

INTRODUCTION
Wilms tumor is the second most common intraabdominal cancer of childhood and the fifth most common pediatric malignancy overall. It represents approximately 6% of all pediatric cancers and accounts for more than 95% of all tumors of the kidney in the pediatric age group [1,2]. In the United States there are approximately eight cases of Wilms tumor per million children less than 15 years of age per year, with the total number of new cases being estimated at about 650 cases per year [3]. Approximately 75% of the cases occur in children less than 5 years of age with a peak incidence at 2 to 3 years of age. Survival for patients with Wilms tumor when considered as a whole, once <30%, is currently greater than 90%, making it one of the real successes of modern medicine. This dramatic improvement in survival is due, in part, to the systematic manner in which the approach to therapy has evolved. Beginning in 1969, the National Wilms Tumor Study (NWTS) Group, which was incorporated into the Children’s Oncology Group (COG) in 2001, evaluated different treatment strategies conducted through multi-institutional trials in which the primary goals were (and remain) to treat patients according to well-defined risk groups to achieve the highest cure rates, decrease the frequency and intensity of acute and late toxicity, and minimize cost. Surgery has always been a critical component of Wilms tumor therapy; the addition and refinement of chemotherapy, and, in certain circumstances, radiation therapy, have also had a significant impact on achieving improved survival rates.
PATHOLOGY
Classic Wilms tumor has a triphasic appearance, with the three cell types being stromal, epithelial, and blastemal. All three elements are not required, however, to have a diagnosis of Wilms tumor. The neoplastic cells can often be seen to be forming primitive tubules and glomeruli. One of the major contributions of the NWTS was a report by Beckwith and Palmer [4] that separated Wilms tumors into distinct histopathologic categories based on prognosis. An analysis of 427 specimens found that 11% of the total patients in this study contributed to 52% of the mortality. Since that study, two distinct histopathologic types of Wilms tumor have become recognized—favorable and unfavorable. The unfavorable group comprises Wilms tumors with anaplasia (extreme nuclear and cytologic atypia). Anaplasia is present in about 5% of Wilms tumors and is more common in older children, reaching a peak at approximately 5 years of age [5]. This histopathologic variant is also more frequent in African American patients. Anaplasia can be focal or diffuse in nature, with the focal subtype being somewhat more favorable.
GENETICS
Childhood tumors, including Wilms tumor, have long been favorite models for unraveling the molecular events of carcinogenesis because the early age of onset suggests that only a few events are required to establish a neoplastic phenotype. As with many cancer genes, the first clue to the location of a Wilms tumor gene came from the cytogenetic analysis of DNA from patients with Wilms tumor in whom there were commonly associated, genetically determined anomalies (Table 1). Because of the association of seemingly unrelated phenotypic abnormalities, a large chromosomal disruption would be predicted. This strategy revealed a constitutional deletion of band 13 of the short arm of chromosome 11 in patients with the rare congenital syndrome consisting of Wilms tumor, aniridia, genitourinary abnormalities or gonadoblastoma, and mental retardation (WAGR syndrome). Loss of genetic material from this region was associated with tumorigenesis, suggesting that the critical loss was of a tumor-suppressor gene. Candidate genes were sought from the minimum deletion region of chromosome 11p13 and, ultimately, the WT1 gene was isolated and cloned [6]. Sequence analysis suggests that WT1 is a transcriptional regulator whose protein product binds to specific DNA motifs. Although the exact function of the WT1 protein has not been clearly defined, the pattern of WT1 expression suggests that it has a role in metanephric stem cell differentiation [7]. This may explain the finding of associated genitourinary abnormalities. In patients with isolated Wilms tumor, however, evidence for WT1 mutation exists in only about 5% to 10% of the cases.
Table 1.
Syndromes related to the WT1 and WT2 loci
| Syndrome | Locus | Genetic lesion | Phenotype | Wilms tumor risk (%) |
|---|---|---|---|---|
| WAGR syndrome | 11p13 | Deletion WT1 gene | Aniridia, genitourinary anomalies, delayed-onset renal failure | 30 |
| Denys-Drash | 11p13 | Point mutation in zinc-finger regions of WT1 gene | Ambiguous genitalia, diffuse mesangial sclerosis | 90 |
| Frasier | 11p13 | Point mutation in WT1 intron 9 donor splice site | Ambiguous genitalia, streak gonads, focal segmental glomerulosclerosis | Low |
| Beckwith-Wiedemann | 11p15 | Precise genetic lesion unclear; loss of imprinting of several genes including IGF2, H19, and p57kip2 implicated | Organomegaly, large birth weight, macroglossia, omphalocele, hemihypertrophy, ear pits and creases, neonatal hypoglycemia | 5 |
Abbreviation: WAGR, Wilms’ tumor, aniridia, genitourinary abnormalities or gonadoblastoma, and mental retardation.
Data from Dome JS, Coppes MF. Recent advances in Wilms tumor genetics. Curr Opin Pediatr 2002;14(1):5–11.
Another syndrome associated with constitutional abnormalities of the WT1 gene is the Denys-Drash syndrome. In this syndrome, patients have severe genitourinary abnormalities (eg, male pseudohermaphroditism) and renal failure secondary to progressive, diffuse glomerular nephropathy. Fifty percent to 90% of these patients will develop Wilms tumor [8]. The alteration of the WT1 gene in patients with this syndrome seems to be a missense mutation within the DNA-binding region of this transcription factor.
The Beckwith-Wiedemann syndrome (BWS) is another syndrome of which Wilms tumor is a common component. This syndrome consists of several abnormalities, including macroglossia, macrosomia, hypoglycemia, visceromegaly, and omphalocele, in addition to a predisposition to several tumors, most commonly Wilms tumor. The locus for BWS seems also to be on the short-arm of chromosome 11 but at a site distinct from WT1, that being 11p15, as identified by large karyotypic abnormalities and linkage analysis of BWS families [9]. In sporadic Wilms tumor, there seems to be a small percentage of patients with loss of one copy of genetic material in the 11p15 region. Nearly always, there has been a loss of the maternal allele, suggesting that genetic imprinting occurs at this locus. In rare patients, uniparental paternal disomy has occurred whereby there is inheritance of two paternal copies of this region. Several genes have been identified within the 11p15 region that demonstrate genomic imprinting; alterations of one of these may be responsible for the development of BWS.
Familial Wilms tumor occurs at an incidence of approximately 5%. Linkage analysis shows, however, that the locus for this predisposition is not WT1 or WT2, suggesting that there are other loci involved in the genesis or predisposition for Wilms tumor. An additional Wilms tumor locus at 16q was suggested by loss of heterozygosity (LOH; loss of one of two copies of a chromosomal region) for chromosome 16q markers in about 20% of Wilms tumors [10]. LOH at a locus mapping to the distal chromosome 1p has also been found in a small group of patients with Wilms tumor (~12%) and at 11p in 33% of Wilms tumor patients [11]. Patients with LOH for chromosome 16q had relapse rates 3.3 times higher and a mortality rate 12 times higher than patients without LOH for chromosome 16q [12], suggesting that a gene within this site may be involved in disease progression to a more aggressive phenotype.
Another gene that seems to have some prognostic importance when activated in Wilms tumors is p53. The p53 gene encodes a protein that, among many functions, seems to play a role in cell cycle arrest in cases in which DNA damage has occurred. When the damage cannot be corrected, p53 induces the cell to apoptose. Although p53 alterations are the most common genetic abnormality detected in adult tumors, they are rare in pediatric malignancies, including Wilms tumor. However, there is a high incidence of p53 gene mutation in the anaplastic histologic subtype of Wilms tumor [13]. This may explain why these tumors do not respond well to chemotherapy—their p53-dependent apoptotic pathway may have become inactivated.
Increasingly, array-based studies are being performed to further investigate the molecular genetics of Wilms tumor. One recent study used array comparative genomic hybridization and complementary DNA microarray expression profiling to compare anaplastic and favorable histology Wilms tumor. Gain of 1q and MYCN, and loss of 16q, were common to both histologies, whereas genome loss of 4q and 14q were specific for anaplastic tumors [14]. A second study evaluated the utility gene expression analysis to predict relapse in favorable histology Wilms tumor and found that stratifying local stage III tumors using a classifier of fewer than 50 genes was feasible and modestly accurate [15]. Several similar analyses are ongoing at a variety of institutions.
Determining additional molecular characteristics of Wilms tumor will be important so that refinements in therapy can continue. Further genetic studies may identify those patients at high risk for relapse so that intensification of therapy can be instituted in this select group of patients. Similarly, genetic characterization of patients with Wilms tumor will likely lead to the identification of subgroups of patients with a more favorable prognosis who can be managed according to a minimal therapeutic regimen.
PRESENTATION
Children with Wilms tumor typically present with an asymptomatic abdominal mass. It is not uncommon for the tumor to be discovered by a parent while bathing the child or by a relative who notices a protuberant abdomen. Associated signs and symptoms such as malaise, pain, and either microscopic or gross hematuria are found in approximately 20% to 30% of the children. Hypertension, presumably because of increased renin activity, is present in approximately 25% of children with Wilms tumor. Occasionally, a child will present with a rapidly enlarging abdominal mass, anemia, hypertension, pain, and fever. These children usually have a subcapsular hemorrhage within the tumor that leads to these symptoms.
EVALUATION
The work-up of a child with an intraabdominal mass suspected of being a Wilms tumor should proceed in a systematic fashion. Real-time ultrasonography is usually the initial study and can determine whether or not the mass is intrarenal or extrarenal, and whether the lesion is cystic or solid. A CT scan of the abdomen and pelvis is generally the imaging study of choice for those patients suspected of having a renal tumor. This will confirm the presence of a solid renal mass and will afford the opportunity to visualize the contralateral kidney to confirm its presence (and function) and to exclude synchronous bilateral disease with a high degree of sensitivity.
Intravenous tumor extension of Wilms tumor occurs in about 11% of cases, with the thrombus extending into the inferior vena cava in about 6% of cases [16]. Therefore, this should be specifically investigated in the preoperative evaluation of all children with a renal mass because it may alter the conduct of surgery. This can generally be done most easily and accurately with Doppler ultrasonography. However, a recent study suggests that contrast-enhanced CT scans using current technology have high sensitivity and specificity for detection of cavoatrial tumor thrombus and may obviate Doppler ultrasonography, given that CT scanning is performed routinely in children with an abdominal mass [17]. A CT scan may, therefore, be sufficient because only cavoatrial tumor extent needs to be identified preoperatively because it might change the treatment approach; thrombus in the renal vein should be identified at the time of surgery by palpation of the renal vein before tying and dividing it. MRI can also be used to assess intravascular tumor extension but usually requires sedation in young children and is not routinely used. However, it may be helpful in defining an extensive tumor thrombus that extends up to the level of the hepatic veins or even into the right atrium. In addition, MRI may be useful in distinguishing Wilms tumor from nephrogenic rests. Finally, echocardiography may be useful in rare circumstances to demonstrate (or exclude) intracardiac tumor extension.
The most common site of metastatic spread of Wilms tumor is the lungs; chest CT scan is the preferred imaging modality for evaluating this site. Other rare sites of metastases, such as the liver, are usually well evaluated with the initial abdominopelvic CT scan.
STAGING
Accurate staging of patients with Wilms tumor is imperative and the staging system developed by the NWTS and currently in use in the COG is a surgicopathologic staging system (Box 1). Because appropriate therapy, as well as prognosis, is based on tumor stage, accurate staging of patients with Wilms tumor at the time of diagnosis is imperative. In particular, careful regional lymph node sampling is important because the presence of nodal involvement is associated with an increased incidence of tumor relapse and a poorer prognosis.
Box 1. Wilms tumor staging system (COG).
Tumor limited to kidney and completely excised. The surface of the renal capsule is intact. Tumor was not ruptured before or during removal. There is no residual tumor apparent beyond the margins of excision.
Tumor extends beyond the kidney, but is completely excised. There is regional extension of the tumor; ie, penetration through the outer surface of the renal capsule into perirenal soft tissues. Vessels outside the kidney substance are infiltrated or contain tumor thrombus. There is no residual tumor apparent at or beyond the margins of excision.
- Residual nonhematogenous tumor confined to abdomen. Any one or more of the following occur:
- Lymph nodes are involved with tumor.
- There has been peritoneal contamination by tumor such as by biopsy or rupture of the tumor before or during surgery, or by tumor growth that has penetrated through the peritoneal surface.
- Implants are found on the peritoneal surfaces.
- The tumor extends beyond the surgical margins either microscopically or grossly.
- The tumor is not completely resectable because of local infiltration into vital structures.
Hematogenous metastases. Deposits beyond stage III; ie, lung, liver, bone, and brain.
Bilateral renal involvement at diagnosis. An attempt should be made to stage each side according to the above criteria on the basis of extent of local disease.
NWTS
Owing to the rarity of Wilms tumor, organized clinical investigation was limited until the establishment of the NWTS in 1969. This represented a cooperative effort among several groups to treat patients in a clearly defined manner so that statistically relevant comparisons of treatment variations could be made. Five sequential trials have been completed, with the basic goal of each successive NWTS trial having been to maintain a high cure rate for patients with Wilms tumor, while reducing the intensity and duration of therapy, based on surgical stage and histologic evaluation. Although the importance of surgery in the treatment of Wilms tumor has long been recognized, the roles for chemotherapy and radiation therapy have evolved based on the results of the NWTS trials.
NWTS-1 showed that postoperative abdominal radiotherapy was not necessary for children who were less than 2 years of age whose tumors were limited to the kidney and completely resected [18]. In addition, the combination of vincristine and dactinomycin was shown to be more effective for the treatment of children with tumors that extended beyond the kidney than either drug alone. NWTS-2 demonstrated that 6 months of combination chemotherapy with vincristine and dactinomycin was effective treatment of children with tumors limited to the kidney and completely resected, none of whom received abdominal radiation. The addition of doxorubicin hydrochloride (Adriamycin) to the combination of vincristine and dactinomycin was found to improve the relapse-free survival of other patients [19]. The separation of Wilms tumor into distinct histopathologic categories based on prognosis was used to stratify patients in NWTS-3 [20]. This study also began to define the low dose of ionizing radiation to be used, when necessary, and showed that the addition of cyclophosphamide did not improve survival over that generated with three-drug therapy. NWTS-4 examined the utility of dose intensive scheduling to cut down on the duration of therapy [21]. The more recently concluded NWTS-5, a single-arm therapeutic trial designed to evaluate the prognostic value of certain biologic markers in Wilms tumor, demonstrated that LOH for genetic material on chromosomes 1p and 16q in stage I and II favorable histology Wilms tumor was associated with a poorer prognosis [22]. This information, LOH of 1p and 16q, is now being used to further stratify patients in the current COG trial for Wilms tumor.
CURRENT PROTOCOLS
Historically, the most important prognostic variables for patients with Wilms tumor have been the histopathologic tumor classification and surgical stage [23]. Survival statistics based on these factors, which have largely guided treatment, are shown in Table 2. The staging system developed by the NWTS and currently in common use, is shown in Box 1. Because appropriate therapy, as well as prognosis, is based on tumor stage, accurate staging of patients with Wilms tumor at the time of diagnosis is imperative and includes histologic assessment of regional lymph node involvement.
Table 2.
Ten-year outcomes for patients with Wilms tumor treated on NWTS-4
| Histology | Stage | 10 y relapse-free survival (%) | 10 y overall survival (%) |
|---|---|---|---|
| Favorable | I | 91 | 96 |
| II | 85 | 93 | |
| III | 84 | 89 | |
| IV | 75 | 81 | |
| V | 65 | 78 | |
| Anaplastic | I | 69 | 82 |
| II–III | 43 | 49 | |
| IV | 18 | 18 |
It has recently been recognized that a Wilms tumor risk-stratification system based on histology and stage alone does not accurately identify all patients at risk for recurrence. New clinical and genetic risk factors for recurrence have been validated and have now been incorporated into the assigning of therapy in the current COG clinical trials for patients with Wilms tumor. These factors include patient age at the time of diagnosis, tumor weight, histologic response to therapy, and the allelic status of chromosomes 1p and 16q in resected tumors.
Since 2006, four clinical trials have opened within COG for the treatment of patients with Wilms tumor. Together these protocols cover the entire spectrum of Wilms tumor. Central to the approach to therapy for these patients is a risk classification scheme, which is defined in Table 3. To facilitate accurate and timely risk assessment, enrollment in an overarching tumor collection and biology classification protocol, AREN03B2: Renal Tumors Classification, Biology, and Banking Study, is a prerequisite (Fig. 1). Patients are then enrolled on one of the therapeutic protocols.
Table 3.
Risk stratification and treatment assignment for patients with favorable histology Wilms tumor (COG)
| Patient age | Tumor weight | Stage | LOH | Rapid response | Final-risk group | Treatment study |
|---|---|---|---|---|---|---|
| <2 y | <550 g | I | Any | N/A | Very Low | AREN0532 |
| Any | ≥550 g | I | None | N/A | Low | None |
| ≥2 y | Any | I | None | N/A | Low | None |
| Any | Any | II | None | N/A | Low | None |
| ≥2 y | Any | I | LOH | N/A | Standard | AREN0532 |
| Any | ≥550 g | I | LOH | N/A | Standard | AREN0532 |
| Any | Any | II | LOH | N/A | Standard | AREN0532 |
| Any | Any | III | None | Any | Standard | AREN0532 |
| Any | Any | III | LOH | Any | Higher | AREN0533 |
| Any | Any | IV | LOH | Any | Higher | AREN0533 |
| Any | Any | IV | None | Yes | Standard | AREN0533 |
| Any | Any | IV | None | No | Higher | AREN0533 |
| Any | Any | V | Any | Any | Bilateral | AREN0534 |
Fig. 1.
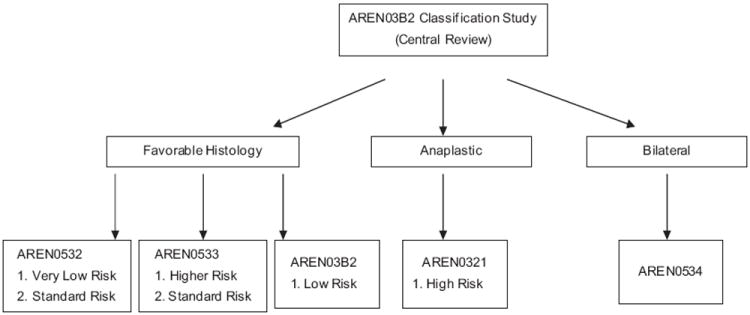
COG protocols for patients with Wilms tumor. Patients being treated for low-risk disease stage I to II, favorable histology, without 1p and 16q LOH are treated with regimen EE-4A and are followed on AREN03B2.
AREN0532: treatment of very low-risk and standard-risk favorable histology Wilms tumor
Only patients with nonmetastatic, favorable histology disease are eligible (Fig. 2). The very low-risk arm of this protocol proscribes surgery alone as definitive treatment of children less than 2 years of age with stage I disease in which the tumor (plus kidney) weight is less than 550 gm. It should be noted that a critical component of the entry criteria is that adequate lymph node sampling be performed at the time of tumor resection to ensure accurate confirmation of stage I disease. These children were thought to be at such low risk for recurrence that the risks of adjuvant chemotherapy might outweigh the risks of recurrence. Therefore, in NWTS-5, these patients were treated with surgery alone. However, because the 2-year event-free survival (EFS), 86.5%, did not meet the required EFS of 90%, this arm of the trial was stopped [24]. Further follow-up has revealed that patients treated with surgery alone had a 5-year EFS of 84% while comparable patients, who also received two-drug chemotherapy, had a 5-year EFS of 97% (P = .002). However, the estimated 5-year overall survival was 98% and 99%, respectively (P = .7) because almost all patients treated with surgery alone who relapsed could be salvaged [25]. Therefore, this approach has been reinstated as a COG protocol to eliminate the potential toxic side-effects of chemotherapy for most patients. Of additional interest are recent studies that suggest that the small subset of patients with very low-risk Wilms tumor with a higher likelihood of recurrence can be identified by molecular analysis of the resected tumors [26]. Specifically, WT1 mutation and 11p15 LOH were found to be associated with relapse in patients who did not receive adjuvant chemotherapy [27]. These biomarkers may be used as part of the entry criteria to future protocols that use a surgery-only approach.
Fig. 2.
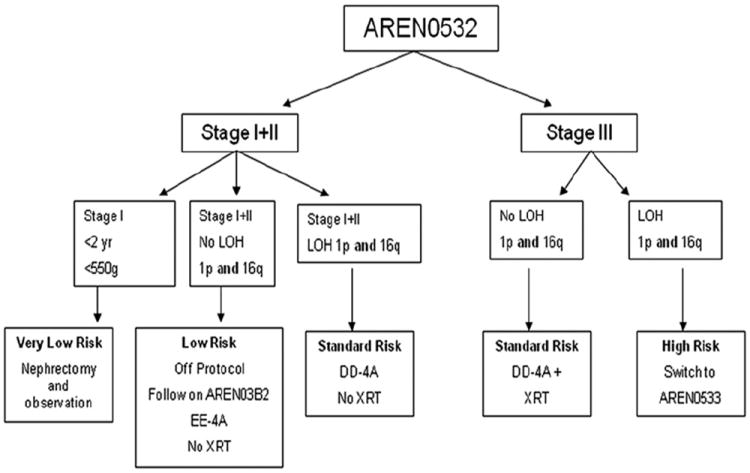
Treatment outline for patients with favorable histology Wilms tumor on AREN0532: Treatment of very low and standard risk favorable histology Wilms tumor.
Children with stage I disease who do not qualify for surgery alone and those with stage II disease are still considered low–risk. However, in addition to surgery, they are started on treatment with 22 weeks (seven cycles) of two-drug chemotherapy (vincristine and dactinomycin) on regimen EE-4A. These patients are technically not treated on a protocol but are followed on AREN03B2. However, if the tumor from these patients is subsequently found to have LOH of both 1p and 16q, these patients are switched to standard-risk therapy consisting of 28 weeks (nine cycles) of three-drug chemotherapy in which doxorubicin is added to vincristine and dactinomycin, on regimen DD-4A. Patients with stage III disease whose tumors do not have 1p and 16q are also treated with standard-risk DD-4A, plus radiation therapy. However, if the tumor is subsequently found to have both 1p and 16q LOH, these patients with stage III disease, are considered higher risk and are switched to AREN0533 (see later discussion).
AREN0533: treatment of newly diagnosed higher risk favorable histology Wilms tumors
Patients eligible for this protocol have favorable histology tumors and either stage III disease that is found to have 1p and 16q LOH, or stage IV (metastatic) disease (Fig. 3). Those with stage III disease and 1p and 16q LOH are treated for 33 weeks (11 cycles) with vincristine, dactinomycin, and doxorubicin, plus cyclophosphamide and etoposide, on regimen M, as well as abdominal radiation. Patients with stage IV disease without 1p and 16q whose pulmonary lesions respond rapidly and completely (see later discussion) are treated with regimen DD-4A chemotherapy and no pulmonary radiation. All other patients with metastatic disease—those with 1p and 16q LOH, those with slow, incomplete response of their pulmonary disease (see later discussion), or those whose metastases are extrapulmonary—are treated with regimen M and radiation to the sites of metastatic disease. Of note, however, is a recent study that has suggested that the presence of hepatic metastases at diagnosis is not an independent adverse prognostic factor [28].
Fig. 3.
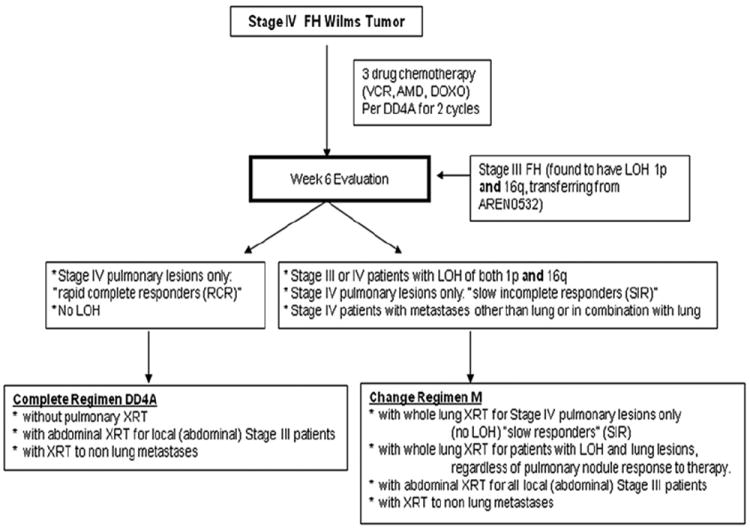
Treatment of patients enrolled on AREN0533: treatment of newly diagnosed, higher risk favorable histology Wilms tumor.
AREN0321: treatment of high-risk renal tumors
All patients with anaplastic Wilms tumor are treated on this protocol. For risk assessment and treatment purposes, a distinction is made between focal (anaplasia confined to one or a few discrete loci within the primary tumor, with no anaplasia or marked nuclear atypia elsewhere) and diffuse anaplasia. Patients whose tumors have focal anaplasia, stage I to III, or diffuse anaplasia stage I, are treated with regimen DD-4A. Patients with stage IV focal anaplasia, stage II to III diffuse anaplasia, and stage IV diffuse anaplasia without measurable disease are treated for 30 weeks with cyclophosphamide-carboplatin-etoposide and vincristine-doxorubicin-cyclophosphamide plus radiation therapy (regimen UH-1). Patients with stage IV diffuse anaplasia with measurable disease are treated with one to two cycles of irinotecan-vincristine as window therapy to evaluate tumor response and determine whether this combination should be added to the backbone treatment with UH-1.
AREN0534: treatment of patients with bilateral, multicentric, or bilaterally predisposed unilateral Wilms tumor
Owing to an increased risk of renal failure in patients with bilateral Wilms tumor, these patients receive neoadjuvant therapy with three-drug chemotherapy of regimen DD-4A in an effort to shrink the tumors before surgery and facilitate the preservation of renal parenchyma, thereby preserving renal function. Also eligible for enrollment on this protocol are patients with Wilms tumor arising in a solitary kidney or those patients less than 1 year of age with a unilateral Wilms tumor who are at an increased risk for a metachronous tumor. Patients with several genetic syndromes, particularly those associated with abnormalities of the Wilms tumor 1 (WT1) and Wilms tumor 2 (WT2) genes on the short arm of chromosome 11, carry this risk (see Table 1). Patients with unilateral Wilms tumor and a Wilms tumor predisposition syndrome are treated with regimen EE-4A if their disease is stage I to II but DD-4A if the disease is stage III to IV. Postoperative chemotherapy (and radiation) are determined by the histopathologic findings at the time of surgery, which should occur after 6 weeks (two cycles) or, at the latest, 12 weeks (four cycles) of preoperative chemotherapy. Protracted courses of chemotherapy in an effort to make the lesions more amenable to nephron-sparing surgery should not be undertaken.
SURGERY FOR WILMS TUMOR
The role of surgery in the therapy of Wilms tumor is paramount because a meticulous and well-performed procedure will accurately determine the stage of the disease and future therapy. A poorly performed procedure can lead to inadequate therapy if the patient’s disease is not appropriately staged or to unnecessarily intensive therapy if operative spill of the tumor occurs or if incomplete resection of the primary tumor is performed. The main responsibility of the surgeon is to remove the primary tumor completely, without spillage, and to accurately assess the extent to which the tumor has spread, with particular attention to adequately assessing lymph node involvement.
Timing of surgery
One of the main controversies in the treatment of children with unilateral Wilms tumor is whether or not to administer preoperative chemotherapy [29,30]. The International Society of Pediatric Oncology (SIOP) recommends giving vincristine and dactinomycin chemotherapy before nephrectomy for localized renal tumors. A core needle biopsy obtained via a posterior approach can be performed without upstaging the patient’s disease but should only be performed in cases of unusual presentation (older age, signs of infection, inflammation) or unusual imaging findings (significant adenopathy, no renal parenchyma seen, intratumoral calcification). Posttreatment histology-based risk classification (Box 2) and stage dictate postoperative chemotherapy (Table 4). Of note, the presence of necrotic tumor or chemotherapy-induced changes in a lymph node or at the resection margin is regarded as proof of previous tumor and, therefore, considered stage III. Proponents of preoperative therapy suggest that the tumor is easier to resect with a decreased incidence of tumor spill (and need for abdominal radiation) [31] and a lower mortality and morbidity. The COG espouses the use of upfront nephrectomy to more accurately assess tumor histology and stage so as to avoid either overtreatment or undertreatment. Evidence to support the dangers of undertreatment comes from a SIOP study that showed an increased incidence of intraabdominal relapses in patients who did not receive postoperative radiation therapy [32]. Likely, patients with lymph node involvement were missed due to preoperative chemotherapy. However, specific patient groups can be identified who would seem to benefit from preoperative chemotherapy. These are patients with bilateral tumors, patients with inferior vena cava and intra-atrial involvement, and patients with massive tumors considered by the operating surgeon to be unre-sectable without undue risk to the patient.
Box 2. Classification of pretreated renal tumors of childhood (SIOP).
Low-risk tumors
Mesoblastic nephroma
Cystic partially differentiated nephroblastoma
Completely necrotic nephroblastoma
Intermediate-risk tumors
Nephroblastoma: epithelial type
Nephroblastoma: stromal type
Nephroblastoma: mixed
Nephroblastoma: regressive type
Nephroblastoma: focal anaplasia
High-risk tumors
Nephroblastoma: blastemal type
Nephroblastoma: diffuse anaplasia
Clear cell sarcoma of the kidney
Rhabdoid tumor of the kidney
Table 4.
Summary of postoperative treatment strategies for localized tumors (SIOP)
| Stage I | Stage II | Stage III | |
|---|---|---|---|
| Low-risk | No further treatment | AV-2a | AV-2 |
| Intermediate-risk | AV-1b | Randomize to AVDc | Randomize to RT or AVD |
| or | or | ||
| AV-2 | RT or AV-2 | ||
| High-risk | AVD | HRd + RT | HR + RT |
Abbreviations: HR, high-risk; RT, radiation therapy.
Vincristine weekly for 8 weeks, then on days 1 and 7 with a 2-week interval between courses for 6 courses, actinomycin D every 3 weeks for 9 doses.
Vincristine weekly for 4 weeks, actinomycin D at day 7.
Vincristine, actinomycin as per AV-2, doxorubicin every 6 weeks for 4 doses.
Cyclophosphamide-doxorubicin alternating with etoposide-carboplatin for 34 weeks.
Surgical exploration
A radical nephrectomy should be performed through a generous transverse, transperitoneal incision. A thoracic extension may be necessary but has been associated with a higher complication rate [33]. Isolation of the hilar vessels before mobilization of the primary tumor is no longer recommended because major vascular injury to the mesenteric arteries, celiac vessels, and aorta have been reported [34]. Palpation of the renal vein before dividing it is recommended to exclude the possibility of a tumor thrombus, which occurs in about 11% of cases. Biopsy of the primary tumor should not be performed before removal and a meticulous dissection to avoid rupture of the tumor capsule with spillage of tumor cells is imperative because tumor spill is strongly associated with recurrence [35]. During tumor resection, the ureter is ligated and divided as low as possible, but complete removal of the ureter down to the bladder is not necessary. Intraoperative inspection of the liver and the contralateral kidney are no longer required, unless lesions were identified on preoperative imaging studies, because of the high accuracy of current imaging modalities. However, lymph node sampling is critically important, despite the absence of abnormal nodes on preoperative imaging or on gross inspection during operative exploration, because a review of lymph node sampling by the NWTS demonstrated a false-negative rate of 31% and a false-positive rate of 18% based on preoperative and intraoperative assessment [36]. Unfortunately, there is currently a fairly high incidence of inadequate intraoperative staging, primarily due to failure to sample lymph nodes [37].
There are reports of surgeons performing partial nephrectomy for unilateral Wilms tumor, particularly in Europe where children routinely receive preoperative chemotherapy [38-40], despite the very low incidence of renal failure after unilateral radical nephrectomy. Although the cumulative incidence of end-stage renal disease due to chronic renal failure 20 years after Wilms tumor diagnosis is less than 1% [41], some studies are suggesting that these patients may have more subtle but significant degrees of renal insufficiency that may be minimized by a nephron-sparing approach to all Wilms tumors [42]. However, other studies have reported an increased risk of local recurrence after partial nephrectomy for unilateral Wilms tumors [38] and survival for patients with intraabdominal recurrence is markedly diminished. One recent meta-analysis did suggest, however, that in highly selected patients, oncologic outcomes of partial versus radical nephrectomy for unilateral Wilms tumor did not differ [43]. However, the percentage of patients whose tumor might be amenable to a partial nephrectomy is low, ranging from 8% to 33% [44], based on criteria, including size, location, and degree of kidney uninvolvement. One additional concern is that upwards of 40% of kidneys with unilateral Wilms tumor will have nephrogenic rests outside of the tumor [45]. The natural history of these lesions that would be left behind with a partial nephrectomy when treated with vincristine and dactinomycin is unknown. Thus, the appropriateness, adequacy, and benefits of a nephron-sparing approach to unilateral Wilms tumor have not been confirmed and partial nephrectomy for unilateral Wilms tumor is not currently endorsed by the COG.
Similarly, the use of a minimally invasive (laparoscopic) approach to nephrectomy (radical or partial) for Wilms tumor is also not currently endorsed by the COG, given the importance of the accurate assessment of the surgical margins and the adverse consequences of tumor spill. Nevertheless, laparoscopic radical nephrectomy is emerging as the treatment of choice for localized renal malignancies in adults and for benign renal disease in infants and children and a few reports have been published describing small series of patients whose tumor has been removed by this approach [46-48]. Of note, however, one recent case report described the consequences of the unsuccessful use of this approach [49]. Thus, this is another aspect of the surgery for Wilms tumor that needs to be carefully investigated.
Bilateral Wilms tumor
Synchronous disease in both kidneys at presentation occurs in approximately 5% of children with Wilms tumor. Unfavorable histology is seen in approximately 10% of the cases and there can be discordant histology between kidneys. However, because tumor biopsy, regardless of the technique, is very unlikely to document anaplastic histology [50] and because there are few other things, other than Wilms tumor, in the differential diagnosis for a child with bilateral renal masses, biopsy at presentation is now discouraged by COG.
Because surgery is a critical component in the treatment of Wilms tumor, the challenge in the management of patients with bilateral disease is to achieve a high cure rate while maintaining adequate long-term renal function. Several studies have demonstrated that patients with bilateral Wilms tumor are at risk for developing renal failure [51-54]. The exact cause of renal failure is not always clear and is likely multifactorial [52,54]. However, because of an increasing appreciation of the potential for renal failure in these patients, the management of synchronous bilateral Wilms tumor has evolved from primary kidney resection to renal-preserving surgical approaches, facilitated by the use of preoperative chemotherapy. A review of the experience at St. Jude Children’s Research Hospital found that bilateral partial nephrectomies could be performed in nearly all patients (>90%) with favorable histology disease, despite occasionally foreboding preoperative imaging [55]. Complications were minimal and long-term renal function and survival have been excellent. Definitive operative intervention should be done early, by 12 weeks after initiation of chemotherapy because little significant further change in tumor size is likely [56] and it is important to determine the exact tumor histology [50,57]. The finding of either anaplastic or blastemal predominant histology would mandate intensification of therapy if it is not stage I disease, whereas certain other circumstances would allow for discontinuation of doxorubicin.
Intravascular tumor extension
Vena cava and intra-atrial extension of Wilms tumor can occur in patients with Wilms tumor, with an incidence of approximately 6%. Survival does not seem to be affected and the prognosis is comparable stage by stage to children without intravascular involvement. Localization of the thrombus should be determined before operation using real-time ultrasonography and/or CT scanning; echocardiography and MRI can sometimes provide additional information. Surgical excision of the primary tumor and thrombus is recommended when technically feasible. An intraabdominal approach is sufficient for infrahepatic lesions with extraction of the caval thrombus after proximal and distal control of the vena cava are obtained. Free-floating thrombi that are easily removed are classified as stage II, but thrombi that invade the vessel or are extremely adherent to the wall of the vessel are classified as stage III. Patients with atrial extension of a tumor thrombus require cardiopulmonary bypass for thrombus removal. In these patients, a midline abdominal incision with a median sternotomy can be used. Alternatively, strong consideration should be given to the use of preoperative chemotherapy [16,58-60].
Metastatic disease
The primary distant site for Wilms tumor metastases is the lungs; hepatic metastases are much less common. Approximately 12% of Wilms tumor patients will have evidence of hematogenous metastases at diagnosis, with 80% having pulmonary metastases. Patients with stage IV favorable histology tumors at diagnosis still have a good prognosis, whereas unfavorable histology patients and patients who relapse with metastatic disease have a grave prognosis. Approximately 20% of favorable histology patients will relapse following therapy with most relapses occurring in the lungs. Patients with pulmonary metastases usually can be managed by combined chemotherapy and radiation therapy [61]; pulmonary resection is rarely indicated because chemotherapy is effective. Although histologic confirmation of pulmonary relapse may be indicated, complete removal of pulmonary metastases at relapse probably does not increase survival.
A new response-based approach is now being taken for patients with stage IV disease in AREN0533. Those patients treated with regimen DD-4A, who have complete radiographic disappearance of their lung metastases (or who have tissue confirmation that residual nodules do not contain viable tumor) at the 6-week reevaluation, are considered rapid responders, continue on DD-4A, and do not receive pulmonary radiation. Patients who do not have complete resolution of pulmonary nodules are considered slow, incomplete responders, are switched to regimen M, and receive whole lung radiation. Those patients who, at the time of diagnosis have pulmonary metastases confirmed histologically and have the lesions completely resected (and, therefore, have no residual disease available for response monitoring) are treated with DD-4A but also are required to receive whole lung radiation. Therefore, consideration should be given to the implications of resecting pulmonary lesions at the time of diagnosis. If the lesions are found to be benign, the patient will have been spared doxorubicin (if local stage I–II) and pulmonary radiation. However, if the lesions are metastatic Wilms tumor and are removed completely, the patient will have lost the opportunity to be considered a rapid responder and avoid whole lung radiation.
Recurrent disease
Approximately 15% of favorable histology and 50% of anaplastic histology Wilms tumor will recur [62], with most relapses occurring early (within 2 years of diagnosis). Relapse occurs most often in the lungs (60%) but can also occur in the abdomen (30%). More rarely, Wilms tumor recurs in the bone or brain. Factors that influence survival after relapse include tumor histology and initial therapy. Survival with favorable histology Wilms tumor is still about 60%, whereas for anaplastic histology it is very poor. A worse outcome is associated with the use of doxorubicin as a part of the initial chemotherapy[62]. Time to recurrence and site of recurrence, once thought to be prognostic, are no longer thought to be so. Therapeutic regimens for relapse include drugs that are not generally used during initial treatment, such as ifosfamide, carboplatin, etoposide, and cyclophosphamide. The roles of surgery and radiation therapy for relapsed Wilms tumor have yet to be precisely defined, although some recent studies suggest that complete surgical resection and radiation of previously unradiated fields may improve survival [63]. The role of high-dose chemotherapy with stem cell rescue for relapsed Wilms tumor is uncertain.
LATE EFFECTS
With the marked improvement in survival for children with Wilms tumor has come the opportunity to observe the long-term complications of the therapy they received. Doxorubicin, used in the treatment of unfavorable histology and high-stage favorable histology Wilms tumor, is associated with an increased risk of cardiac toxicity that is lifelong and dose-dependent, with the greatest risk being to those who received a cumulative dose greater than 250 mg/m2 [64]. NWTS studies suggest that the rate of congestive heart failure 20 years after diagnosis was 4.4% for patients treated with doxorubicin at initial diagnosis [65]; more subtle abnormalities in cardiac function may exist in a much higher percentage of patients. The consequences of ionizing radiation are also significant, particularly when administered to young children. Sequelae include musculoskeletal growth defects, infertility (particularly in females), and, rarely, secondary malignancies, estimated to occur in approximately 3% of children treated for Wilms tumor [66].
The occurrence of renal failure is multifactorial and depends on both genetic predisposition and therapy. The overall incidence of renal failure in children treated for Wilms tumor is less than 1% [41], although, as with cardiac toxicity, the incidence of more subtle abnormalities in renal function is likely higher. Patients with Denys-Drash syndrome have a 20-year cumulative incidence of renal failure of 82.7%, whereas for those with WAGR syndrome it is 43.3%. In those with clinical features consistent with WT1 abnormalities, such as cryptorchidism and hypospadias, the incidence may be as high as 16% [41]. Other contributors to renal failure include the use of abdominal radiation and, of course, surgical resection of the kidneys due to progressive bilateral disease.
Thus, because of these potential late effects in patients treated for Wilms tumor in childhood, there is an increasing effort both to increase the vigilance in the follow-up of these patients and to decrease the toxicity of current therapy.
SUMMARY
Significant improvement has been made in the treatment of children with Wilms tumor. New protocols are in place designed to maintain a high rate of cure for these patients while minimizing toxicity, based on refinement of the risk-stratification system.
Key Points.
The overall survival for children with Wilms tumor is >90%.
Current therapy aims to maintain this high rate of cure while minimizing toxicity.
Histology and stage remain important prognostic factors but the impact molecular genetic factors is becoming increasingly recognized.
Acknowledgments
This work was supported in part by NIH grant CA 021765 and by the American Lebanese Syrian Associated Charities (ALSAC).
References
- 1.Pastore G, Znaor A, Spreafico F, et al. Malignant renal tumours incidence and survival in European children (1978-1997): report from the Automated Childhood Cancer Information System project. Eur J Cancer. 2006;42:2103–14. doi: 10.1016/j.ejca.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 2.Breslow N, Olshan A, Beckwith JB, et al. Epidemiology of Wilms tumor. Med Pediatr Oncol. 1993;21:172–81. doi: 10.1002/mpo.2950210305. [DOI] [PubMed] [Google Scholar]
- 3.Bernstein L, Linet M, Smith M, et al. Renal tumors. National Cancer Institute, SEER Program. Bethesda (MD): National Institutes of Health; 1999. Report No: Publication No 99–4649. [Google Scholar]
- 4.Beckwith JB, Palmer NF. Histopathology and prognosis of Wilms tumors: results from the First National Wilms’ Tumor Study. Cancer. 1978;41:1937–48. doi: 10.1002/1097-0142(197805)41:5<1937::aid-cncr2820410538>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 5.Green DM, Beckwith JB, Breslow NE, et al. Treatment of children with stages II to IV anaplastic Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1994;12:2126–31. doi: 10.1200/JCO.1994.12.10.2126. [DOI] [PubMed] [Google Scholar]
- 6.Call KM, Glaser T, Ito CY, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509–20. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 7.Grubb GR, Yun K, Williams BR, et al. Expression of WT1 protein in fetal kidneys and Wilms tumors. Lab Invest. 1994;71:472–9. [PubMed] [Google Scholar]
- 8.Alomari AI, Tham JC. Denys-Drash syndrome (DDS) Pediatr Nephrol. 2006;21:1237–40. doi: 10.1007/s00467-006-0182-7. [DOI] [PubMed] [Google Scholar]
- 9.Weksberg R, Teshima I, Williams BR, et al. Molecular characterization of cytogenetic alterations associated with the Beckwith-Wiedemann syndrome (BWS) phenotype refines the localization and suggests the gene for BWS is imprinted. Hum Mol Genet. 1993;2:549–56. doi: 10.1093/hmg/2.5.549. [DOI] [PubMed] [Google Scholar]
- 10.Maw MA, Grundy PE, Millow LJ, et al. A third Wilms’ tumor locus on chromosome 16q. Cancer Res. 1992;52:3094–8. [PubMed] [Google Scholar]
- 11.Grundy P, Coppes MJ, Haber D. Molecular genetics of Wilms tumor. Hematol Oncol Clin North Am. 1995;9:1201–15. [PubMed] [Google Scholar]
- 12.Grundy PE, Telzerow PE, Breslow N, et al. Loss of heterozygosity for chromosomes 16q and 1p in Wilms’ tumors predicts an adverse outcome. Cancer Res. 1994;54:2331–3. [PubMed] [Google Scholar]
- 13.Bardeesy N, Falkoff D, Petruzzi MJ, et al. Anaplastic Wilms’ tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet. 1994;7:91–7. doi: 10.1038/ng0594-91. [DOI] [PubMed] [Google Scholar]
- 14.Williams RD, Al-Saadi R, Natrajan R, et al. Molecular profiling reveals frequent gain of MYCN and anaplasia-specific loss of 4q and 14q in Wilms tumor. Genes Chromosomes Cancer. 2011;50:982–95. doi: 10.1002/gcc.20907. [DOI] [PubMed] [Google Scholar]
- 15.Huang CC, Gadd S, Breslow N, et al. Predicting relapse in favorable histology Wilms tumor using gene expression analysis: a report from the Renal Tumor Committee of the Children’s Oncology Group. Clin Cancer Res. 2009;15:1770–8. doi: 10.1158/1078-0432.CCR-08-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shamberger RC, Ritchey ML, Haase GM, et al. Intravascular extension of Wilms tumor. Ann Surg. 2001;234:116–21. doi: 10.1097/00000658-200107000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khanna G, Rosen N, Anderson JR, et al. Evaluation of diagnostic performance of CT for detection of tumor thrombus in children with Wilms tumor: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2012;58(4):551–5. doi: 10.1002/pbc.23222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Angio GJ, Evans AE, Breslow N, et al. The treatment of Wilms’ tumor: Results of the national Wilms’ tumor study. Cancer. 1976;38:633–46. doi: 10.1002/1097-0142(197608)38:2<633::aid-cncr2820380203>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 19.D’Angio GJ, Evans A, Breslow N, et al. The treatment of Wilms’ tumor: results of the Second National Wilms’ Tumor Study. Cancer. 1981;47:2302–11. doi: 10.1002/1097-0142(19810501)47:9<2302::aid-cncr2820470933>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 20.D’Angio GJ, Breslow N, Beckwith JB, et al. Treatment of Wilms’ tumor. Results of the Third National Wilms’ Tumor Study. Cancer. 1989;64:349–60. doi: 10.1002/1097-0142(19890715)64:2<349::aid-cncr2820640202>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 21.Green DM, Breslow N, Beckwith JB, et al. Comparison between single-dose and divided-dose administration of dactinomycin and doxorubicin for patients with Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 1998;16:237–45. doi: 10.1200/JCO.1998.16.1.237. [DOI] [PubMed] [Google Scholar]
- 22.Grundy PE, Breslow NE, Li S, et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol. 2005;23:7312–21. doi: 10.1200/JCO.2005.01.2799. [DOI] [PubMed] [Google Scholar]
- 23.Breslow N, Sharples K, Beckwith JB, et al. Prognostic factors in nonmetastatic, favorable histology Wilms’ tumor. Results of the Third National Wilms’ Tumor Study. Cancer. 1991;68:2345–53. doi: 10.1002/1097-0142(19911201)68:11<2345::aid-cncr2820681103>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 24.Green DM, Breslow NE, Beckwith JB, et al. Treatment with nephrectomy only for small, stage I/favorable histology Wilms’ tumor: a report from the National Wilms’ Tumor Study Group. J Clin Oncol. 2001;19:3719–24. doi: 10.1200/JCO.2001.19.17.3719. [DOI] [PubMed] [Google Scholar]
- 25.Shamberger RC, Anderson JR, Breslow NE, et al. Long-term outcomes for infants with very low risk Wilms tumor treated with surgery alone in National Wilms Tumor Study-5. Ann Surg. 2010;251:555–8. doi: 10.1097/SLA.0b013e3181c0e5d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sredni ST, Gadd S, Huang CC, et al. Subsets of very low risk Wilms tumor show distinctive gene expression, histologic, and clinical features. Clin Cancer Res. 2009;15:6800–9. doi: 10.1158/1078-0432.CCR-09-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perlman EJ, Grundy PE, Anderson JR, et al. WT1 mutation and 11P15 loss of heterozygosity predict relapse in very low-risk Wilms tumors treated with surgery alone: a children’s oncology group study. J Clin Oncol. 2011;29:698–703. doi: 10.1200/JCO.2010.31.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ehrlich PF, Ferrer FA, Ritchey ML, et al. Hepatic metastasis at diagnosis in patients with Wilms tumor is not an independent adverse prognostic factor for stage IV Wilms tumor: a report from the Children’s Oncology Group/National Wilms Tumor Study Group. Ann Surg. 2009;250:642–8. doi: 10.1097/SLA.0b013e3181b76f20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green DM. Controversies in the management of Wilms tumour - immediate nephrectomy or delayed nephrectomy? Eur J Cancer. 2007;43:2453–6. doi: 10.1016/j.ejca.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 30.D’Angio GJ. Pre- or postoperative therapy for Wilms’ tumor? J Clin Oncol. 2008;26:4055–7. doi: 10.1200/JCO.2008.16.5316. [DOI] [PubMed] [Google Scholar]
- 31.Kalapurakal JA, Li SM, Breslow NE, et al. Intraoperative spillage of favorable histology wilms tumor cells: influence of irradiation and chemotherapy regimens on abdominal recurrence. A report from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys. 2010;76:201–6. doi: 10.1016/j.ijrobp.2009.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tournade M, Com-Nougue C, Voute PA, et al. Results of the Sixth International Society of Pediatric Oncology Wilms’ Tumor Trial and Study: a risk-adapted therapeutic approach in Wilms’ tumor. J Clin Oncol. 1993;11:1009–10. doi: 10.1200/JCO.1993.11.6.1014. [DOI] [PubMed] [Google Scholar]
- 33.Ritchey ML, Shamberger RC, Haase G, et al. Surgical complications after primary nephrectomy for Wilms’ tumor: report from the National Wilms’ Tumor Study Group. J Am Coll Surg. 2001;192:63–8. doi: 10.1016/s1072-7515(00)00749-3. [DOI] [PubMed] [Google Scholar]
- 34.Ritchey ML, Lally KP, Haase GM, et al. Superior mesenteric artery injury during nephrectomy for Wilms’ tumor. J Pediatr Surg. 1992;27:612–5. doi: 10.1016/0022-3468(92)90460-o. [DOI] [PubMed] [Google Scholar]
- 35.Shamberger RC, Guthrie KA, Ritchey ML, et al. Surgery-related factors and local recurrence of Wilms tumor in National Wilms Tumor Study 4. Ann Surg. 1999;229:292–7. doi: 10.1097/00000658-199902000-00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otherson HB, deLorimier A, Hrabovsky E, et al. Surgical evaluation of lymph node metastases in Wilms’ tumor. J Pediatr Surg. 1990;25:330–1. doi: 10.1016/0022-3468(90)90079-o. [DOI] [PubMed] [Google Scholar]
- 37.Ehrlich PF, Ritchey ML, Hamilton TE, et al. Quality assessment for Wilms’ tumor: a report from the National Wilms’ Tumor Study-5. J Pediatr Surg. 2005;40:208–12. doi: 10.1016/j.jpedsurg.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 38.Haecker FM, von Schweinitz D, Harms D, et al. Partial nephrectomy for unilateral Wilms tumor: results of study SIOP 93-01/GPOH. J Urol. 2003;170:939–42. doi: 10.1097/01.ju.0000073848.33092.c7. [DOI] [PubMed] [Google Scholar]
- 39.Linni K, Urban C, Lackner H, et al. Nephron-sparing procedures in 11 patients with Wilms’ tumor. Pediatr Surg Int. 2003;19:457–62. doi: 10.1007/s00383-003-0957-x. [DOI] [PubMed] [Google Scholar]
- 40.Moorman-Voestermans CG, Aronson DC, Staalman CR, et al. Is partial nephrectomy appropriate treatment for unilateral Wilms’ tumor? J Pediatr Surg. 1998;33:165–70. doi: 10.1016/s0022-3468(98)90425-0. [DOI] [PubMed] [Google Scholar]
- 41.Lange J, Peterson SM, Takashima JR, et al. Risk factors for end stage renal disease in non-WT1-syndromic Wilms tumor. J Urol. 2011;186:378–86. doi: 10.1016/j.juro.2011.03.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cozzi F, Schiavetti A, Morini F, et al. Renal function adaptation in children with unilateral renal tumors treated with nephron sparing surgery or nephrectomy. J Urol. 2005;174:1404–8. doi: 10.1097/01.ju.0000173132.19010.ff. [DOI] [PubMed] [Google Scholar]
- 43.Cost NG, Lubahn JD, Granberg CF, et al. Oncologic outcomes of partial versus radical nephrectomy for unilateral Wilms Tumor. Pediatr Blood Cancer. 2012;58(6):898–904. doi: 10.1002/pbc.23240. [DOI] [PubMed] [Google Scholar]
- 44.Cozzi DA, Zani A. Nephron-sparing surgery in children with primary renal tumor: indications and results. Semin Pediatr Surg. 2006;15:3–9. doi: 10.1053/j.sempedsurg.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 45.Beckwith JB, Kiviat NB, Bonadio JF. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms’ tumor. Pediatr Pathol. 1990;10:1–36. doi: 10.3109/15513819009067094. [DOI] [PubMed] [Google Scholar]
- 46.Duarte RJ, Denes FT, Cristofani LM, et al. Laparoscopic nephrectomy for Wilms’ tumor. Expert Rev Anticancer Ther. 2009;9:753–61. doi: 10.1586/era.09.44. [DOI] [PubMed] [Google Scholar]
- 47.Duarte RJ, Denes FT, Cristofani LM, et al. Further experience with laparoscopic nephrectomy for Wilms’ tumour after chemotherapy. BJU Int. 2006;98:155–9. doi: 10.1111/j.1464-410X.2006.06214.x. [DOI] [PubMed] [Google Scholar]
- 48.Duarte RJ, Denes FT, Cristofani LM, et al. Laparoscopic nephrectomy for wilms tumor after chemotherapy: initial experience. J Urol. 2004;172:1438–40. doi: 10.1097/01.ju.0000138230.51134.65. [DOI] [PubMed] [Google Scholar]
- 49.Chui CH, Lee AC. Peritoneal metastases after laparoscopic nephron-sparing surgery for localized Wilms tumor. J Pediatr Surg. 2011;46:e19–21. doi: 10.1016/j.jpedsurg.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 50.Hamilton TE, Green DM, Perlman EJ, et al. Bilateral Wilms’ tumor with anaplasia: lessons from the National Wilms’ Tumor Study. J Pediatr Surg. 2006;41:1641–4. doi: 10.1016/j.jpedsurg.2006.05.053. [DOI] [PubMed] [Google Scholar]
- 51.Montgomery BT, Kelalis PP, Blute ML, et al. Extended followup of bilateral Wilms tumor: results of the National Wilms Tumor Study. J Urol. 1991;146:514–8. doi: 10.1016/s0022-5347(17)37840-0. [DOI] [PubMed] [Google Scholar]
- 52.Ritchey ML, Green DM, Thomas PR, et al. Renal failure in Wilms’ tumor patients: a report from the National Wilms’ Tumor Study Group. Med Pediatr Oncol. 1996;26:75–80. doi: 10.1002/(SICI)1096-911X(199602)26:2<75::AID-MPO1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 53.Smith GR, Thomas PR, Ritchey M, et al. Long-term renal function in patients with irradiated bilateral Wilms tumor. National Wilms’ Tumor Study Group. Am J Clin Oncol. 1998;21:58–63. doi: 10.1097/00000421-199802000-00013. [DOI] [PubMed] [Google Scholar]
- 54.Breslow NE, Collins AJ, Ritchey ML, et al. End stage renal disease in patients with Wilms tumor: results from the National Wilms Tumor Study Group and the United States Renal Data System. J Urol. 2005;174:1972–5. doi: 10.1097/01.ju.0000176800.00994.3a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davidoff AM, Giel DW, Jones DP, et al. The feasibility and outcome of nephron-sparing surgery for children with bilateral Wilms tumor. The St Jude Children’s Research Hospital experience: 1999-2006. Cancer. 2008;112:2060–70. doi: 10.1002/cncr.23406. [DOI] [PubMed] [Google Scholar]
- 56.Shamberger RC, Haase GM, Argani P, et al. Bilateral Wilms’ tumors with progressive or nonresponsive disease. J Pediatr Surg. 2006;41:652–7. doi: 10.1016/j.jpedsurg.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 57.Dome JS, Cotton CA, Perlman EJ, et al. Treatment of anaplastic histology Wilms’ tumor: results from the fifth National Wilms’ Tumor Study. J Clin Oncol. 2006;24:2352–8. doi: 10.1200/JCO.2005.04.7852. [DOI] [PubMed] [Google Scholar]
- 58.Lall A, Pritchard-Jones K, Walker J, et al. Wilms’ tumor with intracaval thrombus in the UK Children’s Cancer Study Group UKW3 trial. J Pediatr Surg. 2006;41:382–7. doi: 10.1016/j.jpedsurg.2005.11.016. [DOI] [PubMed] [Google Scholar]
- 59.Murthi GV, Kocyildirim E, Sellathury S, et al. Wilms’ tumour with persistent intravascular extension: a review of the surgical aspects of management. J Pediatr Urol. 2006;2:439–45. doi: 10.1016/j.jpurol.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 60.Szavay P, Luithle T, Semler O, et al. Surgery of cavoatrial tumor thrombus in nephroblastoma: a report of the SIOP/GPOH study. Pediatr Blood Cancer. 2004;43:40–5. doi: 10.1002/pbc.20056. [DOI] [PubMed] [Google Scholar]
- 61.Green DM, Breslow NE, Ii Y, et al. The role of surgical excision in the management of relapsed Wilms’ tumor patients with pulmonary metastases: a report from the National Wilms’ Tumor Study. J Pediatr Surg. 1991;26:728–33. doi: 10.1016/0022-3468(91)90021-k. [DOI] [PubMed] [Google Scholar]
- 62.Spreafico F, Pritchard JK, Malogolowkin MH, et al. Treatment of relapsed Wilms tumors: lessons learned. Expert Rev Anticancer Ther. 2009;9:1807–15. doi: 10.1586/era.09.159. [DOI] [PubMed] [Google Scholar]
- 63.Dome JS, Liu T, Krasin M, et al. Improved survival for patients with recurrent Wilms tumor: the experience at St. Jude Children’s Research Hospital. J Pediatr Hematol Oncol. 2002;24:192–8. doi: 10.1097/00043426-200203000-00007. [DOI] [PubMed] [Google Scholar]
- 64.Sorensen K, Levitt GA, Bull C, et al. Late anthracycline cardiotoxicity after childhood cancer: a prospective longitudinal study. Cancer. 2003;97:1991–8. doi: 10.1002/cncr.11274. [DOI] [PubMed] [Google Scholar]
- 65.Green DM, Grigoriev YA, Nan B, et al. Congestive heart failure after treatment for Wilms’ tumor: a report from the National Wilms’ Tumor Study group. J Clin Oncol. 2001;19:1926–34. doi: 10.1200/JCO.2001.19.7.1926. [DOI] [PubMed] [Google Scholar]
- 66.Termuhlen AM, Tersak JM, Liu Q, et al. Twenty-five year follow-up of childhood Wilms tumor: a report from the Childhood Cancer Survivor Study. Pediatr Blood Cancer. 2011;57:1210–6. doi: 10.1002/pbc.23090. [DOI] [PMC free article] [PubMed] [Google Scholar]