

Abstract
Secreted proteins dictate a range of cellular functions in human health and disease. Due to the high degree of cellular heterogeneity and, more importantly, polyfunctionality of individual cells, there is an unmet need to simultaneously measure an array of proteins from single cells and to rapidly assay a large number of single cells (more than 1000) in parallel. We describe a simple bioanalytical assay platform consisting of a large array of sub-nanoliter microchambers integrated with high-density antibody barcode microarrays for highly multiplexed protein detection from over a thousand single cells in parallel. This platform has been tested for both cell lines and complex biological samples such as primary cells from patients. We observed distinct heterogeneity among the single cell secretomic signatures that, for the first time, can be directly correlated to the cells’ physical behavior such as migration. Compared to the state-of-the-art protein secretion assay such as ELISpot and emerging microtechnology-enabled assays, our approach offers both high throughput and high multiplicity. It also has a number of clinician-friendly features such as ease of operation, low sample consumption and standardized data analysis, representing a potentially transformative tool for informative monitoring of cellular function and immunity in patients.
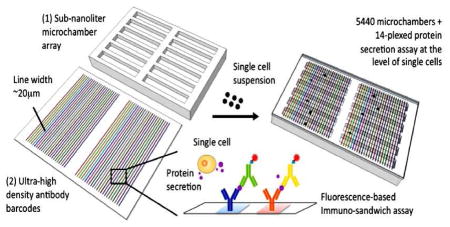
INTRODUCTION
Secreted proteins including cytokines, chemokines and growth factors represent important functional regulators mediating a range of cellular behavior and cell-cell paracrine/autocrine signaling, e.g. in the immunological system1, tumor microenvironment2 or stem cell niche3. Detection of these proteins is of great value not only in basic cell biology but also for disease diagnosis and therapeutic monitoring. However, due to co-production of multiple effector proteins from a single cell, referred to as polyfunctionality, it is biologically informative to measure a panel of secreted proteins, or secretomic signature, at the level of single cells. Recent evidence further indicates that a genetically-identical cell population can give rise to diverse phenotypic differences4. Non-genetic heterogeneity is also emerging as a potential barrier to accurate monitoring of cellular immunity and effective pharmacological therapies5,6, suggesting the need for practical tools for single cell analysis of proteomic signatures.
Fluorescence-activated cell sorting (FACS) represents the state-of-the-art for single cell analysis7. FACS is typically used to detect and sort cell phenotypes by their surface markers. It has been extended to the detection of intracellular proteins7–9, including cytokines within the cytoplasm, by blocking vesicle transport10. However, intracellular cytokine staining is not a true secretion analysis, and it also requires cell fixing, which means the cells are no longer alive after flow cytometric analysis and cannot be recovered for further studies. The mainstay of real single cell secretion analysis to date is a simple approach called ELISpot that detects the secretion footprint of individual cells using an immunosandwich-based assay11. Immune cells are loaded into a microtiter plate that has been pre-coated with a layer of primary antibody. After incubation, secreted proteins are captured by the antibodies located proximal to the cells, giving rise to spots indicative of a single cell secretion footprint 12. Recently, a variant of ELISpot, called FLUOROSpot, which exploits two fluorescent dyes to visualize protein secretion footprints, enabled a simultaneous dual function analysis. Highly multiplexed measurements of proteins secreted from a population of cells can be done using an encoded bead assay such as the Illumina VeraCode system13 or antibody microarrays manufactured using a pin-spotting technique 14,15. However, these highly multiplexed technologies cannot perform single cell measurements. Microfabricated chips have emerged as a new category of single cell analytic technologies16–21. A prototype microchip has demonstrated the feasibility of multiplexed protein secretion assay and revealed significant polyfunctional heterogeneity in phenotypically similar immune cells from patients22,23, pointing to the urgent need for single cell secretion profiling in clinical diagnosis and therapeutic monitoring. However, these microchips either lack sufficient throughput or multiplicity, or require sophisticated operation, precluding widespread application in cell biology and clinical evaluation of cellular functions.
Herein we describe a high-throughput single-cell secretomic analysis platform that integrates a sub-nanoliter microchamber array and high-density antibody barcodes for simultaneous detection of 14 cytokines from more than a thousand single cells in parallel. The chip can be executed in a simple assay “kit” with no need of sophisticated fluid handling or bulky equipment. We demonstrate the utility of this device for analyzing the secretion of human cell lines and primary cell samples dissociated from fresh tumor of patients. The results reveal that there is distinct heterogeneity among the single cell secretomic signatures of a population and that the correlations obtained between the various proteins studied are in agreement with their functional classifications. This technology builds upon prior successes in antibody barcode-based protein secretion measurement technique22, but uses simplified schemes of cell capture24, quantification, automated data analysis, and eliminates bulky fluid handling systems, resulting in a truly practical and informative tool that may find immediate use in both laboratory research and clinical cellular diagnosis.
RESULTS AND DISCUSSION
Design, fabrication and assembly of a single-cell secretomic analysis chip
Our single-cell secretomic analysis device consists of two separate parts (Fig. 1a): a high-density antibody barcode encoded glass substrate for surface-bound immunoassay and a sub-nanoliter microchamber array for capture of single cells. The antibody barcode array slide comprises 30 repeats of barcodes, each of which contains up to 20 stripes of different antibodies, immobilized on a poly-L-lysine-coated surface. The antibody stripes are 20μm in width and the pitch size of a full barcode is 1mm. The microchamber array is a one-layer microchip fabricated by soft lithography25 from polydimethylsiloxane (PDMS)25, an optically transparent silicone elastomer widely used for biological microfluidics. It contains 5440 rectangular microchambers, each of which is 1.8mm, 20μm and 15μm, in length, width and depth, respectively. These two parts were manufactured independently and combined during the assay such that the barcode slide acts as a disposable test strip and the microchamber array as a reusable device. To use this platform, a drop of single cell suspension (~106cells/ml) is directly pipetted onto the surface of the microchamber array chip. The cells fall into the microchambers by gravity, and then the aforementioned antibody barcode array slide is placed antibody-side down on top of the microchambers such that the antibody barcodes are perpendicular to the length of the microchambers. The microchamber is designed to be sufficiently long as to contain at least a full set of barcodes, thereby eliminating the need for precise alignment. Finally this assembly is fixed by two transparent plastic plates with four spring-adjusted screws (Supporting Fig. S1) and placed in a conventional tissue incubator for single-cell secretion measurement. Proteins secreted from individual cells are captured by the antibody barcodes and read out by incubating with biotinylated detection antibodies and then streptavidin conjugated with a fluorescence probe (e.g. Cy5 in our experiments). As compared to the prototype single cell proteomic chip22, this setup does not require a sophisticated microfluidic control system or any bulky equipment to operate and thus is more amenable to widespread use by researchers and clinicians with minimal engineering background.
Figure 1.

High-throughput multiplexed single cell secretomic assay. (a) Schematic illustration showing integration of a high-density antibody barcode array chip and a subnanoliter microchamber array chip for high-throughput multiplexed protein secretion assay at the single cell level. (b) Scanned fluorescence image showing high uniformity of protein loading across the entire barcode microarray (1″ × 2″). Fluorescently labeled bovine serum albumin (FITC-BSA) was used in this test. (c) Photograph stitched from a large number of individual pictures collected by an automated, motorized phase contrast microscope. Together it covers the entire subnanoliter microchamber chip that was loaded with human immune cells (U973). Scale bar 2mm. The first enlarged image shows a column of microchamber array (scale bar 300μm). The second enlarged image shows individual cells loaded in microchambers (scale bar 50μm).
The high-density antibody barcode array is fabricated using a microchannel-guided flow patterning technique (Supporting Methods) modified from the approach reported for patterning DNA barcodes26. The flow-patterning chip is a separate PDMS slab that has inlets leading to 20 individual serpentine microchannels in which individual antibody solutions (1μL each) are precisely metered, added and flowed through all microchannels in parallel to ensure uniform loading of antibodies on the surface. Fluorescein isothiocyanate labeled bovine serum albumin (FITC-BSA) solution was used to evaluate the patterning quality. The result shows successful fabrication of high-density protein array across a large area (1in × 2in) and excellent uniformity (< 5% in fluorescent intensity) as revealed by the fluorescence intensity line profile (Fig. 1b and Supporting Fig. S2). This ensures the observed protein signal variations (> 10%) from the following single cell secretomic assays are attributed to cellular heterogeneity, rather than the non-uniformity of the starting antibody barcode array.
A motorized phase-contrast imaging system has been developed to image all cells in the cell capture chip within 10 minutes (Fig. 1c) and an image analysis algorithm allows for identification of individual cells and their x/y coordinates, and counting of cells in each microchamber. The simple microchamber array chip format was chosen because it is easy to operate, but as a consequence it is not possible to ensure that one cell is captured per chamber. However, optimization of cell density in the stock solution readily gives rise to more than 1000 single cell chambers in a microchip (Supporting Fig. S3), permitting high-throughput analysis of single cells.
Protein panel and validation
The proteins assayed by the antibody barcodes are listed in Figure 2a. Assessment of these particular proteins secreted from single cells is of particular importance due to their functions in a range of cellular processes27–30. They include cytokines, chemokines and growth factors involved in a wide range of immunological or pathophysiological processes. Assessment of these proteins secreted from single cells is of importance in the study of cellular immunity and cell-cell signaling networks. In order to simultaneously measure these proteins from single cells, capture antibodies are immobilized on the substrate as a high-density barcode array. Prior to performing single-cell analysis, we validated the assay using recombinant proteins. Individual recombinant protein was spiked into fresh cell culture medium over a 4-log range of concentrations and exposed to the full panel of antibodies in order to assess cross-reactivity, the limit of detection (LOD) and dynamic range. The antibodies with cross-reactivity over 5% (at 5ng/mL of protein concentration) is eliminated or replaced. Ultimately we obtained a panel of antibody pairs as summarized in Supporting Table S1. The titration curves (Fig. 2b) demonstrate the feasibility of quantitative measurements of these proteins in the multiplexed array, with a typical measurement range of 3 orders of magnitude. The LOD ranged from 400pg/ml to below 10pg/ml depending on the affinity of antibody pairs. Based on the volume of a microchamber (~0.54 nl) and the representative detection sensitivity (~10 pg/ml), the amount of protein that can be detected by antibody barcodes in a microchamber is on the order of 5.4 attograms, which is approximately equal to ~160 molecules. Thus, our platform has the sensitivity to detect proteins secreted from a single cell (typical copy number ~102–5).
Figure 2.

Protein panel. (a) List of all 22 proteins assayed in single cell microchips and their functions in human physiology. (b) Titration curves obtained using recombinant proteins. 18 antibody pairs were validated and 4 others were left out in the titration curves due to the lack of working recombinants. Fluorescence intensity represents the original photon counts averaged from 16 spots for each protein. Error bars indicate 3 × SD.
Single-cell protein secretomic analysis on cell lines
We first used the single-cell secretomic analysis chip to measure 14 proteins from a human glioblastoma multiforme cell line (U87). In this experiment, up to 10 cells were captured in each microchamber, with 1278 of the microchambers capturing single cells. During the flow patterning of antibody barcodes, FITC-BSA (0.5mg/ml) was always flowed in Channel 1 to form a continuous line of fluorescence signal serving as both a position reference and an internal quality/uniformity control. As shown in a representative region of the scanned fluorescence image (Fig. 3a and Supporting Fig. S4a), both the blue FITC-BSA reference line and the red patterned signals corresponding to protein secretion levels are readily visible. Shown in the same figure are a bright field image of 14 microchambers with cells loaded, the corresponding fluorescent barcode image, and an overlay of the two. The major proteins observed after 24 hours of incubation (FGF, VEGF, MIF, IL-6, IL-8, and MCP-1) are mainly pro-inflammatory cytokines or chemoattractant proteins.
Figure 3.

Single-cell secretomic analysis on U87 cell lines. (a) Representative region of the scanned image showing the raw data of single cell secretomic measurement. Three subpanels on the right are optical micrograph, fluorescence image and overlay for 16 microchambers. (b) Heat map that shows the profile of 14 proteins secreted from 1278 single cells (U87). Each row is a single cell and each column corresponds to a protein of interest. (c) Scatter plots showing fluorescence intensity measured for six selected proteins (FGF, VEGF, MIF, IL-6, IL-8, MCP-1) versus the number of cells in a microchamber. (* P < 0.05, ** P < 0.01, *** P < 0.001) (d) Population kinetics for U87 cell line. Control (MEM medium), secretion supernatant from population at different time points (0 hr, 1 hr, 2 hr, 3 hr, 6 hr, 9 hr, 12 hr, 24 hr).
We conducted automated quantitation of the fluorescence intensity of each protein in a microchamber using the image analysis software Genepix 6.0. We extracted the secretomic profile for only those microchambers containing single cells and a heat map of the resulting secretion profiles (Fig. 3b and Supporting Fig. S5) indicates the existence of cell-cell variation. While the majority of cells produce IL-6, and IL8, the level of these proteins varies among individual cells and the secretion of other lower abundance proteins such as MCP-1 and FGF apparently exhibit heterogeneous signatures – only a small fraction of cells express these proteins at high levels. To verify the single cell measurements, a kinetic bulk population secretion measurement was performed in parallel on the supernatant collected from the same cells, incubated over the same time, and measured using a conventional pin-spotted microarray. The result (Fig. 3d and Supporting Fig. S6) also reveals FGF, VEGF, IL-6, IL-8, and MCP-1 as the top five proteins that are all consistent with single-cell analysis although the relative levels are different. However, MIF did not show up in the population assay. Interestingly, we observed that the protein level as measured by fluorescence intensity is not always proportional to the number of cells and sometimes cannot be interpreted by an additive effect (Fig. 3c and Supporting Fig. S7). A secretomic analysis chip was loaded with many more cells, MIF signal decreases with increasing number of cells in the capture chambers, revealing possibility of paracrine signaling and the regulation of MIF with increasing number of cells (Supporting Fig. S7). This small level of discrepancy is expected as the two assays are not biologically identical (for example, the bulk assay detects the end point protein profile while the single cell assay measures accumulated signals over the period of incubation; the population arrays are subjected to paracrine signaling while single cell measurements are not). Overall, these comparative studies are in good agreement with each other and demonstrated the validity of the single cell secretomic analysis microchip. Another advantage of our platform is that it also measures proteins secreted from multiple cells at the same time. While IL-6 and IL-8 secretion increases with increasing number of cells in a microchamber, the amount of MCP-1 or MIF increase does not change significantly when cell number exceeds 2, suggesting the existence of a possible mechanism similar as ‘quorum sensing’ in which the paracrine mechanism in the multi-cellular system controls homeostasis.
The single cell secretomic analysis chip was also used to measure two additional cell lines in order to assess the broad applicability of this platform. The first is an immune cell line (U937). The cells are human monocytes, which can be stimulated with phorbol myristate acetate (PMA) to differentiate into functional macrophage cells and then challenged by cytotoxin lipopolysaccharide (LPS) to stimulate cytokine production. This process emulates the inflammatory immune response of human macrophages to Gram-negative bacteria31. The major proteins observed are RANTES, TNFα, MCP-1, IL6 and IL-8 (Supporting Fig. S4b). While the majority of cells produce RANTES, IL8 and TNFα, the level of these proteins varies among individual cells and the secretion of other lower abundance proteins such as MCP-1 and IL-6 exhibit heterogeneous signatures. A bulk population secretion measurement was performed in parallel on the supernatant collected from the same cells to verify the single cell experiments (Supporting Fig. S8). The result also reveals RANTES, IL8, MCP-1, IL-6 and TNFα as the top five proteins, consistent with the single cell analysis. IL-8 and RANTES secretion increases with increasing number of cells in a microchamber, the amount of MCP-1 or TNFα increases does not change significantly when cell number exceeds 4 (Supporting Fig. S4c). The second is a lung carcinoma cell line (A549) that constitutively produces cytokines or growth factors. Therefore, we measured the basal level secretion from these cells with no stimulation. The major proteins observed are MCP-1, IL-6, IL-8, VEGF and FGF (Supporting Fig. S9) that were also validated by the bulk population assay using standard pin-spotted antibody microarray assays (Supporting Fig. S10). The proteins secreted from A549 cells include both pro-inflammatory cytokines and growth factors, in agreement with the role of lung tumor cells in both maintaining tumor growth and promoting an inflammatory microenvironment. Altogether, the cell line studies demonstrated that our platform is capable of rapid, quantitative and high throughput analysis of protein secretion profiles in single cells compared to current conventional methods such as ELISpot. Validating our platform with cell lines allow us to expand our sample repertoire to include more complex samples such as tissue specimens from patients.
Correlation between secretomic signature and migratory property
Although flow cytometry-based single cell analysis allows for multiplexed protein measure, the measured protein profile cannot be directly correlated to the cell’s behavior and activity such as migratory property. Our platform utilizes live cell imaging to count captured cells, thus permitting simultaneous measurement of cellular behavior and subsequent correlation to the corresponding protein profile of the same cell. Herein we measure the migration of lung cancer cells (A549) loaded in the single cell secretomic analysis chip by measuring the distance of movement before incubation and after 24 hours of incubation (Fig. 4a). These cells were seen to adhere to the channel wall and migrate at varying speeds. The results are summarized in a heatmap showing single-cell secretomic profiles sorted by increasing cell migration distance(Supporting Fig. S11). We performed p-value analysis of cytokine levels in high motility (top 20% over the observed range) vs low motility (bottom 20% of the same range) cells. While the majority of the cells do not migrate, the highly migratory cells are statistically associated with high expression of IL-8 (P < 0.01) (Fig. 4b). the correlation between the secretion of MCP-1 and cell migration was less significant (Fig. 4c, d). While IL-6 appears to be negatively associated with cell motility in the scatter plots, but does not show statistical correlation using the aforementioned test. These proteins have been linked to the increase of motility and metastatic potential in different cancers32–34, and through the investigation of single cell IL-8 secretion, it may be possible to study the secretomic signatures of individual cells linked to metastasis. In brief, our platform for the first time shows simultaneous measurement of protein secretomic signature and phenotypic properties (e.g. migration) of single live cells that can lead to improved understanding of cellular functions and the underlying molecular mechanisms.
Figure 4.
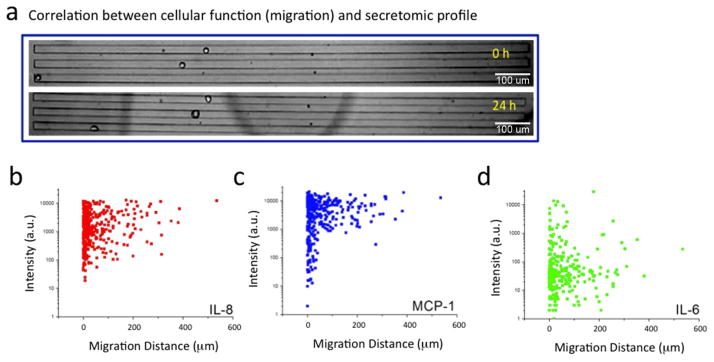
Correlation between protein secretion profiles and cellular migration for A549 cells. (a) Representative optical images showing three single cells (n=384) before (0h) and after (24h) protein secretion assay. (b) Scatter plot showing the fluorescence intensity corresponding to IL-8 secretion versus migration distance of individual cells (P < 0.05). (c) Scatter plot showing a similar analysis on MCP-1 (P = 0.14). (d) Scatter plot showing a similar analysis on IL-6 (P = 0.75). Each dot represents a single cell.
Secretomic profiling of single tumor cells from clinical patient specimens
To expand the utility of our platform to measuring multiplexed secretion in cells derived from complex biospecimens, we also applied our device to the measurement of fresh primary tumor tissue from three patients (Supporting Table S2) with malignant brain tumor, glioblastoma multiforme (Patients 1&2), or meningioma (Patient 3). A portion (<0.2g) of the surgically-resected tumor tissue is washed with ice cold phosphate-buffered saline, minced into smaller fractions and then dissociated into a single cell suspension using collagenase (Fig. 5a and Supporting Methods). The cells were spun down and re-suspended in medium at a density of ~106 cells/mL. Within 1 hour of tissue procurement, the single cell suspension is loaded onto the single-cell secretomic analysis device via pipette. After allowing the cells to secrete cytokines for 12 hr, the pattern on the barcode array is developed with detection antibodies and scanned. A raw fluorescent image (Fig. 5b, Patient 1) shows excellent protein signals and similar background compared to the scanned image from cell lines. The antibody barcode array includes 14 proteins as shown in Figure 5b. In this experiment, between 0 to 22 cells were captured within a microchamber, with 1058 of the microchambers capturing single cells. We quantified the fluorescence intensities of each secreted cytokine from each individual channel, and then generated a heat map of the single cell secretion profiles (Fig 5c). Unsupervised hierarchical clustering of the single cell secretion profiles resolved three separate populations of cells with varying activity. One cluster of cells (Fig. 5c, blue cluster) was generally more active, secreting a wider range of proteins presumably corresponding to more aggressive phenotype, while the cells indicated by green exhibit the lowest level of cytokine production and may represent more quiescent phenotypes such as tumor stem/progenitor cells35. The large fraction indicated by orange are a variety of functional phenotypes. The result from the Patient 2 (Fig. 5d) shows similarities to the results from Patient 1, such as MIF and IL-8 as major proteins, but different pattern in that it has much reduced production of inflammatory cytokines and higher level of EGF. The second tier proteins all show distinct cellular heterogeneity. Supporting Figure S12 and Supporting Figure S13 presents histograms and scatter plots of individual proteins, which show both the relative levels of proteins and the distributions amongst the cell population.
Figure 5.

Single-cell secretomic analysis of primary tumor cells from patients. (a) Procedure for processing tissue specimens, preparation of single cell suspension and application of primary cells to the subnanoliter microchamber array chip. (b) Representative region of the scanned image for Patient 1. (c) and (d) Heat maps showing single-cell secretomic signatures of primary tumor cells from two patients (Patients 1 and 2), respectively. The data are presented as a result of non-supervised hierarchical clustering analysis. (e) Histogram plots of individual proteins measured on the sample from Patient 1. (e) Scatter plot matrices showing protein-protein correlation in single cells. Each subpanel is the scatter plot showing the level of a protein versus the other in all single cells measured. The proteins are indicated at the diagonal line. The correlation coefficient is computed as R via a linear regression analysis. The entire matrix is color-coded by red (positive correlation) and blue (negative correlation). The color intensity is proportional to the R value.
We compiled pseudo-three-dimensional scatter plots of the single cell cytokine measurements for the patient primary tumors in the format of flow cytometric plots and formed a 14×14 mosaic matrix (Fig. 5e). The proteins are shown at the diagonal line and each panel is a pair-wise correlation plot, for each of which we performed a linear regression analysis to yield the R value. Then the whole matrix is color-coded by red (positive correlation) and blue (negative correlation), and the color intensity is proportional to R. In the Patient 1 matrix, all the inflammatory cytokines are apparently associated within one cluster and several growth factors are grouped in a separate cluster, reflecting their functional difference. In the result for Patient 2, the pro-inflammatory cytokines, although generally expressed at low levels, also show inter-correlation. Interesting, the secretion of EGF is negatively correlated to pro-inflammatory and chemoattactant proteins (MCP-1, GMCSF and IL-8). We also analyzed a third sample from a patient (Patient 3) with transitional meningioma, which is considered a more homogeneous and less inflammatory tumor. Indeed our results for Patient 3 (Supporting Fig. S14 and Supporting Fig. S15) show reduced pro-inflammatory cytokine signals. These studies, while still very preliminary, imply the relevance of our results to these cells’ physiological functions or pathological condition. Currently surgical treatment remains the most effective therapy of human glioblastoma. Afterwards, chemotherapy might be carried out systemically or by putting drug-containing wafers to the surgical cavity to further eradicate invasive tumor cells that have diffused to normal brain tissue36. Our platform potentially can distinguish and quantitate invasive cell phenotypes as they generally produce more cytokines as well as different profile of cytokines, which has the clinical value to determine tumor invasiveness and tailor the chemotherapeutic strategy for individual patients. In addition, these proteins that act as the soluble signals to mediate cell-cell communication in tumor microenvironment may be identified as new therapeutic targets for personalized treatment37–39.
CONCLUSIONS
Single cell proteomic analysis has generally been much more challenging than genetic analysis from single cells, due to the lack of equivalent amplification methods for proteins such as polymerase chain reaction (PCR) for nucleic acids. Recent advance in flow cytometric analysis allows for 34-plexed measurement of protein markers from single cells, but most proteins are either surface receptors or cytoplasmic proteins40. Intracellular cytokine staining (ICS) enables indirect assessment of “secreted” proteins, but currently the number of cytokines that can be measured is practically limited to below 10, presumably due to increased non-specific binding from a large number of antibodies in the limited volume of a single cell. Moreover, unlike protein secretion, it is not a direct measurement of cell function. Thus, multiplexed protein secretion measurement is a missing piece of functional characterization of single cells. It has become increasingly evident that even genetically homogeneous cells can be extremely heterogeneous, leading to many unanswered questions in studying their biology41. Studying the secretion profile of single cells can reveal much more about tumor heterogeneity than studying the signaling patterns of cells in population wherein the signals become averaged out and all defining information is lost, emphasizing the need for studying single cell secretion42,43.
We have described a sub-nanoliter multiplexed immunoassay chip that enables high throughput, simultaneous detection of a panel of 14 cytokines secreted from over a thousand single cells in parallel. This platform provides significant advantages specific to the detection of secreted proteins and offers information complementary to that obtained through flow cytometry. An example scenario where this device would offer unique advantages is that when a cell separation tool is used to sort out a phenotypically identical cell population using specific surface markers, these cells can then be placed in our device to further reveal cellular heterogeneity at the functional level. For instance, human T cell lineages often display a number of functions and the complex combinations of multiple functions in a single T cells dictates the “quality” of this cell in response to a specific antigen. Recent studies showed that multifunctional T cells often exhibit greater potency and durability.44 The latest HIV vaccine trials employed the ELISpot technique to count interferon-γ(IFN-γ)-secreting T cells as a means to assess the efficacy of vaccination, but it turns out that most IFN-γ-secreting cells are terminally differentiated effector T cells and have minimal protective effect against viral infection. Our platform represents a promising tool to perform polyfunctional analysis on the cells isolated from flow cytometer or other separation techniques, e.g. magnetically assisted cell sorting45, to bring single-cell protein assay to another level of functional analysis. A potential concern of our platform is that cells are isolated in the sealed micro-chamber and may experience a condition that affects the normal functioning of primary cells ex vivo. As a bioanalytical tool, our microchip was not intended to perform long-term culture of cells and the typical assay time is a few hours to 1 day. It has been reported that ex vivo assay of primary cells in sealed and isolated environment do produce proteins over a long time as anticipated for their intrinsic physiological activity 22,46 and interestingly the cells could gain greater viability in a sealed nL-chamber because it recapitulates the in vivo crowdedness in primary tissue and retains sufficient concentrations of cytokines for more effective autocrine signaling. Thus, our microchip is a promising platform for high-throughput analysis of protein secretion profiles from single primary cells and may assist in differential diagnosis and monitoring of cellular functions in patients.
Supplementary Material
Acknowledgments
This study was supported by the NIH LINCS Program Technology Center Grant (NIH 1 U01 CA164252, MPI: R.F. and K.M.J.), the U.S. National Cancer Institute Howard Temin Pathway to Independence Award (NIH 4R00 CA136759, PI: R.F.), a grant from the Bill & Melinda Gates Foundation through the Grand Challenges Explorations Initiative (PI: R.F.), the Alzheimer Association New Investigator Grant, the NIH grants U54CA143868 (PI: D.W.) and R01 GM084204 (PI: D.W.), and the Single Cell Profiling Core supported by the NIH grant U54 CA143798 (subaward PI: R.F.). We also acknowledge the Yale Institute for Nanoscience and Quantum Engineering (YINQE) and the Yale Nanofabrication Center to allow us to use their facilities.
Footnotes
The authors declare no competing financial interests.
Experimental details for device design, fabrication and use in single cell secretomic analysis are included in Supporting Information.
References
- 1.Rothenberg EV. Nat Immunol. 2007;8:441. doi: 10.1038/ni1461. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Cell. 2011;144:646. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Gnecchi M, Zhang ZP, Ni AG, Dzau VJ. Circ Res. 2008;103:1204. doi: 10.1161/CIRCRESAHA.108.176826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niepel M, Spencer S, Sorger P. Curr Opin Chem Biol. 2009 doi: 10.1016/j.cbpa.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gascoigne KE, Taylor SS. Cancer Cell. 2008;14:111. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Cohen AA, Geva-Zatorsky N, Eden E, Frenkel-Morgenstern M, Issaeva I, Sigal A, Milo R, Cohen-Saidon C, Liron Y, Kam Z, Cohen L, Danon T, Perzov N, Alon U. Science. 2008;322:1511. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 7.Sachs K, Perez O, Pe’er D, Lauffenburger DA, Nolan GP. Science. 2005;308:523. doi: 10.1126/science.1105809. [DOI] [PubMed] [Google Scholar]
- 8.Kotecha N, Flores NJ, Irish JM, Simonds EF, Sakai DS, Archambeault S, Diaz-Flores E, Coram M, Shannon KM, Nolan GP, Loh ML. Cancer Cell. 2008;14:335. doi: 10.1016/j.ccr.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud Ø, Gjertsen BT, Nolan GP. Cell. 2004;118:217. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 10.Prussin C. J Clin Immunol. 1997;17:195. doi: 10.1023/a:1027350226435. [DOI] [PubMed] [Google Scholar]
- 11.Sachdeva N, Asthana D. Front Biosci. 2007;12:4682. doi: 10.2741/2418. [DOI] [PubMed] [Google Scholar]
- 12.Stratov I, DeRose R, Purcell DF, Kent SJ. Curr Drug Targets. 2004;5:71. doi: 10.2174/1389450043490686. [DOI] [PubMed] [Google Scholar]
- 13.Henshall M, Gorfain E. Genet Eng Biotechn N. 2007;27:1. [Google Scholar]
- 14.Chen S, LaRoche T, Hamelinck D, Bergsma D, Brenner D, Simeone D, Brand RE, Haab BB. Nat Methods. 2007;4:437. doi: 10.1038/nmeth1035. [DOI] [PubMed] [Google Scholar]
- 15.Liotta LA, Espina V, Mehta AI, Calvert V, Rosenblatt K, Geho D, Munson PJ, Young L, Wulfkuhle J, Petricoin EF. Cancer Cell. 2003;3:317. doi: 10.1016/s1535-6108(03)00086-2. [DOI] [PubMed] [Google Scholar]
- 16.Wang DJ, Bodovitz S. Trends Biotechnol. 2010;28:281. doi: 10.1016/j.tibtech.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheong R, Wang CJ, Levchenko A. Science Signaling. 2009:2. doi: 10.1126/scisignal.275pl2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Love JC, Ronan JL, Grotenbreg GM, van der Veen AG, Ploegh HL. Nat Biotechnol. 2006;24:703. doi: 10.1038/nbt1210. [DOI] [PubMed] [Google Scholar]
- 19.Lee SS, Horvath P, Pelet S, Hegemann B, Lee LP, Peter M. Integr Biol. 2012;4:381. doi: 10.1039/c2ib00144f. [DOI] [PubMed] [Google Scholar]
- 20.Rowat AC, Bird JC, Agresti JJ, Rando OJ, Weitz DA. P Natl Acad Sci USA. 2009;106:18149. doi: 10.1073/pnas.0903163106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lecault V, VanInsberghe M, Sekulovic S, Knapp DJHF, Wohrer S, Bowden W, Viel F, McLaughlin T, Jarandehei A, Miller M, Falconnet D, White AK, Kent DG, Copley MR, Taghipour F, Eaves CJ, Humphries RK, Piret JM, Hansen CL. Nat Methods. 2011;8:581. doi: 10.1038/nmeth.1614. [DOI] [PubMed] [Google Scholar]
- 22.Ma C, Fan R, Ahmad H, Shi Q, Comin-Anduix B, Chodon T, Koya RC, Liu C-C, Kwong GA, Radu CG, Ribas A, Heath JR. Nat Med. 2011;17:738. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin Young S, Remacle F, Fan R, Hwang K, Wei W, Ahmad H, Levine RD, Heath JR. Biophys J. 2011;100:2378. doi: 10.1016/j.bpj.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Science. 2004;305:1622. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 25.Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Science. 2000;288:113. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 26.Fan R, Vermesh O, Srivastava A, Yen BKH, Qin LD, Ahmad H, Kwong GA, Liu CC, Gould J, Hood L, Heath JR. Nat Biotechnol. 2008;26:1373. doi: 10.1038/nbt.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raman D, Baugher PJ, Thu YM, Richmond A. Cancer Lett. 2007;256:137. doi: 10.1016/j.canlet.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dranoff G. Nat Rev Cancer. 2004;4:11. doi: 10.1038/nrc1252. [DOI] [PubMed] [Google Scholar]
- 29.Zou W. Nat Rev Cancer. 2005;5:263. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- 30.Wu Y, Lu Y, Chen W, Fu J, Fan R. PLoS Comput Biol. 2012;8:e1002355. doi: 10.1371/journal.pcbi.1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aderem A, Ulevitch RJ. Nature. 2000;406:782. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- 32.Singh RK, Gutman M, Radinsky R, Bucana CD, Fidler IJ. Cancer Res. 1994;54:3242. [PubMed] [Google Scholar]
- 33.Li A, Dubey S, Varney ML, Dave BJ, Singh RK. J Immunol. 2003;170:3369. doi: 10.4049/jimmunol.170.6.3369. [DOI] [PubMed] [Google Scholar]
- 34.Waugh DJJ, Wilson C. Clin Cancer Res. 2008;14:6735. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 35.Wicha MS, Liu SL, Dontu G. Cancer Res. 2006;66:1883. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 36.Lesniak MS, Brem H. Nat Rev Drug Discov. 2004;3:499. doi: 10.1038/nrd1414. [DOI] [PubMed] [Google Scholar]
- 37.Dvorak HF. J Clin Oncol. 2002;20:4368. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- 38.Reardon DA, Wen PY. Oncologist. 2006;11:152. doi: 10.1634/theoncologist.11-2-152. [DOI] [PubMed] [Google Scholar]
- 39.Rich JN, Bigner DD. Nat Rev Drug Discov. 2004;3:430. doi: 10.1038/nrd1380. [DOI] [PubMed] [Google Scholar]
- 40.Bendall SC, Simonds EF, Qiu P, Amir EAD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP. Science. 2011;332:687. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhatia S, Frangioni JV, Hoffman RM, Iafrate AJ, Polyak K. Nat Biotechnol. 2012;30:604. doi: 10.1038/nbt.2294. [DOI] [PubMed] [Google Scholar]
- 42.Bendall SC, Nolan GP. Nat Biotechnol. 2012;30:639. doi: 10.1038/nbt.2283. [DOI] [PubMed] [Google Scholar]
- 43.Michor F, Polyak K. Cancer Prev Res. 2010;3:1361. doi: 10.1158/1940-6207.CAPR-10-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seder RA, Darrah PA, Roederer M. Nat Rev Immunol. 2008:8. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 45.Adams JD, Kim U, Soh HT. P Natl Acad Sci USA. 2008;105:18165. doi: 10.1073/pnas.0809795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han Q, Bagheri N, Bradshaw EM, Hafler DA, Lauffenburger DA, Love JC. P Natl Acad Sci USA. 2012;109:1607. doi: 10.1073/pnas.1117194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.