

Abstract
Wolbachia endosymbionts are widespread in arthropods and are generally considered reproductive parasites, inducing various phenotypes including cytoplasmic incompatibility, parthenogenesis, feminization and male killing, which serve to promote their spread through populations. In contrast, Wolbachia infecting filarial nematodes that cause human diseases, including elephantiasis and river blindness, are obligate mutualists. DNA purification methods for efficient genomic sequencing of these unculturable bacteria have proven difficult using a variety of techniques. To efficiently capture endosymbiont DNA for studies that examine the biology of symbiosis, we devised a parallel strategy to an earlier array-based method by creating a set of SureSelect™ (Agilent) 120-mer target enrichment RNA oligonucleotides (“baits”) for solution hybrid selection. These were designed from Wolbachia complete and partial genome sequences in GenBank and were tiled across each genomic sequence with 60 bp overlap. Baits were filtered for homology against host genomes containing Wolbachia using BLAT and sequences with significant host homology were removed from the bait pool. Filarial parasite Brugia malayi DNA was used as a test case, as the complete sequence of both Wolbachia and its host are known. DNA eluted from capture was size selected and sequencing samples were prepared using the NEBNext® Sample Preparation Kit. One-third of a 50 nt paired-end sequencing lane on the HiSeq™ 2000 (Illumina) yielded 53 million reads and the entirety of the Wolbachia genome was captured. We then used the baits to isolate more than 97.1 % of the genome of a distantly related Wolbachia strain from the crustacean Armadillidium vulgare, demonstrating that the method can be used to enrich target DNA from unculturable microbes over large evolutionary distances.
Keywords: Wolbachia, Obligate endosymbiont, Target enrichment, NextGen sequencing, DNA capture, SureSelect™
Introduction
Whole genome sequencing (WGS) using Next-Generation sequencing (NGS) technologies is becoming a cost efficient and effective procedure for many research laboratories. While metagenomic projects (purification and DNA sequencing of mixed populations of bacteria en masse) are becoming common, purification methods for efficient sequencing of selected unculturable bacteria from amongst other DNA populations, such as symbiont hosts, have proven difficult. Purification strategies have generally used a variety of techniques including chemical gradients (Charles and Ishikawa 1999), pulsed-field gel (PFG) purification with or without whole genome amplification, library construction followed by gene walking, etc. (Foster et al. 2005; Iturbe-Ormaetxe et al. 2011; Mavingui et al. 2005; Sun et al. 2001). Informatics methods (Kumar and Blaxter 2011) and cell sorting methods (Santos-Garcia et al. 2012) have also been used.
More recently, DNA capture methods using oligonucleotide probes have been used to isolate symbiont or parasitic DNA away from host DNA, providing significant enrichment for more efficient sequencing (Kent et al. 2011; Melnikov et al. 2011). Our goal was to further assess this capture methodology using SureSelect™ (Agilent) technology that could be applied to purify DNA from widely divergent unculturable bacteria (e.g. symbionts) away from other DNA sources as an initiation point for WGS, when some homologous or similar DNA sequence is known. We used the obligate endobacterium Wolbachia as the target species for capture, to eliminate host DNA from downstream library construction and subsequent DNA sequencing. Wolbachia are obligate alpha-proteobacteria closely related to rickettsial organisms and are present in insects, mites, crustaceans, spiders and parasitic filarial nematodes (Werren et al. 2008; Werren and Windsor 2000; Cordaux et al. 2012; Bouchon et al. 1998). It has been recently estimated that ~40 % of arthropod species are infected with Wolbachia (Zug and Hammerstein 2012), making it the most widespread intracellular bacterial species. Phylogenetic analyses using single genes or multilocus sequence typing currently describe seven well-resolved Wolbachia groups, designated as supergroup lineages (A to H, no group G) together with a number of additional lineages (Lo et al. 2002; Casiraghi et al. 2005; Casiraghi et al. 2003; Lo and Evans 2007; Baldo and Werren 2007; Bordenstein et al. 2009).
In 1999, the Wolbachia Genome Consortium planned to sequence various genomes representing the diversity of Wolbachia (Slatko et al. 1999). At the onset of our study, only four Wolbachia genomes had been completely sequenced and deposited in GENBANK, wMel of Drosophila melanogaster (Wu et al. 2004), wBm of Brugia malayi (Foster et al. 2005), wPip of Culex quinquefasciatus Pel (Klasson et al. 2008), wRi of Drosophila simulans (Klasson et al. 2009). Additional genomes are currently being sequenced/annotated (Werren et al. 2008) and partial sequences for several Wolbachia strains are available, e.g., http://www.ncbi.nlm.nih.gov/nuccore/?term=wolbachia. A common goal of many Wolbachia genome projects is to provide comparative genomic information for understanding mechanisms of genome evolution and mechanisms of host phenotype manipulation by Wolbachia (Cordaux et al. 2011; Saridaki and Bourtzis 2010; Werren 1997). In addition, the evolutionary analysis of Wolbachia prophages is of interest and capturing the symbiont DNA from divergent sources not only provides DNA sequence of Wolbachia but also can capture their prophages (Kent et al. 2011).
To test the selectivity of the designed baits, we used the Wolbachia wBm strain (supergroup D) as a test case for the efficiency of Wolbachia DNA capture. The sequence of the 1.1 Mb Wolbachia genome is already known (Foster et al. 2005) and was included in the bait pool. Using these baits, we then attempted to selectively capture Wolbachia DNA from a phylogenetically distant strain, the feminizing wVulC strain from the isopod Armadillidium vulgare (supergroup B) (Cordaux et al. 2004) for which no genomic sequences were included in the bait design.
In the case of wVulC, DNA sequencing and comparative analysis from related strains with alternate phenotypes (cytoplasmic incompatibility and feminization) (Bouchon et al. 2008) will aid in the identification of the genetic basis of the phenotypic differences induced by Wolbachia within this group of isopod crustaceans. In the case of wBm, it has been shown that this endosymbiont is a novel drug target against human filariasis (Slatko et al. 2010), and identification of worldwide variants will be useful. The described DNA capture technique should find application in drug discovery, evolutionary analysis and populational/ecological studies. For example, a similar approach with Roche Nimblegen arrays was utilized by the Bordenstein lab to isolate and analyze the Wolbachia genome and WO prophages of the strain wVitB from the parasitic wasp Nasonia vitripennis (Kent et al. 2011).
Materials and methods
Wolbachia strains
Two Wolbachia strains were analyzed in this study: the obligate wBm strain from the nematode B. malayi (TRS Labs, Georgia, USA) (Foster et al. 2005) and the feminizing wVulC strain from the terrestrial isopod A. vulgare (maintained in the EES lab) (Cordaux et al. 2004). The 1.1 Mb wBm genome sequence is known and the wVulC genome is in final assembly steps. The size of the wVulC genome has been estimated by PFG electrophoresis at 1.75 Mb (Bouchon et al. 2008) and the current sequence consists of 10 contigs of 1.66 Mb, which agrees with pulsed-field estimates (Liu et al. manuscript in preparation).
RNA bait library design
To design the targeted genome enrichment library, we created a SureSelect™ set of enrichment oligonucleotides for solution hybrid selection. The library of biotinylated complementary RNA baits was designed and synthesized by Agilent (Santa Clara, CA). RNA baits were utilized because of the stability of RNA-DNA hybrids in the selection process and the ease of their removal in subsequent steps. The 120-mer RNA bait library was created based upon compiling 11 Wolbachia complete and partial genome sequences found in GenBank (Wolbachia infections of Muscidifurax uniraptor (NZ_ACFP00000000.1), Wuchereria bancrofti (PRJNA43539), Onchocerca volvulus (PRJNA12625), Drosophila willistoni (PRJNA16739), Drosophila simulans gdsi 540 (AAGC01000629.1), Drosophila simulans (AAGC00000000.1), Drosophila melanogaster (AE017196.1), Drosophila ananassae gdan 143 (AAGB00000000), Culex quinquefasciatus pel (AM999887.1), Culex quinquefasciatus jhb (ABZA00000000), Brugia malayi (AE017321.1)). Bait sequences were tiled across each genomic sequence with 60 bp overlap and pooled, resulting in approximately 215,000 baits with about 207,000 unique sequences. Baits were then filtered for homology against select host genomes that contain Wolbachia (Brugia malayi (GCF_000002995.1), Onchocerca volvulus (ADBW00000000.1), Wuchereria bancrofti (ADBV00000000.1)) using BLAT, a high speed and more accurate BLAST-Like Alignment Tool with the ability to use an internal set of sequences for assembly and rapidly find high similarity sequences of relatively short length (Kent 2002). 6,000 baits with significant host homology were removed from the bait pool. The final bait count was 201,776 after removing BLAT rejects. Baits were also tested for uniqueness against two nematodes that do not harbor the endosymbiont (Caenorhabditis elegans (GCA_000002985.2), Loa loa (GCA_000183805.1)). This BLAT search did not produce significant hits and thus no additional baits were removed.
DNA extraction and preparation
Total DNA from the nematode B. malayi and from the isopod A. vulgare were extracted as described (Sambrook and Russell 2001; Bouchon et al. 1998). Quantification of the DNA samples was performed using a Nanodrop 1,000 spectrophotometer (Thermo Scientific) and the Qubit 2.0 fluorimeter (Invitrogen). DNA samples were normalized to 3 μg of DNA for each sequence capture protocol. DNA samples were sheared by sonication to an average length of 200 bp using a Covaris S1 then end repaired, followed by 3′dA addition using the NEBNext® Sample Preparation Kit (New England Biolabs). Adaptors were ligated onto the ends and following purification the DNA was PCR amplified (6 cycles) using indexed PCR primers and the Illumina InPE1.0 forward PCR primer. After purification, quality assays were performed using the Caliper GX (Life Science) and Bioanalyzer (Agilent) to determine the average fragment sizes and concentrations.
Capture and sequencing
Wolbachia DNA was captured from the prepared total DNA by hybridization to the biotinylated cRNA baits for 24 h at 65 °C, following the Agilent SureSelect™ protocol, but supplemented with custom blocking oligos complementary to the barcoded adaptors. Bound DNA was recovered using magnetic streptavidin beads, PCR amplified (12 cycles) using Illumina forward and reverse primers and purified. Sequencing samples were prepared using the NEBNext® Sample Preparation Kit (New England Biolabs). The library was paired-end sequenced on the Illumina HiSeq 2000 at HudsonAlpha, Inc.
Bioinformatics analysis
Sequence reads provided by HudsonAlpha were quality controlled using FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and mapped against the complete wBm genome and the partial wVulC genome using Bowtie (version 0.12.7) (Langmead et al. 2009) and Bowtie2 (version 2.0.0-beta5) (Langmead and Salzberg 2012). Mapping results were processed by SAMtools (Li et al. 2009) and visualized using Artemis (Carver et al. 2012). Assemblies were performed using Velvet assembler (Version 1.2.03) (Zerbino and Birney 2008). Optimization of the assembly was performed by using different k-mers (from 19 to 49 by steps of 2 bases); the optimal assembly was chosen considering the N50, the length of the longest contig and the total bases in the contig.
Results and discussion
wBm sequence capture and sequencing
Nearly ninety-two million (91,724,826) reads from duplicated one third lanes on an Illumina HiSeq 2000 were produced from DNA captured by the oligo bait set. High quality metrics were obtained from the run (Q scores above 30 for bases 1–13 and above 40 for bases 14–50). Sensitive parameters of Bowtie2 were used to map these reads onto the genome sequence of the Wolbachia endosymbiont from the TRS strain of Brugia malayi (NC_006833). The entire Wolbachia genome was captured at a depth of over 3000×. These results for wBm were anticipated as the capture oligos we designed were based on all Wolbachia genomic sequences in GenBank or RefSeq including those of wBm (Fig. 1).
Fig. 1.
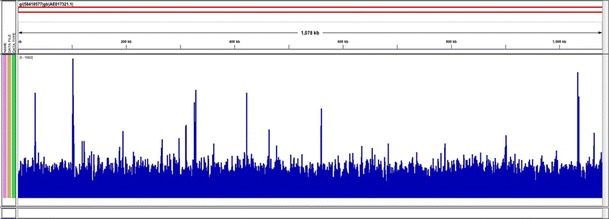
The average number of mapped reads per genomic location averaged over a 25 nt window. The X axis represents a linear map of the Wolbachia genome from B. malayi and the Y axis (1–15,000) represents relative sequence coverage. The count file was generated from a BAM file using IGV Tools (Thorvaldsdottir et al. 2012). 100 % coverage was obtained
Depth of coverage was generally uniform but showed spikes over certain regions (Y axis, Fig. 1). These regions could either be repetitive in the genome or could be regions of duplicated lateral gene transfers into the host genome (Dunning Hotopp et al. 2007).
Only 5.20 % of the reads did not map to the Wolbachia wBm genome. These unmapped reads were assembled by Velvet and the resulting 1,107 contigs were used to query the NCBI database by BLASTn (Fig. 2). 27.8 % of these ~5 % unmapped sequences BLAST to host nematode sequences and 51 % appeared to BLAST to wBm Wolbachia sequence. These represent contigs assembled from reads that do not map to the reference genome (reads with more than 3 mismatches were not mapped) but have significant similarity to wBm by BLAST. Unknown sequences (15.59 %) were investigated for GC content and they appeared to have an average GC content of 28.1 %, a value similar to Wolbachia and the host (Foster et al. 2005; Ghedin et al. 2007). Thus they can not be differentiated as to whether they represent Wolbachia or B. malayi genes or derive from other organisms with similarly low GC content.
Fig. 2.
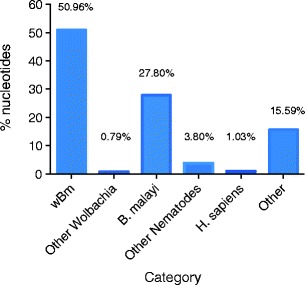
Distribution of the 5.2 % reads not mapped to wBm. Bar graph denotes % of nucleotides out of the total assembled contigs that show significant BLAST scores to wBm genome, other Wolbachia the B. malayi host, other nematodes or H. sapiens. The “Other Nematodes” category corresponds to sequences with a significant BLAST score to other species of nematodes (mainly filarial nematodes). The “H. sapiens” category may represent DNA contamination. The “Other” category corresponds to sequences with no significant BLAST similarity to any sequences in the NCBI database
wVulC sequence capture and sequencing
Over thirty-six million reads (one-third of a 50 nt paired-end sequencing lane on the Illumina HiSeq™ 2000) were produced from DNA capture by the oligo bait set. As with the wBm mapping set, sequences were quality assayed. The entire dataset was of high quality (Q scores above 30 for bases 1–9 and above 40 for bases 10–50).
Different parameters of Bowtie were tested to improve the mapping of the total paired-end read dataset to the incompletely sequenced wVulC genome and the final mapping using Bowtie2 provided a total of 34,524,894 millions reads mapping to the reference genome (94.73 %).
This mapping covered 97.1 % of the partially known wVulC sequence (Fig. 3). Several regions appear to be over-represented in the data set, as with the wBm mapping. These include the single copy DNA-directed RNA polymerase, beta/beta’ subunit gene as well as the phage major capsid protein E gene, which is likely repetitive in the genome.
Fig. 3.
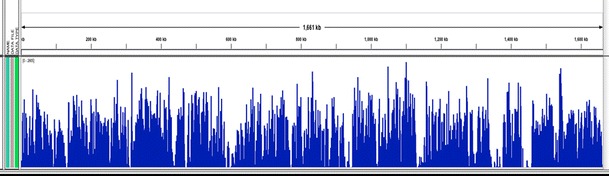
The average number of mapped reads per genomic location averaged over a 25 nt window. The X axis represents a linear map of the artificially concatenated Wolbachia pseudo-contig from A. vulgare and the Y axis (1–3,000) represents relative sequence coverage. The count file was generated from a BAM file using IGV Tools (Thorvaldsdottir et al. 2012). 97.1 % coverage was obtained
The unmapped reads (5.27 % of the total reads) were assembled by Velvet and the resulting 28 contigs were used to query the NCBI database by BLASTn. Of these contigs, 24 (94.78 % of the total unmapped reads) corresponded to other Wolbachia sequences. These sequences may represent wVulC genes not yet identified in the incomplete genome as the average GC% of these sequences is similar to the GC content of Wolbachia strain. None of the remaining 4 contigs had any significant match to any other sequences in the database.
As the wVulC genome is still in draft form, it cannot be excluded that there are errors in the preliminary sequence. To correct the sequence, mapping results were used to call variants and to detect errors, which were manually corrected. By this method, more than 300 base substitutions and over 30 indels have been corrected by use of the oligo bait-captured DNA sequence.
De novo assembly of the wVulC genome sequence
A primary goal of this study was to sequence an unknown genome and thus we performed a de novo assembly of the oligo-captured wVulC sequence data. After optimization of the Velvet parameters (k-mer = 45, insert length = 170, standard deviation for insert length = 63 and minimum contig length = 200 bases), a manual assembly produced 523 contigs with N50 of 5826, a maximum length of 26,288 bases. This used over 87 % of the oligo-captured reads (31.81/36.45 million reads) providing a total length of 1.32 Mb, representing 75.5 % of the estimated length of the genome and 79.5 % of the partially known sequence.
To ensure that all the contigs built from the oligo-captured DNA were wVulC sequences, we aligned and ordered them to the previously sequenced wVulC genome using Mauve (Darling et al. 2010) and r2cat (Husemann and Stoye 2010). Overall, 95.7 % of the bases in contigs built from oligo-captured DNA were wVulC, which represents 76 % of the 1.66 Mb concatenated reference genome. 61 contigs did not match the incomplete wVulC genome (Fig. 4). Several of these contain sequences that show similarities with other Wolbachia strain sequences (0.1 %) while a contig of 16 kb appeared to be a region from the Armadillidium vulgare 18S ribosomal gene (1.22 %), certainly host DNA contamination (Fig. 4). Since the wVulC genome is not finished and gaps remain between the 10 contigs, analysis of the contigs built from oligo-captured DNA that matched other Wolbachia strains or didn’t match any known sequence in NCBI (2.41 %) are being used to complete the reference genome sequence as they may represent yet unknown wVulC sequences.
Fig. 4.
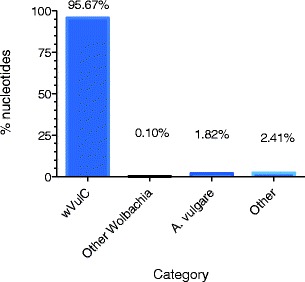
Capture efficiency of the wVulC Wolbachia genome. Bar graph shows the distribution of nucleotides out of the total assembled contigs that show significant BLAST scores to the partial wVulC genome, other Wolbachia or the A. vulgare host. The “Other” category corresponds to sequences with no significant BLAST similarity to any sequences in the NCBI database
Our results confirm and extend the results of Kent et al. (2011) and demonstrate that this method can potentially be used with any Wolbachia strain and on any endobacterium with either a reference genome or a highly similar sequence, providing an approach for isolating a significant fraction of symbiont DNA from host DNA for sequencing and comparative genomic analysis. In this context, of interest is the observation that both wBm and wVulC were successfully purified away from host and mitochondrial DNA with the same bait library even though they are members of phylogenetically distant clades. In filarial nematodes, this approach will enable rapid isolation and analysis of Wolbachia strains from worldwide populations to identify polymorphisms related to drug discovery initiatives and evolutionary analysis of Wolbachia prevalence and distribution. In isopods, this approach will allow DNA isolation for genomic comparison of Wolbachia strains, which induce either various types of feminizing phenotypes or cytoplasmic incompatibility. For example, the isopod A. vulgare may harbor another Wolbachia strain, wVulM, which has a lower feminizing effect (~70 %) than the wVulC strain (~80 %) (Cordaux et al. 2004). In the Porcellionides pruinosus complex of species, 3 distinct feminizing Wolbachia strains have been identified which are present in ~60 % of populations where Wolbachia are present only in females or ~90 % in populations where both males and females are infected (Marcadé et al. 1999; Lefebvre and Marcadé 2005), a situation also encountered in populations of Oniscus asellus (Rigaud et al. 1999). Further, most Wolbachia strains infecting isopods induce a feminizing phenotype (Bouchon et al. 2008), however, 3 of them induce cytoplasmic incompatibility (Legrand et al. 1978; Moret et al. 2001). One of them, wCon, which infects Cylisticus convexus, is closely related to wVulC (Cordaux et al. 2012). Genomic comparisons will thus be useful in helping decipher the evolution of Wolbachia and its various biological manifestations.
Acknowledgments
This work was funded by New England Biolabs and by a grant from the Bill and Melinda Gates Foundation to the Liverpool School of Tropical Medicine as part of the A-WOL consortium (BES). We thank Drs William Jack, Tilde Carlow and Don Comb for their interest and support. Research in Poitiers was funded by the Centre National de la Recherche Scientifique (CNRS) and the French Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche. The authors also thank Seth Bordenstein for helpful discussions in preparation of the manuscript.
References
- Baldo L, Werren JH (2007) Revisiting Wolbachia supergroup typing based on WSP: spurious lineages and discordance with MLST. Curr Microbiol 55:81–87 [DOI] [PubMed]
- Bordenstein SR et al (2009) Parasitism and mutualism in Wolbachia: what the phylogenomic trees can and cannot say. Mol Biol Evol 26:231–241 [DOI] [PMC free article] [PubMed]
- Bouchon D, Rigaud T, Juchault P (1998) Evidence for widespread Wolbachia infection in isopod crustaceans: molecular identification and host feminization. Proc Biol Sci 265:1081–1090 [DOI] [PMC free article] [PubMed]
- Bouchon D, Cordaux R, Grève P (2008) Feminizing Wolbachia and the evolution of sex determination in isopods. In: Bourtzis K, Milller TA (eds) Insect symbiosis. CRC Press, Boca Raton, pp 273–294
- Carver T, et al. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28:464–469. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casiraghi M et al (2003) dnaA gene sequences from Wolbachia pipientis support subdivision into supergroups and provide no evidence for recombination in the lineages infecting nematodes. Parassitologia 45:13–18 [PubMed]
- Casiraghi M et al (2005) Phylogeny of Wolbachia pipientis based on gltA, groEL and ftsZ gene sequences: clustering of arthropod and nematode symbionts in the F supergroup, and evidence for further diversity in the Wolbachia tree. Microbiology 151:4015–4022 [DOI] [PubMed]
- Charles H, Ishikawa H (1999) Physical and genetic map of the genome of Buchnera, the primary endosymbiont of the pea aphid Acyrthosiphon pisum. J Mol Evol 48:142–150 [DOI] [PubMed]
- Cordaux R et al (2004) Evidence for a new feminizing Wolbachia strain in the isopod Armadillidium vulgare: evolutionary implications. Heredity (Edinb) 93:78–84 [DOI] [PubMed]
- Cordaux R, Bouchon D, Greve P. The impact of endosymbionts on the evolution of host sex-determination mechanisms. Trends Genet. 2011;27:332–341. doi: 10.1016/j.tig.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Cordaux R et al. (2012) Widespread Wolbachia infection in terrestrial isopods and other crustaceans. Zookeys 176:123–131 [DOI] [PMC free article] [PubMed]
- Darling AE, Mau B, Perna NT. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning Hotopp JC, et al. Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science. 2007;317:1753–1756. doi: 10.1126/science.1142490. [DOI] [PubMed] [Google Scholar]
- Foster J et al (2005) The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol 3:e121 [DOI] [PMC free article] [PubMed]
- Ghedin E et al (2007) Draft genome of the filarial nematode parasite Brugia malayi. Science 317:1756–1760 [DOI] [PMC free article] [PubMed]
- Husemann P, Stoye J. r2cat: synteny plots and comparative assembly. Bioinformatics. 2010;26:570–571. doi: 10.1093/bioinformatics/btp690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturbe-Ormaetxe I et al (2011) A simple protocol to obtain highly pure Wolbachia endosymbiont DNA for genome sequencing. J Microbiol Methods 84:134–136 [DOI] [PubMed]
- Kent WJ. BLAT-the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent BN et al (2011) Complete bacteriophage transfer in a bacterial endosymbiont (Wolbachia) determined by targeted genome capture. Genome Biol Evol 3:209–218 [DOI] [PMC free article] [PubMed]
- Klasson L et al (2008) Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol Biol Evol 25:1877–1887 [DOI] [PMC free article] [PubMed]
- Klasson L et al (2009) The mosaic genome structure of the Wolbachia wRi strain infecting Drosophila simulans. Proc Natl Acad Sci U S A 106:5725–5730 [DOI] [PMC free article] [PubMed]
- Kumar S, Blaxter ML. Simultaneous genome sequencing of symbionts and their hosts. Symbiosis. 2011;55:119–126. doi: 10.1007/s13199-012-0154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre, F, Marcadé I (2005) New insights in the Porcellionides pruinosus complex (Isopoda, Oniscidea): biological, behavioural, and morphological approaches. Crustacceana 78:465–480
- Legrand JJ, Martin G, Artault JC (1978) Correlation between the presence of a bacterial symbiont in oocytes of Parcellio dilatatus petiti, and the sterility of the cross P.d petiti male x P.d dilatatus female. Arch Inst Pasteur Tunis 55:507–504 [PubMed]
- Li H, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo N, Evans TA (2007) Phylogenetic diversity of the intracellular symbiont Wolbachia in termites. Mol Phylogenet Evol 44:461–466 [DOI] [PubMed]
- Lo N et al (2002) How many Wolbachia supergroups exist? Mol Biol Evol 19:341–346 [DOI] [PubMed]
- Marcadé II et al (1999) Mitochondrial DNA variability and Wolbachia infection in two sibling woodlice species. Heredity (Edinb) 83(Pt 1):71–78 [DOI] [PubMed]
- Mavingui P et al (2005) Efficient procedure for purification of obligate intracellular Wolbachia pipientis and representative amplification of its genome by multiple-displacement amplification. Appl Environ Microbiol 71:6910–6917 [DOI] [PMC free article] [PubMed]
- Melnikov A, et al. Hybrid selection for sequencing pathogen genomes from clinical samples. Genome Biol. 2011;12:R73. doi: 10.1186/gb-2011-12-8-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moret Y, Juchault P, Rigaud T (2001) Wolbachia endosymbiont responsible for cytoplasmic incompatibility in a terrestrial crustacean: effects in natural and foreign hosts. Heredity 86:325–322 [DOI] [PubMed]
- Rigaud T, Moreau J, Juchault P (1999) Wolbachia infection in the terrestrial isopod Oniscus asellus: sex ratio distortion and effect on fecundity. Heredity 83:469–475 [DOI] [PubMed]
- Sambrook J, Russell DW (2001) Isolation of high-molecular-weight DNA from mammalian cells using proteinase K and phenol. In: Sambrook J, Russell DW (eds) Molecular cloning a laboratory manual, Vol 1, 3rd ed. Cold Spring Harbor Press, Cold Spring Harbor, pp 6.4–6.12
- Santos-Garcia D, Peris-Bondia F, D’Auria G, Moya A, Silva F, Latorre A (2012) Enrichment of insect samples with bacterial symbionts using flow cytometry, ABSTRACT 7th International Symbiosis Society Congress
- Saridaki A, Bourtzis K (2010) Wolbachia: more than just a bug in insects genitals. Curr Opin Microbiol 13:67–72 [DOI] [PubMed]
- Slatko BE et al (1999) The Wolbachia genome consortium. Microb Comp Genomics 4:161–165 [DOI] [PubMed]
- Slatko BE, Taylor MJ, Foster JM (2010) The Wolbachia endosymbiont as an anti-filarial nematode target. Symbiosis 51:55–65 [DOI] [PMC free article] [PubMed]
- Sun LV et al (2001) Determination of Wolbachia genome size by pulsed-field gel electrophoresis. J Bacteriol 183:2219–2225 [DOI] [PMC free article] [PubMed]
- Thorvaldsdottir H, Robinson JT, Mesirov JP (2012) Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. doi:10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed]
- Werren JH (1997) Biology of Wolbachia. Annu Rev Entomol 42:587–609 [DOI] [PubMed]
- Werren JH, Windsor DM (2000) Wolbachia infection frequencies in insects: evidence of a global equilibrium? Proc Biol Sci 267:1277–1285 [DOI] [PMC free article] [PubMed]
- Werren JH, Baldo L, Clark ME (2008) Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol 6:741–751 [DOI] [PubMed]
- Wu M et al (2004) Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol 2:E69 [DOI] [PMC free article] [PubMed]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zug R, Hammerstein P (2012) Still a host of hosts for Wolbachia: analysis of recent data suggests that 40 % of terrestrial arthropod species are infected. PLoS One 7:e38544 [DOI] [PMC free article] [PubMed]