

Abstract
Genetic studies in human patients with idiopathic hypogonadotropic hypogonadism (IHH) identified mutations in the genes that encode neurokinin B (NKB) and the neurokinin 3 receptor (NK3R). However, determining the mechanism whereby NKB regulates gonadotropin secretion has been difficult because of conflicting results from in vivo studies investigating the luteinizing hormone (LH) response to senktide, a NK3R agonist. NK3R is expressed in a subset of GnRH neurons and in kisspeptin neurons that are known to regulate GnRH secretion. Thus, one potential source of inconsistency is that NKB could produce opposing direct and indirect effects on GnRH secretion. Here, we employ the GT1-7 cell model to elucidate the direct effects of NKB on GnRH neuron function. We find that GT1-7 cells express NK3R and respond to acute senktide treatment with c-Fos induction and increased GnRH secretion. In contrast, long-term senktide treatment decreased GnRH secretion. Next, we focus on the examination of the mechanism underlying the long-term decrease in secretion and determine that senktide treatment represses transcription of GnRH. We further show that this repression of GnRH transcription may involve enhanced c-Fos protein binding at novel activator protein-1 (AP-1) half-sites identified in enhancer 1 and the promoter, as well as chromatin remodeling at the promoter of the GnRH gene. These data indicate that NKB could directly regulate secretion from NK3R-expressing GnRH neurons. Furthermore, whether the response is inhibitory or stimulatory toward GnRH secretion could depend on the history or length of exposure to NKB because of a repressive effect on GnRH transcription.
Gonadotropin-releasing hormone (GnRH) is the central regulator of reproduction. Pulsatile GnRH secreted from hypothalamic neurons acts on pituitary gonadotrope cells to stimulate the synthesis and secretion of the gonadotropin hormones, LH and FSH. LH and FSH act on receptors in the gonads, testes in males and ovaries in females, to stimulate steroid synthesis and gametogenesis. Throughout the juvenile period, the GnRH neuron network is quiescent, until the onset of puberty when there is a sustained increase in the pulsatile release of GnRH (1). Thus, puberty is thought to occur in response to the awakening of the GnRH neuron network that leads to increased GnRH secretion. In support of this, the administration of pulsatile GnRH was sufficient to induce puberty in the female monkey (2). However, apart from the identification of several permissive factors, the mechanism that underlies the increase in pulsatile GnRH secretion at the onset of puberty remains largely unknown.
Recently, genetic studies identified mutations in TAC3 and TACR3, the genes that encode for neurokinin B (NKB) and its receptor, NK3R, in humans with idiopathic hypogonadotropic hypogonadism (IHH) (3–5). IHH is a disorder that is characterized by a failure to undergo puberty and low circulating levels of LH and FSH. Because the administration of exogenous GnRH rescued LH levels in patients with mutations in TAC3 and TACR3, NKB was suggested to act at the level of the hypothalamus, presumably by stimulating GnRH release (5). However, in vivo studies employing the central administration of senktide, a NK3R agonist, have failed to consistently show stimulatory effects on serum LH (6). Senktide treatment decreased serum LH in ovariectomized female rats (7) and mice treated with low estrogen (8), but increased LH in gonad-intact diestrus female rats (9), follicular phase ewes (10), and prepubertal rhesus monkeys (11). It has been suggested that the inconsistent effects of senktide on serum LH may be because of differences among the animal models employed; including species, age, sex, gonadal status, and circulating hormonal milieu. A recent investigation of Tacr3-null mice reported normal timing for pubertal onset and normal circulating LH levels. However, males were reported to have small testes and low circulating FSH levels and females had decreased uterine weight, abnormal estrous cycles, reduced fertility, and decreased occurrence of corpora lutea, suggesting an ovulatory defect (12). Thus, although in vivo studies have not yet definitively classified NKB as an activator of LH secretion or confirmed its requirement for pubertal onset, they suggest a key role in the regulation of gonadotropin hormone secretion and reproduction.
Mutations in GPR54, the receptor for kisspeptin, were also identified in human IHH patients (13, 14). Moreover, the administration of kisspeptin has consistently been shown to increase serum LH in vivo (15–19). Mice lacking the GPR54 or kisspeptin genes exhibit phenotypes that mimic IHH, including low serum gonadotropin levels and disrupted puberty (13, 14, 20–22). Thus, kisspeptin is thought to activate puberty by stimulating GnRH secretion. Kisspeptin, NKB, and dynorphin are coexpressed in a population of neurons referred to as KNDy neurons within the arcuate nucleus (ARC), a region of the hypothalamus implicated in the control of pulsatile GnRH secretion (8, 23, 24). Axons from KNDy neurons project to GnRH neuron terminals in the median eminence and have bilateral projections to other KNDy neurons (25). Moreover, because KNDy neurons in the ARC express NK3R and exhibit c-Fos induction in response to the central administration of senktide (8), it was suggested that NKB could act autosynaptically or transynaptically to regulate kisspeptin secretion. NK3R immunoreactivity has also been detected in a subset (about 16%) of GnRH neuron somata in the OVLT, and in GnRH axon terminals at the median eminence in the female rat (26). In addition, NK3R expression was detected by RT-PCR in 50% of pools of GnRH neurons from GnRH-GFP mice (27). Thus, we hypothesized that NKB could regulate GnRH secretion from a subset of NK3R-expressing GnRH neurons and, in addition to effects on KNDy neurons in the ARC, this might contribute to the LH response to the central administration of senktide. However, the functional significance of NKB acting directly on NK3R-expressing GnRH neurons is currently unknown.
GnRH neurons are a small population, totaling only about 800 cells in the mouse, and are distributed in a dispersed pattern throughout a large area of the forebrain rather than in a single nucleus. As a result, the in vivo investigation of GnRH neurons is technically challenging. The immortalized GT1-7 cell line has been well characterized and shown to faithfully represent the mature, postmigratory GnRH neuron. Similar to GnRH neurons in vivo, GT1-7 cells exhibit neuronal morphology, form synaptic contacts, synthesize and process mature GnRH, and secrete GnRH in a pulsatile fashion (28, 29). Importantly, the GT1-7 cell line enables the evaluation of the direct effects on GnRH neurons in the absence of many of the confounding variables present in in vivo studies, such as altered kisspeptin release or changes in the circulating hormonal milieu. GT1-7 cells also allow for detailed molecular studies that are not possible in vivo. In this report, GT1-7 cells are shown to express NK3R and respond to senktide treatment with an acute activation and a long-term repression of GnRH secretion. We further examine the mechanism underlying the long-term decrease in GnRH secretion and determine that senktide treatment represses GnRH transcription by a mechanism that involves the induction of c-Fos, novel activator protein 1 (AP-1) half-sites in enhancer 1 and the promoter, and chromatin remodeling of the promoter of the GnRH gene. These results indicate that NKB could directly regulate the synthesis and secretion of GnRH in NK3R-expressing GnRH neurons.
Materials and Methods
Cell culture and treatments
GT1-7 cells (28) were cultured in DMEM with 4.5 g/L glucose, L-glutamine and sodium pyruvate (DMEM; Mediatech, Manassas, Virginia) supplemented with 10% Foundation B fetal bovine serum (FBS; Gemini Bio-Products, West Sacramento, California) in 5% CO2/95% O2 at 37°C. Senktide (Tocris Bioscience, Ellisville, Missouri) was dissolved in Dulbecco's phosphate-buffered saline (PBS) without calcium or magnesium (dPBS; Mediatech) supplemented with 0.1% Fraction V bovine serum albumin (BSA; Sigma, St Louis, Missouri). NK3R inhibitor SB 222200, PKC inhibitor Go6983, and Phorbol 12-myristate 13-acetate (TPA) (Tocris Bioscience) were dissolved in dimethylsulfoxide (DMSO; Sigma). Aliquots of stock solutions were stored at –80°C until use. Compounds that were dissolved in DMSO were further diluted in DMSO as needed before a final dilution of 1:1000 in media (0.1% DMSO final).
Static culture secretion experiments
For secretion studies, GT1-7 cells were seeded into 6-well plates and allowed to adhere overnight. Two hours before treatment, media was changed to serum free media (DMEM + 0.1% BSA). Cells were treated with either vehicle or 50 nM senktide. Conditioned media was collected from cells and transferred to a 1.5-ml microcentrifuge tube and centrifuged for 10 minutes at 2,000 rpm at room temperature to remove contaminating cells. Supernatant was then transferred to a clean 2-ml round-bottom microcentrifuge tube and frozen at –80°C. Conditioned media was lyophilized using a speedvac, and dried samples were stored at –80°C until GnRH content could be assayed by radioimmunoassay (RIA). In brief, 0.2 ml PBS-gel [0.14M NaCl, 0.1M NaPO4, 0.1% gelatin, (pH 7.4)] was added to samples, and they were left on a rocking shaker at 4°C overnight. The next day, samples were manually resuspended by vortexing and pipetting, then 0.1 ml aliquots were prepared in duplicate in glass test tubes. The 1:100 stock of EL-14 GnRH primary antibody was diluted 1:700 (1:70,000 final) in NRS-EDTA [0.14M NaCl, 0.01M NaPO4, 0.1% gelatin, 0.5M EDTA (pH 7.4) supplemented with 1% normal rabbit serum], and 50 μl was added to each sample. After incubating 48 hours at 4°C, 50 μl containing ∼10,000 cpm of [125I] Tyr5-Luteinizing Hormone-Releasing Hormone (PerkinElmer, Boston, Massachusetts) diluted in PBS-gel was added to each sample. After incubating for 48 hours at 4°C, samples were precipitated with 3 ml ice-cold ethanol and centrifuged for 15 minutes at 2500 rpm at 4°C. Supernatant was decanted and dried pellets were counted with a γ-counter (Micromedic, Huntsville, Alabama) to determine CPM for bound radioactivity. Standards were run in triplicate and duplicate standard curves were counted at the beginning and end of the assay. GnRH content for experimental samples was extrapolated from the standard curve. Intra-assay variability was 21.2%.
RNA isolation and RT-PCR
Cells were grown to approximately 90% confluence before harvest for RNA. Media was aspirated and cells were rinsed with cold PBS. One ml of TRIzol (Invitrogen, Carlsbad, California) was used to isolate RNA from each 10-cm dish according to manufacturer's protocol. Five micrograms of total RNA was DNase I treated using DNA-free (Ambion) and reverse transcribed in a total volume of 20 μl using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen) using oligo-(dT)20 (for NK3R, NK1R, NKB expression studies) or random hexamer primers (c-Fos, c-Jun, FosB, JunB, Fra-1, JunD, Fra-2 expression). PCR was performed on the resulting cDNA using a thermocycler [MJ Research, St Bruno (Quebec) Canada] and primers specific for NK3R, NK1R, or NKB (Supplemental Table 1, published on The Endocrine Society's Journals Online web site at http://mend.endojournals.org). Quantitative and semiquantitative PCR was performed using the iQ5 Real-Time PCR Detection System and Software and iQ SYBR Green Supermix (Bio-Rad, Hercules, California) according to the manufacturer's recommendations, including melt curve analysis, with primers for specific for c-Fos, c-Jun, FosB, JunB, Fra-1, JunD, Fra-2, and βActin (Supplemental Table 1). For quantitative PCR, serial dilutions of plasmid DNA, either quantitative PCR (qPCR) amplicons cloned into pCR2.1 (Invitrogen) for βActin, or complete cDNA in pcDNA3.1 for c-Fos and c-Jun, were used to generate standard curves.
Plasmids and cloning
Cloning of the rat NK3R, murine c-Jun and c-Fos and A-FOS expression constructs were previously described (30–32). The –5 Kb rat GnRH (-4984 to +22 relative to the transcription start site), 5′ truncations and GnRH-E1/GnRH-P luciferase reporters have been previously described (33, 34). For the GnRH-E1/TKp luciferase reporter, enhancer 1 was digested from GnRH-E1/GnRH-P using KpnI and NheI and subcloned into a pGL3 vector containing the –81 bp thymidine kinase minimal promoter (GnRH-E1/TKp). For the GnRH-P luciferase reporter, the –173 bp rat GnRH promoter was digested from the –520 bp truncation of the GnRH reporter using KpnI and HindIII and subcloned into the pGL3 vector. Base pair substitutions were introduced into enhancer 1 and promoter luciferase reporter constructs (GnRH-E1/TKp and GnRH-E1/GnRH-P) to disrupt AP-1 half-sites using the QuickChange Site-Directed Mutagenesis Kit (Agilent Technologies, La Jolla, California). The AP-1 cis-Reporting system (Agilent) used for the AP-1 multimer luciferase reporter contains seven copies of an AP-1 binding site (TGACTAA).
Luciferase assays
For transient transfections, approximately 24 hours before transfection, GT1-7 cells were seeded into 24-well plates at a density of 150,000 cells per well. Cells were transfected with plasmid DNA using FuGENE 6 Transfection Reagent (Promega, Madison, Wisconsin) according to the manufacturer's protocol with a 3:1 ratio of FuGENE to DNA. Approximately 24 hours after transfection, cells were switched to serum-free media (DMEM + 0.1% BSA). Cells were treated with pharmacological agents as indicated in the figure legends. Approximately 48 hours post-transfection, cells were rinsed one time with PBS and lysed with 55 μl 100 nM KPO4 + 0.2% Triton X-100. Twenty μl cell lysate was used to measure luciferase activity on a Veritas Microplate luminometer (Turner Biosystems, Sunnyvale, California) by injection with 100 μl luciferase assay buffer (25 mM Tris pH 7.4, 15 mM MgSO4, 10 mM ATP, and 65 μM luciferin) into each well. Twenty μl cell lysate was used to measure β-galactosidase activity using the Tropix Galacto-light β-galactosidase assay (Applied Biosystems, Foster City, California). For each well, RLU for luciferase was normalized to β-galactosidase to control for transfection efficiency. Transfections were performed in triplicate and experiments were repeated at least three independent times.
DNase I sensitivity assay
Approximately 24 hours before transfection, 3.5 × 106 GT1-7 cells were seeded into each 10-cm plate. Cells were transfected with 8.7 μg of rat NK3R expression plasmid using FuGENE 6 Transfection Reagent (Promega) according to the manufacturer's protocol with a 3:1 ratio of FuGENE to DNA. Approximately 24 hours after transfection, cells were switched to serum-free media (DMEM + 0.1% BSA). The next day, cells were treated with pharmacological agents as indicated in the figure legends. Cell nuclei were isolated and DNase I sensitivity assays were performed as previously described (35). In brief, cell nuclei were resuspended in 1× DNase I Reaction Buffer (Promega) containing 2% glycerol, 5 U of DNase I (Promega) and incubated at 37°C for 5 minutes. DNase I was inactivated using DNase I Stop Solution (Promega) and incubated at 65°C for 10 minutes. Treated nuclei were lysed in Nuclei Lysis Buffer [100 mM TrisHCl (pH 8.0), 5 mM EDTA, 200 mM NaCl, 0.2% SDS] followed by RNase A and proteinase K digestion. Genomic DNA was then isolated by extraction twice with phenol/chloroform/isoamyl alcohol and once with chloroform. Samples were handled with wide-mouth pipette tips to minimize DNA shearing. DNA was ethanol precipitated and resuspended in TE buffer followed by 55°C incubation for 1 hour. Samples were analyzed by qPCR using primers (Supplemental Table 1) specifically designed to detect only the endogenous mouse GnRH regulatory elements (33) (GnRH-E1, –1814/−1653; GnRH-P, –173/+53) and not the rat and human transgenes used to generate the GT1-7 (36) cell line. Mouse FSHβ (37) and β-actin promoters (38) were used as negative and positive controls for DNase I sensitivity, respectively. Forty ng DNA from each treatment condition was quantified relative to a standard curve of dilutions of undigested DNA.
Chromatin immunoprecipitation (ChIP)
3.5 × 106 GT1-7 cells were seeded into each 10-cm plate. After 48 hours, cells were switched to serum-free media (DMEM + 0.1% BSA). After 21 hours, cells were treated with vehicle or 100 nM TPA for 3 hours, and ChIP assays were performed as previously described (33, 37). In brief, chromatin was sonicated to an average length of 300–500 bp using a Branson Sonifier 250 (Branson Ultrasonics Corp., Danbury, Connecticut). The sc-253 (rabbit) antibody was used to recognize multiple Fos proteins and normal rabbit IgG, sc-2027, was used as a control (Santa Cruz Biotechnology). Immunoprecipitated DNA and DNA from input chromatin was analyzed by qPCR using primers specific to GnRH regulatory elements (Supplemental Table 1) and quantified relative to a standard curve representing percent of input chromatin as previously described (33). Data are represented as fold versus IgG control with mean ± SEM for four independent experiments.
Electrophoretic mobility shift assays
3.5 × 106 GT1-7 cells were seeded into each 10 cm plate. After 48 hours, cells were switched to serum-free media (DMEM + 0.1% BSA). After 21 hours, cells were treated with vehicle or 100 nM TPA for 3 hours. Nuclear extracts were prepared as previously described (39), and protein concentration was determined by Bio-Rad Protein Assay (Bio-Rad, Hercules, California). Oligonucleotide probes are listed in Supplemental Table 1. Annealed double-stranded oligonucleotides (1 pmol/μl) were end-labeled with T4 Polynucleotide Kinase (New England Biolabs) and [γ32P]ATP (7000 Ci/mmol; MP Biomedicals, Solon, Ohio) then purified using Micro Bio-Spin 6 Chromatography Columns (Bio-Rad). Binding reactions contained 2 μg nuclear protein and 100,000 CPM of labeled probe in 10 mM HEPES (pH 7.9), 25 mM KCl, 2.5 mM MgCl2, 1% glycerol, 0.1% Nonidet P-40, 0.25 mM EDTA, 0.25% BSA, 1 mM dithiothreitol, and 0.0125 μg/μl poly dIdC. For cold competitor, 1000-fold molar excess unlabeled competitor probe was added to the reaction and incubated for 10 minutes on ice. For supershift experiments, 2 μg of an antibody that recognizes all Fos proteins, sc-253X, a c-Fos-specific antibody (K-25), or normal rabbit IgG (Santa Cruz Biotechnology) was added to the reaction and incubated for 20 minutes on ice before the addition of labeled probe. After addition of probe, samples were incubated for 10 minutes at room temperature before loading on a 5% nondenaturing polyacrylamide gel in 0.25X Tris-borate EDTA buffer. Gels were run for 2.5 hours at 200–250 V then dried under vacuum and exposed to film with L-Plus intensifying screens (Fisher Biotech) for times indicated in figure legends.
Statistical analyses
Statistical analysis was performed using JMP version 8 (SAS Institute, Inc., Cary, North Carolina). Data were analyzed either by Student's t test or ANOVA followed by Tukey honestly significant difference (HSD), as indicated in figure legends. When necessary to achieve a normal distribution, data were transformed by the method of Box and Cox. A P value of less than .05 was the cut-off for significance.
Results
GT1-7 cells express NK3R and respond to senktide with altered GnRH secretion
A subset of GnRH neurons was reported to express NK3R in vivo (26, 27). To determine if GT1-7 cells express NK3R, RT-PCR was performed on total RNA using primers specific for Tacr3 (NK3R, Supplemental Table 1). A band of the expected 387-bp size was present in cDNA prepared from GT1-7 cells and whole brain (Figure 1A, RT) and was absent in negative control samples prepared without reverse transcriptase (Figure 1A, No RT). The PCR product was confirmed to be Tacr3 by sequencing. These data confirm that GT1-7 cells express the RNA that encodes for NK3R.
Figure 1.
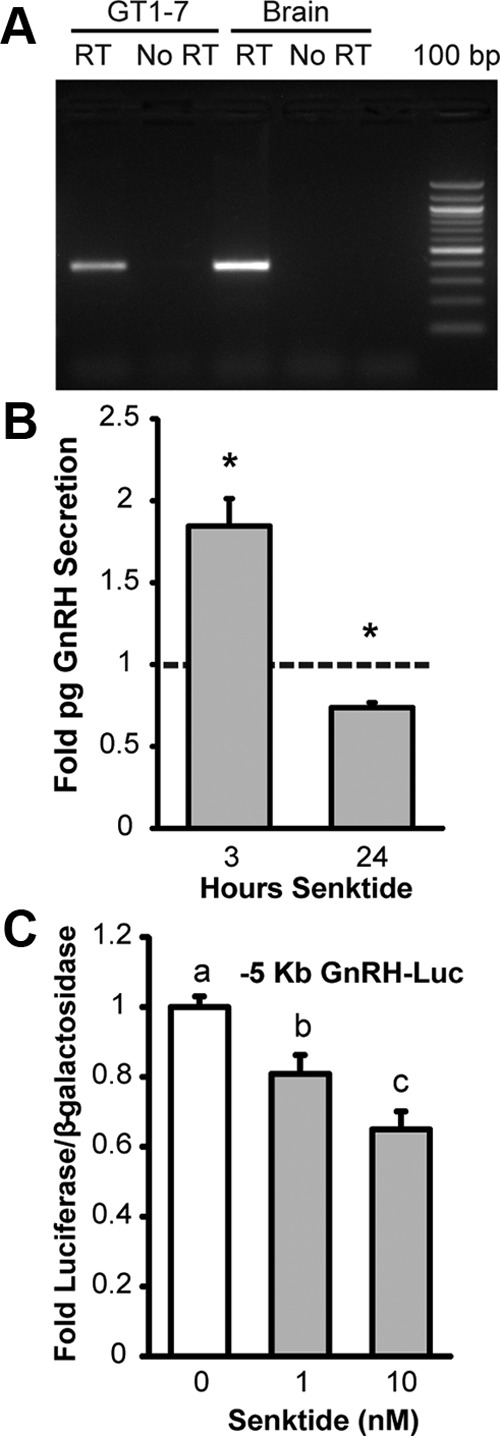
GT1-7 cells express NK3R and respond to agonist with altered GnRH secretion and transcription. A, RT-PCR for NK3R using total RNA from GT1-7 cells and mouse brain. A single band of the expected 387 bp size was observed in cDNA prepared with reverse transcriptase (RT) but not negative controls prepared without reverse transcriptase (No RT). Bands were excised and confirmed to be NK3R by sequencing. B, GnRH peptide accumulation in conditioned media from GT1-7 cells. Cells were treated with 50 nM senktide or vehicle for either three or 24 hours. GnRH peptide content in conditioned media was measured by RIA. Data are normalized to vehicle control and shown as mean fold ± SEM for n = 3 to 5 independent experiments. Comparisons were made using a t-test against the hypothetical mean 1.0. *Significantly different from 1.0 (P < .05). C, Transient transfection of the –5 Kb GnRH gene sequence upstream of a luciferase reporter (-5 Kb GnRH-Luc) into GT1-7 cells treated with vehicle or senktide (1 or 10 nM) for 10 hours. Luciferase values were internally normalized using a cotransfected β-galactosidase reporter to control for transfection efficiency and normalized to an external control. Data are represented as mean ± SEM for n = 3 independent experiments. Statistical analysis was performed using ANOVA with Least squares means differences Tukey HSD (α = .05). Levels not connected by same letter are significantly different.
Next, the functional significance of NK3R signaling on GnRH secretion was examined. Conditioned media from GT1-7 cells grown in static culture and treated with either vehicle or 50 nM senktide was assayed for GnRH content using a RIA. GT1-7 cells given an acute exposure to senktide (3 hours) showed an 84.5% increase in GnRH secretion (Figure 1B). In contrast, long-term exposure to senktide (24 hours) reduced GnRH secretion by 26% (Figure 1B). These data indicate that GnRH secretion from NK3R-expressing GnRH neurons could be directly regulated by NKB and that the response, whether stimulatory or inhibitory, could depend on the duration of the exposure to NKB.
Senktide treatment represses GnRH transcription
Decreased GnRH secretion in response to chronic exposure to senktide may result from reduced GnRH biosynthesis and a failure to replenish the releasable pool. Therefore, we examined the effect of senktide treatment on GnRH gene transcription. GT1-7 cells were transiently transfected with a luciferase reporter containing five kilobases of upstream regulatory sequence of the rat GnRH gene (−5 Kb GnRH) and treated for 10 hours with either 1 or 10 nM senktide. Treatment with either 1 or 10 nM senktide significantly decreased the activity of a –5 Kb GnRH luciferase reporter (Figure 1C). These data indicate that long-term senktide treatment may decrease GnRH secretion, at least in part, by reducing GnRH transcription. It is noteworthy that sensitivity to the 1 nM dose of senktide was lost in later experiments with a concomitant decrease in NK3R expression by RT-PCR (Supplemental Figure 1). To circumvent the variability that could result from fluctuating levels of NK3R expression, GT1-7 cells without endogenous NK3R were transiently transfected with a rat NK3R expression plasmid in remaining studies.
A dose-response experiment was performed to determine the amount of senktide required to maximally repress GnRH transcription in GT1-7 cells transiently transfected with NK3R (Figure 2A). Using 10 hours of treatment, senktide repressed the activity of the –5 Kb GnRH luciferase reporter with an EC50 value of 0.42 ± 0.34 nM (n = 5). A maximal level of repression, around 40%, was consistently achieved by a dose of 30 nM, and this dose was employed in remaining experiments. Alternatively, the EC50 dose of 0.4 nM was used to achieve a submaximal level of repression in studies that employed inhibitors in order to enable the blockade of repression. Next, a time-course experiment was performed to determine the treatment time required to achieve a maximal repression of GnRH transcription. GT1-7 cells were transiently transfected with NK3R, and the luciferase activity of a –5 Kb GnRH reporter was examined after 6, 8, 10, and 24 hours treatment with 30 nM senktide. Luciferase reporter activity was significantly repressed by 6 hours and became maximally repressed by 8, 10, and 24 hours treatment (Figure 2B). As a result, either 8 or 10 hours of senktide treatment was used in remaining studies.
Figure 2.
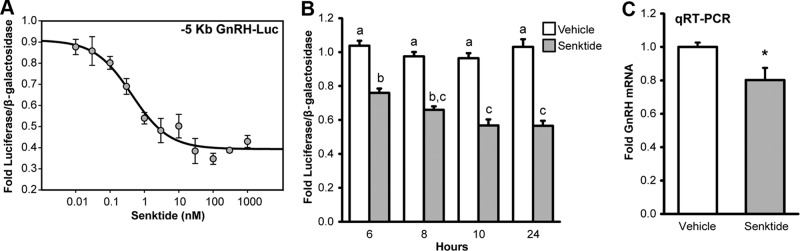
Dose-response curve and time course of senktide-mediated repression of GnRH transcription. A, Dose-response curve. GT1-7 cells without endogenous NK3R expression were transiently transfected with a rat NK3R expression plasmid and a –5 Kb GnRH luciferase reporter. Cells were treated for 10 hours with vehicle or senktide at the indicated doses. Luciferase values were internally normalized using a cotransfected β-galactosidase reporter to control for transfection efficiency, and data were externally normalized to wells transfected with pcDNA3.1 and the –5 Kb GnRH reporter. A representative dose-response curve fitted by nonlinear regression using a four-parameter logistic equation is shown. The mean ± SEM EC50 for 5 independent dose-response curves was 0.42 +/− 0.34 nM. A 30 nM dose was sufficient to achieve maximal repression, a decrease of around 40%. B, Time-course. GT1-7 cells were transiently transfected with rat NK3R and the –5 Kb GnRH Luc reporter. Cells were treated with 30 nM senktide (gray) or vehicle (white) for 6, 8, 10, or 24 hours. Luciferase values were internally normalized using a cotransfected β-galactosidase reporter to control for transfection efficiency, and data were externally normalized to wells transfected with pcDNA3.1 and the –5 Kb GnRH reporter. Data are represented mean ± SEM for n = 3 independent experiments. Statistical analysis was performed using ANOVA with Least squares means Tukey HSD (α = 0.05). Levels not connected by same letter are significantly different. C, qRT-PCR measurement of effect of senktide treatment on endogenous GnRH mRNA GT1-7 cells were transiently transfected with rat NK3R and treated for 24 hours with 30 nM senktide. Total RNA was isolated and qRT-PCR was performed using primers specific for GnRH. Starting quantity of GnRH was normalized using the housekeeping gene β-actin, and data are expressed as mean fold ± SEM versus vehicle treated controls for n = 3 independent experiments. Statistical analysis was performed using a Student's t test. *Significantly different from vehicle control (P < .05).
Next, we sought to determine if senktide treatment is able to repress endogenous GnRH gene transcription. GT1-7 cells were transiently transfected with NK3R and treated for 24 hours with 30 nM senktide. RNA was isolated and used to perform quantitative RT-PCR for GnRH. Twenty-four hours of senktide treatment significantly decreased GnRH mRNA levels by 20% (Figure 2C). Thus, in addition to regulating the activity of a rat GnRH luciferase reporter, senktide treatment is capable of repressing endogenous GnRH gene transcription in GT1-7 cells.
The repression of GnRH transcription by senktide is specific to the activation of NK3R
NK3R is a member of the tachykinin receptor family that also includes NK1R and NK2R. Receptors in this family are known to exhibit cross-reactivity and preference rather than a strict selectivity for ligand binding. In contrast to NKB, senktide was shown to be a selective and potent agonist for NK3R by radioligand binding assays (40, 41). However, because we found that GT1-7 cells also express RNA for NK1R by RT-PCR (Supplemental Figure 2), we thought it necessary to confirm that the response to senktide was specific to the activation of NK3R in our model system. GT1-7 cells were transiently transfected with a rat NK3R expression plasmid, a rat NK1R expression plasmid, or the empty vector control, pcDNA3.1. Again, luciferase activity was repressed by 30 nM senktide treatment in GT1-7 cells transfected with the expression plasmid for rat NK3R (Figure 3A). In contrast, senktide failed to repress the activity of the –5 Kb GnRH luciferase reporter in GT1-7 cells transfected with either NK1R or the pcDNA3.1 vector control (Figure 3A). In addition, the NK3R-specific inhibitor, SB 222200, blocked repression by 0.4 nM senktide in GT1-7 cells transiently transfected with NK3R (Figure 3B). These data confirm that the senktide-mediated repression of GnRH transcription is specific to the activation of NK3R.
Figure 3.
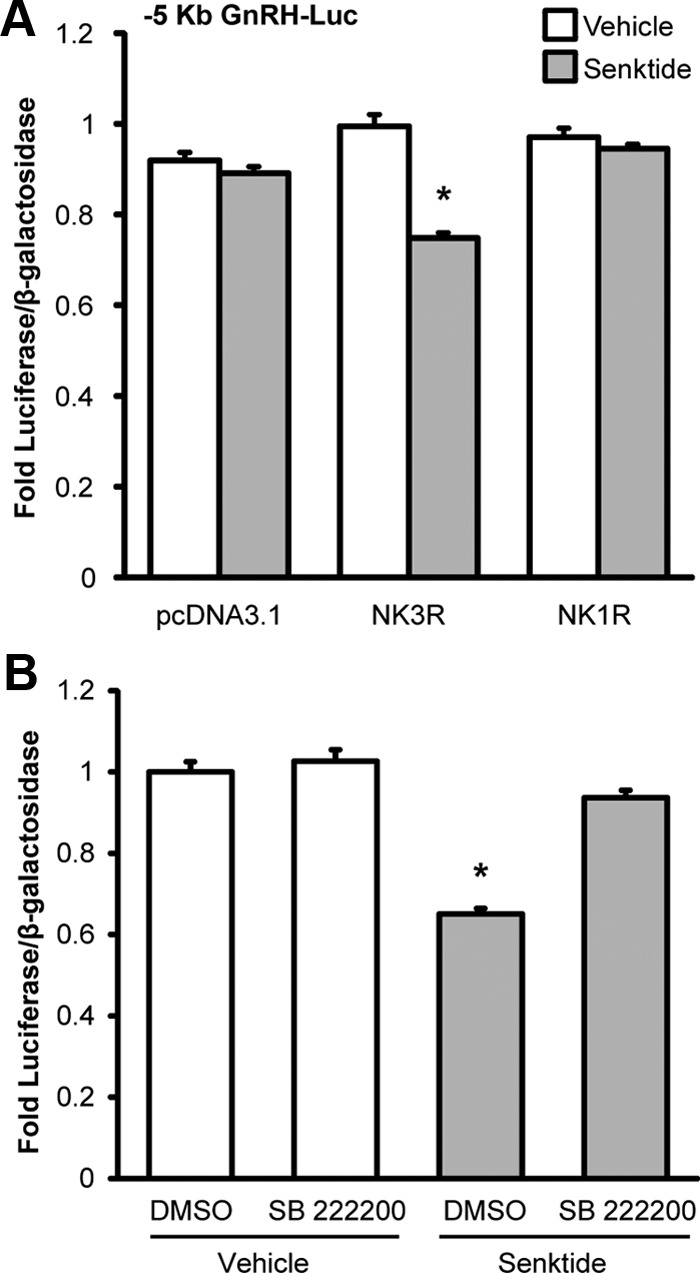
Senktide-mediated repression of GnRH transcription is specific to activation of the neurokinin 3 receptor. A, GT1-7 cells without endogenous NK3R expression were transiently transfected with rat NK3R, rat NK1R or pcDNA3.1 control vector and a –5 Kb GnRH Luc reporter and treated for 10 hours with either 30 nM senktide (gray) or vehicle (white). B, GT1-7 cells were transiently transfected with rat NK3R and a –5 Kb GnRH luciferase reporter. Cells were pretreated for 30 minutes with 2 μM SB 222200, a NK3R-specific inhibitor, and then cotreated with vehicle (0.1% DMSO) or 0.4 nM senktide for 8 hours. Luciferase values were internally normalized using a cotransfected β-galactosidase reporter to control for transfection efficiency and normalized to an external control. Data are represented as mean fold luciferase/β-galactosidase ± SEM for n = 3 independent experiments. Statistical analysis was performed using ANOVA with least squares means Tukey HSD (α = .05). *Significantly different from vehicle control.
Mapping identifies enhancer 1 and the –173 bp promoter as sufficient for the transcriptional repression by senktide
The –5 Kb rat GnRH reporter contains multiple evolutionarily conserved regions, including three enhancers (GnRH-E1, -E2, and -E3) and a promoter (GnRH-P). Luciferase assays with truncations and deletions of the –5 Kb rat regulatory region were used to map the elements that are necessary for senktide-mediated repression. The repression of luciferase activity in response to senktide treatment was maintained after truncation to –2168 bp (Figure 4). In addition to flanking sequences, only two of the known regulatory elements remain within the –2168 bp reporter, enhancer 1 and the promoter. In the absence of other flanking sequence, senktide repressed the activity of a luciferase reporter containing only enhancer 1 and the –173 bp promoter (Figure 4, E1/P). Next, to examine sufficiency for repression, the –173 bp promoter was examined in the absence of the enhancer. Although basal activity was low and essentially equal to that observed for the PGL3 vector control, senktide treatment still produced a significant repression of the –173 bp promoter reporter (Figure 4, P). Next, to examine sufficiency for repression in the absence of the GnRH promoter, enhancer 1 was cloned into a vector containing the thymidine kinase promoter (TKp). Similar to pGL3, the TKp vector control did not respond to senktide (Figure 4, TKp). Also, similar to the –5 Kb GnRH reporter, luciferase activity from the reporter containing only enhancer 1 was significantly repressed by senktide (Figure 4, E1/TKp). Of note, the E1/P and E1/TKp reporters were repressed to a similar degree by senktide as the full-length –5 Kb GnRH reporter, whereas the reporter containing only the –173 bp GnRH promoter was repressed less. These data indicate that enhancer 1 is sufficient for repression by senktide and that there is a potential for additional effects at the promoter.
Figure 4.
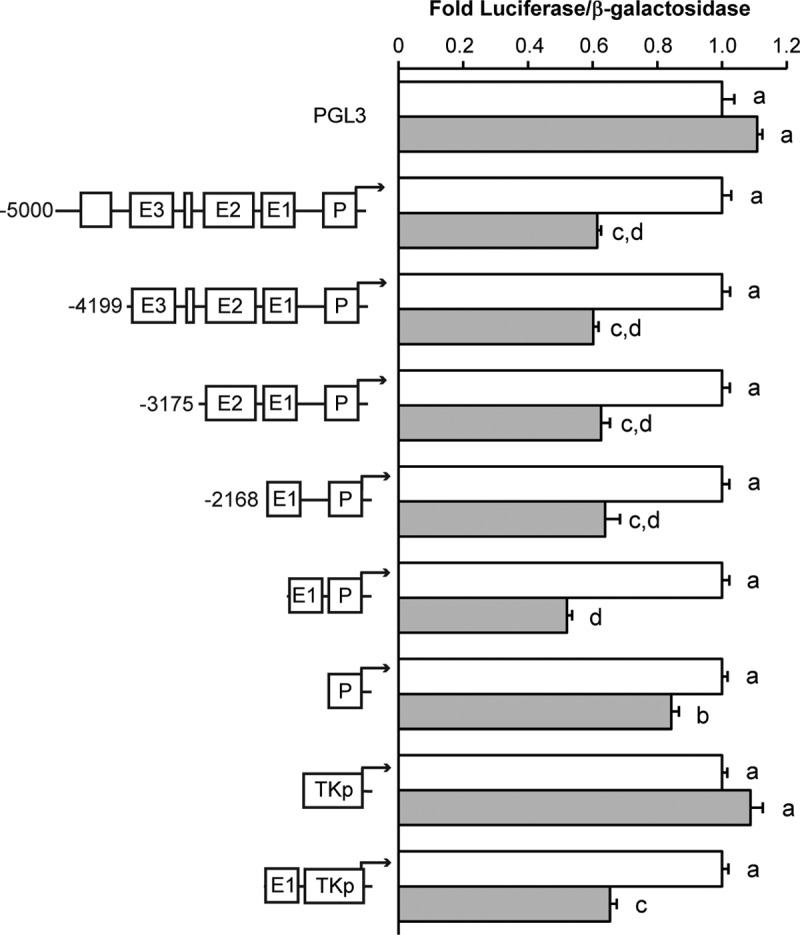
Mapping with truncation and deletion constructs of the rat GnRH gene identifies roles for enhancer 1 and the promoter in repression by senktide treatment. GT1-7 cells were transiently transfected with the NK3R and a luciferase reporters containing truncations of the –5 Kb GnRH regulatory sequence denoted as base-pair positions upstream of the GnRH transcription start site; GnRH-E1/P Luc, enhancer 1 (−1863/−1571) together with the promoter (-173/+112); TKp, a –81 bp thymidine kinase minimal promoter; or GnRH-E1/TKp Luc, enhancer 1 together with a TK promoter. Cells were treated with vehicle or 30 nM senktide for 10 hours. Luciferase values were normalized to a cotransfected β-galactosidase reporter to control for transfection efficiency and normalized to an external control. Data are represented as mean ± SEM for n = 3 independent experiments. Comparisons were made using ANOVA with Least squares means Tukey HSD (α = .05). Levels not connected by same letter are significantly different.
Senktide treatment induces c-Fos and increases AP-1 activity
Senktide treatment was shown to induce the mRNA for c-Fos in neurons expressing NK3R in vivo (9, 42–44). Moreover, previous studies showed treatment of GT1-7 with TPA repressed GnRH gene expression by a mechanism that involved Fos (45, 46). Therefore, we hypothesized that the mechanism whereby senktide treatment caused the repression of GnRH transcription in GT1-7 cells could involve the induction of Fos expression. GT1-7 cells transiently transfected with NK3R and treated for one hour with 30 nM senktide had a 79% increase in c-Fos RNA levels by quantitative RT-PCR (Figure 5A). Next, because Fos family members heterodimerize with Jun family members to form an AP-1 complex and alter AP-1 target gene transcription, we examined the effect of senktide on the expression of other Fos and Jun family members. Senktide treatment reduced FosB expression by 83% (Figure 5C) but had no effect on the expression of the other Fos family members, Fra-1 and Fra-2 (Figure 5E and G). For Jun family members, senktide treatment reduced c-Jun expression by 42% (Figure 5B), but had no effect on JunB or JunD expression (Figure 5D and F).
Figure 5.
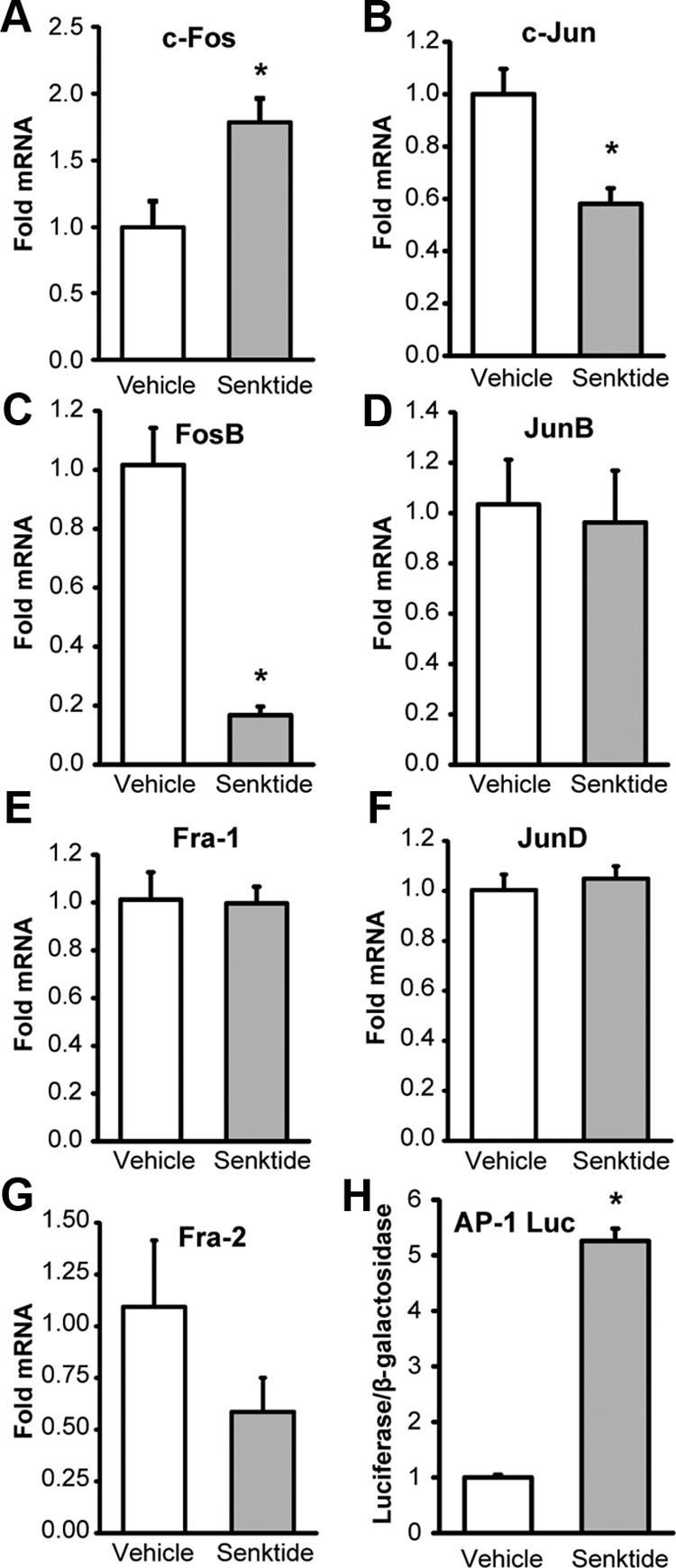
Senktide treatment induces expression of c-Fos and no other AP-1 family member and increases AP-1 transcriptional activity. A-G, RNA was isolated from GT1-7 cells transiently transfected with rat NK3R and treated for 1 hour with vehicle (white) or 30 nM senktide (gray). cDNA was prepared using oligo-dT and quantitative PCR was performed using primers specific for c-Fos (A) and c-Jun (B). Semiquantitative PCR was performed using primers for FosB (C), JunB (D), Fra-1 (E), JunD (F), or Fra-2 (G). Data were normalized to β-actin, a nonresponsive housekeeping gene, and are represented as mean fold mRNA ± SEM versus vehicle-treated controls. H, GT1-7 cells were transiently transfected with rat NK3R and a luciferase reporter containing multiple copies of an AP-1 binding sequence and treated for 6 hours with 30 nM senktide after serum starvation. Luciferase values were normalized to a cotransfected β-galactosidase reporter to control for transfection efficiency, and data were normalized to an external control. Data are represented as mean fold versus vehicle-treated control ± SEM for n = 3 independent experiments. *Significantly different from vehicle control by Student's t test (P < .05).
Although we observed a 79% increase in c-Fos expression in response to senktide, this is a relatively modest increase when compared with the 37-fold increase that we observed in response to TPA treatment (data not shown). However, the induction of c-Fos in response to senktide treatment is limited by NK3R transfection efficiency, and it is possible that the effects were diluted by RNA derived from cells without NK3R transfection. Thus, to determine whether the increase in c-Fos RNA was functionally significant, we employed a luciferase assay to see if senktide treatment could alter AP-1 transcription by using a multimer reporter containing seven copies of an AP-1 binding sequence. After completing a time-course, we found that GT1-7 cells transiently transfected with NK3R and treated for 6 hours with 30 nM senktide showed 5-fold increase AP-1 luciferase reporter activity (Figure 5H). Therefore, the amount of c-Fos induced by senktide treatment in transfected cells could significantly alter the transcription of AP-1 target genes.
Senktide treatment induces c-Fos protein binding to an AP-1 consensus probe
Gel shifts were performed with an AP-1 consensus oligonucleotide probe to determine whether c-Fos induced by senktide treatment in GT1-7 cells transiently transfected with NK3R would be sufficient to detect changes in c-Fos protein binding (Figure 6A). Using a positive control of 3-hour treatment with 100 nM TPA, a dose and time found to maximally induce c-Fos protein in a preliminary experiment (Supplemental Figure 3), we observed increased complex formation (C1) on an AP-1 consensus probe (Figure 6A, lane 1 versus 2). This complex was competed away by preincubation with unlabeled oligonucleotide or “self” (Figure 6A, lane 3) and supershifted by a pan-specific Fos antibody (Figure 6A, lanes 6 and 7) and a c-Fos specific antibody (Figure 6A, lanes 8 and 9) but not IgG control (Figure 6A, lanes 4 and 5). Of note, whereas Fos was present in extracts from vehicle-treated cells, the amount of complex that was supershifted by αFos antibody was increased by TPA treatment (Figure 6A, lane 6 versus 7) with an even greater increase observed with the c-Fos specific antibody (Figure 6A, lane 8 versus 9). These control data indicate that the conditions employed are adequate to observe changes in c-Fos protein binding.
Figure 6.
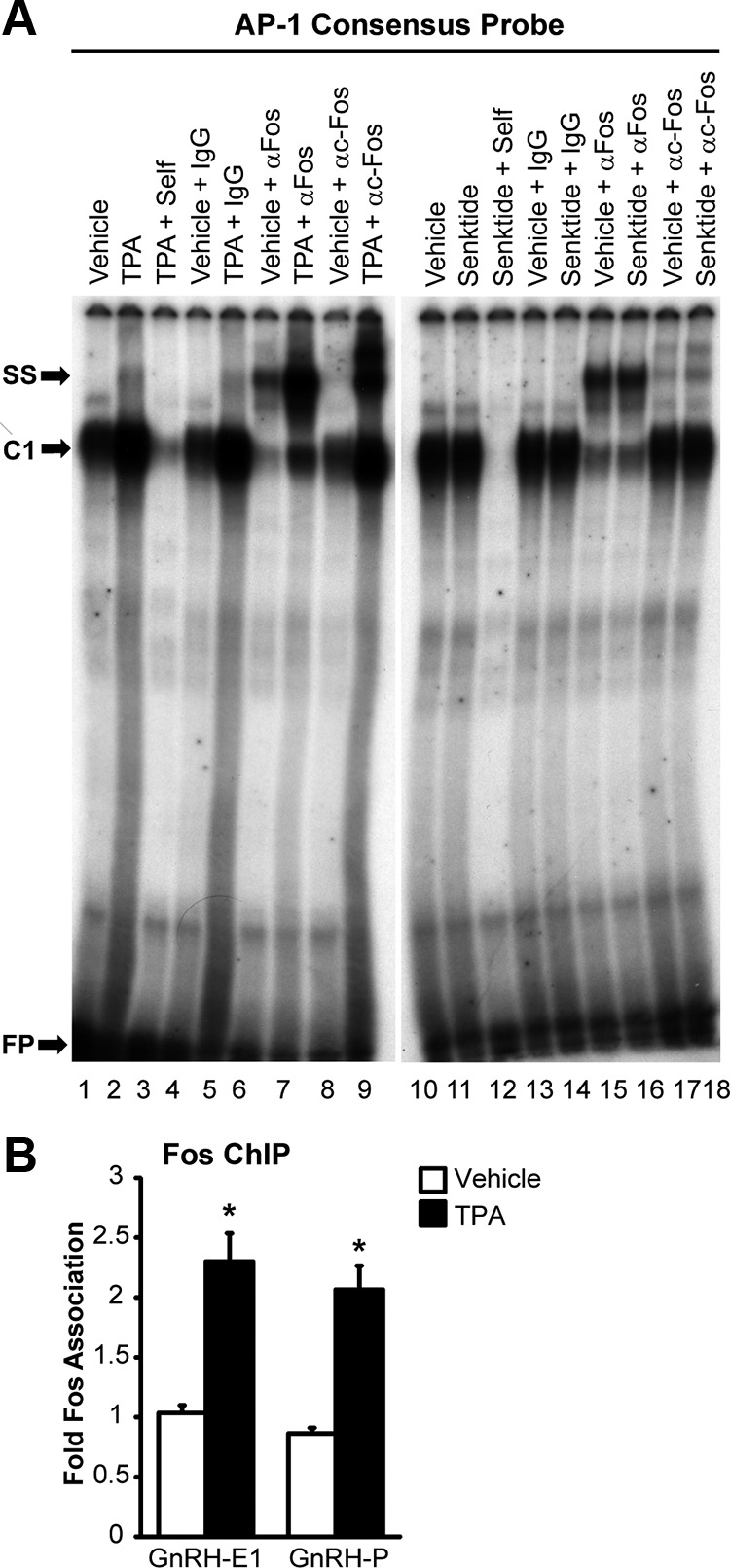
Fos protein binding to an AP-1 consensus probe is induced by senktide, and Fos protein binds to chromatin at enhancer 1 and the promoter of the GnRH gene. A, Nuclear extracts from GT1-7 cells transfected with NK3R and treated for 3 hours with either vehicle or 30 nM Senktide (lanes 1–9) or untransfected GT1-7 cells treated for 3 hours with either vehicle or 100 nM TPA (lanes 10–18) were incubated with radiolabeled probes containing a consensus AP-1 binding sequence. Extracts from senktide and TPA treated cells were further incubated with unlabeled competitors (self), an IgG control, a pan-specific Fos antibody (αFos), or a c-Fos-specific antibody (TPA + αc-Fos). C1 indicates a new protein complex that is formed on oligo probes in response to treatment. SS indicates a supershift of complex C1 by antibody. FP indicates free-probe. Film was exposed for 3.5 days at room temperature. B, GT1-7 cells were treated with either vehicle (0.1% DMSO) or 100 nM TPA for 3 hours. Nuclei were extracted and subjected to ChIP with an antibody against Fos or normal IgG control. The resulting chromatin was analyzed by quantitative PCR with primers to enhancer 1 and the promoter region of GnRH. The data are presented as the mean fold enrichment relative to IgG control ± SEM for n = 4 independent experiments. *Significantly different from vehicle control by Student's t test (P < .05).
Next, the response to senktide treatment was examined. Complex C1 was also observed on the AP-1 consensus probe in extracts from cells treated with vehicle and senktide (Figure 6A, lanes 10 and 11). Again, the C1 band was competed away by preincubation with unlabeled oligo (Figure 6A, lane 12), and supershift was induced by preincubation with pan-specific Fos and c-Fos specific antibodies but not an IgG control (Figure 6A, lanes 13–18). Also, an increase in supershift by the c-Fos specific antibody was observed in response to senktide versus vehicle treatment (Figure 6A, lane 17 versus 18). Importantly, these data suggest that the binding of c-Fos, and no other Fos family member, is induced by senktide treatment. This result is supported by previous data showing the induction of c-Fos and no other Fos mRNA in response to senktide treatment (Figure 5).
The amount of complex supershifted by incubation with a c-Fos specific antibody was quantified by phosphoimager. In comparison to vehicle-treated control, senktide induced a 3-fold increase in supershift with αc-Fos antibody (Figure 6A, lane 17 versus 18). In comparison, TPA treatment induced a 30-fold increase (lane 8 versus 9). The lower amount of c-Fos binding to the consensus AP-1 oligonucleotide probe in response to senktide treatment likely reflects a limitation in c-Fos induction because of NK3R transfection efficiency. Therefore, to avoid false negative results because of an insufficient level of induction, TPA treatment was used to maximally induce c-Fos and examine putative binding sites in enhancer 1 and the promoter of the GnRH gene in several of the following experiments.
c-Fos protein associates with novel AP-1 half-sites identified in enhancer 1 and the promoter
Previously, the repression of GnRH transcription by c-Fos was mapped to a region within –126 to –73 bp in the rat promoter (46). However, despite the presence of an imperfect AP-1 site at –99, gel shifts with an oligonucleotide probe corresponding to –111/−73 bp were unable to confirm the direct binding of Fos protein. As a result, it was suggested that Fos might repress GnRH transcription by tethering to the promoter through protein-protein interactions (46). Chromatin immunoprecipitation (ChIP) assays are able to detect proteins that are either directly or indirectly associated with DNA. Using TPA treatment to maximally induce c-Fos, ChIP assays were performed to examine whether Fos protein associates with chromatin within enhancer 1 and the promoter of the endogenous mouse GnRH gene. The amount of Fos protein associated with enhancer 1 and the promoter was increased by approximately 100% after induction by TPA (Figure 6B). These data indicate that c-Fos could bind, either directly or indirectly, to both the enhancer 1 and the promoter regions to regulate the transcription of GnRH.
Sequence analysis identified several putative AP-1 half-sites containing 5 of 7 bases of the consensus AP-1 in enhancer 1 and the promoter. Gel shifts with oligonucleotide probes shown schematically in Figure 7A (sequences in Supplemental Table 1) were used to examine the direct binding of Fos proteins. Unlike the AP-1 consensus probe, TPA treatment failed to induce any changes in protein complexes bound to E1B or E1D probes (Supplemental Figure 4, lanes 4–6 and 10–12). Also, a very faint TPA-responsive complex (C1) was observed on the E1C and PB probes (Supplemental Figure 4, lanes 7–9 and 13–15). However, Fos binding could not be confirmed because the complex on the E1C probe was not supershifted by αFos antibodies (Supplemental Figure 5, lanes 5 and 6), and the complex on the PB probe was supershifted equally well by IgG and αFos antibodies (Supplemental Figure 5, lanes 10–12). Thus, we are unable to confirm the direct binding of Fos to putative AP-1 half-sites located within –1714/-1692 (E1B), –1691/−1669 (E1C), and –1616/−1594 (E1D) in enhancer 1, and –54/−32 (PB) in the promoter. In contrast, similar to the consensus AP-1 probe, TPA treatment induced the formation of new complex (C1) on the E1A and PA oligonucleotide probes (Supplemental Figure 6, lanes 7–8 and 13–14). Also, C1 bound to E1A and PA probes was competed away by preincubation with excess unlabeled probe (Supplemental Figure 6, lanes 9 and 15). In addition, C1 on E1A and PA probes was supershifted by preincubation with αFos antibody but not IgG control (Supplemental Figure 6, lanes 10–11 and 16–17). Moreover, preincubation with αc-Fos antibody induced a supershift of C1 bound to the E1A element and a very faint supershift of C1 on the PA probe (Supplemental Figure 6, lanes 12 and 18). These data confirm that c-Fos is able to bind directly to novel AP-1 half-sites identified within –1737/−1715 of enhancer 1 and –106/−84 of the promoter. However, the amount of c-Fos induced by senktide in GT1-7 cells transfected with NK3R was not sufficient to detect changes in binding to these lower-affinity AP-1 half-sites in the E1 and the promoter (data not shown).
Figure 7.

Fos protein binds to novel AP-1 half-sites in enhancer 1 and the promoter of the rat GnRH gene. A, A schematic showing the location of putative AP-1 half-sites identified in enhancer 1 and the promoter used for oligonucleotide probes. B, Nuclear extracts from GT1-7 cells treated for 3 hours with either vehicle (0.1% DMSO) or 100 nM TPA were incubated with radiolabeled probes containing sequence corresponding to –1737/−1715 of enhancer 1 (E1A lanes 1–9) or –106/−84 of the promoter (PA lanes 10–18) with and without mutation of the sequence corresponding to the identified AP-1 half-site. Extracts were further incubated with unlabeled competitors (TPA + self), an IgG control (TPA + IgG), a pan-specific Fos antibody (TPA + αFos), or a c-Fos-specific antibody (TPA + αc-Fos). C1 indicates a new protein complex that is formed on oligo probes in response to treatment. SS indicates a supershift of complex C1. FP indicates free-probe. Film was exposed for 4.5 hours at room temperature. C, Site-directed mutagenesis was employed to make mutations in the GnRH-E1/TKp Luc reporter that correspond to those shown to disrupt TPA-mediated complex formation on E1A (-1737/−1715) by gel shift. GT1-7 cells were transiently transfected with rat NK3R and a GnRH-E1/TKp Luc reporter with either a wild-type or a mutant AP-1 element and treated for 8 hours with either vehicle or 100 nM TPA. D, Site-directed mutagenesis was employed to make mutations in the GnRH-E1/GnRH-P Luc reporter corresponding to those shown to disrupt TPA-complex formation on PA (-106/−84) by gel shift (E1 is wild-type). GT1-7 cells were transiently transfected with rat NK3R and a GnRH-E1/P Luc reporter with either a wild-type or a mutant AP-1 element and treated for 8 hours with either vehicle or 100 nM TPA. For C and D, luciferase values were internally normalized using a cotransfected β-galactosidase reporter to control for transfection efficiency, and data were normalized to an external control. Data are presented as mean fold ± SEM for n = 3 independent experiments. Comparisons were made using ANOVA with Least squares means Tukey HSD (α = .05). *Significantly different from vehicle or wild-type control (indicated by bracket).
The AP-1 half-sites within –106/−84 of the promoter and –1737/−1715 of enhancer 1 are involved in the regulation of basal GnRH transcription
Next, gel shifts with 5- to 8-base pair substitutions in AP-1 half-sites of the E1A and PA oligonucleotide probes (Supplemental Table 1) were employed to determine whether the AP-1 half-sites are required for TPA-responsive complex formation and Fos protein binding. Wild-type E1A and PA control probes gave similar results as previously described. In brief, TPA treatment induced formation of a new complex (C1) that was supershifted in response to incubation with αFos antibody (Figure 7B, lanes 1–3 and 10–12). In contrast, the E1A and PA probes with mutated AP-1 half-sites did not show formation of complex C1 in response to TPA or a supershift by αFos antibody (Figure 7B, lanes 4–9 and 13–18). Also, the mutation of E1A induced the formation of several new complexes (Figure 7B, lanes 4–9), and the mutation of PA caused the loss of complexes (C2 and C3) in comparison to wild-type probes (Figure 7B, lanes 13–18). However, these complexes do not migrate the same distance as C1 in the gel or undergo supershifting in response to preincubation with the αFos antibody, so they are unlikely to include Fos. These data indicate that the AP-1 half-sites within the E1A and PA probes are required for TPA-responsive complex formation and the direct binding of Fos protein.
Next, the functional roles of AP-1 half-sites within –1737/−1715 of enhancer 1 and –106/−84 of the promoter in the regulation of GnRH transcription were examined by luciferase assay. Site-directed mutagenesis was used to introduce the base-pair substitutions that disrupted TPA-responsive complex formation and Fos binding by EMSA into luciferase reporters. First, to test the site within –1737/−1715 of enhancer 1, GT1-7 cells were transiently transfected with wild-type and mutant GnRH-E1/TKp luciferase reporters and treated with either 100 nM TPA or DMSO vehicle for 8 hours. Compared with vehicle-treated wild-type E1, mutation of the AP-1 half-site in enhancer 1 caused a significant increase in basal luciferase activity (Figure 7C), indicating that the AP-1 half-site in enhancer 1 could repress the basal transcription of GnRH. However, similar to wild-type, TPA repressed the activity of the GnRH-E1/TKp reporter with mutant AP-1 (Figure 7C), and similar results were obtained using senktide treatment (Figure 8A). These data indicate there may be additional bases other than those mutated that are involved in the repression of enhancer 1 activity by TPA and senktide.
Figure 8.
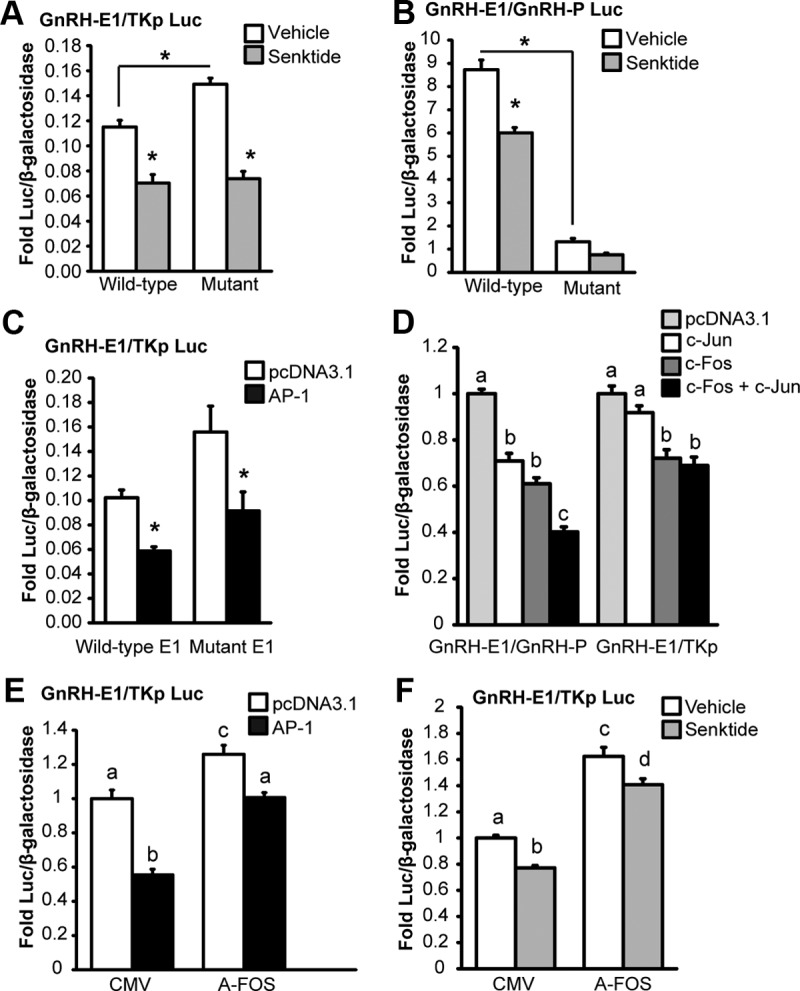
The roles of AP-1 half-sites and AP-1 complex formation in the repression of enhancer 1 and the promoter activity by senktide. Mutation of AP-1 half-sites in enhancer 1 and the promoter of GnRH changes basal transcription but does not block repression by senktide. A, Site-directed mutagenesis was employed to make mutations in the GnRH-E1/TKp Luc reporter that correspond to those shown to disrupt TPA-mediated complex formation on E1A (-1737/−1715) by gel shift. GT1-7 cells were transiently transfected with rat NK3R and a GnRH-E1/TKp Luc reporter with either a wild-type or a mutant AP-1 element and treated for 8 hours with either vehicle or 30 nM senktide. B, Site-directed mutagenesis was employed to make mutations in the GnRH-E1/GnRH-P Luc reporter corresponding to those shown to disrupt TPA-complex formation on PA (-106/−84) by gel shift (E1 is wild-type). GT1-7 cells were transiently transfected with rat NK3R and a GnRH-E1/P Luc reporter with either a wild-type or a mutant AP-1 element and treated for 8 hours with either vehicle or 30 nM senktide. C, GT1-7 cells were transiently transfected with rat NK3R and either a wild-type or a mutant AP-1 GnRH-E1/TKp Luc reporter with AP-1 transcription factors (c-Fos and c-Jun) or empty vector (pcDNA3.1). D, GT1-7 cells were transiently transfected with rat NK3R and either a GnRH-E1/GnRH-P or GnRH-E1/TKp luciferase reporter with empty vector control (pcDNA3.1), c-Fos, or c-Jun individually balanced with pcDNA3.1, or c-Fos and c-Jun in combination. E, GT1-7 cells were transiently transfected with rat NK3R and either A-FOS or CMV (empty vector control) and AP-1 (c-Jun and c-Fos) or pcDNA3.1 (empty vector control). F, C, GT1-7 cells were transiently transfected with rat NK3R and either A-FOS or CMV (empty vector control) and treated for 8 hours with either vehicle or 30 nM senktide. For A through F, luciferase values were internally normalized using a cotransfected β-galactosidase reporter to control for transfection efficiency, and data were normalized to an external control. Data are presented as mean fold ± SEM for n = 3 independent experiments. Comparisons were made using ANOVA with Least squares means Tukey HSD (α = .05). *Significantly different from vehicle or wild-type control (indicated by bracket) or levels not connected by same letter are significantly different.
Likewise, the functional role of the AP-1 half-site within –106/−84 of the promoter was examined. GT1-7 cells were transiently transfected with GnRH-E1/GnRH-P luciferase reporters with either wild-type or mutant AP-1 half-sites within –106/−84 of the promoter (E1 is wild-type) and treated with either 100 nM TPA or DMSO vehicle for 8 hours. The mutation of the AP-1 half-site in the promoter significantly reduced the basal activity of the GnRH-E1/GnRH-P luciferase reporter (Figure 7D). However, similar to wild-type, TPA repressed the activity of the reporter with mutant P (Figure 7D), perhaps because of the presence of wild-type E1. In contrast, the repression of GnRH-E1/GnRH-P luciferase activity by senktide was disrupted by the mutation of P (Figure 8B); however, it is possible that the inability to observe repression is because of the low basal activity of this luciferase reporter. These data indicate that the AP-1 half-site within –106/−84 bp of the promoter may be involved in maintaining basal GnRH transcription as well as the repression of GnRH transcription by senktide.
Next, c-Fos and c-Jun (AP-1) overexpression was used to determine if the repression by TPA and senktide that is present after mutating the AP-1 half-site within –1737/-1715 could be AP-1 dependent or AP-1 independent. Similar to wild-type, the mutant GnRH-E1/TKp reporter was repressed by AP-1 overexpression (Figure 8C). Therefore, at least one other site within enhancer 1 besides that mutated within –1737/-1715 could be involved in repression by c-Fos. Recall that there was a faint TPA-responsive complex on the E1C probe corresponding to –1691/-1669 of enhancer 1 in gel shift, but we were unable to confirm Fos binding. An AP-1 complex can either be formed by heterodimers between Fos (c-Fos, Fra1, FosB) and Jun proteins (c-Jun, JunB, JunD) or by homodimers of Jun proteins (47). Thus, perhaps Fos protein binding was not identified within –1691/-1669 because this site binds to AP-1 composed of a Jun protein homodimer, and this could be sufficient for repression.
Luciferase assays were used to examine the role of c-Jun in the ability of AP-1 to induce repression of enhancer 1 and the promoter. Overexpression of c-Fos and c-Jun in combination (AP-1) repressed the activity of both the GnRH-E1/GnRH-P and GnRH-E1/TKp luciferase reporters in comparison to pcDNA3.1 vector transfected controls (Figure 8D). For the reporter also containing the promoter (GnRH-E1/GnRH-P), overexpression of either c-Jun or c-Fos alone induced an equivalent level of repression, and this repression was increased when both c-Jun and c-Fos were overexpressed in combination (Figure 8D). In contrast, for the reporter containing only enhancer 1 (GnRH-E1/TKp), overexpression of c-Jun alone did not induce repression. Moreover, the overexpression of c-Fos and c-Jun in combination gave no further repression than c-Fos alone (Figure 8D). These data suggest that the promoter is sensitive to repression by c-Jun, but enhancer 1 is not. Thus, although the promoter may bind to an AP-1 complex composed of a c-Jun homodimer, or c-Jun in complex with a Fos member that is already present in the cells without overexpression (i.e., not c-Fos), this complex is not able to repress enhancer 1. Also, because additional repression was observed when c-Jun and c-Fos were overexpressed in combination, these data indicate that the promoter may be more repressed by an AP-1 heterodimer composed of c-Jun and c-Fos. In contrast, enhancer 1 was not repressed by the overexpression of c-Jun in the absence of c-Fos, and when overexpressed in combination with c-Fos, c-Jun gave no more repression that c-Fos alone. These data may indicate that c-Fos binds to enhancer 1 with Jun family members that are present in GT1-7 cells without overexpression, as detected by RT-PCR (Figure 5) and RNAseq (data not shown), including c-Jun, JunB, and JunD. Alternatively, c-Fos may repress enhancer 1 by a mechanism that does not involve AP-1 complex formation, such as by tethering to DNA through protein–protein interactions.
A-FOS is composed of the c-Fos dimerization domain with an artificial acidic extension that interacts with the basic region of dimerization partners and acts as a dominant negative toward AP-1 complex formation (32). Using the overexpression of A-FOS, we examined the role of AP-1 complex formation in repression of enhancer 1 by AP-1. GnRH-E1/TKp luciferase reporter activity was repressed by AP-1 overexpression in cells coexpressing either CMV or A-FOS (Figure 8E). However, the difference between pcDNA3.1 and AP-1 transfected cells was reduced by overexpression of A-FOS in comparison to CMV, indicating that AP-1 complex formation is involved in the repression of enhancer 1 activity by AP-1 overexpression. Of note, the overexpression of A-FOS increased basal activity of the GnRH-E1/TKp luciferase reporter in both pcDNA3.1 and AP-1 transfected cells (Figure 8E). These data indicate that endogenous Fos and/or Jun proteins are present without overexpression and repress the basal activity of enhancer 1 by a mechanism that involves AP-1 complex formation. Importantly, this increase in basal activity in response to A-FOS expression is reminiscent of the increase observed upon mutation of the AP-1 half-site within –1737/−1715 (Figure 8A). Together, these data suggest that an AP-1 complex binds to the AP-1 half-site within –1737/−1715 and acts to repress the basal activity of enhancer 1. However, the amount of repression, i.e., the difference between pcDNA3.1 and AP-1 transfected cells was decreased but not completely blocked by A-FOS (Figure 8E), suggesting that either A-FOS expression was unable to completely block AP-1 complex formation or that the repression induced by c-Fos overexpression may be in part independent of AP-1 complex formation.
Next, A-FOS was employed to determine whether the repression of enhancer 1 by senktide treatment involves AP-1 complex formation. Again, basal activity of the GnRH-E1/TKp reporter was increased in response to A-FOS, indicating a role for AP-1 complex formation in the repression of the basal activity of enhancer 1 (Figure 8F) and again, similar to AP-1 overexpression, senktide treatment significantly repressed GnRH-E1/TKp reporter activity in cells transfected with either CMV or A-FOS. However, in contrast to AP-1 overexpression, the amount of repression, i.e., the difference between vehicle and senktide, was not significantly different in cells transfected with A-FOS versus CMV controls. Thus, coexpression of A-FOS failed to block repression of enhancer 1 by senktide treatment. These data suggest that the repression of enhancer 1 activity by senktide could be, at least in part, independent of AP-1 complex formation or that A-FOS is insufficiently active to completely block the actions of AP-1.
Senktide treatment induces chromatin remodeling in the promoter of the GnRH gene
The tachykinin receptor family has been shown to signal via the activation of PKC (48). Previously, the treatment of GT1-7 cells with TPA, an activator of PKC, was shown to cause an acute increase in GnRH secretion and long-term repression of GnRH transcription by effects that involved enhancer 1 and the promoter (45, 49–51). Because of the similarities in the responses to TPA and senktide treatment, we hypothesized that NK3R signals through the activation of PKC to repress GnRH transcription. GT1-7 cells were transiently transfected with a GnRH-E1/TKp luciferase reporter and given a 30 pretreatment with either Go6983, a broad-spectrum PKC inhibitor, or DMSO, before 8 hours cotreatment with vehicle, 0.4 nM senktide, or 100 nM TPA. Both senktide and TPA treatment significantly repressed GnRH-E1/TKp luciferase reporter activity in DMSO-treated controls (Figure 9A). In contrast, in cells treated with Go6983, neither senktide nor TPA treatment repressed activity of the GnRH-E1/TKp reporter (Figure 9A). Of note, the activity of the GnRH-E1/TKp reporter in vehicle-treated controls was significantly repressed by Go6983. However, no additional repression was observed by TPA or senktide treatment. Thus, these data suggest NK3R could signal through PKC to induce the repression of enhancer 1.
Figure 9.
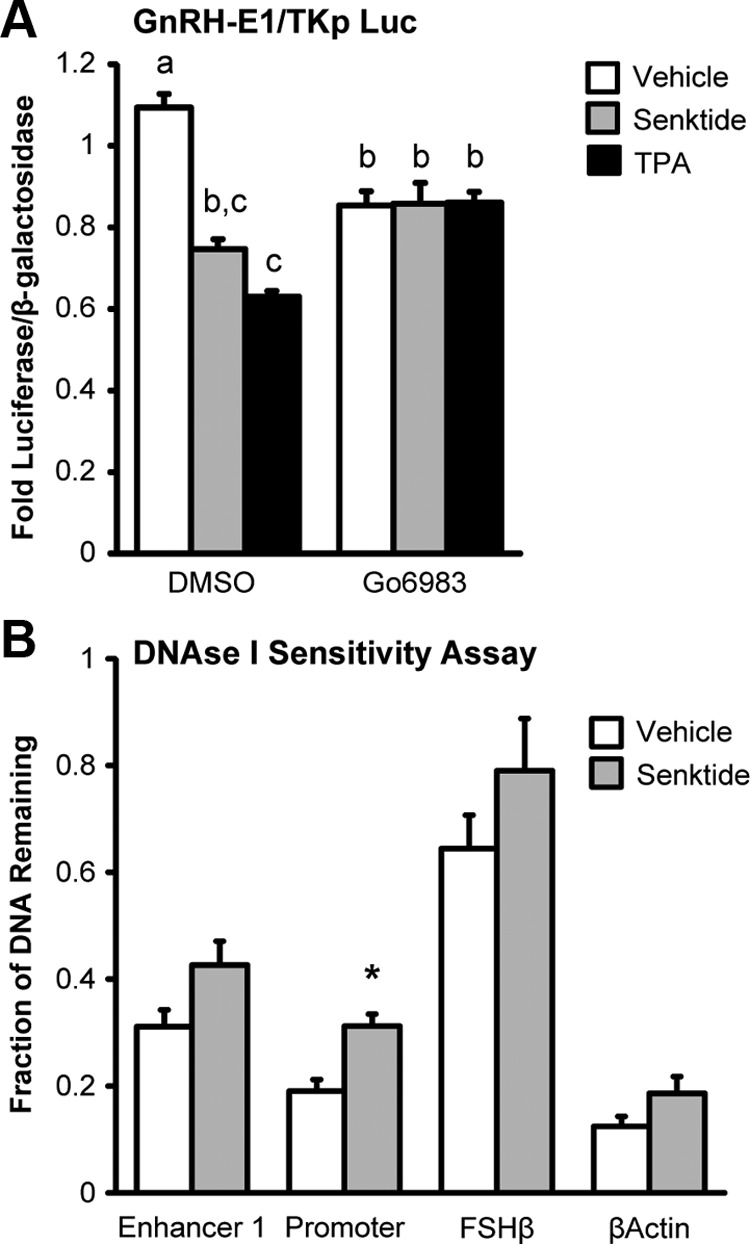
NK3R may induce the long-term repression of GnRH transcription by activation of PKC and a closing of chromatin at the promoter of the GnRH gene. A, GT1-7 cells were transfected with NK3R and pretreated with DMSO or 10 μM of the broad spectrum PKC inhibitor Go6983 for 30 minutes before 8 hours cotreatment with vehicle, 30 nM senktide or 100 nM TPA. Luciferase values were internally normalized using a cotransfected β-galactosidase reporter to control for transfection efficiency, and data were normalized to an external control. Data are presented as mean fold ± SEM for n = 3 independent experiments. Comparisons were made using ANOVA with Least squares means Tukey HSD (α = .05). Levels not connected by same letter are significantly different. B, DNAse I Sensitivity Assay. GT1-7 cells were transfected with NK3R and treated with either vehicle or 30 nM senktide for 10 hours. DNA was isolated from nuclei and treated with 5 U of DNase I, then analyzed by quantitative PCR using primers specific to enhancer 1 or the promoter region of the GnRH gene (Supplemental Table 1). Amplicon quantities were normalized to undigested genomic DNA and are presented as the mean fraction of DNA remaining ± SD for n = 4 independent experiments. *Significantly different from vehicle-treated control by Student's t test (P < .05).
c-Fos protein induced by TPA returns to basal by around 8 hours (Supplemental Figure 3), whereas TPA induces a repression of GnRH mRNA that persists as long as 48 hours (45). Therefore, it seems unlikely that c-Fos induction is solely responsible for the repression of GnRH transcription. Chromatin remodeling is an epigenetic change that is capable of producing lasting effects on gene transcription. Often, the chromatin of active promoters is present in a more open conformation to facilitate the binding of proteins such as those in the initiation complex. In contrast, the chromatin of inactive promoters is in a more closed conformation. As a result, the chromatin of active genes displays a greater sensitivity to digestion with DNase I when compared with the chromatin of inactive genes (52). Previously, we found that TPA treatment induced the closing of chromatin at the GnRH promoter by using a DNAse I sensitivity assay (35). Here, we employ a similar approach to determine whether senktide treatment induces chromatin remodeling at enhancer 1 or the promoter of the GnRH gene. Nuclei were prepared from GT1-7 cells transiently transfected with NK3R and treated for 10 hours with 30 nM senktide then treated with 0 or 5 U DNase I. The remaining fraction of DNA after digestion with DNase I was determined using quantitative PCR (qPCR) using primers specific for enhancer 1 or the promoter of the GnRH gene, in comparison to βActin (open chromatin of an active gene) and FSHβ (closed chromatin of an inactive gene). In this assay, a higher fraction of DNA remaining indicates a greater protection from DNase I and a more closed chromatin structure, whereas a lower fraction of DNA remaining indicates chromatin that is less protected and in a more open conformation. For example, the remaining fraction of DNA for FSHβ, a gene that is not expressed in GT1-7 cells (closed conformation), was only about 30% reduced by digestion with DNase I (Figure 9B). In contrast, βActin, a highly expressed gene in GT1-7 cells (open conformation), was about 80% reduced by digestion with DNase I (Figure 9B). Consistent with GnRH being highly expressed in GT1-7 cells, the fraction remaining of enhancer 1 and the promoter region DNA was reduced between 70 and 80% by digestion with DNase I (Figure 9B). Senktide did not significantly affect the amount of DNA remaining for the FSHβ and βActin control genes or enhancer 1 (Figure 9B). However, senktide treatment caused a significant increase in the fraction of DNA remaining for the promoter region of the GnRH gene (Figure 9B). These data indicate that NKB could cause the closure of chromatin in the promoter of the GnRH gene to extend the repression of GnRH gene transcription beyond that mediated by the acute and transient induction of c-Fos.
Discussion
In this investigation, the immortalized GT1-7 cell line, an in vitro model of the mature, differentiated GnRH neuron, was employed to test the hypothesis that NKB could directly regulate the function of NK3R-expressing GnRH neurons. We report that GT1-7 cells express NK3R and respond to senktide treatment with a time-dependent change in GnRH secretion. In addition, senktide treatment represses GnRH transcription by a mechanism that involves enhancer 1 and the promoter, the induction of c-Fos and chromatin remodeling. In addition, novel AP-1 half-sites are identified in enhancer 1 and the promoter that directly binds to c-Fos and play a role in the regulation of GnRH transcription.
GnRH transcription is regulated during development, aging, across the estrous cycle, and in response to hormones and neurotransmitters (53–59). Here, we show that GnRH transcription is repressed in response to senktide through effects that are mapped to enhancer 1 and the promoter. These two regions are evolutionarily conserved and have been shown to play key roles in the regulation of GnRH gene transcription (60, 61). Together, enhancer 1 and the promoter are sufficient to confer cell-specific GnRH expression to GnRH neurons in vitro and in vivo using transgenic mice (62). In mouse, the deletion of a region containing the conserved enhancer, –2806 to –2078, resulted in reduced GnRH mRNA expression in the hypothalamus and a delay in pubertal onset and disrupted estrous cycles (63). Thus, enhancer 1 and the promoter are required for the proper expression of the GnRH gene, and the regulation of GnRH transcription by these elements could be involved in puberty and reproduction.
Previous studies have shown that the repression of GnRH transcription by TPA involves multiple binding elements that are common to the promoter and enhancer 1 of rat GnRH gene, including sites for pre-B cell leukemia transcription factor (Pbx)/Prep1, Msx1, Dlx2, and octamer-binding transcription factor-1 (Oct-1) (34, 51, 64–68). Previous studies mapped repression of GnRH transcription by TPA and Fos overexpression to –126 to –73 bp (46, 50) but were unable to show the direct binding of Fos protein to an imperfect AP-1 at –99 bp by EMSA with a probe corresponding to –111/−73 bp of the rat GnRH promoter (46, 50). Here, we are able to confirm the direct binding of c-Fos by EMSA using a probe corresponding to –106/−84 bp of the rat GnRH promoter. Perhaps the sensitivity of our assay was increased because of differences in the length of TPA treatment, probe sequences, and/or antibodies used for supershifts. Regardless, this result confirms that c-Fos protein can directly bind to the imperfect AP-1 site at –99 bp in the promoter. Because we also show c-Fos binding to an AP-1 half-site within –1737/−1715 of enhancer 1, we propose that AP-1 half-sites may be yet another common element among enhancer 1 and the promoter that is regulated in response to TPA.
Despite the identification of AP-1 half-sites in both enhancer 1 and the promoter, our data suggest differences for the mechanisms of repression at these two regions. First, the complex bound to the AP-1 half-site within the –1737/−1715 probe for enhancer 1 seems more intense than that present on the –106/−84 probe for the promoter, suggesting a higher affinity for this binding site. Perhaps this indicates a weaker affinity site in the promoter that restricts AP-1 binding to conditions when c-Fos becomes strongly induced. Second, enhancer 1 and the promoter were differentially repressed by c-Jun overexpression. The promoter was repressed by overexpression of c-Fos or c-Jun alone and further repressed by c-Jun and c-Fos overexpressed in combination, whereas enhancer 1 was only repressed by c-Fos alone and not further repressed by c-Fos and c-Jun overexpressed in combination. These data suggest a different requirement for c-Jun in AP-1 complex formation and repression at the enhancer and the promoter. Finally, only the promoter showed a closing of chromatin in response to senktide, a similar result to that which was observed in response to TPA treatment (35).
Our data show that the AP-1 half-site within enhancer 1 is involved in the regulation of GnRH transcription, and at least one additional site is involved. However, although the mutation of the AP-1 half-site at –1737/−1715 increased basal transcription, it did not block repression of enhancer 1 in response to TPA, senktide, or AP-1 overexpression. Initially, four putative AP-1 sites were identified in enhancer 1. In addition to the E1A probe (-1737/−1715), a faint TPA responsive complex was observed by EMSA using the E1C probe (-1691/−1669). However, we were not able to confirm the binding of c-Fos protein to this region by supershift. Perhaps this region is involved in repression and our assay was not sensitive enough to observe supershifting of this faint band. Alternatively, repression could be mediated by AP-1 elsewhere, or because c-Jun was not required for repression, c-Fos may act by indirectly tethering to DNA through protein–protein interactions.
Here, we show that senktide can repress GnRH transcription by effects that are at least in part mediated by the promoter. Previously, repression by PKC was mapped to –126/−73 within the promoter and shown to involve Fos (50). A cluster of Oct-1, Pbx/Prep, and NK2 homeobox 1 (Nkx2.1) binding sites have been identified within the –106/−91 bp region of the promoter and shown to be involved in repression of GnRH transcription by androgen (68, 69). It is noteworthy that the AP-1 half-site identified at –99 bp directly overlaps with this cluster of binding sites and, in addition to C1, the mutation of bases within the AP-1 half-site disrupted the binding of two additional complexes, C2 and C3, by EMSA. Perhaps the loss of basal transcription and senktide-mediated repression that occurred in response to the mutation of bases within AP-1 at –99 was because of a disruption of the binding of Oct-1, Pbx/Prep, and Nkx2.1. If so, perhaps Fos binding to this region acts to displace these activators and coactivators to repress GnRH transcription (70).
Our results suggest that NK3R may signal by the activation of PKC in GnRH neurons. Previous studies that have employed TPA to activate PKC have strongly implicated this pathway in the regulation of GnRH secretion and transcription. TPA treatment was shown to induce an acute increase in GnRH secretion and repress GnRH mRNA by 8 hours. Also like senktide, TPA was shown to induce the expression of c-Fos and the activity of an AP-1 multimer reporter (45) and repress GnRH transcription by effects that involve enhancer 1 and the promoter (50, 51). Finally, like senktide, TPA was shown to induce a long-lasting repression of GnRH mRNA (45) and the closing of chromatin at the promoter (35). These many similarities among the effects observed with TPA and senktide treatment, in addition to our results obtained with Go6983, suggest an activation of PKC in response to NK3R signaling in GnRH neurons. Importantly, by establishing a link between NKB and PKC, our data suggest that NKB may represent a physiological ligand mimicked by TPA in previous studies (35, 45, 50, 51, 71). It is noteworthy that kisspeptin was also recently shown to activate PKC signaling in GT1-7 neurons (72). Thus, physiological ligands that target the PKC signaling pathway in GnRH neurons may be important regulators of puberty and reproduction.
Studies investigating the mechanism whereby NKB regulates GnRH secretion have largely been focused on kisspeptin neurons. These studies have shown that NK3R is expressed in most kisspeptin neurons that coexpress NKB and dynorphin in the ARC, an area known to be involved in the regulation of pulsatile GnRH release (8, 23, 24). The central administration of senktide was shown to induce c-Fos in this population of kisspeptin neurons (73), suggesting increased kisspeptin release could account for the increase in LH secretion by senktide. Moreover, the increased LH response to senktide was shown to require GPR54 signaling in mice and monkeys (74, 75). Finally, kisspeptin neurons were shown by electrophysiology to exhibit an increase in firing in response to senktide (73). However, a recent report showed that the intra-ARC administration of senktide increased the interpulse interval of LH, presumably by reducing pulsatile GnRH secretion. Moreover, a repression of GnRH1 and Kiss1R mRNA expression was observed in micropunches taken from mPOA/AVPV, the location of GnRH and kisspeptin perikarya, when examined 6 hours after senktide treatment (76). Because senktide treatment did not alter Kiss1 expression and repressed Kiss1R mRNA levels, the authors suggested that projections from KNDy neurons in the ARC may regulate GnRH neurons rather than kisspeptin neurons in the mPOA. Our results confirm that NKB could directly repress GnRH secretion and transcription in GnRH neurons in the absence of effects mediated by kisspeptin. However, because KNDy neurons are likely to secrete both kisspeptin and NKB upon depolarization, and kisspeptin has been shown to increase GnRH secretion and transcription in GT1-7 cells (59), it would be interesting to assess the response to senktide and kisspeptin when given in combination. In this regard, the coadministration of senktide did not decrease the kisspeptin-induced increase in LH in the monkey and the rat, suggesting a hierarchy or distinct mechanisms for regulation of GnRH secretion by NKB and kisspeptin (75, 77).
Many in vivo studies have reported an increase in serum LH in response to senktide (9–11, 73–75), and a few have shown a decrease in serum LH in response to senktide (7, 8, 76). We originally hypothesized that opposing direct and indirect effects on GnRH and kisspeptin neurons could explain the inconsistencies in the LH response to senktide obtained in vivo. However, our results indicate that NK3R-expressing GnRH neurons would be acutely activated by NKB, a similar result as would be expected for KNDy neurons in the ARC (73). Thus, it is unlikely that opposing direct and indirect effects account for the inconsistencies reported among in vivo studies. Our data indicate that the duration of exposure to NKB could be a determining factor in the GnRH secretion response. Thus, perhaps differences in the endogenous NKB tone before the administration of senktide treatment could help to explain the differences in the LH response in some in vivo studies. In support of this, ovariectomized (OVX) female rats were shown to have elevated NKB and NK3R mRNA levels in the ARC and respond to senktide with a decrease in serum LH, whereas OVX rats treated with estradiol had lower NKB and NK3R mRNA levels in the ARC and responded to senktide by an increase in serum LH (9). Thus, further studies to elucidate the physiological conditions that regulate the endogenous NKB/NK3R system may help to clarify the role of NKB in regulating GnRH secretion, puberty, and reproduction.
On the basis of the results presented in this report, we propose that NKB may acutely activate and directly induce GnRH secretion from NK3R-expressing GnRH neurons. We find that this response may depend upon the duration or history of exposure to NKB, as chronic treatment with senktide reduced both GnRH secretion and GnRH transcription. We further propose that NKB could induce the long-term repression of GnRH transcription by a mechanism that involves an acute induction of c-Fos, the enhanced binding of c-Fos protein to novel AP-1 half-sites identified within enhancer 1 and the promoter, and the closing of chromatin at the promoter.
Supplementary Material
Acknowledgments
We thank Martha Bosch, Oline Ronnekleiv, and Martin Kelly (University of Oregon Health Sciences Center, Portland, Oregon) for the EL-14 antibody against GnRH. The rat NK3R expression plasmid (30) was kindly provided by Dr. Nigel W. Bunnett (University of California, San Francisco). The murine c-Fos and c-Jun expression vectors were kindly provided by Dr. Djurdjica Coss (University of California, San Diego). The A-FOS expression construct was kindly provided by Dr. Charles Vinson (National Cancer Institute, Center for Cancer Research, Bethesda, Maryland). We also thank Djurdjica Coss, Melissa Brayman, Sunamita Leming, and Alexander S. Kauffman for reading the manuscript.
This work was supported by National Institutes of Health (NIH) Grants R01 DK044838, R01 HD072754, and R01 HD020377 (to P.L.M.) and by National Institute of Child Health and Human Development/NIH through a cooperative agreement (U54 HD012303) as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research (to P.L.M.). P.L.M. was partially supported by P30 DK063491, P30 CA023100, and P42 ES101337. C.G.K. was partially supported by T32 HD007203, the Hartwell Foundation, and T32 DK007044. A.K.I. was partially supported by NIH Grants F32 HD058427 and T32 DK007494.
Disclosure Summary: C.G.-K., P.P.S., A.K.I., A.M.H.G., J.D.M., and P.L.M. have nothing to disclose.
Footnotes
- ARC
- arcuate nucleus
- AP-1
- activator protein-1
- ChIP
- chromatin immunoprecipitation
- DMSO
- dimethylsulfoxide
- IHH
- idiopathic hypogonadotropic hypogonadism
- NKB
- neurokinin B
- NK3R
- neurokinin 3 receptor
- qPCR
- quantitative PCR.
References
- 1. Watanabe G, Terasawa E. In vivo release of luteinizing hormone releasing hormone increases with puberty in the female rhesus monkey. Endocrinology. 1989;125:92–99 [DOI] [PubMed] [Google Scholar]
- 2. Wildt L, Marshall G, Knobil E. Experimental induction of puberty in the infantile female rhesus monkey. Science. 1980;207:1373–1375 [DOI] [PubMed] [Google Scholar]
- 3. Guran T, Tolhurst G, Bereket A, Rocha N, Porter K, Turan S, et al. Hypogonadotropic hypogonadism due to a novel missense mutation in the first extracellular loop of the neurokinin B receptor. J Clin Endocrinol Metab. 2009;94:3633–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Topaloglu AK, Reimann F, Guclu M, Yalin AS, Kotan LD, Porter KM, et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet. 2009;41:354–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Young J, Bouligand J, Francou B, Raffin-Sanson ML, Gaillez S, Jeanpierre M, et al. TAC3 and TACR3 defects cause hypothalamic congenital hypogonadotropic hypogonadism in humans. J Clin Endocrinol Metab. 2010;95:2287–2295 [DOI] [PubMed] [Google Scholar]
- 6. Rance NE, Krajewski SJ, Smith MA, Cholanian M, Dacks PA. Neurokinin B and the hypothalamic regulation of reproduction. Brain Res. 2010;1364:116–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sandoval-Guzman T, Rance NE. Central injection of senktide, an NK3 receptor agonist, or neuropeptide Y inhibits LH secretion and induces different patterns of Fos expression in the rat hypothalamus. Brain Res. 2004;1026:307–312 [DOI] [PubMed] [Google Scholar]
- 8. Navarro VM, Gottsch ML, Chavkin C, Okamura H, Clifton DK, Steiner RA. Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/neurokinin B neurons in the arcuate nucleus of the mouse. J Neurosci. 2009;29:11859–11866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Navarro VM, Castellano JM, McConkey SM, Pineda R, Ruiz-Pino F, Pinilla L, et al. Interactions between kisspeptin and neurokinin B in the control of GnRH secretion in the female rat. Am J Physiol Endocrinol Metab. 2011;300:E202–E210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Billings HJ, Connors JM, Altman SN, Hileman SM, Holaskova I, Lehman MN, et al. Neurokinin B acts via the neurokinin-3 receptor in the retrochiasmatic area to stimulate luteinizing hormone secretion in sheep. Endocrinology. 2010;151:3836–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ramaswamy S, Seminara SB, Ali B, Ciofi P, Amin NA, Plant TM. Neurokinin B stimulates GnRH release in the male monkey (Macaca mulatta) and is colocalized with kisspeptin in the arcuate nucleus. Endocrinology. 2010;151:4494–4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang J, Seminara S. Uncovering novel reproductive defects in neurokinin B receptor null mice: Closing the gap between mice and men. Endocrinology. 2012;153:1498–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr., Shagoury JK, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627 [DOI] [PubMed] [Google Scholar]
- 14. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 2003;100:10972–10976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gottsch ML, Cunningham MJ, Smith JT, Popa SM, Acohido BV, Crowley WF, et al. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology. 2004;145:4073–4077 [DOI] [PubMed] [Google Scholar]
- 16. Thompson EL, Patterson M, Murphy KG, Smith KL, Dhillo WS, Todd JF, et al. Central and peripheral administration of kisspeptin-10 stimulates the hypothalamic-pituitary-gonadal axis. J Neuroendocrinol. 2004;16:850–858 [DOI] [PubMed] [Google Scholar]
- 17. Dhillo WS, Chaudhri OB, Patterson M, Thompson EL, Murphy KG, Badman MK, et al. Kisspeptin-54 stimulates the hypothalamic-pituitary gonadal axis in human males. The Journal of Clinical Endocrinology and Metabolism. 2005;90:6609–6615 [DOI] [PubMed] [Google Scholar]
- 18. Plant TM, Ramaswamy S, Dipietro MJ. Repetitive activation of hypothalamic G protein-coupled receptor 54 with intravenous pulses of kisspeptin in the juvenile monkey (Macaca mulatta) elicits a sustained train of gonadotropin-releasing hormone discharges. Endocrinology. 2006;147:1007–1013 [DOI] [PubMed] [Google Scholar]
- 19. Ezzat Ahmed A, Saito H, Sawada T, Yaegashi T, Yamashita T, Hirata T, et al. Characteristics of the stimulatory effect of kisspeptin-10 on the secretion of luteinizing hormone, follicle-stimulating hormone and growth hormone in prepubertal male and female cattle. J Reprod Dev. 2009;55:650–654 [DOI] [PubMed] [Google Scholar]
- 20. Clarkson J, d'Anglemont de Tassigny X, Moreno AS, Colledge WH, Herbison AE. Kisspeptin-GPR54 signaling is essential for preovulatory gonadotropin-releasing hormone neuron activation and the luteinizing hormone surge. J Neurosci. 2008;28:8691–8697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kauffman AS, Park JH, McPhie-Lalmansingh AA, Gottsch ML, Bodo C, Hohmann JG, et al. The kisspeptin receptor GPR54 is required for sexual differentiation of the brain and behavior. J Neurosci. 2007;27:8826–8835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Messager S, Chatzidaki EE, Ma D, Hendrick AG, Zahn D, Dixon J, et al. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc Natl Acad Sci USA. 2005;102:1761–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wakabayashi Y, Nakada T, Murata K, Ohkura S, Mogi K, Navarro VM, et al. Neurokinin B and dynorphin A in kisspeptin neurons of the arcuate nucleus participate in generation of periodic oscillation of neural activity driving pulsatile gonadotropin-releasing hormone secretion in the goat. J Neurosci. 2010;30:3124–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goodman RL, Lehman MN, Smith JT, Coolen LM, de Oliveira CV, Jafarzadehshirazi MR, et al. Kisspeptin neurons in the arcuate nucleus of the ewe express both dynorphin A and neurokinin B. Endocrinology. 2007;148:5752–5760 [DOI] [PubMed] [Google Scholar]
- 25. Krajewski SJ, Burke MC, Anderson MJ, McMullen NT, Rance NE. Forebrain projections of arcuate neurokinin B neurons demonstrated by anterograde tract-tracing and monosodium glutamate lesions in the rat. Neuroscience. 2010;166:680–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krajewski SJ, Anderson MJ, Iles-Shih L, Chen KJ, Urbanski HF, Rance NE. Morphologic evidence that neurokinin B modulates gonadotropin-releasing hormone secretion via neurokinin 3 receptors in the rat median eminence. J Comp Neurol. 2005;489:372–386 [DOI] [PubMed] [Google Scholar]
- 27. Todman MG, Han SK, Herbison AE. Profiling neurotransmitter receptor expression in mouse gonadotropin-releasing hormone neurons using green fluorescent protein-promoter transgenics and microarrays. Neuroscience. 2005;132:703–712 [DOI] [PubMed] [Google Scholar]
- 28. Mellon PL, Wetsel WC, Windle JJ, Valenca MM, Goldsmith PC, Whyte DB, et al. Immortalized hypothalamic gonadotropin-releasing hormone neurons. Ciba Found Symp. 1992;168:104–117 [DOI] [PubMed] [Google Scholar]
- 29. Wetsel WC, Valenca MM, Merchenthaler I, Liposits Z, Lopez FJ, Weiner RI, et al. Intrinsic pulsatile secretory activity of immortalized luteinizing hormone-releasing hormone-secreting neurons. Proc Natl Acad Sci U S A. 1992;89:4149–4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmidlin F, Roosterman D, Bunnett NW. The third intracellular loop and carboxyl tail of neurokinin 1 and 3 receptors determine interactions with beta-arrestins. Am J Physiol Cell Physiol. 2003;285:C945–C958 [DOI] [PubMed] [Google Scholar]
- 31. Ely HA, Mellon PL, Coss D. GnRH Induces the c-Fos gene via phosphorylation of SRF by the calcium/calmodulin kinase II pathway. Mol Endocrinol. 2011;25:669–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Olive M, Krylov D, Echlin DR, Gardner K, Taparowsky E, Vinson C. A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis. J Biol Chem. 1997;272:18586–18594 [DOI] [PubMed] [Google Scholar]
- 33. Iyer AK, Miller NL, Yip K, Tran BH, Mellon PL. Enhancers of GnRH transcription embedded in an upstream gene use homeodomain proteins to specify hypothalamic expression. Mol Endocrinol. 2010;24:1949–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kelley CG, Givens ML, Rave-Harel N, Nelson SB, Anderson S, Mellon PL. Neuron-restricted expression of the rat gonadotropin-releasing hormone gene is conferred by a cell-specific protein complex that binds repeated CAATT elements. Mol Endocrinol. 2002;16:2413–2425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iyer AK, Brayman MJ, Mellon PL. Dynamic chromatin modifications control GnRH gene expression during neuronal differentiation and protein kinase C signal transduction. Mol Endocrinol. 2011;25:460–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mellon PL, Windle JJ, Goldsmith P, Padula C, Roberts J, Weiner RI. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron. 1990;5:1–10 [DOI] [PubMed] [Google Scholar]
- 37. Coss D, Jacobs SB, Bender CE, Mellon PL. A novel AP-1 site is critical for maximal induction of the follicle-stimulating hormone beta gene by gonadotropin-releasing hormone. J Biol Chem. 2004;279:152–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huh YH, Ryu JH, Chun JS. Regulation of type II collagen expression by histone deacetylase in articular chondrocytes. J Biol Chem. 2007;282:17123–17131 [DOI] [PubMed] [Google Scholar]
- 39. Schreiber E, Matthias P, Müller M, Schaffner W. Rapid detection of octamer binding proteins with mini-extracts prepared from a small number of cells. Nucl Acids Res. 1989;17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nakanishi S. Mammalian tachykinin receptors. Annu Rev Neurosci. 1991;14:123–136 [DOI] [PubMed] [Google Scholar]
- 41. Ingi T, Kitajima Y, Minamitake Y, Nakanishi S. Characterization of ligand-binding properties and selectivities of three rat tachykinin receptors by transfection and functional expression of their cloned cDNAs in mammalian cells. J Pharmacol Exp Ther. 1991;259:968–975 [PubMed] [Google Scholar]
- 42. Ding YD, Shi J, Su LY, Xu JQ, Su CJ, Guo XE, et al. Intracerebroventricular injection of senktide-induced Fos expression in vasopressin-containing hypothalamic neurons in the rat. Brain Res. 2000;882:95–102 [DOI] [PubMed] [Google Scholar]
- 43. Yip J, Chahl LA. Localization of Fos-like immunoreactivity induced by the NK3 tachykinin receptor agonist, senktide, in the guinea-pig brain. Br J Pharmacol. 1997;122:715–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kawasaki M, Ponzio TA, Yue C, Fields RL, Gainer H. Neurotransmitter regulation of c-fos and vasopressin gene expression in the rat supraoptic nucleus. Exp Neurol. 2009;219:212–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wetsel WC, Eraly SA, Whyte DB, Mellon PL. Regulation of gonadotropin-releasing hormone by protein kinases A and C in immortalized hypothalamic neurons. Endocrinology. 1993;132:2360–2370 [DOI] [PubMed] [Google Scholar]
- 46. Bruder JM, Spaulding AJ, Wierman ME. Phorbol ester inhibition of rat gonadotropin-releasing hormone promoter activity: role of fos and jun in the repression of transcription. Mol Endocrinol. 1996;10:35–44 [DOI] [PubMed] [Google Scholar]
- 47. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868 [DOI] [PubMed] [Google Scholar]
- 48. Khawaja AM, Rogers DF. Tachykinins: receptor to effector. Int J Biochem Cell Biol. 1996;28:721–738 [DOI] [PubMed] [Google Scholar]
- 49. Bruder JM, Drebs WD, Nett TM, Wierman ME. Phorbol ester activation of the protein kinase C pathway inhibits gonadotropin-releasing hormone gene expression. Endocrinology. 1992;131:2552–2558 [DOI] [PubMed] [Google Scholar]
- 50. Bruder JM, Wierman ME. Evidence for transcriptional inhibition of GnRH gene expression by phorbol ester at a proximal promoter region. Mol Cell Endocrinol. 1994;99:177–182 [DOI] [PubMed] [Google Scholar]
- 51. Tang Q, Mazur M, Mellon PL. The protein kinase C pathway acts through multiple transcription factors to repress gonadotropin-releasing hormone gene expression in hypothalamic GT1-7 neuronal cells. Mol Endocrinol. 2005;19:2769–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988;57:159–197 [DOI] [PubMed] [Google Scholar]
- 53. Gore AC, Roberts JL, Gibson MJ. Mechanisms for the regulation of gonadotropin-releasing hormone gene expression in the developing mouse. Endocrinology. 1999;140:2280–2287 [DOI] [PubMed] [Google Scholar]
- 54. Gore AC, Oung T, Yung S, Flagg RA, Woller MJ. Neuroendocrine mechanisms for reproductive senescence in the female rat: gonadotropin-releasing hormone neurons. Endocrine. 2000;13:315–323 [DOI] [PubMed] [Google Scholar]
- 55. Zoeller RT, Young WS., 3rd Changes in cellular levels of messenger ribonucleic acid encoding gonadotropin-releasing hormone in the anterior hypothalamus of female rats during the estrous cycle. Endocrinology. 1988;123:1688–1689 [DOI] [PubMed] [Google Scholar]
- 56. Selmanoff M, Shu C, Petersen SL, Barraclough CA, Zoeller RT. Single cell levels of hypothalamic messenger ribonucleic acid encoding luteinizing hormone-releasing hormone in intact, castrated, and hyperprolactinemic male rats. Endocrinology. 1991;128:459–466 [DOI] [PubMed] [Google Scholar]
- 57. He JR, Molnar J, Barraclough CA. Morphine amplifies norepinephrine (NE)-induced LH release but blocks NE-stimulated increases in LHRH mRNA levels: comparison of responses obtained in ovariectomized, estrogen-treated normal and androgen-sterilized rats. Brain Res Mol Brain Res. 1993;20:71–78 [DOI] [PubMed] [Google Scholar]
- 58. Liaw JJ, Barraclough CA. N-methyl-D,L-aspartic acid differentially affects LH release and LHRH mRNA levels in estrogen-treated ovariectomized control and androgen-sterilized rats. Brain Res Mol Brain Res. 1993;17:112–118 [DOI] [PubMed] [Google Scholar]
- 59. Novaira HJ, Ng Y, Wolfe A, Radovick S. Kisspeptin increases GnRH mRNA expression and secretion in GnRH secreting neuronal cell lines. Mol Cell Endocrinol. 2009;311:126–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nelson SB, Eraly SA, Mellon PL. The GnRH promoter: Target of transcription factors, hormones, and signaling pathways. Mol Cell Endocrinol. 1998;140:151–155 [DOI] [PubMed] [Google Scholar]
- 61. Whyte DB, Lawson MA, Belsham DD, Eraly SA, Bond CT, Adelman JP, et al. A neuron-specific enhancer targets expression of the gonadotropin-releasing hormone gene to hypothalamic neurosecretory neurons. Mol Endocrinol. 1995;9:467–477 [DOI] [PubMed] [Google Scholar]
- 62. Lawson MA, MacConell LA, Kim J, Powl BT, Nelson SB, Mellon PL. Neuron-specific expression in vivo by defined transcription regulatory elements of the gonadotropin-releasing hormone gene. Endocrinology. 2002;143:1404–1412 [DOI] [PubMed] [Google Scholar]
- 63. Novaira HJ, Yates M, Diaczok D, Kim H, Wolfe A, Radovick S. The gonadotropin-releasing hormone cell-specific element is required for normal puberty and estrous cyclicity. J Neurosci. 2011;31:3336–3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Eraly SA, Nelson SB, Huang KM, Mellon PL. Oct-1 binds promoter elements required for transcription of the gonadotropin-releasing hormone gene. Mol Endocrinol. 1998;12:469–481 [DOI] [PubMed] [Google Scholar]
- 65. Clark ME, Mellon PL. The POU homeodomain transcription factor Oct-1 is essential for activity of the gonadotropin-releasing hormone neuron-specific enhancer. Mol Cell Biol. 1995;15:6169–6177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nelson SB, Lawson MA, Kelley CG, Mellon PL. Neuron-specific expression of the rat gonadotropin-releasing hormone gene is conferred by interactions of a defined promoter element with the enhancer in GT1-7 cells. Mol Endocrinol. 2000;14:1509–1522 [DOI] [PubMed] [Google Scholar]
- 67. Givens ML, Rave-Harel N, Goonewardena VD, Kurotani R, Berdy SE, Swan CH, et al. Developmental regulation of gonadotropin-releasing hormone gene expression by the MSX and DLX homeodomain protein families. J Biol Chem. 2005;280:19156–19165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee VH, Lee LT, Chow BK. Gonadotropin-releasing hormone: regulation of the GnRH gene. FEBS J. 2008;275:5458–5478 [DOI] [PubMed] [Google Scholar]
- 69. Brayman MJ, Pepa PA, Berdy SE, Mellon PL. Androgen receptor repression of GnRH gene transcription. Mol Endocrinol. 2012;26:2–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mastronardi C, Smiley GG, Raber J, Kusakabe T, Kawaguchi A, Matagne V, et al. Deletion of the Ttf1 gene in differentiated neurons disrupts female reproduction without impairing basal ganglia function. J Neurosci. 2006;26:13167–13179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Eraly SA, Mellon PL. Regulation of GnRH transcription by protein kinase C is mediated by evolutionarily conserved, promoter-proximal elements. Mol Endocrinol. 1995;9:848–859 [DOI] [PubMed] [Google Scholar]
- 72. Ozcan M, Alcin E, Ayar A, Yilmaz B, Sandal S, Kelestimur H. Kisspeptin-10 elicits triphasic cytosolic calcium responses in immortalized GT1-7 GnRH neurones. Neurosci Lett. 2011;492:55–58 [DOI] [PubMed] [Google Scholar]
- 73. Navarro VM, Gottsch ML, Wu M, Garcia-Galiano D, Hobbs SJ, Bosch MA, et al. Regulation of NKB pathways and their roles in the control of Kiss1 neurons in the arcuate nucleus of the male mouse. Endocrinology. 2011;152:4265–4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Garcia-Galiano D, van Ingen Schenau D, Leon S, Krajnc-Franken MA, Manfredi-Lozano M, Romero-Ruiz A, et al. Kisspeptin signaling is indispensable for neurokinin B, but not glutamate, stimulation of gonadotropin secretion in mice. Endocrinology. 2011;153:316–328 [DOI] [PubMed] [Google Scholar]
- 75. Ramaswamy S, Seminara SB, Plant TM. Evidence from the agonadal juvenile male rhesus monkey (Macaca mulatta) for the view that the action of Neurokinin B to trigger gonadotropin-releasing hormone release is upstream from the kisspeptin receptor. Neuroendocrinology. 2011;94:237–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Grachev P, Li XF, Kinsey-Jones JS, di Domenico AL, Millar RP, Lightman SL, et al. Suppression of the GnRH pulse generator by neurokinin B involves a kappa-opioid receptor-dependent mechanism. Endocrinology. 2012;153:4894–4904 [DOI] [PubMed] [Google Scholar]
- 77. Kinsey-Jones JS, Grachev P, Li XF, Lin YS, Milligan SR, Lightman SL, et al. The inhibitory effects of neurokinin B on GnRH pulse generator frequency in the female rat. Endocrinology. 2012;153:307–315 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.