

Abstract
Recent evidence indicates that both the phosphatidylinositol 3-kinase (PI3K)/AKT and the MEK/ERK pathways are strictly regulated by epidermal growth factor receptor in non-small cell lung cancer (NSCLC) that responds to Gefitinib. Gefitinib resistance is partly owing to the activation of two major downstream signaling pathways PI3K/AKT or MEK/ERK. In this study, we found that in Gefitinib-sensitive cell lines, Gefitinib could induce tumor cell apoptosis via upregulation of a proapoptotic protein BIM. Small interfering RNA results showed that silencing of BIM could alleviate apoptosis induced by Gefitinib. We adopted a combination of PI3K inhibitor (LY294002) and MEK inhibitor (U0126) against Gefitinib resistance in cell lines. As expected, the combination substantially induced apoptosis and restored the sensitivity to Gefitinib by increasing the expression of BIM. Our studies provided a theoretical basis for overcoming drug resistance in NSCLC via combination therapy.
Key words: BIM, Gefitinib resistance, MEK/ERK signaling pathway, PI3K/AKT signaling pathway
Introduction
Lung cancer is the most common cause of cancer-related death in the world with a poor 5-year survival rate of 15%.1,2 With the development of targeted therapy, some kinases were identified to be involved in the pathogenesis of lung cancer. Somatic gain-of-function mutations in exons encoding the epidermal growth factor receptor (EGFR) tyrosine kinase domain were found in about 10% of non-small cell lung cancer (NSCLC).3,4 Gefitinib, an EGFR tyrosine kinase inhibitor (TKI), showed substantial efficacy on patients harboring active EGFR mutations. Unfortunately, nearly all patients who experienced remarkable response to Gefitinib will develop progressive disease eventually. This kind of resistance observed in clinical trials includes primary and acquired resistance. Primary resistance usually happened in EGFR wild-type patients with other gene mutation in the downstream of EGFR signaling pathway, such as the KRAS mutation.5 The acquired mutation is mainly due to the mutation in exon 20 after sustained treatment, which is known as the replacement of threonine at position 790 by methionine (T790M) in the tyrosine kinase functional domain of EGFR. T790M mutation located in the ATP-/drug-binding cleft. It can trigger the resistance by blocking the binding of Gefitinib and kinase domain.6 In addition toT790M, other types of acquired mutations, such as D761Y, L747S, and T854A, were also founded in Gefitinib-resistant patients.7 Except for the T790M mutation, c-Met amplification, which was detected in 22% lung cancer specimens, is another major cause of EGFR-TKI resistance. It can lead to the transactivation of the ERBB3-signaling pathway and hamper the inhibition of EGFR.8
The hyperactivation of phosphatidylinositol 3-kinase (PI3K)/AKT and MEK/ERK signaling pathways is frequent in tumors and observed in many human malignant diseases.9 The activation is usually associated with malignant transformation and drug resistance after chemotherapy. The PI3K/AKT and MEK/ERK pathways are regulated by EGFR in cancers responding to ERFG TKI. One of the major effects of EGFR TKI in sensitive EGFR mutant cell lines is the induction of cell apoptosis through the PI3K/AKT and/or MEK/ERK signaling block. Therefore, it suggested that the hyperactivation of these signaling pathways may have an association with resistance to Gefitinib in NSCLC.10–14
Recently, the role of BIM, a BH3 domain protein that is capable of apoptosis induction has been extensively studied. Previous publications demonstrated the involvement of BIM was in imatinib-induced apoptosis of BCR-ABL leukemic cells and gastric cancer cells.15–17 Moreover, upregulation of BIM expression was observed in Paclitaxel-induced apoptosis in NSCLC cells, which did not occur in other Bcl-2 family members.18 Furthermore, many studies have confirmed the close connection between BIM and activation of the PI3K/AKT and MEK/ERK signaling pathways. ERK activation can decrease the expression of BIM by downregulating FOXO3a through MDM2-mediated proteasome degradation of phosphorylated FOXO3a, promoting the survival of tumor cells.19 Some studies also showed that ERK and AKT could phosphorylate BIM and regulated proapoptosis activity of BIM.20,21
In this study, we adopted a combination of PI3K inhibitor (LY294002) and MEK inhibitor (U0126) against Gefitinib resistance in cell lines, investigating their effects on resistant cells. Results had shown that this combination could substantially inhibit the proliferation of Gefitinib-resistant NSCLC cells by inducing apoptosis through elevating the expression of BIM, which is a result of the PI3K/AKT and MEK/ERK signaling pathway inhibition. This study was aimed to provide a theoretical basis to overcome drug resistance in NSCLC by combination therapy in clinical practice.
Materials and Methods
Cell lines and reagents
Human adenocarcinoma cell line NCI-H1975 was obtained from Biocat; 95-D, SPC-A1, H1299, A549, and PC-9 were purchased from Fulengen Company. NCI-H1975, 95-D, and SPC-A1 were cultured in a 1640 culture solution containing 10% fetal bovine serum with 1% penicillin/streptomycin, and H1299, A549, and PC-9 in the DMEM supplemented 10% fetal bovine serum with 1% penicillin/streptomycin. All cells were cultured at 37°C, 5% CO2. Gefitinib was purchased from LC laboratories (Cat No. G-4408); PI3K inhibitor (LY294002) and MEK inhibitor (U0126) were products of Sigma. All compounds were dissolved with DMSO to 20 mM as a stock solution and stored at a temperature of −20°C.
Cell proliferation assay
Tumor cells in a logarithmic growth phase were seeded in 96-well tissue culture plates at appropriate concentration and cultured at 37°C with 5% CO2 at the first night. On the second day, cells were exposed to various concentrations of compounds and then cultured at a temperature of 37°C, under 5% CO2 for 72 hours. About 100 μL/well Cell Titer-Glo (Promega) was added into the plate and incubated at room temperature for about 15 minutes. The luminescence was measured on TECAN infinite 1000. The inhibition rate was calculated as (RLUcontrol−RLUcompound)/(RLUcontrol−RLUblank)×100%
Apoptosis assay
Tumor cells in the logarithmic growth phase were seeded in 96-well tissue culture plates at the concentration of 4×103/well and cultured at 37°C with 5% CO2 at the first night. On the second day, cells exposed to various concentrations of compounds were cultured for another 24 hours, and then caspase3/caspase7 luminescent assay kit Caspase-Glo™ (Promega) was applied to measure the apoptosis according to the manufacturer's instruction.
Western blotting
Tumor cells were seeded and grown to half confluence in six-well plates and then induced with compounds for 24 hours. Cells were washed with cold phosphate-buffered saline (PBS) and lysed. The lysates were then subjected to SDS-PAGE, transferred to nitrocellulose membranes, blocked with 5% milk-PBST, and probed with antibodies against phospho-EGFR (Try1068), phospho-AKT (Ser473), AKT, phospho-ERK1/2, ERK1/2, BIM, and Actin (Cell Signaling Technology).
Flow cytometry
Cells were plated at 2×105/well in six-well plates, and then induced with different drugs for 48 hours. All cells, including the supernatant, were collected and centrifuged at 1000 g for 5 minutes. After two washes with PBS, cells were resuspended with a binding buffer at the concentration of 1×105/mL. Cells then were incubated with 5 μL Annexin V-FITC (Biovision) at room temperature in the dark for 10 minutes. Then, the cells were centrifuged at 1000 g for 5 minutes, and the supernatant was aspirated. Cells were resuspended with 200 μL binding buffer, and 10 μL propidium iodide (PI; Biovision) was added and incubated at room temperature for 5 minutes, and then cells were detected by flow cytometry. The results were analyzed using the FlowJo software.
RNA interference
BIM-specific and negative control small interfering RNAs (siRNA) were purchased from Cell Signaling Technology. Cells were transfected with the Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacture's protocol in the presence of siRNA. After 24 hours of transfection, cells were lysed with radioimmunoprecipitation assay and subjected to SDS-PAGE to detect the transfection efficiency. For flow cytometric analysis, after 24 hours of transfection, cells were washed with the DMEM twice and incubated with the DMEM containing 10% FBS in the presence of DMSO, or Gefitinib for 48 hours.
Immunocytochemistry (SP method)
Sterile coverslip was placed into 24-well culture plates, and the cell suspension 2×104/well cells was seeded into the plates and cultured at 37°C with 5% CO2. Next day, cells were induced with different drugs and exposed to compound for 48 hours, and the coverslip from each well was washed with PBS twice and fixed with 4% paraformaldehyde for 10 minutes at 4°C. After two washes with PBS, cells were penetrated with 0.1% Triton X-100 and then incubated with 3% H2O2 to block endogenous peroxidase. The coverslip was incubated with the normal goat serum working solution at room temperature for 10 minutes, and then appropriate amounts of anti-BIM primary antibody (1:100 dilution) were added and incubated in a moisture box at 4°C overnight, and the biotinylated secondary antibodies were added at a 1:100 dilution and incubated at room temperature for 10 minutes, and then a solution containing HRP-labeled streptavidin was added and incubated at room temperature for 60 minutes; the binding of antibodies was visualized with 3,3-diaminobenzidine (DAB) solution at last. The coverslips were then subjected to hematoxylin staining and differentiated with 1% acid alcohol for several seconds and washed with tap water for 5 minutes. Slides were then dried and mounted.
Statistical analysis
The results were analyzed using SPSS15.0 software. Data were presented as the mean±SEM of three independent experiments. One-way ANOVA was used to analyze data among groups. p<0.05 was used for the significance threshold.
Results
Sensitivity of different NSCLC cell lines to Gefitinib
First, we tested the sensitivity of six different NSCLC cell lines to Gefitinib, as shown in Figure 1A. We found that PC-9 was extremely sensitive to Gefitinib, and the IC50 of Gefitinib against PC-9 was about 0.016 μM. However, other five NSCLC cells lines, including NCI-H1975, 95-D, SPC-A1, H1299, and A549, were resistant to Gefitinib, at the concentration of 10 μM. Gefitinib did not show substantial inhibitory effects on the proliferation of these tumor cell lines. We then used the caspase3/caspase7 luminescent assay kit to investigate the apoptosis induced by Gefitinib in NSCLC cell lines. As shown in Figure 1B, in Gefitinib-sensitive NSCLC cell PC-9, induced with 0.1 μM Gefitinib for 24 hours, caspase3/7 activity in PC-9 increased about 3-fold in contrast with the control group, which implies the occurrence of apoptosis after the treatment of Gefitinib in PC-9 cells, but the activity of caspase3/7 had no change in Gefitinib-resistant tumor cells.
FIG. 1.

Sensitivity of different non-small cell lung cancer (NSCLC) cell lines to Gefitinib. (A) The inhibitory effects of Gefitinib against different NSCLC cell lines. Tumor cells were seeded in 96-well plates and treated with different Gefitinib concentrations for 72 hours, and then cell viability was measured with CellTiter-Glo. (B) Caspase3/7 activity in different NSCLC cell lines after the treatment of Gefitinib. Tumor cells were seeded in 96-well plates and treated with Gefitinib at indicated concentration for 24 hours. The caspase3/7 activity was measured with a caspase3/7 luminescent assay kit Caspase-Glo™.
Gefitinib substantially induced BIM expression in sensitive cell lines
Western blotting was used to detect the effects of Gefitinib on EGFR phosphorylation and its downstream signaling pathways. As shown in Figure 2, in PC-9 cells, 0.1 μM Gefitinib could substantially inhibit the phosphorylation of EGFR. With the inhibition of EGFR phosphorylation, the phosphorylation of AKT and ERK was also inhibited simultaneously. Furthermore, we also investigated the effects of Gefitinib through the expression of BIM, a proapoptotic protein. In a sensitive cell line, after the induction of Gefitinib, BIM expression was obviously increased. However, in Gefitinib-resistant cell lines (A549, SPC-A1, and NCI-H1975), Gefitinib had little effect on the phosphorylation of EGFR, AKT, and ERK as well as the expression of BIM.
FIG. 2.

The effects of Gefitinib on the phosphorylation of epidermal growth factor receptor (EGFR), AKT, and ERK and expression of BIM in NSCLC cell lines. (A) PC-9 cell. (B) A549 cell. (C) SPC-A1 cell. (D) NCI-H1975 cell. Tumor cells were seeded in six-well plates and treated with Gefitinib for 24 hours, and then cells were lysed, and the lysate was subjected to SDS-PAGE, and probed with indicated antibodies.
Gefitinib-induced apoptosis is closely correlated with BIM
To further confirm the role that BIM plays in Gefitinib-induced apoptosis in PC-9 cells, BIM siRNA was adopted to block the expression of BIM in PC-9 cells. As shown in Figure 3A, after 24 hours of induction of BIM siRNA, BIM expression is obviously decreased. Next, we used Annexin V-PI double staining to investigate the activity of Gefitinib on BIM siRNA PC-9 cells. We found that after 48-hour treatment of Gefitinib, apoptosis was happened in about 70% PC-9 cells. However, once the BIM expression was silenced in PC-9 cells, the apoptosis induced by Gefitinib was substantially decreased, and just about 40% cells undergo apoptosis (Fig. 3B). The results clarified that the apoptosis induced by Gefitinib in PC-9 cells was closely correlated with BIM expression in Gefitinib-sensitive cells.
FIG. 3.
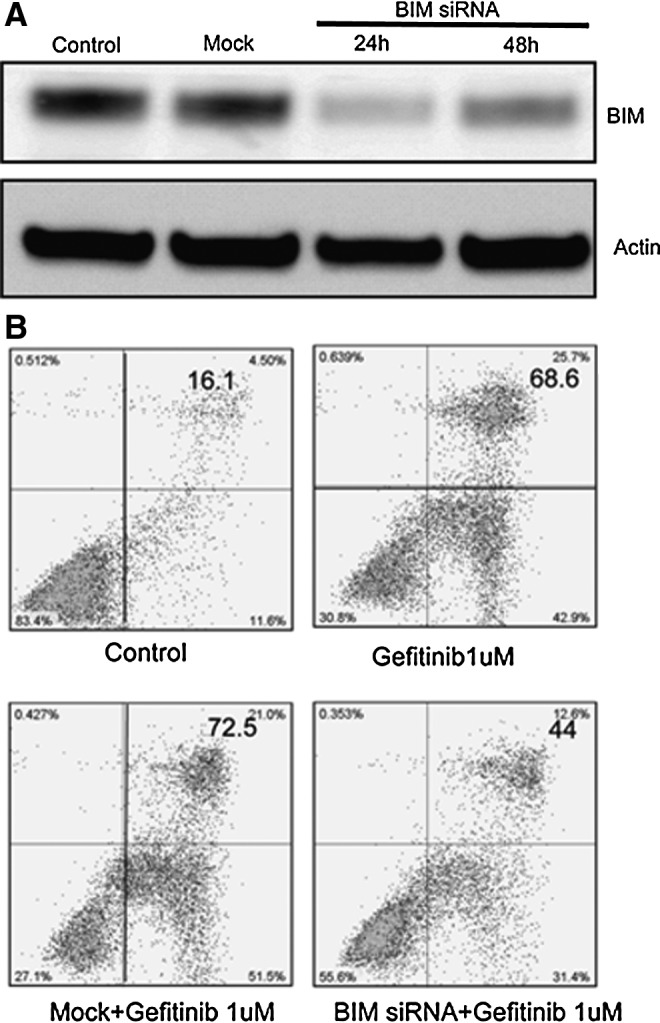
The role of BIM in the the apoptosis induced by Gefitinib in PC-9 cells. (A) BIM silencing in PC-9 cells. PC-9 cells were seeded in 6-well plate, BIM-specific, and control small interfering RNAs (siRNA) were transfected with Lipofectamine 2000. After transfection, cells were lysed, and the lysate was subjected to SDS-PAGE, and probed with BIM antibody. (B) AnnexinV–propidium iodide (PI) staining of PC-9 cells after the BIM was silenced. Cells were seeded in a six-well plate, transfected with BIM-specific or control siRNA. 24 hours after the transfection, and the cells were treated with 1 μM Gefitinib for 48 hours, and then the cells were collected and apoptosis was analyzed by AnnexinV–PI staining.
PI3K inhibitor and MEK inhibitor combination restores the sensitivity to Gefitinib in resistant cell lines
Given the importance of PI3K and MEK signaling pathways in cell proliferation, survival, as well as chemotherapy resistance, we adopted the combination of PI3K inhibitor and MEK inhibitor to study their impact on Gefitinib-resistant NSCLC cell lines. As shown in Figure 4, when this combination was used with Gefitinib together, the apoptosis was obviously increased in Gefitinib-resistant cell lines. In 95-D, NCI-H1975, and H1299 cell lines, the apoptosis increased by about 2.1, 3.3, and 2.7 times, respectively. The level of apoptosis induced in the A549 and SPC-A1 cell lines was 1.2 times and 1.6 times, respectively, in a moderate way. The difference between cell lines may be owing to different cellular context.
FIG. 4.

The apoptosis induced by different drug combinations in Gefitinib-resistant NSCLC cell lines. Cells were seeded in 96-well plates, and different drug combinations as indicated were added into the plates for 24 hours, and then the activity of caspase3/7 was assayed with caspase3/7 luminescent assay kit Caspase-Glo. Concentration of compound is as follows: Gefitinib: 10 μM, LY294002:20 μM, and U0126:10 μM.
PI3K inhibitor and MEK inhibitor combination increases BIM expression in EGFR-resistant cell lines
We tested the expression of BIM after the drug treatment. As shown in Figure 5, PI3K inhibitor (LY294002) and MEK inhibitor (U0126) could, respectively, inhibit the phosphorylation of AKT and ERK in Gefitinib-resistant NSCLC cells. With the inhibition of AKT and ERK, the expression of BIM was increased. Moreover, when the combination of LY294002 and U0126 was used with Gefitinib together, the BIM expression was substantially increased in contrast with these inhibitors were used alone. This result demonstrated that to restore the sensitivity to Gefitinib in Gefitinib-resistant cell lines, it is better to inhibit the activity of AKT and ERK simultaneously.
FIG. 5.

Phosphorylation of AKT and ERK and expression of AKT, ERK, and BIM in Gefitinib-resistant NSCLC cell lines after the treatment of different drug combinations. Tumor cells were seeded in six-well plates and treated with different drug combinations as indicated for 24 hours, then cells were lysed, and the lysate was subjected to SDS-PAGE, and probed with indicated antibodies.
We also confirmed the BIM expression by immunocytochemisty. BIM was mainly expressed in the outer membrane of mitochondria. As shown in Figure 6, the cytoplasm in the control group has shown blue color and a little yellow color. After drug treatment, the cytoplasm showed different extents of yellow color ranging from light yellow to brown; after triple treatment with Gefitinib plus PI3K and MEK inhibitors, the color of the cytoplasm in all cell lines become darker compared with that in the single Gefitinib group.
FIG. 6.

BIM expression in NSCLC cell lines after the treatment of different drug combinations detected by immnocytochemistry (SP). Approximately 2×104/well cells were seeded on the sterile coverslip placed in a 24-well plate and treated with different drug combinations as indicated for 24 hours. The expression of BIM in different groups was performed immmocytochemistry analysis by a stained BIM antibody. Representative photograph per group was shown. (A) H1975 cell. (B) H1299 cell.
Discussion
In this study, we investigated the effects of a combination of PI3K inhibitor and MEK inhibitor on Gefitinib-resistant NSCLC tumor cells. We found that this combination could substantially inhibit the proliferation of resistant cell lines. Moreover, it could increase BIM protein to inducing tumor cell apoptosis.
Gefitinib has shown prospective and exciting efficacy in NSCLC patients with EGFR mutation in the beginning. However, after a sustained treatment, the resistance to Gefitinib was found in clinical trials because of different mechanisms, including EGFR second mutation in the kinase domain, c-Met amplification, as well as mutations in the EGFR downstream signaling pathways. From the results of previous studies, we have learned that the sensitivity of tumor cells to Gefitinib is largely determined by EGFR kinase activity of tumor cells. In tumor cells with EGFR mutations, such as L858R and deletion of exon 19, EFGR hyperactivation plays an important role in the tumor cell proliferation and survival, and the inhibition of EGFR activity can be substantially inhibited tumor growth. Just as in PC-9 cells shown in Figure 1, Gefitinib can potently inhibit tumor cell proliferation via suppressing the autophosphorylation of EGFR, which leads to the inhibition of downstream signaling pathways AKT and ERK, and the induction of proapoptotic protein BIM.
As the two important signaling pathways of EGFR activation, PI3K/AKT and MEK/ERK play important roles in tumor cell survival and proliferation, and their aberrant activation is often related to chemotherapy resistance. Previous studies have demonstrated, in NSCLC cells with EGFR mutation that, when the PI3K/AKT and MEK/ERK pathways were blocked together, the level of cell apoptosis was similar to that treated with EGFR TKI alone.22 We found that in Gefitinib-resistant NSCLC cells, treatment of Gefitinib had little effect on the phosphorylation of AKT and ERK, which probably attribute to their resistance to Gefitinib. Based on the view above, LY294002 and U0126 were combined to treat Gefitinib-resistant NSCLC cells. In our study, we have shown that the combination of PI3K and MEK inhibitors can restore the sensitivity to Gefitinib in Gefitinib-resistant NSCLC cells and induce tumor cells to apoptosis via inducing the expression of BIM in resistant cells.
BIM, a proapoptotic protein, plays an important role in the regulation of tumor cell survival. Our study demonstrated that BIM expression is closely correlated with the sensitivity of NSCLC tumor cells to Gefitinib. In sensitive tumor cells, Gefitinib alone can promote the expression of BIM, but has little effect on BIM expression in resistant cells. The reason that single Gefitinib cannot increase the expression of BIM is due to the activation of downstream signaling pathways such as AKT and ERK. Therefore, inhibition of the activity of these two downstream molecules is important for restoring the sensitivity. Given the complexity of the cellular signaling network and the offset mechanism in tumor cells, inhibition of one signaling pathway cannot effectively potentiate the antitumor activity of Gefitinib. It is necessary to inhibit these two signaling pathways at the same time. As shown in our study, when Gefitinib was combined with LY294002 and U0126, the apoptosis in resistant cells is the most obvious, which is consistent with the BIM expression in resistant cell lines. In conclusion, the combination therapy offers a promise to drug resistance in NSCLC.
Acknowledgment
This work was supported by grants of the National Natural Science Foundation (Grant No. 30873023 and No. 81071922).
Disclosure Statement
There are no conflicts of interest about this article.
References
- 1.Jemal A. Siegel R. Ward E, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Parkin DM. Bray F. Ferlay J, et al. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 3.Kosaka T. Yatabe Y. Endoh H, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12:5746. doi: 10.1158/1078-0432.CCR-06-0714. [DOI] [PubMed] [Google Scholar]
- 4.Lynch TJ. Bell DW. Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 5.Pao W. Miller VA. Politi PA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLos Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mok TS. Wu YL. Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen KS. Kobayashi S. Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer. 2009;10:281. doi: 10.3816/CLC.2009.n.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelman JA. Zejnullahu K. Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 9.Sarkar S. Mazumdar A. Dash R, et al. ZD6474, a dual tyrosine kinase inhibitor of EGFR and VEGFR-2, inhibits MAPK/ERK and AKT/PI3-K and induces apoptosis in breast cancer cells. Cancer Biol Ther. 2010;9:592. doi: 10.4161/cbt.9.8.11103. [DOI] [PubMed] [Google Scholar]
- 10.Janmaat ML. Kruyt FA. Rodriguez JA, et al. Response to epidermal growth factor receptor inhibitors in non-small cell lung cancer cells: Limited antiproliferative effects and absence of apoptosis associated with persistent activity of extracellular signal-regulated kinase or AKT kinase pathways. Clin Cancer Res. 2003;9:2316. [PubMed] [Google Scholar]
- 11.Cragg MS. Kuroda J. Puthalakath H, et al. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med. 2007;4:1681. doi: 10.1371/journal.pmed.0040316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng J. Shimamura T. Perera S, et al. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res. 2007;67:11867. doi: 10.1158/0008-5472.CAN-07-1961. [DOI] [PubMed] [Google Scholar]
- 13.Gong Y. Somwar R. Politi K, et al. Induction of BIM is essential for apoptosis triggered by EGFR kinase inhibitors in mutant EGFR-dependent lung adenocarcinomas. PLoS Med. 2007;4:e294. doi: 10.1371/journal.pmed.0040294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engelman JA. Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev. 2008;18:73. doi: 10.1016/j.gde.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Sharma SV. Gajowniczek P. Way IP, et al. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10:425. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuroda J. Puthalakath H. Cragg MS, et al. BIM and Bad mediate imatinib-induced killing of Bcr/Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103:14907. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gordon PM. Fisher DE. Role for the proapoptotic factor BIM in mediating imatinib-induced apoptosis in a c-KIT-dependent gastrointestinal stromal tumor cell line. J Biol Chem. 2010;285:14109. doi: 10.1074/jbc.M109.078592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li R. Moudgil T. Ross HJ, et al. Apoptosis of non-small-cell lung cancer cell lines after paclitaxel treatment involves the BH3-only proapoptotic protein BIM. Cell Death Differ. 2005;12:292. doi: 10.1038/sj.cdd.4401554. [DOI] [PubMed] [Google Scholar]
- 19.Yang JY. Zong CS. Xia W, et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10:138. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luciano F. Jacquel A. Colosetti P, et al. Phosphorylation of BIM-EL by Erk1/2 on serine69promotes its degradation via the proteasome pathway and regulates its proapoptotic function. Oncogene. 2003;22:6785. doi: 10.1038/sj.onc.1206792. [DOI] [PubMed] [Google Scholar]
- 21.Qi XJ. Wildey GM. Howe PH. Evidence that Ser87 of BIMEL is phosphorylated by AKT and regulates BIMEL apoptotic function. J Biol Chem. 2006;281:813. doi: 10.1074/jbc.M505546200. [DOI] [PubMed] [Google Scholar]
- 22.Faber AC. Li D. Song Y, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Nat1 Acad Sci U S A. 2009;106:19503. doi: 10.1073/pnas.0905056106. [DOI] [PMC free article] [PubMed] [Google Scholar]