

Abstract
Recessive mutations in the TK2 gene typically cause fatal infantile mitochondrial DNA (mtDNA) depletion syndromes (MDS).1–3 However, the progression of weakness may vary,4 as shown by recently described adult patients with late-onset myopathy.5,6 To date, only 5 adult patients with TK2-related MDS have been reported. Herein, we describe a man who had several unusual features. Clinically, he was weak as a child but sought medical attention as an adult. At the molecular level, multiple mtDNA deletions in muscle were more prominent than mtDNA depletion.
Recessive mutations in the TK2 gene typically cause fatal infantile mitochondrial DNA (mtDNA) depletion syndromes (MDS).1–3 However, the progression of weakness may vary,4 as shown by recently described adult patients with late-onset myopathy.5,6 To date, only 5 adult patients with TK2-related MDS have been reported. Herein, we describe a man who had several unusual features. Clinically, he was weak as a child but sought medical attention as an adult. At the molecular level, multiple mtDNA deletions in muscle were more prominent than mtDNA depletion.
Case report
A 22-year-old man was referred to our neuromuscular unit because of muscle weakness. His parents were consanguineous and a 3-year-old sister had died of respiratory failure due to “muscular dystrophy.” There had been 2 more infantile deaths in previous generations in children born of consanguineous parents (figure 1A). The patient was born at term after an uneventful pregnancy and developed normally until 24 months of age, when his mother noticed that he “climbed up” his thighs to stand up from the floor and walked on his toes. At age 12 years, he walked with a waddle but was fully active and independent. At age 20, he had a nasal voice and mild proximal arm weakness. An episode of aspiration pneumonia led to respiratory arrest and mechanical ventilation with tracheostomy, after which the weakness worsened rapidly. The patient is now wheelchair-bound, has dysphagia, nasal dysarthria, and needs nocturnal noninvasive ventilator support. Physical examination shows severe axial and proximal limb weakness sparing distal muscles, facial weakness, pectoral atrophy, scapular winging, ankle contractures, and striking gynecomastia (figure 1C). There is no ptosis and external ocular movements are full.
Figure 1. Clinical, pathologic, and molecular features of the patient.
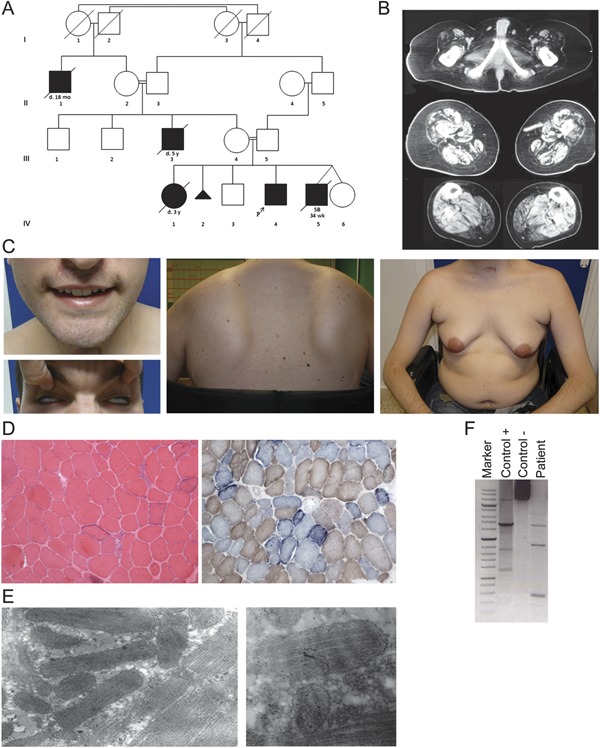
(A) Pedigree (IV-4 is the index case). SB 34 wk under patient IV-5 indicates stillbirth at 34 weeks' gestation. (B) Muscle tomography of lower limbs. (C) Facial involvement with weakness of the orbicularis oculi (Bell phenomenon) and oris, pectoral atrophy, gynecomastia, and scapular winging. (D) The muscle biopsy shows a dystrophic pattern and 50% cytochrome c oxidase–negative fibers, most of which are “ragged-blue” with the succinate dehydrogenase stain (×10). (E) Electron microscopy shows enlarged bizarre mitochondria and paracrystalline inclusions (×39,000; ×46,000). (F) The long-range PCR reveals multiple deletions in mitochondrial DNA from muscle.
The ethics committee approved this study. Creatine kinase level was 757 IU/L (normal <200) and venous lactic acid was normal. Endocrine and cardiologic workups were normal. Nerve conduction studies were normal and EMG showed small motor unit potentials in most muscles but no spontaneous activity. Muscle tomography showed severe atrophy and fatty degeneration of the gluteus, quadriceps, semimembranosus, and semitendinosus muscles in the thighs and of both gastrocnemii in the legs (figure 1B). A biopsy from the tibialis anterior showed dystrophic features with necrotic fibers and endomysial fibrosis. Immunohistochemical studies excluded most forms of muscular dystrophies (figure e-1 on the Neurology® Web site at www.neurology.org). Up to 50% of fibers were cytochrome c oxidase–negative, and most had increased staining for succinic dehydrogenase (figure 1D). Electron microscopy showed groups of abnormally shaped mitochondria, some containing paracrystalline inclusions (figure 1E). The activity of acid α-glucosidase was normal in a dried blood spot. Citrate synthase activity was increased whereas the activity of complex I was 35% of normal when referred to citrate synthase. Multiple mtDNA deletions were evident in muscle with long-range PCR (figure 1F). Quantification by real-time PCR showed a mild reduction of the mtDNA/nuclear DNA ratio in muscle, with 55% mtDNA depletion (i.e., 45% residual mtDNA). Sequencing of the TK2 gene disclosed a previously reported homozygous c.323C>T, p.T108M mutation. Sequencing of the POLG1, SBMA (Kennedy disease), and DMD genes was normal.
Discussion
This adult case of indolent myopathy expands the clinical spectrum of TK2-related diseases. Five recently published adult cases revealed that TK2-related MDS can have early onset and slow progression worsening in adulthood,6 as in our case, or it can present in adulthood.5 All patients showed muscle involvement, including bulbar, facial, and axial weakness, whereas respiratory insufficiency seemed confined to the early-onset cases. Our patient also showed prominent gynecomastia, which was unexplained by endocrine dysfunction. The normal serum lactate is not too surprising, because normal or only slightly increased lactate values have been reported in many patients with TK2 mutations, especially in adult cases.
A review of the literature shows that the degree of mtDNA depletion tends to be higher (more than 80%) in pediatric1,2 than in adult cases (approximately 60%),5,6 an observation confirmed in our patient. Notably, the p.Thr108Met mutation found in our patient has been reported in both pediatric and adult cases.2,4,6 Why the same mutation should produce different degrees of mtDNA depletion remains unclear.
Three of our patient's relatives, also born of consanguineous parents, had died in infancy of respiratory insufficiency and his sister's muscle biopsy had shown dystrophic features. Although we did not have biological samples from those patients, it is plausible that they carried the same mutation in the TK2 gene, a remarkable example of intrafamilial phenotypic heterogeneity.
This report emphasizes that TK2 mutations can cause dystrophic features in adult patients with mitochondrial myopathy, which should alert neurologists to the correct molecular diagnosis.
Supplementary Material
Footnotes
Author contributions: Drs. C. Paradas, E. Rivas, and P. Carbonell managed patient care and designed the study. Dr. P. Gutiérrez Ríos performed the molecular assays. Drs. M. Hirano and S. DiMauro directed and supervised biochemical and molecular studies and edited the manuscript.
Disclosure: C. Paradas, P. Gutiérrez Ríos, E. Rivas, and P. Carbonell report no disclosures. M. Hirano has received honoraria as a member of the Athena Diagnostics Speakers' Bureau. S. DiMauro receives compensation as a member of the editorial board of MedLink Neurology®. Go to Neurology.org for full disclosures.
Supplemental data at www.neurology.org
References
- 1.Gotz A, Isohanni P, Pihko H, et al. Thymidine kinase 2 defects can cause multi-tissue mtDNA depletion syndrome. Brain 2008;131:2841–2850 [DOI] [PubMed] [Google Scholar]
- 2.Mancuso M, Filosto M, Bonilla E, et al. Mitochondrial myopathy of childhood associated with mitochondrial DNA depletion and a homozygous mutation (T77M) in the TK2 gene. Arch Neurol 2003;60:1007–1009 [DOI] [PubMed] [Google Scholar]
- 3.Saada A, Shaag A, Mandel H, Nevo Y, Eriksson S, Elpeleg O. Mutant mitochondrial thymidine kinase in mitochondrial DNA depletion myopathy. Nat Genet 2001;29:342–344 [DOI] [PubMed] [Google Scholar]
- 4.Oskoui M, Davidzon G, Pascual J, et al. Clinical spectrum of mitochondrial DNA depletion due to mutations in the thymidine kinase 2 gene. Arch Neurol 2006;63:1122–1126 [DOI] [PubMed] [Google Scholar]
- 5.Tyynismaa H, Sun R, Ahola-Erkkila S, et al. Thymidine kinase 2 mutations in autosomal recessive progressive external ophthalmoplegia with multiple mitochondrial DNA deletions. Hum Mol Genet 2012;21:66–75 [DOI] [PubMed] [Google Scholar]
- 6.Behin A, Jardel C, Claeys KG, et al. Adult cases of mitochondrial DNA depletion due to TK2 defect: an expanding spectrum. Neurology 2012;78:644–648 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.