

Abstract
The complete and unambiguous 1H NMR assignments of ten marker constituents of Ginkgo biloba are described. The comprehensive 1H NMR profiles (fingerprints) of ginkgolide A, ginkgolide B, ginkgolide C, ginkgolide J, bilobalide, quercetin, kaempferol, isorhamnetin, isoquercetin, and rutin in DMSO-d6 were obtained through the examination of 1D 1H NMR and 2D 1H,1H-COSY data, in combination with 1H iterative Full Spin Analysis (HiFSA). The computational analysis of discrete spin systems allowed a detailed characterization of all the 1H NMR signals in terms of chemical shifts (δH) and spin-spin coupling constants (JHH), regardless of signal overlap and higher order coupling effects. The capability of the HiFSA-generated 1H fingerprints to reproduce experimental 1H NMR spectra at different field strengths was also evaluated. As a result of this analysis, a revised set of 1H NMR parameters for all ten phytoconstituents was assembled. Furthermore, precise 1H NMR assignments of the sugar moieties of isoquercetin and rutin are reported for the first time.
Keywords: NMR, 1H, 1H fingerprint, HiFSA, Full spin analysis, QMTLS, Higher order spin systems, 2D 1H, 1H-COSY, Ginkgo biloba, Ginkgolides
INTRODUCTION
The Ginkgo tree (Ginkgo biloba L.) is the sole remaining species of the Ginkgoaceae family. Originally found in China and Korea, Ginkgo has been widely planted as an ornamental tree in urban areas of North America, Europe and Japan due to its beauty, disease resistance, and longevity. In addition to its decorative use, Ginkgo preparations have been used for centuries as a component of traditional Chinese medicine. The dried Ginkgo leaves were typically used to treat asthma and circulatory diseases. During the last two decades, Ginkgo leaf preparations have been extensively studied for its antioxidant and neuroprotective properties.[1,2] These beneficial effects make Ginkgo biloba one of the largest selling herbal products worldwide.
In this report, the complete 1H NMR assignments of ten standardization markers for the quality assessment of Ginkgo biloba preparations are presented. The markers selected for this study (Figure 1) are representative of two classes of phytoconstituents occurring in Ginkgo leaves: the species-specific terpene trilactones and the ubiquitous flavonoids. The first group includes platelet-activating factor inhibitors such as ginkgolides A, B, C and J (1–4), as well as the neuroprotective agent bilobalide (5). The second group contains a wide variety of flavonols and flavonol glycosides, including quercetin (6), kaempferol (7), isorhamnetin (8), isoquercetin (quercetin-3-O-β-D-glucopyranoside, 9), and rutin [quercetin-3-O-α-L-rhamnopyranosyl-(1→6)-β-D-glucopyranoside, 10].
Figure 1.
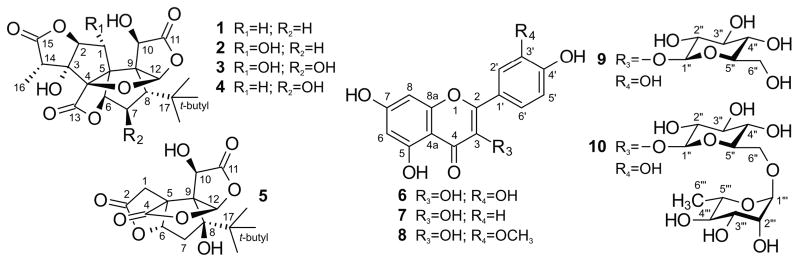
Chemical structures of compounds 1–10.
In order to enable the assembly of comprehensive, reproducible 1H NMR profiles (i.e., 1H fingerprints) of these chemical markers in DMSO-d6, conventional NMR data examination was complemented by computer-assisted spectral analysis with PERCH NMR software.[3,4] This approach, named 1H iterative Full Spin Analysis (HiFSA), permitted a detailed description of the basic 1H NMR parameters based on both structural and line shape considerations.[5–7] As a result, a complete and precise determination of all the 1H chemical shifts (δH) and 1H,1H spin-spin coupling constants (JHH) was accomplished. This information will serve for all subsequent NMR-based studies of these botanical markers, such as quantitative NMR (qNMR) analysis,[8] and can be adapted by any research laboratory even across different field strengths.
EXPERIMENTAL
Materials
The Ginkgo chemical marker compounds 1–10 were obtained from Indofine Chemical Company Inc. (Hillsborough, NJ, USA). All samples were subjected to NMR analysis without further purification. Hexadeuterodimethyl sulfoxide (DMSO-d6, 99.9% D) and deuterium oxide (D2O, 99.9% D) were obtained from Cambridge Isotope Laboratories Inc. (Andover, MA, USA) and Sigma-Aldrich Inc. (St. Louis, MO, USA), respectively. Standard 5 mm, 7″ NMR tubes (XR-55 series) from Norell Inc. (Landisville, NJ, USA) were used.
Sample Preparation
NMR samples were prepared by precisely weighing 1–10 mg (±0.01 mg) of each compound directly into the NMR tubes using a Mettler Toledo XS105 Dual Range analytical balance, followed by the addition of 600 μL of DMSO-d6 using a Pressure-Lok gas syringe (VICI Precision Sampling Inc., Baton Rouge, LA, USA). The samples were prepared at the following concentrations: 1: 6.0 mg·ml−1; 2: 4.7 mg·ml−1; 3: 5.8 mg·ml−1; 4: 5.1 mg·ml−1; 5: 2.7 mg·ml−1; 6: 10.1 mg·ml−1; 7: 4.1 mg·ml−1; 8: 7.6 mg·ml−1; 9: 14.0 mg·ml−1; and 10: 4.8 mg·ml−1.
NMR Spectroscopy
NMR measurements were recorded at 600.13 MHz and 899.94 MHz on Bruker spectrometers with AVANCE and AVANCE II consoles, respectively, each equipped with 5 mm TXI and TCI triple resonance inverse detection cryoprobes with z-axis pulse field gradient. All NMR experiments were acquired at 298 K (25°C) using standard Bruker pulse sequences, and the probes were frequency tuned and impedance matched prior to each sample run. Chemical shifts (δH) are expressed in ppm with reference to the residual protonated solvent signal (DMSO-d5, 2.500 ppm) and internal TMS (0.000 ppm). Coupling constants (JHH) are given in Hertz.
The 1D 1H NMR spectra were acquired using a 90° single-pulse experiment (Bruker pulse sequence zg). The 90° pulse width (p90) was calculated by evaluating the 360° pulse width (p90 = ¼ × p360). NMR experiments recorded at 600 MHz used the following acquisition parameters: a spectral width of 17,985.6 Hz, 143,882 data points, an acquisition time of 4.0 s, a relaxation delay of 60 s, and a 90° pulse width of 9.25 μs. A total of 64 transients were collected with a receiver gain of 16. For NMR experiments recorded at 900 MHz, the following acquisition parameters were used: a spectral width of 27,100.3 Hz, 216,798 data points, an acquisition time of 4.0 s, a relaxation delay of 60 s, and a 90° pulse width of 12.0 μs. A total of 16 transients were collected with a receiver gain of 64. The 1D NMR data were processed with NUTS software (v.201004, Acorn NMR Inc., Las Positas, CA, USA) using a Lorentzian/Gaussian window function for resolution enhancement (line broadening = −1.0 Hz, Gaussian factor = 0.10), followed by zero filling in order to increase two-fold the number of acquired data points before Fourier transformation. The digital resolution after zero-filling was 0.069 Hz/pt (0.13 ppb/pt at 600 MHz). Considering the occurrence of higher order spin systems and the magnitude of small J-couplings, δH and JHH values are reported to a precision of 0.001 ppm and 0.01 Hz, respectively. After manual phasing, a fifth order polynomial baseline correction was applied.
The 2D 1H,1H-COSY experiments were acquired in magnitude mode (Bruker pulse sequence cosygpqf) at 600 MHz with 2k data points in F2 and 256 increments in F1, using a spectral width of 8401.6 Hz in each dimension. A total of eight transients were collected with an acquisition time of 0.25 s. The relaxation delay was 1.0 s, the 90° pulse width was 9.25 μs, and the receiver gain 16. Subsequent 2D NMR data processing was carried out with Mnova software (v.7.1.1, Mestrelab Research S.L., A Coruña, Spain). The data were zero filled to 4k data points in F2, linear predicted to 2k and zero filled to 4k data points in F1. Non-shifted sine-bell window functions were applied to both dimensions before double Fourier transformation, followed by a third order polynomial baseline correction.
1H iterative Full Spin Analysis (HiFSA)
Full spin analysis was performed with PERCH NMR software (v.2011.1, PERCH Solutions Ltd., Kuopio, Finland). The resolution-enhanced 1H NMR spectra of 1–10 were imported into the PERCH shell as JCAMP-DX files and subjected to baseline correction, peak picking and integration in the PAC module. The X-ray crystal structures of 4 (CCDC ID 183040) and 6 (as a complex with bovine xanthine oxidase, PDB ID 3NVY) were used as templates to build molecular models of compounds 1–10. After geometry optimization (GO) and molecular dynamics (MD) simulations, basic 1H NMR parameters (δH, JHH) in DMSO-d6 were predicted using PERCH’s Molecular Modeling Software (MMS). Next, the predicted δH, JHH and line width values were optimized against the observed 1H NMR spectra using the program PERCHit and Quantum Mechanical Total Line Shape (QMTLS) iterators. The subsequent optimization of spectral parameters was carried out in three steps: (i) analysis of discrete spin systems using the Integral-transform (D) mode; (ii) evaluation of the 1H NMR spectra using the Total-line-fitting (T) mode; and (iii) optimization of Gaussian and dispersion contributions to the line shape, also using the T mode. Iterative optimization was performed until an excellent agreement between the experimental and calculated spectra was reached, i.e., convergence with a total intensity root mean square deviation (rmsd) below 0.1%. The low rmsd values and the small, symmetrical residuals support the validity of the iteration results. Although it is still possible that some small long-range couplings (|4,6JHH| ≤ 0.2 Hz) remain undetected,[9] the iterative optimization of the “effective” line widths (see Supporting Information) includes the contribution of these small couplings to the overall line shape and intensity of the NMR signals.[5] Finally, the optimized NMR parameters were stored in individual PERCH PMS text files, transcribed to Mnova, and spectral simulations were performed using the Spin Simulation module in order to generate the 1H NMR spectra at different field strengths.
RESULTS and DISCUSSION
Table 1 summarizes the 1H chemical shifts (δH) and 1H,1H scalar coupling constants (JHH) of compounds 1–5. Despite the intricate, cage-like structure of Ginkgo terpene trilactones, the large number of quaternary carbons (i.e., 8 out of 20 carbons in the ginkgolides), in combination with the presence of one methyl and one tert-butyl group, gives rise to relatively simple 1H NMR spectra. Taking into account the specific characteristics of the 1H,1H spin systems in 1–5, a preliminary assignment of the individual proton resonances was made by 1D and 2D NMR techniques, and signals belonging to hydroxyl protons were identified by D2O exchange studies. However, in some cases, spectral overlap hinders the detailed analysis of specific NMR resonances. This situation was especially noticeable during the analysis of compound 1, where the signals of protons H-6 and H-10, as well as those of the methylene protons H-7α and H-7β, were severely overlapped. Nevertheless, the iterative full spin analysis was able to decipher the observed multiplets, thereby allowing a thorough interpretation of all the overlapped signals (Figure 2A). As a result, all the δH and JHH values were obtained.
Table 1.
Calculated 1H chemical shifts (δH, ppm) and 1H,1H spin-spin coupling constants (JHH, Hz) of compounds 1–5a
| Proton | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
|
| |||||
| δH (ppm), multiplicity | |||||
| H-1α | 2.750, dd | – | – | 2.717, dd | 2.897, d |
| H-1β | 1.806, dd | 4.042, dd | 3.986, dd | 1.753, dd | 2.774, d |
| OH-1 | –b | 4.919, d | 4.972, d | – | – |
| H-2 | 4.834, dd/t | 4.643, d | 4.627, d | 4.832, dd/t | – |
| OH-3 | 6.379, s | 6.472, s | 6.479, s | 6.393, s | – |
| H-6 | 4.946, ps-ddc | 5.304, dd/br d | 4.966, d | 4.646, d | 4.917, dd/t |
| H-7α | 2.027, ps-ddd | 1.932, ddd | 4.054, ddd | 4.171, ddd | 2.562, dd |
| H-7β | 2.044, ps-ddd | 2.137, ddd | – | – | 2.077, dd |
| OH-7 | – | – | 5.658, d | 5.546, d | – |
| H-8 | 1.710, ps-dd | 1.724, dd | 1.551, d | 1.538, d | – |
| OH-8 | – | – | – | – | 5.403, s |
| H-10 | 4.941, d | 5.020, d | 4.998, d | 4.928, d | 5.150, d |
| OH-10 | 6.814, d | 7.463, d | 7.539, d | 6.908, d | 7.253, d |
| H-12 | 6.023, s | 6.074, s | 6.099, s | 6.047, s | 6.274, s |
| H-14 | 2.938, q | 2.843, q | 2.814, q | 2.907, q | – |
| H3-16 | 1.118, d | 1.107, d | 1.108, d | 1.118, d | – |
| t-butyl | 1.017, s | 1.028, s | 1.091, s | 1.082, s | 1.034, s |
| Coupling | JHH (Hz) | ||||
|
| |||||
| H-1α, H-1β | −15.17 | – | – | −15.14 | −18.14 |
| H-1α, H-2 | 7.29 | – | – | 7.25 | – |
| H-1β, H-2 | 8.26 | 7.40 | 7.22 | 8.17 | – |
| H-1β, OH-1 | – | 3.56 | 3.68 | – | – |
| H-6, H-7α | 4.09 | 4.14 | 4.20 | 4.18 | 7.10 |
| H-6, H-7β | 0.68 | 0.83 | – | – | 6.85 |
| H-7α, H-7β | −13.64 | −13.75 | – | – | −13.33 |
| H-7α, OH-7 | – | – | 6.27 | 6.86 | – |
| H-7α, H-8 | 14.33 | 14.35 | 12.40 | 12.30 | – |
| H-7β, H-8 | 4.79 | 4.65 | – | – | – |
| H-10, OH-10 | 5.16 | 5.56 | 5.65 | 5.35 | 5.12 |
| H-14, H3-16 | 7.19 | 7.08 | 7.09 | 7.20 | – |
The δH and JHH values were generated via 1H iterative Full Spin Analysis (HiFSA) using experimental NMR data acquired in DMSO-d6 at 600 MHz and 298 K.
A hyphen (–) denotes δH or JHH not present.
The “pseudo” (ps-) prefix indicates proton signals belonging to a higher order spin system.
Figure 2.
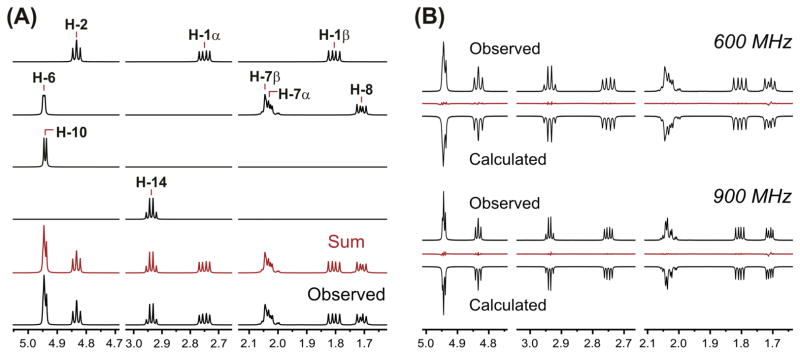
The 1H iterative Full Spin Analysis of 1 (6.0 mg·ml−1, DMSO-d6): (A) Sections of the simulated spin system subspectra, arithmetic sum (in red), and observed 1H NMR spectrum (600 MHz, 298 K). (B) Comparison of the observed and calculated 1H NMR spectra at different applied field strengths, including residuals in red.
In addition, the above-described HiFSA methodology demonstrated the diastereotopicity of the methylene protons H-7α (δH 2.027 ppm) and H-7β (δH 2.044). Because the small chemical shift difference between H-7α and H-7β is of the same order of magnitude as the scalar coupling constant between these geminal protons [|ΔνH| = 10.05 Hz at 600 MHz; 2J(H-7α, H-7β) = −13.64 Hz], a complex, yet distinctive, signal pattern that defies first-order interpretation is observed (Figure 2A). This higher order spin coupling relationship also affects the signals of neighboring protons H-6 (δH 4.946) and H-8 (δH 1.710), which exhibit characteristic sidebands that should not be mistaken for impurities.[10]
As the computational study involves both the structural analysis of the molecule of interest and the total line shape analysis of the 1H NMR spectrum, HiFSA is able to predict signal distortion in higher order coupled spin networks and reproduce complex signal patterns (see Figure 2A). Furthermore, because the HiFSA-generated 1H NMR profiles consist of field-independent parameters, δH and JHH, these high-resolution 1H fingerprints can be efficiently used to simulate 1H NMR data acquired at different field strengths. As an example, Figure 2B shows a comparison between sections of the calculated and observed 1H NMR spectra of 1 at 600 and 900 MHz. In both cases, the simulated spectra are in excellent agreement with the experimental NMR data.
In addition, this computer-aided approach allows for a detailed analysis of spin systems with small scalar coupling constants (<2 Hz). For example, the 3J(H-6, H-7β) couplings in compounds 1 and 2 were calculated by HiFSA as 0.68 and 0.83 Hz, respectively, even if these JHH values are below the intrinsic line width of the corresponding 1H NMR signals in DMSO-d6 (≥1.5 Hz). Interestingly, these small coupling constants are further evidence that vicinal protons H-6 and H-7β are located on opposite faces of the five-membered ring.[11]
In the case of the Ginkgo flavonols 6–10, a thorough compilation of δH and JHH values for the aglycones is presented in Table 2. The 1H NMR assignments were established by inspection of the 1D and 2D NMR data in combination with HiFSA, which simultaneously computed all the 1H,1H coupling constants in the aromatic rings. As a result of the total line shape fitting, small para-couplings such as 5J(H-2′, H-5′) were readily determined, thus producing a complete map of J-couplings for the AA′XX′ spin system of 7, as well as the AMX spin systems in the 3′,4′-disubstituted flavonols. Moreover, HiFSA led to the determination of accurate δH values for the overlapped signals of protons H-2′ and H-6′ in compounds 9 and 10.
Table 2.
Calculated 1H chemical shifts (δH, ppm) and 1H,1H spin-spin coupling constants (JHH, Hz) of 6–8 and the aglycones of 9, 10a
| Proton | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|
|
| |||||
| δH (ppm), multiplicity | |||||
| OH-3 | 9.598, br sb | 10.110, br s | 9.747, s | – | – |
| OH-5 | 12.494, s | 12.484, s | 12.467, s | 12.646, s | 12.600, s |
| H-6 | 6.183, d | 6.187, d | 6.192, d | 6.199, d | 6.190, d |
| OH-7 | 10.784, s | 10.788, br s | 10.774, br s | 10.860, br s | 10.829, br s |
| H-8 | 6.403, d | 6.436, d | 6.477, d | 6.402, d | 6.381, d |
| H-2′ | 7.675, dd/d | 8.042, AA′-type | 7.752, dd/d | 7.572, ps-dd/dd | 7.526, ps-dd/d |
| H-3′ | –c | 6.923, AA′-type | – | – | – |
| OH-3′ | 9.374, s | – | – | 9.729, br s | 9.672, br s |
| OH-4′ | 9.312, br s | 9.409, br s | 9.445, s | 9.224, br s | 9.183, br s |
| H-5′ | 6.881, dd/d | 6.923, AA′-type | 6.937, dd/d | 6.840, ps-dd/d | 6.836, ps-dd/d |
| H-6′ | 7.537, dd | 8.042, AA′-type | 7.687, dd | 7.577, ps-dd | 7.538, ps-dd |
| 3′-OCH3 | – | – | 3.840, s | – | – |
| Coupling | JHH (Hz) | ||||
|
| |||||
| H-6, H-8 | 2.03 | 2.03 | 2.01 | 2.04 | 2.06 |
| H-2′, H-3′ | – | 8.78 | – | – | – |
| H-2′, H-5′ | 0.46 | 0.27 | 0.29 | 0.34 | 0.22 |
| H-2′, H-6′ | 2.20 | 2.48 | 2.06 | 2.13 | 2.23 |
| H-3′, H-5′ | – | 2.48 | – | – | – |
| H-3′, H-6′ | – | 0.27 | – | – | – |
| H-5′, H-6′ | 8.47 | 8.78 | 8.47 | 8.36 | 8.46 |
The δH and JHH values were generated via 1H iterative Full Spin Analysis (HiFSA) using experimental NMR data acquired in DMSO-d6 at 600 MHz and 298 K.
Broad singlets (br s) have a line width ≥ 3.0 Hz.
A hyphen (–) denotes δH or JHH not present.
The “pseudo” (ps-) prefix indicates proton signals belonging to a higher order spin system.
Once the 1H fingerprints of the aglycones were assembled, detailed analyses of the glucose and rutinose moieties of 9 and 10, respectively, were carried out. In order to overcome problems related to potential signal overlap during the 1H NMR analysis of flavonol glycosides, the assignment of individual proton resonances was performed in three steps. First, a preliminary assessment of the δH values was conducted using 2D 1H,1H-COSY experiments and exchange studies. Next, HiFSA was performed using the simplified 1H NMR spectra in DMSO-d6 after D2O exchange to determine the exact position of all the non-exchangeable protons and evaluate the corresponding scalar coupling constants (Figure 3A). Finally, a second full spin analysis was carried out with the 1H NMR spectra acquired before D2O exchange to establish the chemical shifts of all hydroxyl protons and obtain the remaining JHH values (Figure 3B). Although hydroxyl protons are frequently ignored during structural elucidation due to their broad absorptions and their higher sensitivity to changes in concentration, pH, and temperature, these exchangeable protons can play an important role in the NMR analysis of carbohydrates. Freshly prepared, dilute solutions of the analyte in polar aprotic solvents, such as DMSO-d6, exhibit key COSY correlations between the hydroxyl protons and their neighboring non-exchangeable protons (Figure 3C), thus offering new paths to access relatively complex spin systems and analyze proton resonances in crowded regions of the 1H NMR spectrum.
Figure 3.

The 1H iterative Full Spin Analysis of 10 (4.8 mg·ml−1, DMSO-d6): (A) Comparison of the calculated and observed 1H NMR spectra (600 MHz, 298 K) after addition of 25 μL of D2O, including residuals in red. (B) Comparison of the calculated and observed 1H NMR spectra prior to D2O exchange, including residuals in red. Asterisks (*) indicate the presence of residual ethanol in the sample. (C) Identification of individual hydroxyl proton resonances from key cross-peaks in a 2D magnitude-mode 1H,1H-COSY experiment.
When applying the described methodology to the 1H NMR analysis of 9, a complete fingerprint of the glucose moiety was obtained (Table 3), and precise chemical shifts of protons H-2″ (δH 3.236) and H-3″ (δH 3.213), as well as H-4″ (δH 3.084) and H-5″ (δH 3.079), were extracted. The resonance proximity of these coupled protons causes severe higher order effects that are evident in the distorted signals of both the anomeric proton H-1″ (δH 5.466) and the exchangeable proton OH-4″ (δH 4.956, see Supporting Information). A similar situation was observed during the analysis of compound 10, where overlapped protons H-2″ (δH 3.220) and H-3″ (δH 3.205) cause substantial distortion of the signals belonging to H-1″ (δH 5.342), OH-2″ (δH 5.286), OH-3″ (δH 5.116), and H-4″ (δH 3.048). Despite these higher order effects and heavy spectral overlap, with six of the glycosidic protons located in a narrow 100 ppb range (δH 3.300–3.200), a full 1H NMR assignment of the rutinose moiety of 10 was achieved via HiFSA (Table 4).
Table 3.
Calculated 1H chemical shifts (δH, ppm) and 1H,1H spin-spin coupling constants (JHH, Hz) of the 3-O-β-glucopyranosyl moiety of 9a
| Proton | δH (ppm), multiplicity | Coupling | JHH (Hz) |
|---|---|---|---|
| H-1″ | 5.466, ps-db | H-1″, H-2″ | 7.73 |
| H-2″ | 3.236, ps-ddd | H-2″, OH-2″ | 4.76 |
| OH-2″ | 5.293, ps-d | H-2″, H-3″ | 9.10 |
| H-3″ | 3.213, ps-ddd | H-3″, OH-3″ | 4.82 |
| OH-3″ | 5.079, ps-d | H-3″, H-4″ | 8.21 |
| H-4″ | 3.084, ps-ddd | H-4″, OH-4″ | 5.54 |
| OH-4″ | 4.956, ps-d | H-4″, H-5″ | 10.01 |
| H-5″ | 3.079, ps-ddd | H-5″, H-6″a | 2.13 |
| H-6″a | 3.577, ps-ddd | H-5″, H-6″b | 5.83 |
| H-6″b | 3.317, ps-ddd | H-6″a, H-6″b | −11.71 |
| OH-6″ | 4.269, dd/t | H-6″a, OH-6″ | 5.45 |
| H-6″b, OH-6″ | 5.75 |
The δH and JHH values were generated via 1H iterative Full Spin Analysis (HiFSA) using experimental NMR data acquired in DMSO-d6 at 600 MHz and 298 K.
The “pseudo” (ps-) prefix indicates proton signals belonging to a higher order spin system.
Table 4.
Calculated 1H chemical shifts (δH, ppm) and 1H,1H spin-spin coupling constants (JHH, Hz) of the 3-O-α-rhamnopyranosyl-(1→6)-β-glucopyranosyl moiety of 10a
| Proton | δH (ppm), multiplicity | Coupling | JHH (Hz) |
|---|---|---|---|
| β-Glucopyranosyl | |||
| H-1″ | 5.342, ps-db | H-1″, H-2″ | 7.73 |
| H-2″ | 3.220, ps-ddd | H-2″, OH-2″ | 4.71 |
| OH-2″ | 5.286, ps-d | H-2″, H-3″ | 9.02 |
| H-3″ | 3.205, ps-ddd | H-3″, OH-3″ | 4.89 |
| OH-3″ | 5.116, ps-d | H-3″, H-4″ | 8.72 |
| H-4″ | 3.048, ps-ddd | H-4″, OH-4″ | 5.83 |
| OH-4″ | 5.075, d | H-4″, H-5″ | 9.81 |
| H-5″ | 3.238, ddd | H-5″, H-6″a | 1.75 |
| H-6″a | 3.701, dd/br d | H-5″, H-6″b | 6.92 |
| H-6″b | 3.281, dd | H-6″a, H-6″b | −11.24 |
| α-Rhamnopyranosyl | |||
| H-1‴ | 4.376, d | H-1‴, H-2‴ | 1.59 |
| H-2‴ | 3.384, ddd/br t | H-2‴, OH-2‴ | 4.09 |
| OH-2‴ | 4.351, br d | H-2‴, H-3‴ | 3.01 |
| H-3‴ | 3.275, ddd | H-3‴, OH-3‴ | 5.69 |
| OH-3‴ | 4.400, br d | H-3‴, H-4‴ | 9.50 |
| H-4‴ | 3.067, ddd/dt | H-4‴, OH-4‴ | 3.62 |
| OH-4‴ | 4.535, br d | H-4‴, H-5‴ | 9.28 |
| H-5‴ | 3.265, dq | H-5‴, H3-6‴ | 6.21 |
| H3-6‴ | 0.987, d |
The δH and JHH values were generated via 1H iterative Full Spin Analysis (HiFSA) using experimental NMR data acquired in DMSO-d6 at 600 MHz and 298 K.
The “pseudo” (ps-) prefix indicates proton signals belonging to a higher order spin system.
CONCLUSIONS
Given that rapid identification of known chemical entities by NMR relies on the availability of precise NMR data for comparison, this study focused on the development of 1H NMR profiles of ten diagnostic chemical constituents of Ginkgo biloba leaves. A complete set of 1H fingerprints of compounds 1–10 was obtained by means of HiFSA, which leads to a structural analysis strategy that combines the computational prediction of NMR parameters (δH, JHH) and iterative lineshape fitting. This process allows continuous experimental verification of the theoretical model, providing a unique platform to combine computational analysis of spin systems with the user’s structural elucidation skills.
The 1H fingerprints generated by HiFSA not only are in excellent agreement with the NMR parameters reported in previous studies,[12–21] but also include a number of δH and JHH values that only have been estimated or have not been measured at all. For example, the precise chemical shifts of protons H-7α and H-7β of 1 are here established, as are all the J-couplings of the AA′XX′ spin system of 7. Furthermore, the exhaustive investigation of the flavonol glycosides 9 and 10 allowed a complete interpretation of their 1H NMR data for the first time. The relative configuration of all stereogenic centers of the sugar moieties was verified through the analysis of the complex resonance patterns, as was the unambiguous description of each proton present in their structures.
Overall, this study shows the high level of detail that can be accomplished through computer-assisted spectral analysis. Simulated 1H NMR spectra of the chemical markers can be created in a wide variety of NMR processing software (e.g., Mnova, iNMR, TopSpin, NUTS, SpinWorks) using the δH and JHH values summarized in Tables 1–4 and the effective line width values collected in the Supporting Information. In addition, most software packages allow the user to specify additional parameters such as field strength, spectral width, and the number of data points, thereby allowing a better comparison between the simulated spectrum and the experimental NMR data.
It is important to keep in mind that basic NMR parameters, especially chemical shifts, might be affected by a number of experimental conditions. The use of DMSO-d6 as the NMR solvent ensures a high stability of the δH values at different concentrations of the analyte, with typical fluctuations in the order of 0.01–0.02 ppm. Large deviations from the temperature at which the NMR data were acquired (298 K) might require additional corrections, particularly for exchangeable protons. Ultimately, the 1H NMR assignments reported herein will facilitate the identification of these ten important marker constituents of Ginkgo biloba materials, some of which (6–10) also occur in other plants. Moreover, the production of these high-resolution 1H fingerprints is a critical step in the development of qNMR methodology for simultaneous characterization of known constituents in complex plant materials.[22]
Supplementary Material
Acknowledgments
Funded by the National Institutes of Health (NIH), National Center for Complementary and Alternative Medicine (NCCAM), grant RC2 AT005899.
The authors thank Drs. J. B. McAlpine and K. Riihinen for their very valuable comments during the manuscript preparation and B. Ramirez for his assistance in the NMR facility at the UIC Center for Structural Biology (CSB). The kind support and hospitality of M. Niemitz and Dr. S.-P. Korhonen (PERCH Solutions Ltd.) during a training visit of J.G.N to their facilities is gratefully acknowledged. The present research work was financially supported by the National Institutes of Health (NIH) through grant RC2 AT005899 awarded by the National Center for Complementary and Alternative Medicine (NCCAM). The purchase of the 900 MHz NMR spectrometer and construction of the UIC CSB was funded via the NIH grant P41 GM068944, awarded to Dr. P. G. W. Gettins by the National Institute of General Medical Sciences (NIGMS).
References
- 1.Mahadevan S, Park Y. J Food Sci. 2008;73:R14–R19. doi: 10.1111/j.1750-3841.2007.00597.x. [DOI] [PubMed] [Google Scholar]
- 2.Chan P-C, Xia Q, Fu PP. J Environ Sci Heal C. 2007;25:211–244. doi: 10.1080/10590500701569414. [DOI] [PubMed] [Google Scholar]
- 3.Laatikainen R, Niemitz M, Malaisse WJ, Biesemans M, Willem R. Magn Reson Med. 1996;36:359–365. doi: 10.1002/mrm.1910360306. [DOI] [PubMed] [Google Scholar]
- 4.Laatikainen R, Niemitz M, Weber U, Sundelin J, Hassinen T, Vepsäläinen J. J Magn Reson A. 1996;120:1–10. [Google Scholar]
- 5.Laatikainen R, Tiainen M, Korhonen S-P, Niemitz M. Computerized Analysis of High-resolution Solution-state Spectra. In: Harris RK, Wasylishen RE, editors. Encyclopedia of Magnetic Resonance. John Wiley & Sons; Chichester: Published online 15th December 2011. [DOI] [Google Scholar]
- 6.Niemitz M, Laatikainen R, Chen S-N, Kleps R, Kozikowski AP, Pauli GF. Magn Reson Chem. 2007;45:878–882. doi: 10.1002/mrc.2061. [DOI] [PubMed] [Google Scholar]
- 7.Kolehmainen E, Laihia K, Laatikainen R, Vepsäläinen J, Niemitz M, Suontamo R. Magn Reson Chem. 1997;35:463–467. [Google Scholar]
- 8.Pauli GF, Jaki BU, Lankin DC. J Nat Prod. 2005;68:133–149. doi: 10.1021/np0497301. [DOI] [PubMed] [Google Scholar]
- 9.Tiainen M, Maaheimo H, Soininen P, Laatikainen R. Magn Reson Chem. 2010;48:117–122. doi: 10.1002/mrc.2553. [DOI] [PubMed] [Google Scholar]
- 10.Pauli GF. J Nat Prod. 2000;63:834–838. doi: 10.1021/np990527t. [DOI] [PubMed] [Google Scholar]
- 11.Napolitano JG, Gavín JA, García C, Norte M, Fernández JJ, Hernández Daranas A. Chem Eur J. 2011;17:6338–6347. doi: 10.1002/chem.201100412. [DOI] [PubMed] [Google Scholar]
- 12.van Beek TA. Bioorg Med Chem. 2005;13:5001–5012. doi: 10.1016/j.bmc.2005.05.056. [DOI] [PubMed] [Google Scholar]
- 13.Llabres G, Baiwir M, Sbit M, Dupont L. Spectrochim Acta A. 1989;45:1037–1045. [Google Scholar]
- 14.Roumestand C, Perly B, Hosford D, Braquet P. Tetrahedron. 1989;45:1975–1983. [Google Scholar]
- 15.Weinges K, Hepp M, Jaggy H. Liebigs Ann Chem. 1987:521–526. [Google Scholar]
- 16.Nakanishi K. Pure Appl Chem. 1967;14:89–113. doi: 10.1351/pac196714010089. [DOI] [PubMed] [Google Scholar]
- 17.Bernard F-X, Sablé S, Cameron B, Provost J, Desnottes J-F, Crouzet J, Blanche F. Antimicrob Agents Chemother. 1997;41:992–998. doi: 10.1128/aac.41.5.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Markham KR, Geiger H. In: The Flavonoids: Advances in Research since 1986. Harborne JB, editor. Chapman & Hall; London: 1994. pp. 441–497. [Google Scholar]
- 19.Shen C-C, Chang Y-S, Ho L-K. Phytochemistry. 1993;34:843–845. [Google Scholar]
- 20.Khalifa TI, Muhtadi FJ, Hassan MMA. Zbl Pharm. 1983;122:809–813. [Google Scholar]
- 21.Batterham TJ, Highet J. Aust J Chem. 1964;17:428–439. [Google Scholar]
- 22.Napolitano JG, Gödecke T, Rodríguez-Brasco MF, Jaki BU, Chen SN, Lankin DC, Pauli GF. J Nat Prod. 2012;75:238–248. doi: 10.1021/np200949v. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.