

Abstract
The p21-activated serine-threonine kinase (PAK1) is activated by small GTPase-dependent and -independent mechanisms and regulates cell motility. Both PAK1 and the hormone prolactin (PRL) have been implicated in breast cancer by numerous studies. We have previously shown that the PRL-activated tyrosine kinase JAK2 (Janus tyrosine kinase 2) phosphorylates PAK1 in vivo and identified tyrosines (Tyr) 153, 201, and 285 in the PAK1 molecule as sites of JAK2 tyrosyl phosphorylation. Here, we have used human breast cancer T47D cells stably overexpressing PAK1 wild type or PAK1 Y3F mutant in which Tyr(s) 153, 201, and 285 were mutated to phenylalanines to demonstrate that phosphorylation of these three tyrosines are required for maximal PRL-dependent ruffling. In addition, phosphorylation of these three tyrosines is required for increased migration of T47D cells in response to PRL as assessed by two independent motility assays. Finally, we show that PAK1 phosphorylates serine (Ser) 2152 of the actin-binding protein filamin A to a greater extent when PAK1 is tyrosyl phosphorylated by JAK2. Down-regulation of PAK1 or filamin A abolishes the effect of PRL on cell migration. Thus, our data presented here bring some insight into the mechanism of PRL-stimulated motility of breast cancer cells.
Prolactin (PRL), a hormone utilized at both the endocrine and autocrine levels, regulates the differentiation of secretory glands, including the mammary gland, ovary, prostate, submaxillary and lacrimal glands, pancreas, and liver (for review see Refs. 1 and 2). PRL binding to its receptor activates tyrosine kinase JAK2 (Janus tyrosine kinase 2), PRL receptor phosphorylation, and phosphorylation of signal transducer and activator of transcription (STAT)5A and 5B, STAT3, and STAT 1 (3–5). This triggers STAT dimerization, nuclear translocation, and DNA binding, which leads to events necessary for PRL-triggered responses. PRL also activates other pathways including the Src/Grb2/MAPK (6, 7), protein kinase C (8, 9), Src kinase (10, 11), and phosphatidylinositol 3-kinase (12). Increasing evidence supports the involvement of PRL in breast cancer [Refs. 13 and 14); for review see Refs. 15–21]. PRL has been shown to increase cell motility in breast cancer cells (22–24). These data, combined with animal studies reporting increased metastases with PRL administration (25), suggest that PRL is involved in the development of metastasis and tumor progression. On the other hand, PRL has also been reported to act as a suppressor of breast cancer cell invasion (26, 27), suggesting that the role of PRL in breast cancer must be explored further.
Cell motility is a critical rate-limiting step in the invasive growth program under physiological and pathophysiological conditions. Little is known about the mechanisms that underlie the process of PRL-induced cell motility and its putative role in tumor progression. PRL was previously shown to act as a chemoattractant for human breast carcinoma (22), and activation of NIMA-related kinase 3 (Nek3 kinase) and Vav1/Rac1 as well as paxillin phosphorylation have been proposed as a PRL-dependent mechanism to regulate motility of breast cancer cells (23, 24, 28). Another small GTPase Cdc42 is also activated by PRL in mammary epithelia (29).
We have found that the p21-activated serine-threonine kinase (PAK1), a downstream effector for both Cdc42 and Rac1, participates in PRL-dependent signaling (30). PAK1 plays a key role in coordinating dynamic reorganization of the actin and microtubule cytoskeletons and is implicated in breast cancer (for review see Ref. 31). Heregulin-activated PAK1 increased invasiveness of breast cancer cells (32), whereas expression of a kinase-dead PAK1 mutant in highly invasive breast cancer cells led to stabilization of stress fibers, enhanced cell spreading, and reduction in invasiveness (33). Conversely, hyperactivation of the PAK1 pathway in the noninvasive breast cancer MCF-7 cell line promotes cell migration and anchorage-independent growth (34) and suppresses anoikis in MCF10A breast epithelial cells (35). Additionally, the constitutive activation of PAK1 in breast cancer cells is the result of mislocalization of PAK1 to focal adhesions (36). PAK1 regulates the actin cytoskeleton through stimulation of LIM kinase 1 activity, which in turn increases the phosphorylation and inactivation of cofilin, leading to a reduction in the depolymerization of actin filaments (37). PAK1 also directly phosphorylates other cytoskeletal proteins, including myosin light chain kinase (38), paxillin (39), filamin A (FLNa), p41-Arc, and merlin (40–42).
We have previously shown that PAK1 is a novel substrate of the JAK2 tyrosine kinase and that PRL-activated JAK2 phosphorylates PAK1 in vivo. PAK1 tyrosines [Tyr(s) 153, 201, and 285] were identified as sites of JAK2 tyrosyl phosphorylation by mass spectrometry and two-dimensional peptide mapping. Our findings indicated that JAK2 phosphorylates PAK1 at these specific tyrosines and that this phosphorylation plays an important role in cell survival and in the regulation of cyclin D1 promoter activity (30, 43). We have recently demonstrated that phosphorylation of these three tyrosines of PAK1 by JAK2, as well as the presence of FLNa and adapter protein Src homology 2 SH2B1β (actin-binding protein and substrate of JAK2) play a role in PRL-dependent changes of the actin cytoskeleton (44, 45).
FLNa is one of the best characterized PAK1 substrates (40). FLNa is a 280-kDa actin cross-linking protein containing an N-terminal actin-binding domain and a rod region containing 24 Ig-like repeats (reviewed in Ref. 46). The last repeat of the rod region enables the filamin molecules to dimerize, allowing for a flexible structure mediating the actin gelation activity of filamins. Filamins have more than 90 interacting partners, including adapter proteins, small GTPases, transmembrane receptors, and membrane channels (47). FLNa participates in the activation of various kinases as well as being regulated by kinases itself. FLNa binding to PAK1 enhances the kinase activity of PAK1, which subsequently phosphorylates FLNa at serine (Ser) 2152, resulting in PAK1-dependent membrane ruffling (40). FLNa also stimulates PAK1 by interacting with sphingosine kinase 1, which phosphorylates sphingosine, leading to the direct activation of PAK1 (48).
Here, we extend our findings suggesting a role for tyrosyl-phosphorylated PAK1 in breast cancer cell motility. We use human breast cancer T47D cell lines that stably overexpress PAK1 wild type (WT) or PAK1 Y3F mutant in which the three JAK2 phosphorylation sites [Tyr(s) 153, 201, and 285] were mutated to phenylalanine. We have demonstrated that Tyr(s) 153, 201, and 285 of PAK1 are required for maximal PRL-dependent cell ruffling and cell migration. Finally, we have demonstrated that PAK1 phosphorylates Ser 2152 of the actin-binding protein FLNa to a greater extent when PAK1 is tyrosyl phosphorylated by JAK2.
Results
Characterization of T47D clones expressing PAK1 WT and PAK1 Y3F mutant
We have previously shown that JAK2 tyrosine kinase phosphorylates Tyr(s) 153, 201, and 285 on PAK1 (30). To determine whether JAK2-dependent phosphorylation of PAK1 promotes changes in the actin cytoskeleton and cell motility in response to PRL, we established T47D cell lines that stably express green fluorescent protein (GFP) either alone (as vector control) or with either myc-tagged PAK1 WT or PAK1 Y3F mutant in which the three JAK2 phosphorylation sites [Tyr(s) 153, 201, and 285] were mutated to phenylalanine. These retroviral constructs include internal ribosome entry site (IRES) elements that allow the transcription of a single bicistronic mRNA of myc-PAK1-IRES2-enhanced GFP (EGFP), and so produce PAK1 with N-terminal myc tag together with EGFP as a reporter for expression of PAK1. We isolated several clones expressing a relatively modest level of PAK1 WT or PAK1 Y3F expression over the levels seen in vector-transfected or in parental cells, as judged by the differences in the expression of myc-tagged PAK1s and endogenous PAK1 (Figure 1A). For subsequent experiments we used clone 10 of PAK1 WT and clone 3 of PAK1 Y3F.
Figure 1.

Characterization of T47D cell lines stably expressing GFP alone, or GFP with myc-tagged PAK1 WT and PAK1 Y3F. A, T47D cells stably expressing GFP (lane 1), two clones stably expressing myc-PAK1 WT (lanes 2 and 3), two clones stably expressing myc-PAK1 Y3F (lanes 4 and 5), and parental T47D cells (lane 6) were lysed, and proteins were resolved by SDS-PAGE. Overexpressed proteins were visualized by immunoblotting with antimyc Ab (upper panel). The same membrane was stripped and reblotted with anti-PAK1 Ab to visualize endogenous (lanes 1 and 6) and overexpressed PAK1 (lanes 2–5) (middle panel). B, myc-PAK1 was IP'd with antimyc Ab from T47D PAK1 WT (lanes 1–2) or T47D PAK1 Y3F cells (lanes 3–4) transfected with vector (lanes 1 and 3) or constitutive active Rac1 V12 (lanes 2 and 4). IP'd myc-PAK1 was subjected to an in vitro kinase assay with H4 histone as a substrate (32P incorporation into H4 histone indicated in upper panel), and probed with anti-PAK1 (middle panel) and anti-Rac1 (lower panel) Ab. All blots are representative of at least three experiments. C, T47D PAK1 WT and T47D PAK1 Y3F cells were treated with or without PRL. Myc-PAK1 was IP'd with antimyc Ab and subjected to the in vitro kinase assay as described in panel B. 32P incorporation into PAK1 molecule (PAK1 autophosphorylation) is indicated in the middle panel. Relative PAK1 kinase activity was then normalized by the amount of IP'd PAK1 for each lane and plotted (right plot). Bars represent mean ± SE * P < 0 .05. n = 3. ctrl, Control.
To confirm that the expressed PAK1 Y3F protein was functional and activated by active Rac1, myc-tagged PAK1 WT and PAK1 Y3F were immunoprecipitated (IP'd) from cell lines transiently transfected with constitutively active Rac1 V12. The kinase activities of the IP'd PAK1 WT and PAK1 Y3F were measured in an in vitro kinase assay with H4 histone as a substrate. Both PAK1 WT and PAK1 Y3F were similarly activated by Rac1 V12 (Figure 1B). Next, we set out to determine whether PRL treatment altered PAK1 kinase activity. T47D cell clones were treated without or with PRL (200 ng/ml, 20 min), and PAK1 kinase activity was measured. PRL treatment more than doubled the kinase activity of PAK1 WT as compared with untreated control (Figure 1C). PRL activated both PAK1 WT and PAK1 Y3F through Rac1 [activation of Rac1 by PRL has been shown previously (23)]. However, in the presence of PRL, the kinase activity of PAK1 WT was significantly stronger than the kinase activity of PAK1 Y3F (Figure 1C). These data indicated that PRL further increased PAK1 kinase activity and suggested that Tyr(s) 153, 201, and 285 were responsible for this activation.
Tyr(s) 153, 201, and 285 of PAK1 are required for maximal prolactin-dependent ruffling
We sought to determine whether tyrosyl-phosphorylated (pTyr)-PAK1 participates in PRL-dependent actin reorganization. PRL is a potent activator of JAK2 (49–51) and rapidly induced ruffling in T47D cells (22, 45). PAK1 is tyrosyl phosphorylated by JAK2 in response to PRL (30). However, a potential role for pTyr-PAK1 in PRL-dependent regulation of the actin cytoskeleton remains to be determined.
We tested the effect of the three phosphorylated tyrosines of PAK1 on PRL-dependent ruffling. T47D cells stably expressing either GFP alone (vector control), or GFP plus PAK1 WT or PAK1 Y3F mutant, were serum deprived, treated with or without PRL, and actin ruffling was assayed. Overexpression of WT and Y3F PAK1 had no effect on the number of ruffles in the unstimulated cells (Figure 2, A and B). In contrast, whereas overexpression of PAK1 WT strongly enhanced cell ruffling in response to PRL, cells expressing mutant PAK1 Y3F failed to enhance ruffling in response to PRL. We confirmed these data in MCF-7 cells, transiently overexpressing either GFP, PAK1 WT, or PAK1 Y3F mutant and treated with or without PRL (Figure 2, C and D)
Figure 2.

PAK1 WT enhances and PAK1 Y3F inhibits PRL-induced membrane ruffling. A, T47D cells stably expressing GFP alone [vector (vctr)], or GFP plus myc-tagged PAK1 WT or PAK1 Y3F were treated with or without PRL. Filamentous actin was visualized by staining with Texas Red-phalloidin (shown). Scale bar, 50 μm. C, MCF-7 cells were transfected with plasmid encoding GFP (vctr), myc-tagged PAK1 WT, or PAK1 Y3F and treated as in panel A. Scale bar, 50 μm. Arrows indicate representative ruffles in both transfected and untransfected cells, and asterisks denote transfected cells. B and D, Cells positive for GFP or antimyc were scored for the presence of ruffles for each experimental condition. The ruffling index as a number of ruffles per cell was counted. Bars represent mean ± SE. # P < 0.05 compared with cells expressing GFP (vctr) with the same treatment. Each experiment was repeated three times with 100 cells each time. n = 3 for each experimental condition.
These results suggest that the three phosphorylated tyrosines of PAK1 are required for maximal PRL-induced actin ruffling.
PRL-activated PAK1 stimulates cell migration
The data in Figure 2 suggested that pTyr-PAK1 could play a role in cell motility. We employed several strategies to examine this hypothesis. Confluent monolayers of T47D cells stably overexpressing either GFP, PAK1 WT, or PAK1 Y3F mutant were serum deprived, wounded, and cultured without or with PRL for 18 h. The distance of wound closure was monitored by phase microscopy and plotted (Figure 3). The cells migrated into the wound by cell movement, not cell division, because PRL does not increase cell proliferation during 18 h in the absence of serum (data not shown). Without PRL, all cell lines migrated into the area of the wound relatively slowly. However, in the presence of PRL, the migration of T47D PAK1 WT cells was significantly faster than T47D GFP and T47D PAK1 Y3F cells, and this led to a more rapid healing of wounded monolayers. T47D PAK1 WT cells reduced the width of the wound by 31–34% for 18 h whereas T47D GFP cells migrated twice slower in response to PRL (15% wound closure). In contrast, T47D PAK1 Y3F cells failed to demonstrate PRL-induced motility in the same period of time (5–6% wound closure; ie, the same percentage as in the absence of PRL).
Figure 3.
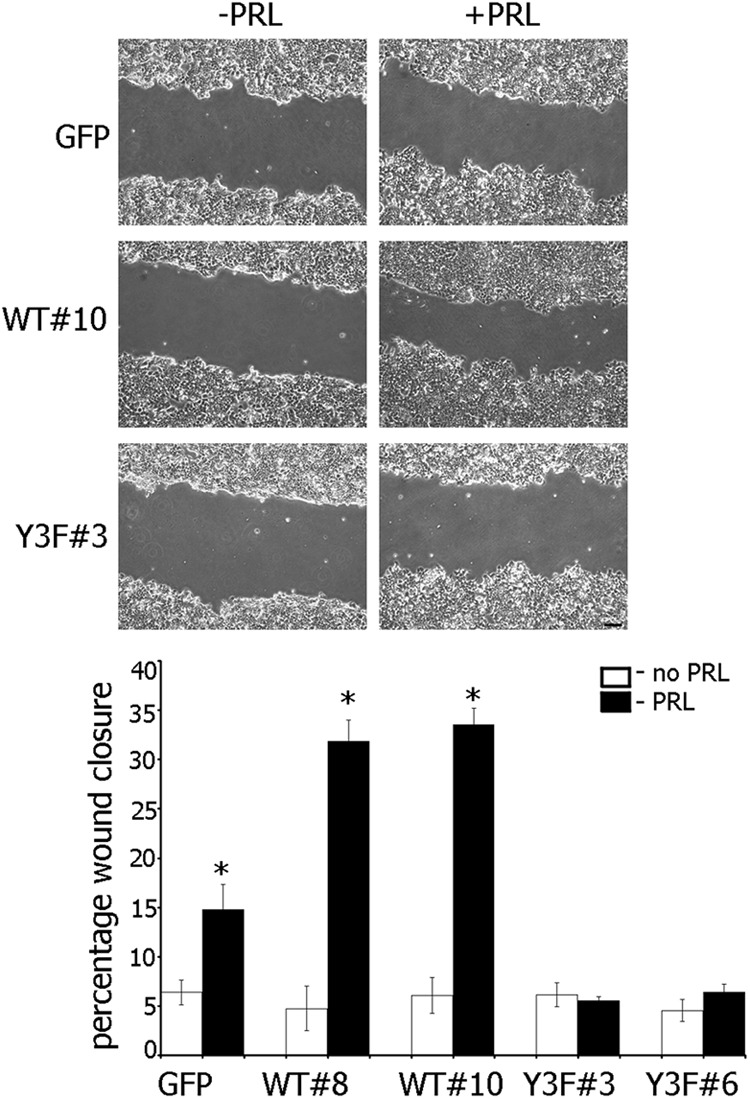
Effect of PRL-dependent pTyr-PAK1 on closure of wounded monolayer. Monolayers of T47D cells stably overexpressing GFP, PAK1 WT, or PAK1 Y3F were wounded and cultured without (white bar) or with (black bar) PRL. Bars represent mean ± SE. * P < .05 compared with the same cells untreated with PRL. Each experiment was repeated three times with 10 measurements each time. n = 3 for each experimental condition. Scale bar, 200 μm.
The effect of pTyr-PAK1 WT on PRL-induced cell motility was confirmed by evaluating migration through Transwell pores (Figure 4). T47D GFP, PAK1 WT, or PAK1 Y3F cells were serum deprived, and equal amounts of cells were loaded into the upper part of the Boyden chamber. The number of cells that migrated to the lower surface of the chamber toward PRL after 48 h were counted and plotted. Similar to the wounding assay, overexpression of PAK1 WT accelerated migration in response to PRL (>50 cells migrated through the pores) as compared with the cells overexpressing GFP (30 migrated cells) whereas overexpression of PAK1 Y3F (<20 migrated cells) significantly inhibited cell migration (Figure 4, A and B).
Figure 4.

Maximal migration of T47D cells in response to PRL requires tyrosyl phosphorylation of PAK1. A, Equal amount of T47D cells stably overexpressing GFP, PAK1 WT, or PAK1 Y3F were loaded into the upper part of the Boyden chamber. The number of cells that migrated to the lower surface of the chamber toward PRL (black bar) or buffer control (white bar) after 48 h were counted and plotted (B). Scale bar, 300 μm. C and D, T47D cells (C) or MCF-7 cells (D) were transfected with control or PAK1 siRNA, stimulated (black bars) or not (white bars) with PRL, and assessed for migration using the Boyden chamber assay as in panel A. Silencing efficiency was judged by immunoblotting with anti-PAK1 Ab 48 and 72 h after transfection. The expression levels of γ-tubulin (Tu) were used as an internal control (Ctrl). Bars represent mean ± SE. * P < 0.05 compared with the same cells untreated with PRL. n = 3 for each experimental condition.
To directly establish the significance of PAK1 signaling on PRL-dependent cell migration, we next examined the effect of knockdown of endogenous PAK1 by PAK1-specific small interfering RNA (siRNA) in T47D and MCF-7 cells. We found that in both types of cells, PAK1-depleted cells maintained basal migration (white bars in plots in Figure 4, C and D), but demonstrated ablation of PRL-induced migration (black bars in plots in Figure 4, C and D).
JAK2-dependent phosphorylation of PAK1 increases FLNa Ser2152 phosphorylation
It has been demonstrated that PAK1 phosphorylates actin-binding protein FLNa on Ser 2152, leading to actin cytoskeletal reorganization and cell ruffling (40). In an attempt to understand the pTyr-PAK1-dependent mechanism that regulates cell ruffling and cell migration, we decided to determine whether JAK2-dependent tyrosyl phosphorylation of PAK1 changes phosphorylation of Ser 2152 of FLNa. To provide evidence for that, myc-tagged FLNa was coexpressed in 293T cells with or without HA-tagged PAK1 and with either WT JAK2 or a kinase-inactive JAK2 mutant K882E. Transient overexpression of WT JAK2 in 293T cells produced constitutively Tyr-phosphorylated active JAK2 (52, 53). When myc-FLNa was IP'd from the cell lysates with antimyc Ab and immunoblotted with anti-phospho-Ser 2152 Ab, Ser-phosphorylation of FLNa was increased when FLNa was coexpressed with PAK1 and JAK2 (Figure 5A, lane 3) as compared with coexpression of FLNa with either kinase-inactive JAK2 mutant K882E (lane 2) or vector control (lane 4). The anti-phosho-Ser 2152 signal in lanes 2 and 4 represents phosphorylation of FLNa by overexpressed nontyrosyl-phosphorylated PAK1, which is kinase active. However, tyrosyl phosphorylation of PAK1 by JAK2 leads to increased PAK1 activity toward FLNa. The faint phospho-Ser 2152 FLNa signal in lane 1 likely represents phosphorylation of overexpressed FLNa by endogenous PAK1. The fine band in lane 4 detected by anti-JAK2 represents endogenous JAK2. The same blot was stripped and reprobed with antimyc Ab. The same amount of FLNa was IP'd with antimyc in all lanes (lanes 1–4), indicating that differences in the amount of Ser 2152-phosphorylated FLNa were not due to differences in the levels of FLNa expression. Next, we repeated the same experiments with T47D cells with one exception. We overexpressed constitutively active JAK2 V617F instead of JAK2 WT. Although the levels of overexpression of all transfected proteins were significantly lower than in 293T cells, the quantification of Ser-phosphorylated FLNa and FLNa bands from lane 1 vs. 3 showed that Ser phosphorylation of FLNa was maximal when FLNa was coexpressed with PAK1 and JAK2 V617F (Figure 5B). These findings suggest that tyrosyl phosphorylation of PAK1 by JAK2 enhances phosphorylation of FLNa by PAK1. Finally, we overexpressed FLNa in T47D clones, deprived the cells from serum, treated them with or without PRL, and assessed phosphorylation of FLNa by PAK1 as described above. The quantification of Ser-phosphorylated FLNa and FLNa bands showed that Ser phosphorylation of FLNa was increased maximally when T47D PAK WT cells were treated with PRL (Figure 5C).
Figure 5.
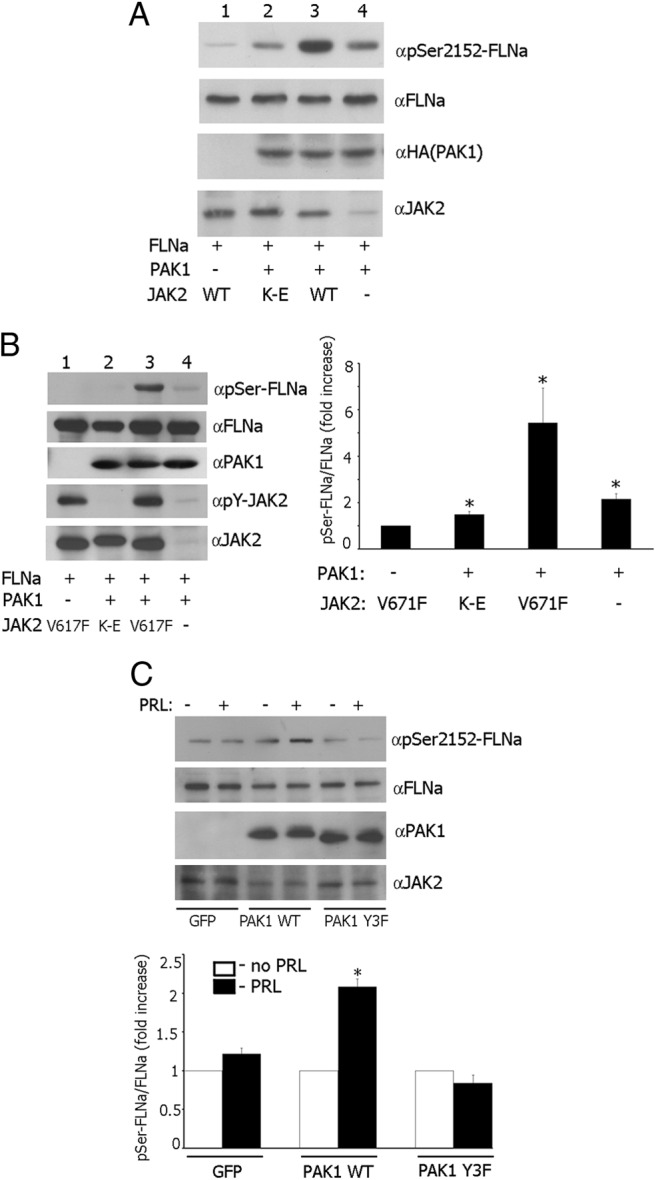
JAK2-dependent phosphorylation of PAK1 increases FLNa Ser2152 phosphorylation. A, 293T cells were cotransfected with plasmid encoding myc-FLNa, HA-PAK1 WT or vector, and JAK2 WT or kinase-inactive JAK2 K882E mutant (K–E). Myc-FLNa was IP'd from cell lysates with antimyc Ab. Proteins were resolved by SDS-PAGE followed by immunoblotting with the indicated Abs. B, T47D cells were transfected with plasmid encoding myc-FLNa, HA-PAK1 WT or vector, and constitutively active JAK2 V617F or kinase-inactive JAK2 K882E mutant (K–E) and processed as in panel A. The graph represents the densiometric analysis of the band obtained for phosphorylated FLNa normalized with total FLNa for each lane. Bars represent mean ± SE. * P < 0.05 compared with cells without overexpressed PAK1. n = 5. C, FLNa was overexpressed in T47D GFP cells, T47D PAK1 WT, or T47D PAK1 Y3F cells. The cells were treated with or without PRL. FLNa was IP'd and immunobloted with anti-pSer 2152 and reblotted with anti-FLNa. The graph represents densiometric analysis of the band obtained for phosphorylated FLNa normalized with total FLNa for each lane. Bars represent mean ± SE. * P < 0.05 compared with the same cells untreated with PRL. n = 3.
To show a mechanistic role of phopshorylated FLNa in the PRL-PAK1-dependent cell migration, we knocked down FLNa in T47D clones using FLNa-specific siRNA and evaluated cell migration through Transwell pores (Figure 6). Depletion of FLNa increased basal migration of all cell clones (compare white bars in control siRNA with white bars in FLNa siRNA in Figure 6B); however, PRL-induced cell migration was abrogated in all cell clones (compare white and black bars in FLNa siRNA in Figure 6B), suggesting that phosphorylated FLNa participates in PRL-induced breast cancer cell migration.
Figure 6.
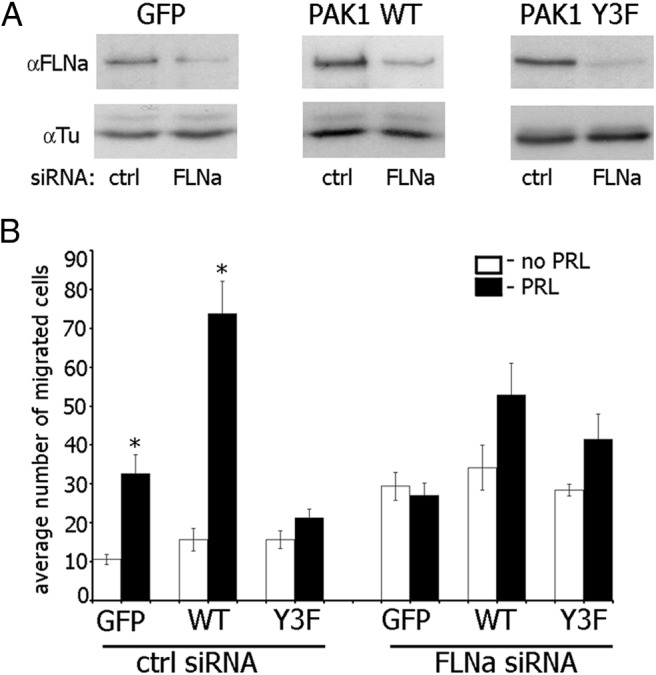
FLNa is required for PAK1 function in PRL-induced cell migration. A, T47D GFP cells, T47D PAK1 WT, or T47D PAK1 Y3F cells were transfected with control (ctrl) or FLNa siRNA. Endogenous FLNa was immunoblotted with anti-FLNa in 72 h after transfection. The expression levels of γ-tubulin (Tu) were used as an internal control. B, FLNa was depleted from the indicated cells as described in panel A, and the cells were assessed for migration using the Boyden chamber assay with (black bars) or without (white bars) PRL. Bars represent mean ± SE. * P < 0.05 compared with the same cells untreated with PRL. n = 3 for each experimental condition.
Discussion
PAK1 is an effector kinase for the small Rho GTPases Cdc42 and Rac 1 (54), and its activity is regulated by several pathways both dependent on and independent of small GTPases (32, 55–60). The role of PAK1 in actin-dependent cell functions is well documented (for review, please see Refs. 31 and 61–63). PAK1 is localized in areas of the cortical actin cytoskeleton and regulates it (64, 65); PAK1 kinase activity participates in directional motility (66–68), and PAK1 directly phosphorylates cytoskeletal proteins, including LIM kinase (37), myosin light chain kinase (38), paxillin (39), FLNa, p41-Arc, and merlin (40–42). Some of the effects of PAK1 on the actin cytoskeleton appear to be independent of PAK1 kinase activity but depend on protein-protein interactions (39, 40, 68).
We have previously implicated tyrosyl phosphorylation of PAK1 in the regulation of unstimulated phagokinesis, which is a combination of two processes that are dependent upon changes in the actin cytoskeleton: cellular movement and phagocytosis (30). Here, we have demonstrated that overexpression of WT PAK1 enhanced the ability of PRL to induce cell ruffling. In contrast, overexpression of PAK1 Y3F failed to increase ruffling. Membrane ruffling has been observed in many cell types in response to certain extracellular factors, and on motile cells where it is believed to be required for directed cell migration. Thus, the formation of membrane ruffles may be considered as a sign of increased response to external stimuli and of elevated cell migration (for review, see Refs. 69 and 70). Here we extend our findings and demonstrate that overexpression of PAK1 WT strongly enhances cell migration in response to PRL in both cell wounding and Boyden chamber assays.
In an attempt to understand the mechanism of the amplifying effect of tyrosyl-phosphorylated PAK1 on cell motility, we focused on FLNa for several reasons. First, the actin-binding protein FLNa is a binding partner of PAK1 (40). Second, we have previously implicated FLNa in PRL-dependent signaling through adapter protein SH2B1β (44). As a potent actin cross-linking protein, FLNa regulates cell migration, although the role of FLNa in this process is controversial. Thus, multiple studies have demonstrated a positive impact of FLNa on the migration of different cell types (eg, Refs. 71–74) One of the first noted defects of FLNa-deficient melanoma cells (M2 cells) was the inability to migrate due to inefficient polarization and continuous blebbing, which was rescued once FLNa was stably expressed (A7 cells) (71). In contrast, FLNa overexpression inhibits neuronal migration (75) and down-regulation of FLNa stimulates cancer cell migration, invasion, and metastatic formation (76). In support of the latter finding, we demonstrated that depletion of FLNa increased basal nonstimulated migration of T47D cells. However, PRL-induced cell migration was suppressed by FLNa knockdown. These previously published and our current results suggest that at normal expression levels, FLNa activity should be strongly regulated to coordinate cell migration.
FLNa works with other proteins to mediate cytoskeletal rearrangements in order to regulate cell adhesion, spreading, and migration. FLNaA binds to Rho, Rac, Cdc42, and ROCK to modulate the cytoskeleton in various cell types (77). FLNa interaction with CD28 is necessary for T-cell cytoskeletal remodeling, along with engagement of lipid microdomains and signaling molecules into the immunological synapse (78). FLNa also assists in HIV infection by connecting HIV-1 receptors to the actin cytoskeleton-regulating network (79). FLNa binding to PAK1 enhances the kinase activity of PAK1, which subsequently phosphorylates FLNa at Ser 2152, resulting in PAK1-dependent membrane ruffling (40). FLNa also stimulates PAK1 by interacting with sphingosine kinase 1, which phosphorylates sphingosine, leading to the direct activation of PAK1 (48). We have recently shown that FLNa directly binds to adapter protein SH2B1β, which is a JAK2 substrate and proposed a model for PRL-dependent regulation of the actin cytoskeleton (44). According to this model, upon ligation of PRL receptor and activation of JAK2, SH2B1β translocates to activated PRL receptor-JAK2 complexes, where it cross-links actin filaments via its two actin-binding domains and binds to FLNa (44, 45). PRL activation of JAK2 also leads to tyrosyl phosphorylation of PAK1, thereby increasing PAK1's activities (both the serine /threonine kinase activity and ability to create potential protein-protein interactions) and stimulating phosphorylation of FLNa. FLNa, in turn, activates PAK1, binds to SH2B1β, and relocates more SH2B1β to the JAK2-PAK1-FLNa complex. Because SH2B1β enhances the tyrosine kinase activity of JAK2 (80), the formation of this multiprotein complex results in enhancement of JAK2 activation and further activation of the JAK2-PAK1-FLNa-actin complex, leading to actin cytoskeleton reorganization. To support this model, our current data have demonstrated that JAK2-dependent tyrosyl phosphorylation of PAK1 enhances Ser 2152 phosphorylation of FLNa by PAK1 (Figure 7). Thus, our current data bring insight into the mechanism of PRL-stimulated motility of breast cancer cells.
Figure 7.
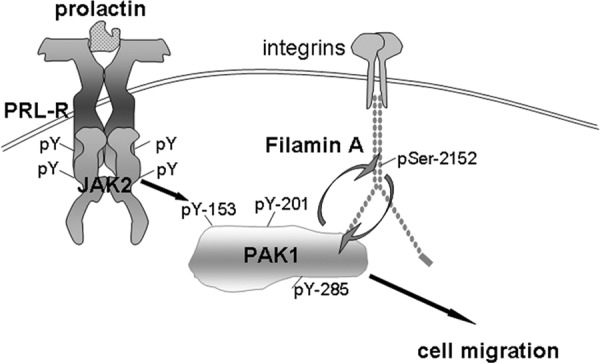
Schematic representation of the proposed working model. Initiation of PRL signaling involves PRL binding to PRL receptor (PRL-R) at the plasma membrane and activation of the tyrosine kinase JAK2, which, in turn, phosphorylates PRL-R. Phosphorylated tyrosines within the receptor and JAK2 recruit an array of signaling proteins, including PAK1. JAK2 tyrosyl phosphorylates PAK1 on Tyr(s) 153, 201, and 285, thereby increases PAK1 activities (both the serine/threonine kinase activity and ability to create potential protein-protein interactions) and stimulates phosphorylation of FLNa on Ser 2152. Phosphorylated FLNa has increased actin-regulating activity to stimulate cell migration and enhances PAK1 kinase activity by a positive feedback loop.
Materials and Methods
Plasmids and antibodies
Construction of PAK1 WT in the pLNCX2 retroviral vector containing the IRES2-EGFP element was described previously (81). Tyrosines 153, 201, and 285 in PAK1 WT were mutated to phenylalanines using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, California) (PAK1 Y3F). Mutations were confirmed by sequencing by the University of Michigan DNA Sequencing Core. The final plasmids PAK1 WT and PAK1 Y3F with N-terminal myc tags were expressed from retroviral constructs that include IRES elements that allow the transcription of a single bicistronic mRNA of myc-PAK1-IRES2-EGFP, and so produce myc-PAK1 together with EGFP as a reporter for expression of PAK1. Hemagglutinin (HA)-tagged PAK1 WT and PAK1 Y3F were described previously (30). Myc-tagged FLNa was from Addgene, Inc (Cambridge, Massachusetts). cDNAs encoding the mutant Rac1 V12 were used with the permission of Dr Hall (Memorial Sloan-Kettering Cancer Center, New York, New York). cDNAs encoding JAK2 WT and kinase-inactive JAK2 K882E mutant were provided by Dr Carter-Su and described earlier (University of Michigan, Ann Arbor, Michigan) (80). JAK2 V617F/pCDNA3 (described in Refs. 82 and 83) was gift of Dr Mayers (University of Michigan). Monoclonal antimyc (9E10) antibody (Ab) and polyclonal anti-PAK1 Ab (N-20) were from Santa Cruz Biotechnology, Inc (Santa Cruz, California). Monoclonal anti-HA Abs was from Roche Applied Science (Indianapolis, Indiana). Monoclonal anti-JAK2 Ab (no. AHO1352), goat antimouse-AlexaFluor 488 Ab, and Texas Red-phalloidin were from Life Technologies, Inc (Gaithersburg, Maryland). Polyclonal anti-phospho-Ser 2152 FLNa Ab was from Cell Signaling Technology, Inc (Danvers, Massachusetts). Monoclonal antiactin Ab (pan Ab-5, clone ACTN05) was from Thermo Scientific (Rockford, Illinois), and monoclonal anti-γ-tubulin (GTU-88) was from Sigma-Aldrich (St Louis, Missouri).
Myc-PAK1/pLNCX2, HA-PAK1/pCMV6, FLNa/pCDNA3, Rac1 V12/pRK5, JAK2 WT/pRK5, JAK2 K882E/pRK5, and JAK2 V617F/pCDNA3 all express their respective cDNAs under the control of a cytomegalovirus promoter.
Cell cultures
T47D cells and their PAK1 clones were maintained in RPMI (Mediatech, Inc, Manassas, Virginia) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich), bovine insulin (0.2 U/ml; Sigma-Aldrich) and antibiotics. HEK 293T cells were maintained in DMEM (Mediatech, Inc) containing 10% calf serum and antibiotics. MCF-7 cells were maintained in DMEM with sodium pyruvate (110 mg/liter) (Mediatech, Inc) supplemented with 10% FBS, bovine insulin, and antibiotics.
To generate stable T47D clones overexpressing GFP, PAK1 WT, or PAK1 Y3F mutant, we used Phoenix cells (gift of Dr. Taylor, University of Toledo, Toledo, Ohio) for virus production. Phoenix cells were maintained at 37°C in DMEM containing 10% FBS, 50 U/ml penicillin, 50 μg/ml streptomycin and supplemented with 4 mM l-glutamine, until 90% confluent. The cells were transfected with pLNCX2-IRES2-EGFP, pLNCX2-myc-PAK1 WT-IRES2-EGFP, or pLNCX2-myc-PAK1 Y3F-IRES2-EGFP using a modification of the polyethylenimine method (84). In 48 hours the virus broth was collected, supplemented with polybrene at a final concentration of 12 μg/ml, and added to the T47D cells at a ratio of 1:3 (viral broth to fresh medium) and cultured at 37°C. The next day, the medium was removed and fresh complete medium was added to the cells. Clonal cell lines were isolated by dilution and expanded, and at least six clonal lines were examined for exogenous expression by antimyc immunoblot.
Transient transfections
For immunofluorescence, MCF-7 cells were transfected using the Fugene 6 kit according to the manufacturer's instruction (Roche Molecular Biochemicals, Indianapolis, Indiana). For coimmunoprecipitation, T47D clones, parental T47D cells, or 293T cells were transfected using the polyethylenimine (84), PolyJet (SignaGen Laboratories, Rockville, Maryland), or calcium phosphate precipitation (85), respectively.
For siRNA transfection, PAK1 siRNA (Cell Signaling Technology), FLNa siRNA (Santa Cruz Biotechnology, Inc), and negative control nontargeting siRNA (Cell Signaling Technology) were transfected using the Lipofectamine RNAiMAX (Invitrogen, Carlsbad, California) reagent according to the manufacturer's instructions. The final concentration of the siRNA was 100 nM.
In vitro kinase assay
To assess PAK1 WT and PAK1 Y3F in vitro kinase activity, myc-tagged WT or mutated PAK1 were IP'd with antimyc Ab from T47D cell lines deprived of serum for 72 hours and treated with or without PRL (200 ng/ml, 20 min) and subjected to an in vitro kinase assay in the presence of 10 μCi of [γ-32P]ATP (MP Biomedicals, Irvine, California), and 5 μg of histone H4 (substrate of PAK1; New England Biolabs, Beverly, Massachusetts). Relative levels of incorporation of 32P into histone H4, an indicator of phosphorylation, were assessed by autoradiography and estimated by a phosphorimager. The same membrane was blotted with antimyc to assess the amount of PAK1 for each condition. Nitrocellulose patterns were scanned and the amount of PAK1 was quantified using Multi-Analyst (Bio-Rad Laboratories, Hercules, California) software. Relative PAK1 kinase activity was then normalized by the amount of IP'd PAK1 for each lane.
Assessment of membrane ruffling
To measure the effect of PAK1 mutants on membrane ruffling, cells expressing the indicated proteins were deprived of serum and treated as indicated in the figure legends. Cells were rapidly rinsed three times with PBS and fixed for 30 minutes at room temperature in 4% formaldehyde. Cell were permeabilized with 0.1% Triton X-100 in PBS for 15 minutes, rinsed three times with PBS, and incubated with antimyc followed by goat antimouse-AlexaFluor 488 (MCF-7 cells). F-actin was stained with Texas Red-phalloidin. Transfected cells expressing myc-tagged forms of PAK1s were located with a fluorescein isothiocyanate filter set using a Zeiss Axiovert 200 microscope (Carl Zeiss, Thornwood, New York). T47D cells stably expressing GFP, WT PAK1, or PAK1 Y3F were stained with only Texas Red-phalloidin and located with a tetramethylrhodamine isothiocyanate filter set using a Zeiss Axiovert 200 microscope. Ruffling index was calculated as the number of total ruffles, counted for 100 transfected cells, divided by the total number of assessed cells (100 cells for each experimental condition). Each transfection was repeated at least three times with similar results.
Cell migration assays
Boyden chamber assay.
In order to assess the effect of PAK1 on cell migration, T47D cells stably expressing GFP, PAK1 WT, or PAK1 Y3F were serum deprived, trypsinized, washed in PBS to remove trypsin, and counted on a hemacytometer. Equal numbers of cells for each condition were placed in deprivation media in the upper chamber of a Boyden chamber (Corning, Inc, Corning, New York). Each chamber was coated with collagen IV (Sigma-Aldrich; 1 μg/ml) on the underside of the filter. Deprivation media with or without 500 ng/ml PRL was placed in the lower chamber. Cells were allowed to migrate for 48 hours, after which nonmigrating cells were removed from the upper chamber by a cotton swab. Cells from five separate fields that had migrated through the pores of the membrane to the underside of the filter were counted after fixation and staining with propidium iodide and visualization by fluorescent microscopy.
Wounding assay.
T47D cells stably expressing GFP, PAK1, or PAK1 Y3F were plated at high density onto tissue culture dishes coated with collagen IV (Sigma-Aldrich; 1 μg/ml). The next day, cells were deprived of serum for 24 hours. After deprivation, monolayers of cells were scarified using a plastic pipette tip, washed extensively, and incubated in deprivation media with or without 200 ng/ml PRL. Ten measurements along each wound were taken using ImageJ software at the initial time of wounding and 18 hours later, in order to calculate the percentage wound closure in 18 hours for each condition.
Coimmunoprecipitation and immunoblotting
Parental T47D cells and T47D clones were transfected using the Amaxa method (Lonza Cologne AG, Cologne, Germany) according to the manufacturer's protocol. 293T cells were transiently transfected using calcium phosphate precipitation. Transfected cells were serum deprived for 72 hours (T47D clones), 24 h (parental T47D cells), or overnight (293T cells) before the assay and treated with (200 ng/ml, 15 min) or without PRL. The cells were rinsed three times with 10 mM sodium phosphate, pH 7.4, 150 mM NaCl, 1 mM Na orthovanadate. Cells were then solubilized in lysis buffer [50 mM Tris (pH 7.5), 0.1% Triton X-100, 150 mM NaCl, 2 mM EGTA, 1 mM Na orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin] and centrifuged at 14,000 × g for 10 minutes at 4° C. The supernatant (cell lysate) was incubated with affinity-purified antimyc antibody on ice for 2 hours. The immune complexes were rotated with protein A/G PLUS-agarose (Santa Cruz Biotechnology, Inc.) for 5 hours at 4°C. The beads were washed three times with washing buffer (50 mM Tris (pH 7.5), 0.1% Triton X-100, 150 mM NaCl, 2 mM EGTA) and boiled for 5 minutes in a mixture (80:20) of lysis buffer and SDS-PAGE sample buffer [250 mM Tris-HCl (pH 6.8), 10% sodium dodecyl sulfate, 10% β-mercaptoethanol, 40% glycerol, 0.01% bromphenol blue]. The solubilized proteins were separated by SDS-PAGE followed by immunoblotting with the indicated antibodies and visualization with the ECL detection system.
Acknowledgments
We thank Drs Mayers and Carter-Su (University of Michigan, Ann Arbor, Michigan) for providing JAK2 V617F, JAK2 WT, and JAK2 K882E constructs and Dr Taylor (University of Toledo, Toledo, Ohio) for providing the Phoenix cells. We thank Diana Suen (University of Toledo, Toledo, Ohio) for help with assessment of cell ruffling.
This work was supported by Grants from the National Institutes of Health (R01 DK88127 to M.D.) and R01 CA131990 (to R.R.M.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Ab
- antibody
- EGFP
- enhanced green fluorescent protein
- FBS
- fetal bovine serum
- FLNa
- filamin A
- GFP
- green fluorescent protein
- HA
- hemagglutinin
- IP'd
- immunoprecipitated
- IRES
- internal ribosome entry site
- JAK2
- Janus tyrosine kinase
- Nek3
- NIMA-related kinase 3
- PAK1
- p21-activated serine-threonine kinase
- PRL
- prolactin
- pTyr
- tyrosyl phosphorylated
- Ser
- serine
- siRNA
- small interfering RNA
- STAT
- signal transducer and activator of transcription
- Tyr
- tyrosine
- WT
- wild type.
References
- 1. Goffin V, Bernichtein S, Touraine P, Kelly PA. Development and potential clinical uses of human prolactin receptor antagonists. Endocr Rev. 2005;26:400–422 [DOI] [PubMed] [Google Scholar]
- 2. Bernichtein S, Touraine P, Goffin V. New concepts in prolactin biology. J Endocrinol. 2010;206:1–11 [DOI] [PubMed] [Google Scholar]
- 3. Ball RK, Friis RR, Schoenenberger CA, Doppler W, Groner B. Prolactin regulation of β-casein gene expression and of a cytosolic 120-kd protein in a cloned mouse mammary epithelial cell line. Embo J. 1988;7:2089–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. DaSilva L, Rui H, Erwin RA, Howard OM, Kirken RA, Malabarba MG, Hackett RH, Larner AC, Farrar WL. Prolactin recruits STAT1, STAT3 and STAT5 independent of conserved receptor tyrosines TYR402, TYR479, TYR515 and TYR580. Mol Cell Endocrinol. 1996;117:131–140 [DOI] [PubMed] [Google Scholar]
- 5. Schaber JD, Fang H, Xu J, Grimley PM, Rui H. Prolactin activates Stat1 but does not antagonize Stat1 activation and growth inhibition by type I interferons in human breast cancer cells. Cancer Res. 1998;58:1914–1919 [PubMed] [Google Scholar]
- 6. Das R, Vonderhaar BK. Involvement of SHC, GRB2, SOS and RAS in prolactin signal transduction in mammary epithelial cells. Oncogene. 1996;13:1139–1145 [PubMed] [Google Scholar]
- 7. Das R, Vonderhaar BK. Activation of raf-1, MEK, and MAP kinase in prolactin responsive mammary cells. Breast Cancer Res Treat. 1996;40:141–149 [DOI] [PubMed] [Google Scholar]
- 8. Banerjee R, Vonderhaar BK. Prolactin-induced protein kinase C activity in a mouse mammary cell line (NOG-8). Mol Cell Endocrinol. 1992;90:61–67 [DOI] [PubMed] [Google Scholar]
- 9. Waters SB, Rillema JA. Role of protein kinase C in the prolactin-induced responses in mouse mammary gland explants. Mol Cell Endocrinol. 1989;63:159–166 [DOI] [PubMed] [Google Scholar]
- 10. Berlanga JJ, Fresno Vara JA, Martin-Perez J, Garcia-Ruiz JP. Prolactin receptor is associated with c-src kinase in rat liver. Mol Endocrinol. 1995;9:1461–1467 [DOI] [PubMed] [Google Scholar]
- 11. Clevenger CV, Medaglia MV. The protein tyrosine kinase P59fyn is associated with prolactin (PRL) receptor and is activated by PRL stimulation of T-lymphocytes. Mol Endocrinol. 1994;8:674–681 [DOI] [PubMed] [Google Scholar]
- 12. Berlanga JJ, Gualillo O, Buteau H, Applanat M, Kelly PA, Edery M. Prolactin activates tyrosyl phosphorylation of insulin receptor substrate 1 and phosphatidylinositol-3-OH kinase. J Biol Chem. 1997;272:2050–2052 [DOI] [PubMed] [Google Scholar]
- 13. Clevenger CV, Chang WP, Ngo W, Pasha TL, Montone KT, Tomaszewski JE. Expression of prolactin and prolactin receptor in human breast carcinoma. Evidence for an autocrine/paracrine loop. Am J Pathol. 1995;146:695–705 [PMC free article] [PubMed] [Google Scholar]
- 14. Nagasawa H, Nozaki D, Miura K, Niki K, Namiki H. Correlation of plasma prolactin and growth hormone with normal and preneoplastic mammary gland growth in virgin SHN mice. Eur J Cancer Clin Oncol. 1985;21:1109–1111 [DOI] [PubMed] [Google Scholar]
- 15. Clevenger CV, Furth PA, Hankinson SE, Schuler LA. The role of prolactin in mammary carcinoma. Endocr Rev. 2003;24:1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clevenger CV, Gadd SL, Zheng J. New mechanisms for PRLr action in breast cancer. Trends Endocrinol Metab. 2009;20:223–229 [DOI] [PubMed] [Google Scholar]
- 17. Harvey PW, Everett DJ, Springall CJ. Adverse effects of prolactin in rodents and humans: breast and prostate cancer. J Psychopharmacol. 2008;22:20–27 [DOI] [PubMed] [Google Scholar]
- 18. Tworoger SS, Hankinson SE. Prolactin and breast cancer risk. Cancer Lett. 2006;243:160–169 [DOI] [PubMed] [Google Scholar]
- 19. Vonderhaar BK. Prolactin: the forgotten hormone of human breast cancer. Pharmacol Ther. 1998;79:169–178 [DOI] [PubMed] [Google Scholar]
- 20. Vonderhaar BK. Prolactin involvement in breast cancer. Endocr Relat Cancer. 1999;6:389–404 [DOI] [PubMed] [Google Scholar]
- 21. Wagner KU, Rui H. Jak2/Stat5 signaling in mammogenesis, breast cancer initiation and progression. J Mammary Gland Biol Neoplasia. 2008;13:93–103 [DOI] [PubMed] [Google Scholar]
- 22. Maus MV, Reilly SC, Clevenger CV. Prolactin as a chemoattractant for human breast carcinoma. Endocrinology. 1999;140:5447–5450 [DOI] [PubMed] [Google Scholar]
- 23. Miller SL, DeMaria JE, Freier DO, Riegel AM, Clevenger CV. Novel association of Vav2 and Nek3 modulates signaling through the human prolactin receptor. Mol Endocrinol. 2005;19:939–949 [DOI] [PubMed] [Google Scholar]
- 24. Miller SL, Antico G, Raghunath PN, Tomaszewski JE, Clevenger CV. Nek3 kinase regulates prolactin-mediated cytoskeletal reorganization and motility of breast cancer cells. Oncogene. 2007;26:4668–4678 [DOI] [PubMed] [Google Scholar]
- 25. Liby K, Neltner B, Mohamet L, Menchen L, Ben-Jonathan N. Prolactin overexpression by MDA-MB-435 human breast cancer cells accelerates tumor growth. Breast Cancer Res Treat. 2003;79:241–252 [DOI] [PubMed] [Google Scholar]
- 26. Nouhi Z, Chughtai N, Hartley S, Cocolakis E, Lebrun JJ, Ali S. Defining the role of prolactin as an invasion suppressor hormone in breast cancer cells. Cancer Res. 2006;66:1824–1832 [DOI] [PubMed] [Google Scholar]
- 27. Sultan AS, Xie J, LeBaron MJ, Ealley EL, Nevalainen MT, Rui H. Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene. 2005;24:746–760 [DOI] [PubMed] [Google Scholar]
- 28. Kline JB, Moore DJ, Clevenger CV. Activation and association of the Tec tyrosine kinase with the human prolactin receptor: mapping of a Tec/Vav1-receptor binding site. Mol Endocrinol. 2001;15:832–841 [DOI] [PubMed] [Google Scholar]
- 29. Akhtar N, Streuli CH. Rac1 links integrin-mediated adhesion to the control of lactational differentiation in mammary epithelia. J Cell Biol. 2006;173:781–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rider L, Shatrova A, Feener EP, Webb L, Diakonova M. JAK2 tyrosine kinase phosphorylates PAK1 and regulates PAK1 activity and functions. J Biol Chem. 2007;282:30985–30996 [DOI] [PubMed] [Google Scholar]
- 31. Vadlamudi RK, Kumar R. P21-activated kinases in human cancer. Cancer Metastasis Rev. 2003;22:385–393 [DOI] [PubMed] [Google Scholar]
- 32. Adam L, Vadlamudi R, Kondapaka SB, Chernoff J, Mendelsohn J, Kumar R. Heregulin regulates cytoskeletal reorganization and cell migration through the p21-activated kinase-1 via phosphatidylinositol-3 kinase. J Biol Chem. 1998;273:28238–28246 [DOI] [PubMed] [Google Scholar]
- 33. Adam L, Vadlamudi R, Mandal M, Chernoff J, Kumar R. Regulation of microfilament reorganization and invasiveness of breast cancer cells by kinase dead p21-activated kinase-1. J Biol Chem. 2000;275:12041–12050 [DOI] [PubMed] [Google Scholar]
- 34. Vadlamudi RK, Adam L, Wang RA, Mandal M, Nguyen D, Sahin A, Chernoff J, Hung MC, Kumar R. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem. 2000;275:36238–36244 [DOI] [PubMed] [Google Scholar]
- 35. Menard RE, Jovanovski AP, Mattingly RR. Active p21-activated kinase 1 rescues MCF10A breast epithelial cells from undergoing anoikis. Neoplasia. 2005;7:638–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stofega MR, Sanders LC, Gardiner EM, Bokoch GM. Constitutive p21-activated kinase (PAK) activation in breast cancer cells as a result of mislocalization of PAK to focal adhesions. Mol Biol Cell. 2004;15:2965–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259 [DOI] [PubMed] [Google Scholar]
- 38. Sanders LC, Matsumura F, Bokoch GM, de Lanerolle P. Inhibition of myosin light chain kinase by p21-activated kinase. Science. 1999;283:2083–2085 [DOI] [PubMed] [Google Scholar]
- 39. Turner CE, Brown MC, Perrotta JA, Riedy MC, Nikolopoulos SN, McDonald AR, Bagrodia S, Thomas S, Leventhal PS. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: A role in cytoskeletal remodeling. J Cell Biol. 1999;145:851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vadlamudi RK, Li F, Adam L, Nguyen D, Ohta Y, Stossel TP, Kumar R. Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat Cell Biol. 2002;4:681–690 [DOI] [PubMed] [Google Scholar]
- 41. Vadlamudi RK, Li F, Barnes CJ, Bagheri-Yarmand R, Kumar R. p41-Arc subunit of human Arp2/3 complex is a p21-activated kinase-1-interacting substrate. EMBO Rep. 2004;5:154–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xiao GH, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem. 2002;277:883–886 [DOI] [PubMed] [Google Scholar]
- 43. Tao J, Oladimeji P, Rider L, Diakonova M. PAK1-Nck regulates cyclin D1 promoter activity in response to prolactin. Mol Endocrinol. 2011;25:1565–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rider L, Diakonova M. Adapter protein SH2B1β binds filamin A to regulate prolactin-dependent cytoskeletal reorganization and cell motility. Mol Endocrinol. 2011;25:1231–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rider L, Tao J, Snyder S, Brinley B, Lu J, Diakonova M. Adapter protein SH2B1β cross-links actin filaments and regulates actin cytoskeleton. Mol Endocrinol. 2009;23:1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stossel TP, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS. Filamins as integrators of cell mechanics and signalling. Nat Rev Mol Cell Biol. 2001;2:138–145 [DOI] [PubMed] [Google Scholar]
- 47. Nakamura F, Stossel TP, Hartwig JH. The filamins: organizers of cell structure and function. Cell Adh Migr. 2011;5:160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maceyka M, Alvarez SE, Milstien S, Spiegel S. Filamin A links sphingosine kinase 1 and sphingosine-1-phosphate receptor 1 at lamellipodia to orchestrate cell migration. Mol Cell Biol. 2008;28:5687–5697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Campbell GS, Argetsinger LS, Ihle JN, Kelly PA, Rillema JA, Carter-Su C. Activation of JAK2 tyrosine kinase by prolactin receptors in Nb2 cells and mouse mammary gland explants. Proc Natl Acad Sci USA. 1994;91:5232–5236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lebrun JJ, Ali S, Sofer L, Ullrich A, Kelly PA. Prolactin-induced proliferation of Nb2 cells involves tyrosine phosphorylation of the prolactin receptor and its associated tyrosine kinase JAK2. J Biol Chem. 1994;269:14021–14026 [PubMed] [Google Scholar]
- 51. Rui H, Lebrun JJ, Kirken RA, Kelly PA, Farrar WL. JAK2 activation and cell proliferation induced by antibody-mediated prolactin receptor dimerization. Endocrinology. 1994;135:1299–1306 [DOI] [PubMed] [Google Scholar]
- 52. Rui L, Herrington J, Carter-Su C. SH2-B is required for nerve growth factor-induced neuronal differentiation. J Biol Chem. 1999;274:10590–10594 [DOI] [PubMed] [Google Scholar]
- 53. Jiao H, Berrada K, Yang W, Tabrizi M, Platanias LC, Yi T. Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1. Mol Cell Biol. 1996;16:6985–6992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Manser E, Leung T, Salihuddin H, Zhao ZS, Lim L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 1994;367:40–46 [DOI] [PubMed] [Google Scholar]
- 55. Bokoch GM, Reilly AM, Daniels RH, King CC, Olivera A, Spiegel S, Knaus UG. A GTPase-independent mechanism of p21-activated kinase activation. Regulation by sphingosine and other biologically active lipids. J Biol Chem. 1998;273:8137–8144 [DOI] [PubMed] [Google Scholar]
- 56. King CC, Gardiner EM, Zenke FT, Bohl BP, Newton AC, Hemmings BA, Bokoch GM. p21-activated kinase (PAK1) is phosphorylated and activated by 3-phosphoinositide-dependent kinase-1 (PDK1). J Biol Chem. 2000;275:41201–41209 [DOI] [PubMed] [Google Scholar]
- 57. Zhao ZS, Manser E, Loo TH, Lim L. Coupling of PAK-interacting exchange factor PIX to GIT1 promotes focal complex disassembly. Mol Cell Biol. 2000;20:6354–6363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Menard RE, Mattingly RR. Cell surface receptors activate p21-activated kinase 1 via multiple Ras and PI3-kinase-dependent pathways. Cell Signal. 2003;15:1099–1109 [DOI] [PubMed] [Google Scholar]
- 59. Menard RE, Mattingly RR. Gbetagamma subunits stimulate p21-activated kinase 1 (PAK1) through activation of PI3-kinase and Akt but act independently of Rac1/Cdc42. FEBS Lett. 2004;556:187–192 [DOI] [PubMed] [Google Scholar]
- 60. Bokoch GM, Wang Y, Bohl BP, Sells MA, Quilliam LA, Knaus UG. Interaction of the Nck adapter protein with p21-activated kinase (PAK1). J Biol Chem. 1996;271:25746–25749 [DOI] [PubMed] [Google Scholar]
- 61. Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781 [DOI] [PubMed] [Google Scholar]
- 62. Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009;28:51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kumar R, Gururaj AE, Barnes CJ. p21-activated kinases in cancer. Nat Rev Cancer. 2006;6:459–471 [DOI] [PubMed] [Google Scholar]
- 64. Dharmawardhane S, Sanders LC, Martin SS, Daniels RH, Bokoch GM. Localization of p21-activated kinase 1 (PAK1) to pinocytic vesicles and cortical actin structures in stimulated cells. J Cell Biol. 1997;138:1265–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210 [DOI] [PubMed] [Google Scholar]
- 66. Sells MA, Boyd JT, Chernoff J. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J Cell Biol. 1999;145:837–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sells MA, Pfaff A, Chernoff J. Temporal and spatial distribution of activated Pak1 in fibroblasts. J Cell Biol. 2000;151:1449–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Manser E, Huang HY, Loo TH, Chen XQ, Dong JM, Leung T, Lim L. Expression of constitutively active α-PAK reveals effects of the kinase on actin and focal complexes. Mol Cell Biol. 1997;17:1129–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Borm B, Requardt RP, Herzog V, Kirfel G. Membrane ruffles in cell migration: indicators of inefficient lamellipodia adhesion and compartments of actin filament reorganization. Exp Cell Res. 2005;302:83–95 [DOI] [PubMed] [Google Scholar]
- 70. Ridley AJ. Membrane ruffling and signal transduction. Bioessays. 1994;16:321–327 [DOI] [PubMed] [Google Scholar]
- 71. Cunningham CC, Gorlin JB, Kwiatkowski DJ, Hartwig JH, Janmey PA, Byers HR, Stossel TP. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 1992;255:325–327 [DOI] [PubMed] [Google Scholar]
- 72. Woo MS, Ohta Y, Rabinovitz I, Stossel TP, Blenis J. Ribosomal S6 kinase (RSK) regulates phosphorylation of filamin A on an important regulatory site. Mol Cell Biol. 2004;24:3025–3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Feng Y, Chen MH, Moskowitz IP, Mendonza AM, Vidali L, Nakamura F, Kwiatkowski DJ, Walsh CA. Filamin A (FLNA) is required for cell-cell contact in vascular development and cardiac morphogenesis. Proc Natl Acad Sci USA. 2006;103:19836–19841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Johansen LD, Naumanen T, Knudsen A, Westerlund N, Gromova I, Junttila M, Nielsen C, Bottzauw T, Tolkovsky A, Westermarck J, Coffey ET, Jaattela M, Kallunki T. IKAP localizes to membrane ruffles with filamin A and regulates actin cytoskeleton organization and cell migration. J Cell Sci. 2008;121:854–864 [DOI] [PubMed] [Google Scholar]
- 75. Sarkisian MR, Bartley CM, Chi H, Nakamura F, Hashimoto-Torii K, Torii M, Flavell RA, Rakic P. MEKK4 signaling regulates filamin expression and neuronal migration. Neuron. 2006;52:789–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xu Y, Bismar TA, Su J, Xu B, Kristiansen G, Varga Z, Teng L, Ingber DE, Mammoto A, Kumar R, Alaoui-Jamali MA. Filamin A regulates focal adhesion disassembly and suppresses breast cancer cell migration and invasion. J Exp Med. 2010;207:2421–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ohta Y, Suzuki N, Nakamura S, Hartwig JH, Stossel TP. The small GTPase RalA targets filamin to induce filopodia. Proc Natl Acad Sci USA. 1999;96:2122–2128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tavano R, Contento RL, Baranda SJ, Soligo M, Tuosto L, Manes S, Viola A. CD28 interaction with filamin-A controls lipid raft accumulation at the T-cell immunological synapse. Nat Cell Biol. 2006;8:1270–1276 [DOI] [PubMed] [Google Scholar]
- 79. Jimenez-Baranda S, Gomez-Mouton C, Rojas A, Martinez-Prats L, Mira E, Ana Lacalle R, Valencia A, Dimitrov DS, Viola A, Delgado R, Martinez AC, Manes S. Filamin-A regulates actin-dependent clustering of HIV receptors. Nat Cell Biol. 2007;9:838–846 [DOI] [PubMed] [Google Scholar]
- 80. Rui L, Carter-Su C. Identification of SH2-bbeta as a potent cytoplasmic activator of the tyrosine kinase Janus kinase 2. Proc Natl Acad Sci USA. 1999;96:7172–7177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li Q, Mullins SR, Sloane BF, Mattingly RR. p21-Activated kinase 1 coordinates aberrant cell survival and pericellular proteolysis in a three-dimensional culture model for premalignant progression of human breast cancer. Neoplasia. 2008;10:314–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, Myers MG., Jr Regulation of Jak kinases by intracellular leptin receptor sequences. J Biol Chem. 2002;277:41547–41555 [DOI] [PubMed] [Google Scholar]
- 83. Robertson SA, Koleva RI, Argetsinger LS, Carter-Su C, Marto JA, Feener EP, Myers MG., Jr Regulation of Jak2 function by phosphorylation of Tyr317 and Tyr637 during cytokine signaling. Mol Cell Biol. 2009;29:3367–3378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]