006). Data presented are from at least three independent experiments, and all values are corrected for the efficiency of the primer pairs and normalized to ama-1 as internal control as well as to transcript levels in same age untreated, wild-type, or nontransgenic sibling animals. All quantitative RT–PCR (qRT–PCR) reactions were performed at least in triplicate. Median ± SEM of ΔΔCt is reported (Pfaffl 2001).
FAME analysis
LIPL-4Ox transgenic hatchlings were selected based on fluorescent signal with a COPAS Biosort and seeded on OP50 plates 6 h earlier than their nontransgenic siblings, and all strains were incubated at 20°C. FAMEs were extracted from young fertile adults as previously reported (Watts and Browse 2002) with modifications. Lipids from an equal number of worms (see the Supplemental Material) were extracted with chloroform:methanol 2:1. FAMEs were generated by incubating in 5% sulfuric acid in methanol for 1 h at 80°C. FAMEs were resuspended in hexane and run in a C20 wax Supelco column. Methyl-fatty acid standards were run to determine retention times. The area under the peaks for the methyl-fatty acids of interest is presented as average percentage of total FAMEs from three independent experiments ± SD.
Starvation resistance
LIPL-4Ox transgenic hatchlings were selected based on fluorescent signal with a COPAS Biosort (Union Biometrica). LIPL-4Ox animals were seeded on OP50 plates 6 h earlier than their nontransgenic siblings. For all starvation resistance experiments, animals were incubated at 20°C. Approximately 10,000 young L4s were harvested for each strain or treatment and extensively washed through a 20-μm mesh. Larvae were resuspended in 10 mL of S-basal, to which we added 2.5 μL of 5 mM Sytox green or red (Molecular Probes) and, when indicated, 10 μM AA (Cayman, catalog no. 90010), 10 μM DGLA (Cayman, catalog no. 90230), or 20 μM EPA (Cayman, catalog no. 90110) or equal volume of carrier (50% ethanol). The worm suspensions were maintained by rocking at 20°C. Approximately 500 worms were run through a Biosort at the indicated times. Animals with TOF (time of flight) between 100 and 500 (arbitrary units) and EXT (extinction) between 60 and 300 (arbitrary units) were analyzed. Animals with green fluorescent signal ≥50 or red signal ≥25 were dead, as established by lack of movement in a liquid drop. Percent survival = [1 − (worms with green or red signal ≥50 or 25/total worms)] × 100. Survival estimates were not necessarily monotonically decreasing, since random samples of each population were taken at each time point. When worms were sampled to determine survival, some number of worms died before the point of sampling, but time of death is unknown. Consequently, our data are left censored. Traditional Kaplan-Meier analysis of survival could not be performed with this experimental setup. Instead, we compared the median and standard deviations at the time point when wild-type untreated worm populations were closest to 50% alive and performed t-test analysis to compare the percentage of animals alive at that particular time point.
Longevity
Wild-type, transgenic (multiple lines), or nontransgenic sibling L4 larvae, selected based on fluorescent signal, were transferred in duplicate to sextuplicate (50–150 total worms) to fresh NGM OP50, RNAi, ω-3/6 PUFA, or mock ethanol-supplemented plates every 2 d until no progeny were observed and then once a week for RNAi, ω-3/6 PUFAs, and their respective controls in order to keep the strength of the treatments. Two to six independent assays were carried out. Life span experiments were conducted at 20°C unless otherwise stated. Animals that were missing or burst were censored and statistically incorporated into the life span analysis. Kaplan-Meier survival analysis was done using SSPS.17 software; significance was determined using log-rank statistics.
Autophagy assessment
LIPL-4∷TagRFP;LGG-1∷GFP transgenic hatchlings were separated from their LGG-1∷GFP-only siblings based on fluorescent signal with a COPAS Biosort and then seeded on OP50 plates. LIPL-4∷TagRFP; LGG-1∷GFP animals were seeded 6 h earlier than worms carrying only LGG-1∷GFP. Animals were harvested as L3–L4 larvae, immobilized with 2,3-Butanedione monoxamine (30 mg/mL; Sigma-Aldrich) for 10 min, mounted, and documented. For supplementation experiments, LGG-1∷GFP hatchlings were seeded in plates containing 50 μM fatty acids or equal volume of 50% ethanol. Mock or supplemented animals were harvested as L3–L4 larvae and treated for documentation as described above. No “autophagic flux” assay or reporter has been described for C. elegans (Klionsky et al. 2008).
For Western-blotting, HeLa cells were grown in complete IMDM medium (Invitrogen) containing 10% fetal bovine serum and supplemented with 100 μM ω3/6 fatty acids or equal volume of ethanol. (Note: Effective concentration of the fatty acids is unclear given the capacity of LBPs in the serum to sequester fatty acids.) Cells were harvested after 16, 24, or 30 h of treatment and immediately frozen for later processing.
HeLa cells and ATG16L1-proficient and ATG16L1-deficient mouse embryonic fibroblasts (RJ Xavier and HW Virgin, in prep.) stably expressing GFP-hLC3B fusion proteins were generated as described before (Kuballa et al. 2008). Inactive LC3 mutant, GFP-hLC3B-G120A, lentiviral construct was generated by site-directed mutagenesis and transduced at least three passages before the first experiment was made. For quantification of GFP-hLC3B punctae, cells were grown in culture-rich medium (complete IMDM medium containing 10% fetal bovine serum); supplemented with 100 μM AA, DGLA, or equal volume of ethanol; and monitored using direct fluorescent microscopy. For inhibitor and substrate degradation experiments monitored by immunoblotting, 5 μg/mL E64d (Sigma) and 10 μg/mL pepstatin (Sigma) were added after 10 h of treatment with fatty acids, and samples were harvested and frozen at 16 h of total treatment (a time point within the dynamic range of LC3II signal). For inhibitor or substrate degradation experiments monitored by direct fluorescence microscopy, 5 μg/mL E64d (Sigma) and 10 μg/mL pepstatin (Sigma) and/or 10 μg/mL DQ-BSA (Molecular Probes) were added after 24 h of treatment with fatty acids, and images were taken after 30 h of total treatment (a time point within the dynamic range of large punctae formation; for AA plus inhibitors, saturation was reached at 48 h of treatment) using a Nikon Eclipse TE2000-S microscope; the number of dots >2 μm per cell was quantified from at least 10 different fields using SPOT software (Diagnostics Instruments, Inc.).
Acknowledgments
We thank members of the Ruvkun, Ausubel, and Kaplan laboratories for helpful comments, especially A. Lee-Conery for help with longevity experiments; J. Larkins-Ford for performing Biosort analyses; J. Rapley, J. Avruch, R. Niedra, and B. Seed for sharing expertise and reagents for mammalian experiments; and H.Y. Mak, A. Soukas, M. Van Gilst, M. Freeman, A. Saghatelian, and A. Tyler for protocols, access to equipment, and discussions on lipid measurements. We thank Dr. Mitani and the National Bioresource Project for strains. We also thank C. Danna for helpful suggestions and comments on the manuscript. This work was supported by grants R01DK070147 (to G.R.) and K99DK087928 (to E.J.O'R.). E.J.O'R. designed the overall studies, performed the experiments, and wrote the manuscript. G.R. revised the manuscript. P.K. and R.X. provided GFP-hLC3B and GFP-hLC3B-G120A HeLa cells and ATG16L1-proficient and ATG16L1-deficient mouse embryonic fibroblasts and revised the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.205294.112.
References
- Apfeld J, O'Connor G, McDonagh T, DiStefano PS, Curtis R 2004. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes Dev 18: 3004–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass TM, Weinkove D, Houthoofd K, Gems D, Partridge L 2007. Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mech Ageing Dev 128: 546–552 [DOI] [PubMed] [Google Scholar]
- Batetta B, Griinari M, Carta G, Murru E, Ligresti A, Cordeddu L, Giordano E, Sanna F, Bisogno T, Uda S, et al. 2009. Endocannabinoids may mediate the ability of (n-3) fatty acids to reduce ectopic fat and inflammatory mediators in obese Zucker rats. J Nutr 139: 1495–1501 [DOI] [PubMed] [Google Scholar]
- Berdichevsky A, Viswanathan M, Horvitz HR, Guarente L 2006. C. elegans SIR-2.1 interacts with 14-3-3 proteins to activate DAF-16 and extend life span. Cell 125: 1165–1177 [DOI] [PubMed] [Google Scholar]
- Berryman DE, List EO, Coschigano KT, Behar K, Kim JK, Kopchick JJ 2004. Comparing adiposity profiles in three mouse models with altered GH signaling. Growth Horm IGF Res 14: 309–318 [DOI] [PubMed] [Google Scholar]
- Cao Y, Pearman AT, Zimmerman GA, McIntyre TM, Prescott SM 2000. Intracellular unesterified arachidonic acid signals apoptosis. Proc Natl Acad Sci 97: 11280–11285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coschigano KT, Clemmons D, Bellush LL, Kopchick JJ 2000. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology 141: 2608–2613 [DOI] [PubMed] [Google Scholar]
- Cuervo AM 2008. Autophagy and aging: Keeping that old broom working. Trends Genet 24: 604–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czaja MJ, Cuervo AM 2009. Lipases in lysosomes, what for? Autophagy 5: 866–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das UN 2008. Folic acid and polyunsaturated fatty acids improve cognitive function and prevent depression, dementia, and Alzheimer's disease—but how and why? Prostaglandins Leukot Essent Fatty Acids 78: 11–19 [DOI] [PubMed] [Google Scholar]
- de Alaniz MJ, de Gomez Dumm IN, Brenner RR 1980. Effect of fasting on δ 5 desaturation activity in rat liver microsomes and HTC cells. Mol Cell Biochem 33: 165–170 [DOI] [PubMed] [Google Scholar]
- Deckelbaum RJ, Calder PC 2010. Dietary n-3 and n-6 fatty acids: Are there ‘bad' polyunsaturated fatty acids? Curr Opin Clin Nutr Metab Care, 13: 123–124 [DOI] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Miller RA, Harrison DE 2001. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci 98: 6736–6741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche KL 2008. Too much linoleic acid promotes inflammation—doesn't it? Prostaglandins Leukot Essent Fatty Acids 79: 173–175 [DOI] [PubMed] [Google Scholar]
- Garg ML, Keelan M, Thomson AB, Clandinin MT 1992. Desaturation of linoleic acid in the small bowel is increased by short-term fasting and by dietary content of linoleic acid. Biochim Biophys Acta 1126: 17–25 [DOI] [PubMed] [Google Scholar]
- Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C 2008. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet 4: e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris WS, Mozaffarian D, Rimm E, Kris-Etherton P, Rudel LL, Appel LJ, Engler MM, Engler MB, Sacks F 2009. ω-6 Fatty acids and risk for cardiovascular disease: A science advisory from the American Heart Association Nutrition Subcommittee of the Council on Nutrition, Physical Activity, and Metabolism; Council on Cardiovascular Nursing; and Council on Epidemiology and Prevention. Circulation 119: 902–907 [DOI] [PubMed] [Google Scholar]
- Harrison DE, Archer JR, Astle CM 1984. Effects of food restriction on aging: Separation of food intake and adiposity. Proc Natl Acad Sci 81: 1835–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M 2000. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem 275: 16023–16029 [DOI] [PubMed] [Google Scholar]
- Jia K, Chen D, Riddle DL 2004. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131: 3897–3906 [DOI] [PubMed] [Google Scholar]
- Kahn-Kirby AH, Dantzker JL, Apicella AJ, Schafer WR, Browse J, Bargmann CI, Watts JL 2004. Specific polyunsaturated fatty acids drive TRPV-dependent sensory signaling in vivo. Cell 119: 889–900 [DOI] [PubMed] [Google Scholar]
- Kang C, You YJ, Avery L 2007. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev 21: 2161–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W 1999. Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J Clin Invest 103: 1489–1498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G 1997. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277: 942–946 [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. 2008. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 4: 151–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuballa P, Xavier RJ 2010. Failure and exploitation of autophagy in human pathologies-cellular integrity between inflammation, infection, and cell survival. Semin Immunopathol 32: 319–322 [DOI] [PubMed] [Google Scholar]
- Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ 2008. Impaired autophagy of an intracellular pathogen induced by a Crohn's disease associated ATG16L1 variant. PLoS ONE 3: e3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuusisto E, Suuronen T, Salminen A 2001. Ubiquitin-binding protein p62 expression is induced during apoptosis and proteasomal inhibition in neuronal cells. Biochem Biophys Res Commun 280: 223–228 [DOI] [PubMed] [Google Scholar]
- Lakowski B, Hekimi S 1998. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci 95: 13091–13096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre LR, Gelino S, Melendez A, Hansen M 2011. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr Biol 21: 1507–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Ashrafi K 2008. A TRPV channel modulates C. elegans neurosecretion, larval starvation survival, and adult lifespan. PLoS Genet 4: e1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SS, Chan WY, Lo CK, Wan DC, Tsang DS, Cheung WT 2004. Requirement of PPARα in maintaining phospholipid and triacylglycerol homeostasis during energy deprivation. J Lipid Res 45: 2025–2037 [DOI] [PubMed] [Google Scholar]
- Leone TC, Weinheimer CJ, Kelly DP 1999. A critical role for the peroxisome proliferator-activated receptor α (PPARα) in the cellular fasting response: The PPARα-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci 96: 7473–7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucanic M, Held JM, Vantipalli MC, Klang IM, Graham JB, Gibson BW, Lithgow GJ, Gill MS 2011. N-acylethanolamine signalling mediates the effect of diet on lifespan in Caenorhabditis elegans. Nature 473: 226–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B 2003. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301: 1387–1391 [DOI] [PubMed] [Google Scholar]
- Melendez A, Hall DH, Hansen M 2008. Monitoring the role of autophagy in C. elegans aging. Methods Enzymol 451: 493–520 [DOI] [PubMed] [Google Scholar]
- Miller RA, Birnbaum MJ 2010. An energetic tale of AMPK-independent effects of metformin. J Clin Invest 120: 2267–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, et al. 2011. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 66: 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T 2007. How to interpret LC3 immunoblotting. Autophagy 3: 542–545 [DOI] [PubMed] [Google Scholar]
- Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, Janette J, Chen C, Alves P, Preston E, et al. 2011. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res 21: 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke EJ, Soukas AA, Carr CE, Ruvkun G 2009. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab 10: 430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P 2007. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell 6: 111–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW 2001. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plenefisch J, Xiao H, Mei B, Geng J, Komuniecki PR, Komuniecki R 2000. Secretion of a novel class of iFABPs in nematodes: Coordinate use of the Ascaris/Caenorhabditis model systems. Mol Biochem Parasitol 105: 223–236 [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. 2010. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90: 1383–1435 [DOI] [PubMed] [Google Scholar]
- Rogers AN, Chen D, McColl G, Czerwieniec G, Felkey K, Gibson BW, Hubbard A, Melov S, Lithgow GJ, Kapahi P 2011. Life span extension via eIF4G inhibition is mediated by posttranscriptional remodeling of stress response gene expression in C. elegans. Cell Metab 14: 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Marino G, Kroemer G 2011. Autophagy and aging. Cell 146: 682–695 [DOI] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ 2009. Autophagy regulates lipid metabolism. Nature 458: 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K, Meisinger C, Doring A, Altmaier E, Belcredi P, Gieger C, Chang D, Milburn MV, Gall WE, Weinberger KM, et al. 2010. Metabolic footprint of diabetes: A multiplatform metabolomics study in an epidemiological setting. PLoS ONE 5: e13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh Y, Das UN 2001. Protective action of arachidonic acid against alloxan-induced cytotoxicity and diabetes mellitus. Prostaglandins Leukot Essent Fatty Acids 64: 37–52 [DOI] [PubMed] [Google Scholar]
- Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH 2006. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet 2: e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gilst MR, Hadjivassiliou H, Jolly A, Yamamoto KR 2005a. Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. elegans. PLoS Biol 3: e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gilst MR, Hadjivassiliou H, Yamamoto KR 2005b. A Caenorhabditis elegans nutrient response system partially dependent on nuclear receptor NHR-49. Proc Natl Acad Sci 102: 13496–13501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez CL, Colombo MI 2009. Assays to assess autophagy induction and fusion of autophagic vacuoles with a degradative compartment, using monodansylcadaverine (MDC) and DQ-BSA. Methods Enzymol 452: 85–95 [DOI] [PubMed] [Google Scholar]
- Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F 2003. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 426: 620. [DOI] [PubMed] [Google Scholar]
- Vrablik TL, Watts JL 2012. Emerging roles for specific fatty acids in developmental processes. Genes Dev 26: 631–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MC, O'Rourke EJ, Ruvkun G 2008. Fat metabolism links germline stem cells and longevity in C. elegans. Science 322: 957–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JL, Browse J 2002. Genetic dissection of polyunsaturated fatty acid synthesis in Caenorhabditis elegans. Proc Natl Acad Sci 99: 5854–5859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JL, Phillips E, Griffing KR, Browse J 2003. Deficiencies in C20 polyunsaturated fatty acids cause behavioral and developmental defects in Caenorhabditis elegans fat-3 mutants. Genetics 163: 581–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Joo HJ, Paik YK 2011. Novel functions of lipid-binding protein 5 in Caenorhabditis elegans fat metabolism. J Biol Chem 286: 28111–28118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Niu W, Lu ZJ, Sarov M, Murray JI, Janette J, Raha D, Sheaffer KL, Lam HY, Preston E, et al. 2010. Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet 6: e1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Nilsson A 1999. Fasting increases tissue uptake and interconversion of plasma unesterified linoleic acid in guinea pigs. Biochim Biophys Acta 1436: 499–508 [DOI] [PubMed] [Google Scholar]
- Zinke I, Schutz CS, Katzenberger JD, Bauer M, Pankratz MJ 2002. Nutrient control of gene expression in Drosophila: Microarray analysis of starvation and sugar-dependent response. EMBO J 21: 6162–6173 [DOI] [PMC free article] [PubMed] [Google Scholar]