

Abstract
Purpose
EGFR-mutant lung cancer was first described as a new clinical entity in 2004. Here, we present an update on new controversies and conclusions regarding the disease.
Methods
This article reviews the clinical implications of EGFR mutations in lung cancer with a focus on epidermal growth factor receptor tyrosine kinase inhibitor resistance.
Results
The discovery of EGFR mutations has altered the ways in which we consider and treat non–small-cell lung cancer (NSCLC). Patients whose metastatic tumors harbor EGFR mutations are expected to live longer than 2 years, more than double the previous survival rates for lung cancer.
Conclusion
The information presented in this review can guide practitioners and help them inform their patients about EGFR mutations and their impact on the treatment of NSCLC. Efforts should now concentrate on making EGFR-mutant lung cancer a chronic rather than fatal disease.
INTRODUCTION
EGFR-mutant lung cancer was first described as a potential distinct clinical entity in 2004.1–3 Eight years later, multiple studies have undisputedly validated the disease as a unique subset of lung cancer, with its own clinical features, natural history, and clinical course. EGFR-mutant lung cancer also serves as a paradigm for an oncogene-addicted solid tumor that can be effectively treated with specific targeted therapy (ie, first-generation epidermal growth factor receptor [EGFR] tyrosine kinase inhibitors [TKIs], gefitinib [Iressa; AstraZeneca, London, United Kingdom] and erlotinib [Tarceva; Genentech, South San Francisco, CA]).4–9 Multiple reviews have been published on the rationale for targeting EGFR in cancer and the subsequent discovery of EGFR mutations in lung cancer.10–12 Here, we review new controversies and conclusions regarding the clinical implications of EGFR mutations in lung cancer, with a focus on EGFR TKI resistance.
DRUG-SENSITIVE EGFR MUTATIONS IN LUNG CANCER
EGFR is a receptor tyrosine kinase that belongs to the EGFR family, consisting of four members: EGFR, ERBB2, ERBB3, and ERBB4. Under normal circumstances, binding of ligands (eg, epidermal growth factor, transforming growth factor-alpha) activates the intracellular tyrosine kinase activity of EGFR via homo- or heterodimerization with EGFR family members.13 In lung cancer, EGFR mutations occur in exons encoding the ATP-binding pocket of the kinase domain (exons 18 to 21; Fig 1). In a cohort of nearly 1,200 patients with EGFR mutations linked to clinical outcomes, more than 145 different types of nucleotide changes have been reported within the EGFR kinase domain.14
Fig 1.

Distribution of EGFR mutations in lung cancer. Schematic of the kinase domain of epidermal growth factor receptor showing exons 18 to 21. Activating drug-sensitive mutations are shown on the top, and tyrosine kinase inhibitor (TKI) –resistant mutations are depicted on the bottom (red: acquired resistant mutations). The most common activating mutations in EGFR are a point mutation in exon 21, which substitutes an arginine for a leucine (L858R), and a small deletion in exon 19 that removes four amino acids (LREA). Together, these account for approximately 85% of the TKI-sensitive mutations observed in EGFR-mutant tumors. Many rare mutations have also been reported.14
The most clinically relevant and extensively studied drug-sensitive mutations are deletions in exon 19 that eliminate a common amino acid motif (LREA) and point mutations in exon 21 that lead to a substitution of arginine for leucine at position 858 (L858R). Together, these two classes of mutations account for approximately 85% of EGFR mutations in the disease. They are constitutively active and oncogenic15,16 as a result of a disruption of autoinhibitory interactions.17 Biochemical studies indicate that these mutants preferentially bind to drugs like gefitinib and erlotinib over ATP.17,18 Other potential drug-sensitive mutations occur at much lower frequency: G719X (3%), L861X (2%),14 and exon 19 insertions (1%).19 The former two were associated with drug sensitivity in the original reports on EGFR mutations,1,2 whereas the exon 19 insertions were just recently reported as drug sensitive.19 The rarity of clinical data associated with these less frequent mutants has made it more difficult to determine how drug sensitive they are in patients, but new data are emerging.20,21
CLINICAL FEATURES ASSOCIATED WITH EGFR MUTATIONS
EGFR mutations can be found in all histologic subtypes of non–small-cell lung cancer (NSCLC), including adenocarcinoma, large-cell carcinoma, and squamous cell carcinoma.14 In North American/European and East Asian countries, EGFR mutations are found in 10% and 30% of unselected NSCLCs,22,23 respectively. Clinical features likely to be associated with EGFR mutations include adenocarcinoma histology, history of never smoking cigarettes (ie, fewer than 100 cigarettes in a lifetime),3,22 and East Asian ethnicity.22 Female sex was originally thought to be correlated with EGFR mutations, but data suggest that this association was made because more women are likely to be never-smokers,24 not necessarily because of a true sex bias. Sixty percent to 80% of tumors from East Asian never-smokers with lung adenocarcinoma harbor EGFR mutations,25,26 whereas only 30% to 50% of tumors from North American/European counterparts have such mutations.3,22 The reason for this discrepancy is unclear; as of yet, no study has determined if US citizens of East Asian descent diagnosed with lung cancer have the same prevalence of EGFR mutations as East Asians themselves. Such a finding would suggest a genetic rather than environmental cause of EGFR alterations.
Most importantly, EGFR mutations (mostly exon 19 deletions and L858R point mutations) are associated with a clinical benefit from gefitinib and erlotinib. In early phase III trials, these drugs were tested in unselected patients with NSCLC and showed less than 10% radiographic response rates (RRs) with short (≤ 3 months) progression-free survival (PFS) rates27–29 (Table 1). After the discovery of EGFR mutations, several prospective single-arm first-line studies enrolling only patients with EGFR-mutant tumors reported unprecedented RRs (73% to 91%) and prolonged PFS (7.7 to 13.3 months).33 Thereafter, five large prospective phase III first-line trials directly compared an EGFR TKI versus platinum doublet chemotherapy in patients with NSCLC harboring EGFR mutations. These trials strongly confirmed the benefit of gefitinib or erlotinib in EGFR-mutant lung cancer, regardless of ethnic background (Table 1).4,6–9,30–32 By comparison, patients with EGFR wild-type tumors displayed 1% RRs and improved PFS with chemotherapy rather than a TKI.4 To receive EGFR TKIs in many regions, such as Canada and the European Union, patients must now have a documented EGFR mutation. In the United States, mutation testing is available in multiple molecular diagnostics laboratories certified by the College of American Pathologists and Certified Laboratory Improvements Amendment of 1988, but the US Food and Drug Administration (FDA) has never required that only patients with EGFR mutations should be treated with an EGFR TKI. The rationale behind this was that the BR.21 trial, which compared survival rates in unselected patients with NSCLC treated with erlotinib versus placebo, showed a statistically significant survival benefit for patients taking the drug, even though the absolute difference was a mere 2 months (6.7 v 4.7 months; P < .001).28 However, consistent with the notion that erlotinib is more effective against EGFR-mutant tumors, a recent study reported that in patients with NSCLC harboring wild-type EGFR, docetaxel induced a higher RR (13.9% v 2.2%; P = .004) and longer PFS (3.4 v 2.4 months; hazard ratio [HR], 0.69; P = .014) in the second-line setting than erlotinib.34
Table 1.
Select Phase III Clinical Trials in Lung Cancer Involving EGFR TKIs
| Trial | Year | Line | No. of Participants | Race | EGFR Mutant (%) | EGFR TKI | Reference Arm | TKI v Reference |
|||
|---|---|---|---|---|---|---|---|---|---|---|---|
| RR (%) | CR (%) | PFS (months) | OS (months) | ||||||||
| ISEL27 | 2005 | Second to third | 1,692 | White, 75%; Asian, 21%* | 12.1† | Gefitinib | Placebo | 8.0 v 1.3 | NA | 3.0 v 2.6 | 5.6 v 5.1 |
| BR.2128 | 2005 | Second to third | 731 | Asian, 12%; other, 88% | 23‡ | Erlotinib | Placebo | 8.9 v < 1 | 0.7 v 0 | 2.2 v 1.8 | 6.7 v 4.7 |
| INTEREST29 | 2008 | Second | 1,433 | White, 75%; Asian, 21%* | 14.8§ | Gefitinib | Docetaxel | 9.1 v 7.6 | NA | 2.2 v 2.2 | 7.6 v 8.0 |
| IPASS4,30 | 2009 | First | 1,217 | East Asian, 100% | 59.7‖ | Gefitinib | Platinum doublet | 43.0 v 32.2 | NA | 5.7 v 5.8 | 18.8 v 17.4 |
| IPASS subgroup4,30 | 2009 | First | 261 | East Asian, 100% | 100 | Gefitinib | Platinum doublet | 71.2 v 47.3 | NA | 9.5 v 6.3 | 21.6 v 21.9 |
| WJTOG34056,31 | 2009 | First | 172 | East Asian, 100% | 100 | Gefitinib | Platinum doublet | 62.1 v 32.2 | NA | 9.2 v 6.3 | 35.5 v 38.8 |
| NEJ0027 | 2009 | First | 224 | East Asian, 100% | 100 | Gefitinib | Platinum doublet | 73.7 v 30.7 | 4.4 v 0 | 10.8 v 5.4 | 30.5 v 23.6 |
| OPTIMAL8,32 | 2011 | First | 165 | East Asian, 100% | 100 | Erlotinib | Platinum doublet | 82 v 36 | 2 v 0 | 13.1 v 4.6 | 22.7 v 28.9 |
| EURTAC9 | 2012 | First | 174 | White, 100% (Hispanic) | 100 | Erlotinib | Platinum doublet | 64 v 18 | 3 v 0 | 9.7 v 5.2 | 19.3 v 19.5 |
Abbreviations: CR, complete response; EGFR, epidermal growth factor receptor; EURTAC, European Tarceva Versus Chemotherapy; INTEREST, IRESSA Non-Small-Cell Lung Cancer Trial Evaluating Response and Survival Against Taxotere; IPASS, Iressa Pan-Asia Study; ISEL, IRESSA Survival Evaluation in Lung Cancer; NA, not applicable; OPTIMAL, Open Label, Phase III Study Comparing First Line Tarceva vs Cisplatin Plus Gemcitabine in Chinese Advanced/Metastatic Non-Small-Cell Lung Cancer Patients With EGFR Activating Mutations; OS, overall survival; PFS, progression-free survival; RR, response rate; TKI, tyrosine kinase inhibitor
Excludes people of Indian origin.
26 positive in 215 tested samples.
40 positive in 177 tested samples.
44 positive in 297 tested samples.
261 positive in 437 tested samples.
IMPACT OF EGFR TKIS ON EGFR-MUTANT LUNG CANCER
No randomized prospective studies have yet officially shown that EGFR TKIs prolong overall survival (OS) compared with chemotherapy (Table 1). One explanation for this discrepancy is that once patients in the chemotherapy arm experience disease progression, they can still display high RRs and prolonged PFS after switching to an EGFR TKI.7 This crossover confounds subsequent survival analyses.
However, several lines of evidence clearly show that patients with EGFR-mutant tumors and treated with TKIs experience historically high survival rates. Multiple prospective first-line clinical trials have now demonstrated that such patients live longer than 2 years.6,7 Such long OS was not routinely observed before the approval of EGFR TKIs (Table 1; Fig 2A). Consistent with these data, a retrospective analysis recently showed that OS in Japanese patients who started first-line chemotherapy after gefitinib was approved was much longer than the OS in patients who started chemotherapy before gefitinib approval (27.2 v 13.6 months; P < .001).41 Furthermore, although gefitinib was withdrawn from the market in the United States in 2005, approximately 250 patients with NSCLC were still alive as of 2011 in the AstraZeneca Iressa Expanded Access Program (NCT00034879), demonstrating that long-term survival of patients with NSCLC is possible while they receive EGFR TKIs.42
Fig 2.

Impact of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) on survival in patients with lung cancers harboring EGFR mutations. (A) The graph depicts the overall survival (OS) rates achieved in various prospective trials conducted in the United States/Europe (left panel) and East Asia (right panel) before and after the introduction of EGFR TKIs. After 2006, OS was consistently longer than 18 months in patients with EGFR-mutant tumors.6,9,35,40 Five trials35–37,39,40 were conducted in unselected patients with non–small-cell lung cancer (NSCLC), and the other trials were performed in patients with EGFR-mutant tumors.9,6,7 One trial38 was for never- or light former smokers; the OS data were from the subgroup of patients with EGFR-mutated lung cancers treated with erlotinib. (B) Age-adjusted 12-month survival rates in patients with metastatic NSCLC in the SEER database. The rate increases especially in the Asian population from 2003 to 2004 onward. The thick lines are the trends for the years 1998 to 2003, and the thin lines are the predicted improvement in 12-month survival, given the 1998 to 2003 trends. (C) Age-adjusted incidence and prevalence (per 100,000 people in the United States) of NSCLC in the SEER database. The incidence decreases starting from 2002, whereas the prevalence displays a small increase. (A to C) Arrows indicate years in which drugs were approved.
The impact of the introduction of EGFR TKIs on the treatment of lung cancer can be further gleaned from an analysis of the US Surveillance Epidemiology and End Results (SEER) program database. Population-based resources like the SEER database do not include detailed information about patients by tumor mutation status. However, inferences can be made based on the frequency of patients with EGFR-mutant NSCLC among specific ethnic groups. For example, the 12-month survival rates for metastatic NSCLC from 1997 to 2008 of the three different ethnic groups represented in the SEER database (ie, white; African American; and Asian-Pacific Islander, Alaskan, and Native American) show that compared with predicted rates, there has been an increase in the survival rates of all groups. For Asians, the growth was larger than that of the other groups. This increase coincides with the widespread introduction of EGFR TKIs into the clinic; gefitinib was FDA approved for use in NSCLC in May 2003, and erlotinib was FDA approved in November 2004 (Fig 2B). As we have stated, lung tumors from Asians are more likely to harbor EGFR mutations and therefore to benefit the most from EGFR TKIs.
In another example, analysis of incidence and 3-year prevalence data in SEER from 2002 to 2008 shows that the incidence of metastatic NSCLC cases has decreased from 26.5 to 23.5 cases per 100,000, whereas prevalence has increased from 14.8 to 15.1 cases per 100,000 (Fig 2C). Incidence represents the number of patients diagnosed with metastatic NSCLC per year (of whom < 10% harbor EGFR mutations), whereas 3-year prevalence represents those patients who were diagnosed within 3 years and are still alive at the end of this time period. The 10% decline in incidence is attributable to lower smoking rates in the US population,43 and the 2% increase in prevalence occurred concurrently with the introduction of EGFR TKIs into the population (Fig 2C). If there is no change in the average survival rate of patients during a period of observation, then the prevalence exactly mimics the behavior of the incidence curve; however, if the survival rate increases during a period of observation, then the prevalence may increase even when the incidence decreases, if the increase of survival rates is sufficiently large. Of course, other drugs (ie, pemetrexed and bevacizumab) were also approved by the FDA for NSCLC treatment during this time (2004 and 2006, respectively); thus, the changes in prevalence cannot be entirely attributed to EGFR TKIs. More dramatic observations have been made in other oncogene-driven cancers treated with TKIs, such as BCR-ABL–driven chronic myelogenous leukemia treated with imatinib,44,45 but that disease is defined by the BCR-ABL translocation and therefore more easily analyzed using population-based databases.
EGFR MUTATIONS AND DRUG RESISTANCE
Unfortunately, approximately 30% of patients still do not experience disease responses despite harboring EGFR-mutant disease,4,6–9 and less than 5% experience a complete response7–9 (Table 1). Acquired resistance to EGFR TKIs in the metastatic setting is inevitable. Moreover, although the average PFS is 10 to 16 months, treatment duration can last as short as 1 month.7 Thus, drug resistance remains a major problem in the clinic. Until new therapies and strategies are developed to overcome such resistance, the new prevalence rate of lung cancer (Fig 2C) will remain flat. Here, we focus on mechanisms of primary and secondary resistance to EGFR TKIs.
Primary Resistance
De novo resistant EGFR mutations.
Tumors with mostly EGFR exon 20 insertions, which account for 4% of EGFR mutations, are associated with a lack of drug sensitivity in preclinical models and in patients.46 Another mutation in exon 20 conferring resistance involves substitution of methionine for threonine at position 790 (T790M). This alteration is found as a heterozygous germline variant in 0.5% of never-smokers with lung adenocarcinoma47 and may confer genetic susceptibility to EGFR-mutant lung cancer.48 Efforts are being made to create an online registry of patients with germline EGFR T790M.49 When the T790M mutation occurs somatically, its frequency in EGFR TKI–naive disease is somewhat controversial. Multiple studies have reported rarely detecting it pretreatment,50,51 and mathematic modeling studies have suggested that pre-existing resistance may be present at a low frequency.51 Others have found frequencies as high as 35%.52–54 By contrast, the data are much more consistent in showing that more than half of patients with acquired resistance to gefitinib or erlotinib develop the T790M mutation. Patients whose tumors harbor somatic T790M mutations before treatment experience a shorter PFS.52,54
Suboptimal drug exposure.
Suboptimal drug exposure may result in a lack of antitumor effect. In an interesting case report, disease in a patient with EGFR-mutant lung cancer (exon 19 deletion) progressed after only 2 months of erlotinib at the standard dose (150 mg orally once per day). The patient was found to have a low plasma concentration of drug, so the dose was increased. At 300 mg orally once per day, a significant response was achieved. Further investigation implicated a drug-drug interaction with fenofibrate. Erlotinib is extensively metabolized by the monooxygenase, cytochrome P450 3A4 (CYP3A4), which can be induced by fenofibrate. Subsequent withdrawal of fenofibrate led to suprahigh levels of erlotinib along with concomitant adverse effects, necessitating a reduction of erlotinib back to 150 mg orally once per day.55 How often such suboptimal dosing occurs is unknown. In an analogous manner, smoking has also been shown to affect erlotinib dosing through the upregulation of CYP1A1.56 Polymorphisms in the genes involved in erlotinib metabolism could further influence drug concentrations in individual patients, as seen with sunitinib in patients with renal cell carcinoma.57
Failure of apoptosis induction.
Induction of the proapoptotic BH-3–only molecule, BIM, is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR–dependent lung adenocarcinomas both in vitro and in vivo.58 Low expression levels of BIM in primary tumors have been associated with shorter PFS in patients treated with EGFR TKIs.59 Functional variants of BIM that impair its function could also explain variability of response.60
Other potential mechanisms.
Other cell intrinsic factors may affect TKI sensitivity. Approximately 50% of NSCLCs, especially adenocarcinomas, harbor recurrent somatic alterations in genes that encode components of major signaling pathways, including ALK, ROS1, RET, HER2, KRAS, NRAS, PIK3CA, AKT1, BRAF, and MEK1.61–63 Among these, only PIK3CA mutations thus far have been shown to commonly co-occur with EGFR mutations.64 Introduction of PIK3CA into EGFR-mutant cells confers resistance to EGFR TKIs,65 and PIK3CA mutations have been shown to be acquired after patients develop resistance (Fig 3). The full spectrum of genome-wide genetic alterations associated with untreated EGFR-mutant lung cancer remains to be established. In another example, activation of the FAS/NFκB signaling pathway may modulate EGFR dependence in lung cancer cells (Fig 3).66 In this study, high expression of NFκB correlated with a significantly shorter PFS in patients treated with EGFR TKIs.
Fig 3.

Schematic representation of the epidermal growth factor receptor (EGFR) signaling pathway and molecules that may affect drug resistance. Gold star indicates mutations in EGFR. Red star indicates genes found to be mutated in tumor samples from patients with acquired resistance to EGFR tyrosine kinase inhibitors. HGF, hepatocyte growth factor; mTOR, mammalian target of rapamycin.
Exogenous factors may also affect EGFR TKI resistance in EGFR-mutant tumors. For example, in one study, hepatocyte growth factor (HGF), the ligand of the MET receptor tyrosine kinase, was found to be overexpressed in 29% of primary resistant lung tumors with drug-sensitive EGFR mutations (13 of 45).67 This result suggests that activation of the MET signaling pathway through HGF stimulation might be associated with primary resistance as well as acquired resistance (Fig 3). In another example, inflammation has been implicated as a resistance mechanism via activation of the interleukin-6/JAK2/STAT3 pathway (Fig 3).68 In xenograft models, administration of an anti–interleukin-6 antibody restores drug sensitivity.
Secondary Resistance
As we have stated, all patients with metastatic EGFR-mutant lung cancer will eventually develop disease progression. For more than 60% of patients, a plausible mechanism of resistance has been identified (Fig 4). The key to these studies has been analysis of new tumor tissue after patients develop resistance,69,70 a practice which should be considered standard to help guide therapy. Here, we review known mechanisms observed in human lung tumors as well as potential mechanisms found in preclinical models.
Fig 4.
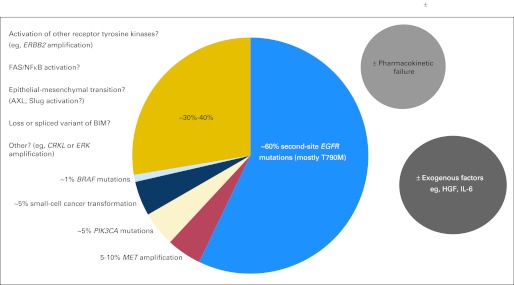
Mechanisms of acquired resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. Multiple mechanisms have been elucidated in human samples and preclinical models. Some factors may overlap. HGF, hepatocyte growth factor; IL-6, interleukin-6.
Second-site EGFR mutations.
Second-site EGFR mutations are the most frequent mechanism of acquired resistance to EGFR TKIs in lung cancer, found in more than 50% of patients. Among the reported mutations—L747S, D761Y, T790M, and T854A—more than 90% are composed of the T790M gatekeeper mutation11,69–72 (Figs 1 and 4). The T790M substitution alters proper binding of the drug to the ATP pocket of EGFR and/or restores the affinity for ATP versus drug back to the level of wild-type EGFR.18
Suboptimal drug exposure in the brain.
Up to 33% of patients with EGFR-mutant lung cancer treated with EGFR TKIs will experience disease progression in the CNS.73,74 Thus far, the second-site EGFR T790M mutation has been found in only four (13%) of 30 examined, a frequency far lower than that seen in peripheral organs.74–79 Concurrently, multiple studies have shown that the concentration of drug achievable in the brain is approximately 1% to 5% of the level found in the plasma.75,77,80 Presumably, the selection pressure for second-site mutations is thus different in the brain versus the periphery. In those patients who experience disease progression only in the brain but not in the rest of the body, brain metastasis treatment should be administered, but erlotinib at standard daily doses can be resumed after completion of the radiation course. In the setting of leptomeningeal disease, which has been historically difficult to treat with conventional chemotherapy, high-dose EGFR TKI has been shown to be clinically tolerable and potentially of some benefit with no dose-limiting toxicity.75,77,78,81 Such a dosing regimen leads to higher levels of drug in the cerebrospinal fluid (CSF; gefitinib 1,250 mg once per day: serum, 3,730 nmol/L and CSF, 39.4 nmol/L; erlotinib 1,500 mg orally once per week: plasma, 11,300 nmol/L and CSF, 130 nmol/L).75,77 Whether plasma drug concentrations decrease over time when EGFR TKIs are taken at standard doses remains unclear. In chronic myelogenous leukemia, studies have shown that imatinib levels can decrease the longer patients receive the drug.82
Activation of EGFR signaling pathways via other aberrant molecules.
Another 5% to 10% of cases of acquired resistance will display amplification of the gene encoding MET.69,70,83,84 Overexpression of MET activates the PI3K/AKT pathway via interaction with ERBB3, rendering cells less solely dependent on mutant EGFR for survival.83 In a tumor cell population, HGF may help expand pre-existing minor populations of cells harboring MET amplification (Figs 3 and 4).85,86
Other mutant signaling proteins may also confer resistance. Up to 5% of patients with acquired resistance examined in one study developed new PIK3CA mutations.70 In another study of nearly 200 tumor samples from patients with acquired resistance, we recently found that approximately 1% of patients harbor BRAF mutations.87 No common mutations in KRAS, NRAS, or MEK1 signaling genes were detected.87 We have also recently shown that ERBB2 amplification may be associated with acquired resistance, especially in patients without detected T790M mutations.88 Other studies have suggested that CRKL or ERK amplification may be a mediator of disease progression.89,89a
Histologic transformation.
In a handful of cases, rebiopsy of growing tumors has obtained cells that no longer display adenocarcinoma histology. Although they still harbor a drug-sensitive EGFR mutation, the cells display features of small-cell lung cancer70,90 or epithelial-to-mesenchymal transition (EMT).70,91 How cells transform to a different histology is not well understood. The RB and p53 pathways may have a role in the transformation to small-cell cancer,92 and the TGFβ pathway may play a role in EMT.68 In preclinical models, cells with EMT features seem to be no longer dependent on EGFR signaling for survival, but drug sensitivity may be restored by treatment with histone deacetylase inhibitors93 or inhibition of the kinase AXL or the zinc finger protein Slug.91,94
Strategies to Overcome Resistance
To overcome EGFR TKI resistance, several new drugs or drug combinations are being developed. To date, however, no agents have been approved for either the first-line setting or patients with acquired resistance.
Second- and third-generation EGFR TKIs.
Second-generation EGFR TKIs include canertinib, neratinib, afatinib, and dacomitinib (Table 2). These irreversible ATP-competitive agents make covalent bonds with a cysteine residue at position 797 in EGFR. They are more potent than gefitinib and erlotinib and also affect other EGFR family members (eg, ERBB2, ERBB4). However, they still inhibit EGFR drug-sensitive mutations at lower concentrations of drug as compared with the common T790M mutant and therefore eventually select for cancer cells with EGFR T790M in preclinical models.51,98 In humans, the concentration of drug needed to overcome T790M-mediated resistance may be not achievable in the absence of significant toxicity.
Table 2.
Small-Molecule EGFR TKIs Clinically Available or in Development
| Drug Name | Generic Name | Trade Name | Manufacturer | Target | Recommended Dose | MTD | Status |
|---|---|---|---|---|---|---|---|
| Reversible | |||||||
| ZD1839 | Gefitinib | Iressa | AstraZeneca, Wilmington, DE | EGFR | 250 mg once per day | 750 mg once per day | Approved (Asia/EU) |
| OSI-776 | Erlotinib | Tarceva | Genentech, South San Francisco, CA | EGFR | 150 mg once per day | 150 mg once per day | Approved |
| BPI-2009H | Icotinib | Conmana | BetaPharma, Branford, CT | EGFR | 150 mg once every 8 hours | Not reached | Approved (China) |
| TAK-165 | Mubritinib | NA | Takeda, Osaka, Japan | EGFR/ERBB2 | NA | NA | Phase I* |
| XL647 | NA | NA | Kadmon, New York, NY | EGFR/ERBB2† | 300 mg once per day | 300 mg once per day | Phase II* |
| ZD6474 | Vandetanib | Zactima | AstraZeneca | EGFR/VEGFR2/RET | 300 mg once per day | 300 mg once per day | Phase III*‡ |
| GW572016 | Lapatinib | Tykerb | GlaxoSmithKline, Philadelphia, PA | EGFR/ERBB2 | 1,250-1,500 mg once per day | Not reached | Preclinical*§ |
| Irreversible | |||||||
| EKB-569 | Pelitinib | NA | Wyeth/Pfizer, New York, NY | EGFR | 50 mg once per day | 75 mg once per day | Phase I* |
| CI-1033 | Canertinib | NA | Pfizer, New York, NY | EGFR/ERBB2/ERBB4 | 150 mg once per day | 150 mg once per day | Phase II* |
| HKI-272 | Neratinib | NA | Puma Biotechnology, Los Angeles, CA | EGFR/ERBB2 | 320 mg once per day2016; | 320 mg once per day | Phase II* |
| BIBW2992 | Afatinib | Tomtovok | Boehringer Ingelheim, Ingelheim, Germany | EGFR/ERBB2/ERBB4 | 50 mg once per day | 50 mg once per day | Phase III |
| PF-00299804 | Dacomitinib | NA | Pfizer | EGFR/ERBB2/ERBB4 | 45 mg once per day | 45 mg once per day | Phase III |
| Third generation | |||||||
| CO-1686 | NA | NA | Clovis/Avila, Boulder, CO | EGFR T790M | NA | NA | Phase I/II |
| WZ4002 | NA | NA | NA | EGFR T790M | NA | NA | Preclinical |
| Other | |||||||
| AP26113 | NA | NA | Ariad Pharmaceuticals, Cambridge, MA | ALK/EGFR¶ | NA | NA | Phase I/II |
Abbreviations: EGFR, epidermal growth factor receptor; EU, European Union; FDA, US Food and Drug Administration; MTD, maximum tolerated dose; NA, not applicable; NSCLC, non–small-cell lung cancer; TKI, tyrosine kinase inhibitor; VEGFR2, vascular endothelial growth factor receptor 2
Currently, no additional trials are being planned for lung cancer.
XL647 inhibits EGFR, ERBB2, VEGFR2, FLT-4, and EPHB4.
Vandetanib failed to show a survival benefit v placebo in unselected patients after prior therapy with EGFR TKI,95 showed efficacy equivalent to that of erlotinib in unselected patients treated with one prior anticancer therapy for advanced NSCLC,96 and showed an additive effect with docetaxel in unselected patients treated with one prior anticancer therapy for advanced NSCLC.97 The FDA has approved vandetanib as an RET inhibitor for medullary thyroid cancers under the trade name Caprelsa (AstraZeneca, London, United Kingdom).
The FDA has approved lapatinib as an ERBB2 inhibitor for breast cancers.
Dose was reduced to 240 mg orally once per day during trial because of toxicity.
AP26113 is reported to reversibly inhibit ALK and EGFR mutant proteins, including EGFR T790M.
Among these four drugs, afatinib has progressed the farthest in development. In a phase III trial for TKI-naive patients with EGFR-mutant tumors, afatinib was superior to platinum doublet chemotherapy in terms of RR (56.1% v 22.6%; P < .001) and PFS (11.1 v 6.9 months; P < .001).99 In a separate randomized phase IIb/III trial of afatinib versus placebo for patients who “had disease progression after at least 12 weeks of treatment with erlotinib or gefitinib”100(p529) (not necessarily with EGFR-mutant tumors), RR and PFS were 7% versus < 1% and 3.3 versus 1.1 months, respectively.100 OS was 10.8 months in the afatinib arm and 12.0 months in the placebo arm.100 In a phase II trial, dacomitinib also has shown promising results in patients with untreated EGFR-mutant tumors (RR, 74%; preliminary median PFS, 17 months).101
Third-generation EGFR inhibitors include WZ4002102 and CO-1686.103 Whereas first- or second-generation EGFR TKIs have a quinazoline core, WZ4002 has an anilinopyrimidine core, which fits better into the ATP pocket of EGFR T790M. The structure of CO-1686 has not yet been released publicly. Notably, these reagents were designed to specifically inhibit the EGFR T790M mutant. To date, no clinical trials for WZ4002 have been initiated, but a phase I/II clinical trial for CO-1686 started in January 2012 (NCT01526928). Another new EGFR inhibitor is AP26113. This drug was originally characterized as an ALK inhibitor but has also been shown in preclinical models to inhibit EGFR mutants including EGFR T790M.104 A phase I/II clinical trial for AP26113 began in September 2011 (NCT01449461).
Drug combinations.
Several studies have examined the addition of an anti-EGFR antibody to an EGFR TKI to overcome resistance. The combination of erlotinib with cetuximab showed no effect in patients who acquired resistance to EGFR TKIs (RR, 0%).12 However, the combination of afatinib and cetuximab has led to highly promising results (RR, 36% in eight of 22 patients).105 Results from this trial importantly demonstrate in patients that tumors remain dependent on EGFR signaling for survival even after developing resistance. Interestingly, 50% of the responders to this combination did not harbor secondary EGFR T790M mutations, suggesting that there exist EGFR signaling pathway–dependent but EGFR T790M–independent mechanisms of resistance.
Other trials have assessed the combination of EGFR TKIs with other classes of inhibitors (eg, mammalian target of rapamycin inhibitors, SRC inhibitors, HSP90 inhibitors, and so on), but results have been disappointing.12 If there is no universal Achilles' heel in tumors, specific combinations may need to be directed against the genetic makeup of individual tumors (eg, adding a MET inhibitor for tumors displaying no EGFR T790M but MET amplification, or adding a PI3K inhibitor for tumors displaying a secondary PI3KCA mutation, and so on65,83).
Treatment beyond progression.
Finally, recent preclinical and clinical studies have revealed interesting properties of EGFR TKI–resistant tumors. In standard practice, cytotoxic cancer drugs are usually discontinued when acquired resistance develops. However, many reports have demonstrated that patients who acquire resistance can re-respond to EGFR TKIs after a drug holiday.106 Multiple studies have now shown that patients who acquire EGFR T790M may have a favorable clinical outcome as compared with those who do not.79,107,108 Consistent with these findings, preclinical studies have demonstrated that cells that acquire EGFR T790M can actually grow slower than parental cells, and after several passages in the absence of TKIs, they become resensitized.51 These findings indicate that resistant tumors are composed of mixed populations of sensitive and resistant cancer cells and suggest a benefit of continued EGFR TKI administration even after the acquisition of resistance. Several clinical studies support this hypothesis,109–112 and prospective trials to test whether an EGFR TKI beyond progression is better than stopping the drug at the time of resistance are currently in progress (A Study of IRESSA Treatment Beyond Progression in Addition to Chemotherapy Versus Chemotherapy Alone [IMPRESS]) as well as in development.
Novel combinations.
Evolutionary mathematic cancer modeling in conjunction with in vitro experimental data was recently used to predict alternative dosing strategies that delay the outgrowth of pre-existing resistance using currently available EGFR TKIs.51 The most promising strategy administers low-dose continuous EGFR TKI (eg, erlotinib) in combination with high-dose pulsed doses (eg, afatinib) with the goal of preventing both the replication of EGFR TKI–sensitive cells as well as optimally delaying the emergence of resistant clones. Such modeling also suggests that pulsed high doses alone will not be sufficient to suppress resistance. The clinical utility of this strategy should be validated. This integrated mathematic modeling and experimental approach represents a new roadmap for the rational design of clinical trials that can also be applied to other cancer types treated with targeted therapy.
Other novel combinations could involve TKIs plus immunologic therapies. BMS-936558 (MDX-1106), an antibody against programmed death-1, a T-cell inhibitory receptor, has shown promising activity in lung cancer with minimal toxicity.113 Addition of agents like BMS-936558 to erlotinib in TKI-naive patients or potentially with more potent TKIs after the development of resistance should be explored.
DISCUSSION
In conclusion, EGFR mutations define a distinct clinical entity of lung cancer. EGFR TKIs such as gefitinib and erlotinib have increased the OS of patients with EGFR-mutant lung cancer. However, despite rapid progress over the past 8 years, a new plateau for survival of patients with EGFR-mutant lung cancer already seems to have been reached. Greater efforts need to be made not only to overcome acquired resistance but also to induce greater responses and longer PFS in the first-line setting. The goal should now be to become as creative as possible with new strategies and therapies to make EGFR-mutant lung cancer a chronic rather than fatal disease.
Appendix
Database Source
Survival – Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence – SEER 17 Regs Research Data + Hurricane Katrina Impacted Louisiana Cases, Nov 2009 Sub (1973-2007 varying) – Linked to County Attributes – Total US, 1969-2007 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Cancer Statistics Branch, released April 2010, based on the November 2009 submission.
Incidence – Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence – SEER 13 Regs Research Data, Nov 2010 Sub (1992-2008) less than Single Ages to 85+, Katrina/Rita Population Adjustment more than – Linked to County Attributes – Total US, 1969 to 2009 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Cancer Statistics Branch, released April 2011 (updated 10/28/2011), based on the November 2010 submission.
Prevalence – Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence – SEER 13 Regs Research Data, Nov 2010 Sub (1992-2008) less than Single Ages to 85+, Katrina/Rita Population Adjustment more than – Linked to County Attributes – Total US, 1969 to 2009 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Cancer Statistics Branch, released April 2011 (updated 10/28/2011), based on the November 2010 submission.
Footnotes
Supported by Grants No. R01-CA121210, P01-CA129243, U54-CA143798, and P30-CA68485 from the National Cancer Institute, National Institutes of Health.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships markedwith a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors
Employment or Leadership Position: None Consultant or Advisory Role: William Pao, Bristol-Myers Squibb (C), Clovis (C), Symphony Evolution (C), AstraZeneca (C) Stock Ownership: None Honoraria: None Research Funding: William Pao, AstraZeneca, Clovis, Bristol-Myers Squibb Expert Testimony: None Other Remuneration: William Pao, MolecularMD
AUTHOR CONTRIBUTIONS
Financial support: William Pao
Administrative support: William Pao
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 5.Rosell R, Moran T, Queralt C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009;361:958–967. doi: 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- 6.Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 7.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 8.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 9.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 10.Hynes NE, Lane HA. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 11.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oxnard GR, Arcila ME, Chmielecki J, et al. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17:5530–5537. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schlessinger J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell. 2002;110:669–672. doi: 10.1016/s0092-8674(02)00966-2. [DOI] [PubMed] [Google Scholar]
- 14.Horn L, Chen H, Lovly CM, et al. DIRECT: DNA-mutation Inventory to Refine and Enhance Cancer Treatment—A catalogue of clinically relevant somatic mutations in lung cancer. J Clin Oncol. 2011;29:494s. (suppl; abstr 7575) [Google Scholar]
- 15.Greulich H, Chen TH, Feng W, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Politi K, Zakowski MF, Fan PD, et al. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yun CH, Boggon TJ, Li Y, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11:217–227. doi: 10.1016/j.ccr.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He M, Capelletti M, Nafa K, et al. EGFR exon 19 insertions: A new family of sensitizing EGFR mutations in lung adenocarcinoma. Clin Cancer Res. 2012;18:1790–1797. doi: 10.1158/1078-0432.CCR-11-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu JY, Yu CJ, Chang YC, et al. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res. 2011;17:3812–3821. doi: 10.1158/1078-0432.CCR-10-3408. [DOI] [PubMed] [Google Scholar]
- 21.Beau-Faller M, Prim N, Ruppert AM, et al. Rare epidermal growth factor receptor (EGFR) mutations in 10,117 patients with non-small cell lung cancer (NSCLC) evaluated by the French ERMETIC IFCT network: Clinical, molecular, and survival data. J Clin Oncol. 2012;30:657s. (suppl; abstr 10507) [Google Scholar]
- 22.Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–346. doi: 10.1093/jnci/dji055. [DOI] [PubMed] [Google Scholar]
- 23.Bell DW, Lynch TJ, Haserlat SM, et al. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: Molecular analysis of the IDEAL/INTACT gefitinib trials. J Clin Oncol. 2005;23:8081–8092. doi: 10.1200/JCO.2005.02.7078. [DOI] [PubMed] [Google Scholar]
- 24.Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers: A different disease. Nat Rev Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- 25.Kosaka T, Yatabe Y, Endoh H, et al. Mutations of the epidermal growth factor receptor gene in lung cancer: Biological and clinical implications. Cancer Res. 2004;64:8919–8923. doi: 10.1158/0008-5472.CAN-04-2818. [DOI] [PubMed] [Google Scholar]
- 26.Sun Y, Ren Y, Fang Z, et al. Lung adenocarcinoma from East Asian never-smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol. 2010;28:4616–4620. doi: 10.1200/JCO.2010.29.6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: Results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer) Lancet. 2005;366:1527–1537. doi: 10.1016/S0140-6736(05)67625-8. [DOI] [PubMed] [Google Scholar]
- 28.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 29.Kim ES, Hirsh V, Mok T, et al. Gefitinib versus docetaxel in previously treated non-small-cell lung cancer (INTEREST): A randomised phase III trial. Lancet. 2008;372:1809–1818. doi: 10.1016/S0140-6736(08)61758-4. [DOI] [PubMed] [Google Scholar]
- 30.Fukuoka M, Wu YL, Thongprasert S, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non–small-cell lung cancer in Asia (IPASS) J Clin Oncol. 2011;29:2866–2874. doi: 10.1200/JCO.2010.33.4235. [DOI] [PubMed] [Google Scholar]
- 31.Mitsudomi T, Morita S, Yatabe Y. Updated overall survival results of WJTOG 3405, a randomized phase III trial comparing gefitinib (G) with cisplatin plus docetaxel (CD) as the first-line treatment for patients with non-small cell lung cancer harboring mutations of the epidermal growth factor receptor (EGFR) J Clin Oncol. 2012;30:485s. (suppl; abstr 7521) [Google Scholar]
- 32.Zhou C, Wu YL, Liu X. Overall survival (OS) results from OPTIMAL (CTONG0802), a phase III trial of erlotinib (E) versus carboplatin plus gemcitabine (GC) as first-line treatment for Chinese patients with EGFR mutation-positive advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2012;30:485s. (suppl; abstr 7520) [Google Scholar]
- 33.Nguyen KS, Kobayashi S, Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer. 2009;10:281–289. doi: 10.3816/CLC.2009.n.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garassino MC, Martelli O, Bettini A, et al. TAILOR: A phase III trial comparing erlotinib with docetaxel as the second-line treatment of NSCLC patients with wild-type (wt) EGFR. J Clin Oncol. 2012;30:480s. (suppl; abstr LBA7501) [Google Scholar]
- 35.Schiller JH, Harrington D, Belani CP, et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N Engl J Med. 2002;346:92–98. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 36.Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 37.Scagliotti GV, Parikh P, von Pawel J, et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non–small-cell lung cancer. J Clin Oncol. 2008;26:3543–3551. doi: 10.1200/JCO.2007.15.0375. [DOI] [PubMed] [Google Scholar]
- 38.Jänne PA, Wang X, Socinski MA, et al. Randomized phase II trial of erlotinib alone or with carboplatin and paclitaxel in patients who were never or light former smokers with advanced lung adenocarcinoma: CALGB 30406 trial. J Clin Oncol. 2012;30:2063–2069. doi: 10.1200/JCO.2011.40.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kubota K, Watanabe K, Kunitoh H, et al. Phase III randomized trial of docetaxel plus cisplatin versus vindesine plus cisplatin in patients with stage IV non–small-cell lung cancer: The Japanese Taxotere Lung Cancer Study Group. J Clin Oncol. 2004;22:254–261. doi: 10.1200/JCO.2004.06.114. [DOI] [PubMed] [Google Scholar]
- 40.Ohe Y, Ohashi Y, Kubota K, et al. Randomized phase III study of cisplatin plus irinotecan versus carboplatin plus paclitaxel, cisplatin plus gemcitabine, and cisplatin plus vinorelbine for advanced non-small-cell lung cancer: Four-arm cooperative study in Japan. Ann Oncol. 2007;18:317–323. doi: 10.1093/annonc/mdl377. [DOI] [PubMed] [Google Scholar]
- 41.Takano T, Fukui T, Ohe Y, et al. EGFR mutations predict survival benefit from gefitinib in patients with advanced lung adenocarcinoma: A historical comparison of patients treated before and after gefitinib approval in Japan. J Clin Oncol. 2008;26:5589–5595. doi: 10.1200/JCO.2008.16.7254. [DOI] [PubMed] [Google Scholar]
- 42.Woodcock J. AstraZeneca Pharmaceuticals LP: Withdrawal of approval of a new drug Application for IRESSA. Federal Register. 2012;77:24723–24724. [Google Scholar]
- 43.Cokkinides V, Bandi P, McMahon C, et al. Tobacco control in the United States: Recent progress and opportunities. CA Cancer J Clin. 2009;59:352–365. doi: 10.3322/caac.20037. [DOI] [PubMed] [Google Scholar]
- 44.Huang X, Cortes J, Kantarjian H. Estimations of the increasing prevalence and plateau prevalence of chronic myeloid leukemia in the era of tyrosine kinase inhibitor therapy. Cancer. 2012;118:3123–3127. doi: 10.1002/cncr.26679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kantarjian H, O'Brien S, Jabbour E, et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: A single-institution historical experience. Blood. 2012;119:1981–1987. doi: 10.1182/blood-2011-08-358135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non-small-cell lung cancer: Preclinical data and clinical implications. Lancet Oncol. 2012;13:e23–e31. doi: 10.1016/S1470-2045(11)70129-2. [DOI] [PubMed] [Google Scholar]
- 47.Girard N, Lou E, Azzoli CG, et al. Analysis of genetic variants in never-smokers with lung cancer facilitated by an Internet-based blood collection protocol: A preliminary report. Clin Cancer Res. 2010;16:755–763. doi: 10.1158/1078-0432.CCR-09-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bell DW, Gore I, Okimoto RA, et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat Genet. 2005;37:1315–1316. doi: 10.1038/ng1671. [DOI] [PubMed] [Google Scholar]
- 49.Oxnard GR, Miller VA, Robson ME, et al. Screening for germline EGFR T790M mutations through lung cancer genotyping. J Thorac Oncol. 2012;7:1049–1052. doi: 10.1097/JTO.0b013e318250ed9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Inukai M, Toyooka S, Ito S, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006;66:7854–7858. doi: 10.1158/0008-5472.CAN-06-1951. [DOI] [PubMed] [Google Scholar]
- 51.Chmielecki J, Foo J, Oxnard GR, et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med. 2011;3 doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maheswaran S, Sequist LV, Nagrath S, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359:366–377. doi: 10.1056/NEJMoa0800668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rosell R, Molina MA, Costa C, et al. Pretreatment EGFR T790M mutation and BRCA1 mRNA expression in erlotinib-treated advanced non-small-cell lung cancer patients with EGFR mutations. Clin Cancer Res. 2011;17:1160–1168. doi: 10.1158/1078-0432.CCR-10-2158. [DOI] [PubMed] [Google Scholar]
- 54.Su KY, Chen HY, Li KC, et al. Pretreatment epidermal growth factor receptor (EGFR) T790M mutation predicts shorter EGFR tyrosine kinase inhibitor response duration in patients with non–small-cell lung cancer. J Clin Oncol. 2012;30:433–440. doi: 10.1200/JCO.2011.38.3224. [DOI] [PubMed] [Google Scholar]
- 55.Mir O, Blanchet B, Goldwasser F. Drug-induced effects on erlotinib metabolism. N Engl J Med. 2011;365:379–380. doi: 10.1056/NEJMc1105083. [DOI] [PubMed] [Google Scholar]
- 56.Hamilton M, Wolf JL, Rusk J, et al. Effects of smoking on the pharmacokinetics of erlotinib. Clin Cancer Res. 2006;12:2166–2171. doi: 10.1158/1078-0432.CCR-05-2235. [DOI] [PubMed] [Google Scholar]
- 57.Garcia-Donas J, Esteban E, Leandro-García LJ, et al. Single nucleotide polymorphism associations with response and toxic effects in patients with advanced renal-cell carcinoma treated with first-line sunitinib: A multicentre, observational, prospective study. Lancet Oncol. 2011;12:1143–1150. doi: 10.1016/S1470-2045(11)70266-2. [DOI] [PubMed] [Google Scholar]
- 58.Gong Y, Somwar R, Politi K, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4:e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Faber AC, Corcoran RB, Ebi H, et al. BIM expression in treatment naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1:352–365. doi: 10.1158/2159-8290.CD-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ng KP, Hillmer AM, Chuah CT, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012;18:521–528. doi: 10.1038/nm.2713. [DOI] [PubMed] [Google Scholar]
- 61.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12:175–180. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 62.Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–870. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pao W, Hutchinson KE. Chipping away at the lung cancer genome. Nat Med. 2012;18:349–351. doi: 10.1038/nm.2697. [DOI] [PubMed] [Google Scholar]
- 64.Yamamoto H, Shigematsu H, Nomura M, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008;68:6913–6921. doi: 10.1158/0008-5472.CAN-07-5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Engelman JA, Mukohara T, Zejnullahu K, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–2706. doi: 10.1172/JCI28656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bivona TG, Hieronymus H, Parker J, et al. FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature. 2011;471:523–526. doi: 10.1038/nature09870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yano S, Yamada T, Takeuchi S, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6:2011–2017. doi: 10.1097/JTO.0b013e31823ab0dd. [DOI] [PubMed] [Google Scholar]
- 68.Yao Z, Fenoglio S, Gao DC, et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A. 2010;107:15535–15540. doi: 10.1073/pnas.1009472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arcila ME, Oxnard GR, Nafa K, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17:1169–1180. doi: 10.1158/1078-0432.CCR-10-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3 doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 72.Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Omuro AM, Kris MG, Miller VA, et al. High incidence of disease recurrence in the brain and leptomeninges in patients with nonsmall cell lung carcinoma after response to gefitinib. Cancer. 2005;103:2344–2348. doi: 10.1002/cncr.21033. [DOI] [PubMed] [Google Scholar]
- 74.Heon S, Yeap BY, Britt GJ, et al. Development of central nervous system metastases in patients with advanced non-small cell lung cancer and somatic EGFR mutations treated with gefitinib or erlotinib. Clin Cancer Res. 2010;16:5873–5882. doi: 10.1158/1078-0432.CCR-10-1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jackman DM, Holmes AJ, Lindeman N, et al. Response and resistance in a non–small-cell lung cancer patient with an epidermal growth factor receptor mutation and leptomeningeal metastases treated with high-dose gefitinib. J Clin Oncol. 2006;24:4517–4520. doi: 10.1200/JCO.2006.06.6126. [DOI] [PubMed] [Google Scholar]
- 76.Katayama T, Shimizu J, Suda K, et al. Efficacy of erlotinib for brain and leptomeningeal metastases in patients with lung adenocarcinoma who showed initial good response to gefitinib. J Thorac Oncol. 2009;4:1415–1419. doi: 10.1097/JTO.0b013e3181b62572. [DOI] [PubMed] [Google Scholar]
- 77.Clarke JL, Pao W, Wu N, et al. High dose weekly erlotinib achieves therapeutic concentrations in CSF and is effective in leptomeningeal metastases from epidermal growth factor receptor mutant lung cancer. J Neurooncol. 2010;99:283–286. doi: 10.1007/s11060-010-0128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Grommes C, Oxnard GR, Kris MG, et al. “Pulsatile” high-dose weekly erlotinib for CNS metastases from EGFR mutant non-small cell lung cancer. Neuro Oncol. 2011;13:1364–1369. doi: 10.1093/neuonc/nor121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hata A, Ktakami N, Yoshioka H, et al. Rebiopsy of non-small cell lung cancer patients with acquired resistance to EGFR-TKI: Comparison between T790M mutation-positive and -negative populations. J Clin Oncol. 2012;30:487s. doi: 10.1002/cncr.28364. (suppl; abstr 7528) [DOI] [PubMed] [Google Scholar]
- 80.Togashi Y, Masago K, Fukudo M, et al. Efficacy of increased-dose erlotinib for central nervous system metastases in non-small cell lung cancer patients with epidermal growth factor receptor mutation. Cancer Chemother Pharmacol. 2011;68:1089–1092. doi: 10.1007/s00280-011-1691-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Milton DT, Azzoli CG, Heelan RT, et al. A phase I/II study of weekly high-dose erlotinib in previously treated patients with nonsmall cell lung cancer. Cancer. 2006;107:1034–1041. doi: 10.1002/cncr.22088. [DOI] [PubMed] [Google Scholar]
- 82.Picard S, Titier K, Etienne G, et al. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2007;109:3496–3499. doi: 10.1182/blood-2006-07-036012. [DOI] [PubMed] [Google Scholar]
- 83.Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 84.Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007;104:20932–20937. doi: 10.1073/pnas.0710370104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yano S, Wang W, Li Q, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479–9487. doi: 10.1158/0008-5472.CAN-08-1643. [DOI] [PubMed] [Google Scholar]
- 86.Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ohashi K, Sequist LV, Arcila ME, et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF mutations but lack mutations in KRAS, NRAS, or MEK1. Proc Natl Acad Sci U S A. 2012;109:E2127–E2133. doi: 10.1073/pnas.1203530109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012;2:922–933. doi: 10.1158/2159-8290.CD-12-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheung HW, Du J, Boehm JS, et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011;1:608–625. doi: 10.1158/2159-8290.CD-11-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89a.Ercan D, Xu C, Yanagita M, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012;2:934–947. doi: 10.1158/2159-8290.CD-12-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zakowski MF, Ladanyi M, Kris MG. EGFR mutations in small-cell lung cancers in patients who have never smoked. N Engl J Med. 2006;355:213–215. doi: 10.1056/NEJMc053610. [DOI] [PubMed] [Google Scholar]
- 91.Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44:852–860. doi: 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meuwissen R, Linn SC, Linnoila RI, et al. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–189. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- 93.Suda K, Tomizawa K, Fujii M, et al. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J Thorac Oncol. 2011;6:1152–1161. doi: 10.1097/JTO.0b013e318216ee52. [DOI] [PubMed] [Google Scholar]
- 94.Chang TH, Tsai MF, Su KY, et al. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am J Respir Crit Care Med. 2011;183:1071–1079. doi: 10.1164/rccm.201009-1440OC. [DOI] [PubMed] [Google Scholar]
- 95.Lee JS, Hirsh V, Park K, et al. Vandetanib versus placebo in patients with advanced non–small-cell lung cancer after prior therapy with an epidermal growth factor receptor tyrosine kinase inhibitor: A randomized, double-blind phase III trial (ZEPHYR) J Clin Oncol. 2012;30:1114–1121. doi: 10.1200/JCO.2011.36.1709. [DOI] [PubMed] [Google Scholar]
- 96.Natale RB, Thongprasert S, Greco FA, et al. Phase III trial of vandetanib compared with erlotinib in patients with previously treated advanced non–small-cell lung cancer. J Clin Oncol. 2011;29:1059–1066. doi: 10.1200/JCO.2010.28.5981. [DOI] [PubMed] [Google Scholar]
- 97.Herbst RS, Sun Y, Eberhardt WE, et al. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer (ZODIAC): A double-blind, randomised, phase 3 trial. Lancet Oncol. 2010;11:619–626. doi: 10.1016/S1470-2045(10)70132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ercan D, Zejnullahu K, Yonesaka K, et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene. 2010;29:2346–2356. doi: 10.1038/onc.2009.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yang JCH, Schuler MH, Yamamoto N, et al. LUX-Lung 3: A randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations. J Clin Oncol. 2012;30:480s. (suppl; abstr LBA7500) [Google Scholar]
- 100.Miller VA, Hirsh V, Cadranel J, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomised trial. Lancet Oncol. 2012;13:528–538. doi: 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- 101.Kris MG, Mok T, Ou SHI, et al. First-line dacomitinib (PF-00299804), an irreversible pan-HER tyrosine kinase inhibitor, for patients with EGFR-mutant lung cancers. J Clin Oncol. 2012;30:487s. (suppl; abstr 7530) [Google Scholar]
- 102.Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Walter AO, Tjin R, Haringsma H, et al. CO-1686, an orally available, mutant-selective inhibitor of the epidermal growth factor receptor (EGFR), causes tumor shrinkage in non-small cell lung cancer (NSCLC) with T790M mutations. Mol Cancer Ther. 2011;10 (suppl 1; abstr C189) [Google Scholar]
- 104.Rivera VM, Wang F, Anjum R, et al. AP26113 is a dual ALK/EGFR inhibitor: Characterization against EGFR T790M in cell and mouse models of NSCLC. Cancer Res. 2012;72 (suppl 1; abstr 1794) [Google Scholar]
- 105.Janjigian YY, Groen HJ, Horn L, et al. Activity and tolerability of afatinib (BIBW 2992) and cetuximab in NSCLC patients with acquired resistance to erlotinib or gefitinib. J Clin Oncol. 2011;29:486s. (suppl; abstr 7525) [Google Scholar]
- 106.Watanabe S, Tanaka J, Ota T, et al. Clinical responses to EGFR-tyrosine kinase inhibitor retreatment in non-small cell lung cancer patients who benefited from prior effective gefitinib therapy: A retrospective analysis. BMC Cancer. 2011;11:1. doi: 10.1186/1471-2407-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Oxnard GR, Arcila ME, Sima CS, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: Distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011;17:1616–1622. doi: 10.1158/1078-0432.CCR-10-2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rosell R, Molina-Vila MA, Taron M, et al. EGFR compound mutants and survival on erlotinib in non-small cell lung cancer (NSCLC) patients (p) in the EURTAC study. J Clin Oncol. 2012;30:485s. (suppl; abstr 7522) [Google Scholar]
- 109.Riely GJ, Kris MG, Zhao B, et al. Prospective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimus. Clin Cancer Res. 2007;13:5150–5155. doi: 10.1158/1078-0432.CCR-07-0560. [DOI] [PubMed] [Google Scholar]
- 110.Faehling M, Eckert R, Kamp TG, et al. Treatment with EGFR tyrosine kinase inhibitors beyond progression in long-term responders to erlotinib in advanced non-small cell lung cancer: A case-control study of overall survival. J Clin Oncol. 2012;30:497s. doi: 10.1016/j.lungcan.2013.02.010. (suppl; abstr 7572) [DOI] [PubMed] [Google Scholar]
- 111.Goldberg SB, Oxnard GR, Digumarthy S, et al. Chemotherapy with erlotinib or chemotherapy alone in advanced NSCLC with acquired resistance to EGFR tyrosine kinase inhibitors (TKI) J Clin Oncol. 2012;30:486s. doi: 10.1634/theoncologist.2013-0168. (suppl; abstr 7524) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Oxnard GR, Lo P, Jackman DM, et al. Delay of chemotherapy through use of post-progression erlotinib in patients with EGFR-mutant lung cancer. J Clin Oncol. 2012;30:491s. (suppl; abstr 7547) [Google Scholar]
- 113.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]