

Abstract
Congenital dyserythropoietic anemias belong to a group of inherited conditions characterized by a maturation arrest during erythropoiesis with a reduced reticulocyte production in contrast with erythroid hyperplasia in bone marrow. The latter shows specific morphological abnormalities that allowed for a morphological classification of these conditions mainly represented by congenital dyserythropoietic anemias types I and II. The identification of their causative genes provided evidence that these conditions have different molecular mechanisms that induce abnormal cell maturation and division. Some altered proteins seem to be involved in the chromatin assembly, such as codanin-1 in congenital dyserythropoietic anemia I. The gene involved in congenital dyserythropoietic anemia II, the most frequent form, is SEC23B. This condition seems to belong to a group of diseases attributable to defects in the transport of newly synthesized proteins from endoplasmic reticulum to the Golgi. This review will analyze recent insights in congenital dyserythropoietic anemias types I and II. It will also attempt to clarify the relationship between mutations in causative genes and the clinical phenotype of these conditions.
Key words: dyserythropoiesis, congenital dyserythropoietic anemia, red blood cells, inherited anemias
Introduction
Every day, approximately 150 billion red blood cells are produced by the erythroid compartment of bone marrow. During this dynamic multistep process, called erythropoiesis, cell amplification and differentiation are inversely co-ordinated. Dyserythropoiesis is due to a derangement of this process caused by a maturation arrest and a consequent reduction of daily red cell production. If this phenomenon is severe it could result in anemia.
Dyserythropoiesis is a subtype of bone marrow failure syndromes characterized by monolineage involvement and morphological abnormalities in erythroid precursor cells. The microscopic morphological evaluation justifies the heterogeneity of these syndromes (Figure 1). The term ‘dyserythropoiesis’ was first used by Crookston,1 for cases later classified as congenital dyserythropoietic anemia (CDA) type II, and by Wendt and Heimpel2, for cases later classified as CDA I. The three classical types of CDAs have been defined on the basis of bone marrow morphology. This working classification is still used in clinical practice. CDA types I and II are autosomal recessive diseases. Whereas CDA I displays abnormalities in chromatin structure, CDA II patients have a marked increase in bi- and multi-nucleated erythroblasts in their bone marrow (Table 1). Conversely, CDA type III is an autosomal dominant disease with giant multi-nucleated erythroblasts in bone marrow. CDA type III was first reported in 1962 in a large Swedish family accounting for 34 patients.3 This allowed for the mapping of the gene on chromosome 15.4,5 Very few additional cases have been published and these do not provide any further molecular information. There are, however, families that fall within the general definition of the CDAs, but do not conform to any of the three classical types. CDA type IV, a CDA type II with negative serum tests, sharing similar bone marrow morphology to CDA type III (multi-nucleated erythroblasts), was originally listed in the group of the CDA variants, as proposed by Wickramasinghe. This group also comprises: CDA with prominent erythroblastosis after splenectomy, CDA with intraerythrocytic inclusions, CDA with thrombocytopenia and, finally, the very rare form of CDA without dysplasia.6
Figure 1.

Flow diagram for differential diagnosis of CDAs. This diagram shows the main steps that allow the differential diagnosis of CDA subtypes among the hypoproliferative anemias, based on clinical, biochemical and molecular findings. The key stages to the diagnosis of CDAs are highlighted in dark blue. CBC: complete blood count; BMF: bone marrow failure; FA: Fanconi's anemia; AA: aplastic anemia; PNH: paroxysmal nocturnal hemoglobinuria; MDS: myelodysplastic syndromes; DBA: Diamond-Blackfan anemia.
Table 1.
Characteristic features of different types of congenital dyserythropoietic anemias.
Dyserythropoiesis appears to be a morphological phenomenon common to several inherited and acquired conditions, and this could account for the difficulties in diagnosing these syndromes. Furthermore, in many families there is only one single affected individual, which makes determination of the inheritance pattern very difficult. Indeed recessive inheritance, de novo mutation and low expression alleles are difficult to distinguish without molecular evaluation.7,8
Recent identification of several causative genes could help reclassify these disorders (Table 1). Variant forms are very rare and this, up to now, has compromised further molecular studies. Linkage studies had previously been the main tool to clarify the genetics of Mendelian disorders; however, extremely rare disorders or sporadic cases caused by de novo variants are not appropriate for this type of study design. Exome sequencing is now becoming technically feasible and more cost-effective due to the recent advances in high-throughput sequence capture methods and next-generation sequencing technologies that have offered new opportunities for research into Mendelian disorders. 9 We suggest that the use of these new technologies will lead to the identification of new causative genes in CDA variants in the near future. In particular, this review will deal with new insights on the two most frequent forms of CDAs: types I and II.
Epidemiology of CDAs
Until December 2011, 712 cases from 614 families were included in the German CDA Registry,10,11 whereas 206 cases from 183 families were enrolled in the Italian CDA registry (A Iolascon, unpublished data, 2011). In particular, 169 cases from 143 families with CDA I, and 454 cases from 356 families with CDA II worldwide were recorded in the literature. Hence, CDA I appears to be approximately 3 times less frequent than CDA II. Most families were from Western European and Middle-East countries, but single cases were also reported from the USA, India, Japan and China.10,12-14 The estimated cumulative incidence of CDA I and CDA II cases in Europe is approximately 0.24 and 0.71 cases/million, respectively. No significant differences according to ethnic origin or sex ratios have been observed. There are large differences in frequency of both types of CDAs between European countries, with an average of approximately one case per million inhabitants. Italy shows a much higher incidence compared to all other European countries, with approximately 2.49 cases⁄million reported; this is only because of the large number of cases with CDA II.10,15 Although CDA II patients are to be found right across the Italian peninsula, the majority of ancestors came from Southern Italy, where a founder effect has been observed.13 The wide variation in CDA incidence among European regions could be due to genetic reasons and also to the presence of reference centers with advanced diagnostic procedures available for the diagnosis of CDAs.10 The true frequency of CDA types I and II is most probably higher than that estimated. This could be due to clinical heterogeneity and diagnostic difficulties, as demonstrated by the observation that in these congenital diseases the correct diagnosis is often delayed until adulthood.
Clinical and laboratory findings
Dyserythropoietic anemia could be suspected in the presence of symptoms and signs of increased hemoglobin (Hb) turnover, such as mild jaundice, and low or absent haptoglobin, as in hemolytic anemias, with a reticulocytosis that does not correspond to the degree of anemia. The bone marrow is always hypercellular, exclusively due to a pronounced increase of erythroblasts, with increased erythropoietic/ granulopoietic ratio. Extramedullary hematopoiesis presenting as paravertebral bulks may be observed in all types of CDAs.7,8
The clinical picture of CDA I includes variable degrees of anemia, sometimes with neonatal symptoms, jaundice, splenomegaly, hepatomegaly, frequent and diverse dysmorphisms (4-14% of cases), predominantly affecting the digits (syndactyly in hands or feet, absence of nails or supernumerary toes),7,8,16 and a progressive build up of iron overload. Retinal angioid streaks and macular abnormalities are also reported.17 Most patients have life-long anemia with Hb concentration between 7-11 g/dL. Occasionally, there are severe cases requiring transfusion in utero18 or immediately after birth, and regular blood transfusions during childhood and adolescence.16 Anemia is usually macrocytic with mean cell volume (MCV) between 100-120 fL, but may be normocytic during childhood. 16,19,20 Bone marrow examination shows 30-60% of early and late polychromatic erythroblasts with extreme abnormalities of nuclear shape and size, but proerythroblasts and immature basophilic erythroblasts usually appear normal. There are large polyploid cells and a small number of cells are bi- or multi-nucleated; in contrast to CDA II, nuclei are of different sizes and stains. The hallmark of CDA I is incompletely divided cells with thin chromatin bridges between pairs of erythroblasts, which may also be seen between two nuclei in a single cell. These abnormalities are the most specific changes, usually present in more than 20% of these cells.2 Electron microscopy (EM) studies demonstrated that heterochromatin is denser than normal and forms sharply delineated clumps with small translucent vacuoles, giving rise to the description of a ‘Swiss cheese’ appearance, and cytoplasm may penetrate through widened pores of the nuclear envelope.21
CDA II is an autosomal recessive disorder in which the severity of anemia varies from mild to severe, and approximately 7% of CDA II cases are transfusion-dependent. In several of these cases there could be co-inheritance of another intra-erythrocyte defect (such as beta- or alphathalassemia) (A Iolascon, personal communication, 2012) and this could account for the severity of the clinical outcome. 22 Very few cases are characterized by clinical manifestations during intrauterine life. Hydrops fetalis has been described in 6 atypical cases which were characterized by erythroblastic multinuclearity but did not fulfil the CDA II diagnostic criteria.7,8,23 Severe molecular defects in the causative gene SEC23B seem to be the possible cause of this condition, but further studies are requested to demonstrate this.23 Although this disorder is considered to be congenital, it is interesting that this type of anemia can be diagnosed in all age groups. Diagnosis of CDA II is usually made later in life compared with CDA I, because the symptoms can be milder (Figure 2).24 Furthermore, this condition could be misdiagnosed as hereditary spherocytosis (HS) because of the similarity in clinical findings. A correct evaluation of reticulocyte number versus Hb level and soluble transferrin receptor (usually higher in CDAs than HS) could resolve the problem of suspected diagnosis. CDA II patients may come to medical attention because of anemia combined with jaundice (90% of cases), splenomegaly (70%) or hepatomegaly (45%). The presence of posterior mediastinal or paravertebral masses consisting of extramedullary hemopoietic tissue can be observed.25-31 The anemia of CDA II is normocytic, Hb levels are somewhat lower in children than in later life, ranging between 8-11 g/dL with a normal or slightly increased MCV (Figure 2), and peripheral blood smear shows anisopoikilocytosis without specific types of poikilocytes, with basophilic stippling of cells and few occasionally binucleated mature erythroblasts. Relative reticulocyte counts are normal or moderately increased (Figure 2). The bone marrow is hypercellular with erythroid hyperplasia but, contrary to CDA I, is of normoblastic appearance with a large number of binucleate (10-35%) and rarely multinucleate late polychromatic erythroblasts (Figure 1).32 On electron microscopy (EM) examination, vesicles loaded with proteins of the endoplasmic reticulum (ER), such as calreticulin, glucose regulated protein (GRP78) and protein disulfide isomerase (PDI),33-36 appear to be running beneath the plasma membrane. Erythrocytes of CDA II patients lyse in acidified serum (Ham test) because of an IgM class antibody that recognizes an antigen present on CDA II cells but that is absent on normal cells. So, the acronym HEMPAS (hemolytic anemia with a positive acidified serum test) was commonly used as a synonym for CDA II. The technical difficulty of this test, and the fact that crosstesting of more than 30 normal sera is needed to obtain a reliable result, has undermined its usefulness.7 The diagnostic hallmark of CDA II is the analysis of erythrocyte membrane proteins by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) identifying the narrower band size and faster migration of erythrocyte anion transporter (EA1), or band 3, and band-4.5 proteins. 37-40 The increased destruction of red blood cells in CDA II has been associated to hypoglycosylation of band 3, which causes an increased clusterization of this protein on the red cell surface and amplified destruction in the spleen.41 Exceptional cases that do not show the characteristic SDS-PAGE pattern have been reported, but it is recommended that these should be considered as CDA II-like conditions. Research on the abnormalities found in CDA II red blood cells has yielded several additional tests, such as Western blotting analysis of ER proteins of red blood cells (GRP-78, calreticulin, PDI).33
Figure 2.
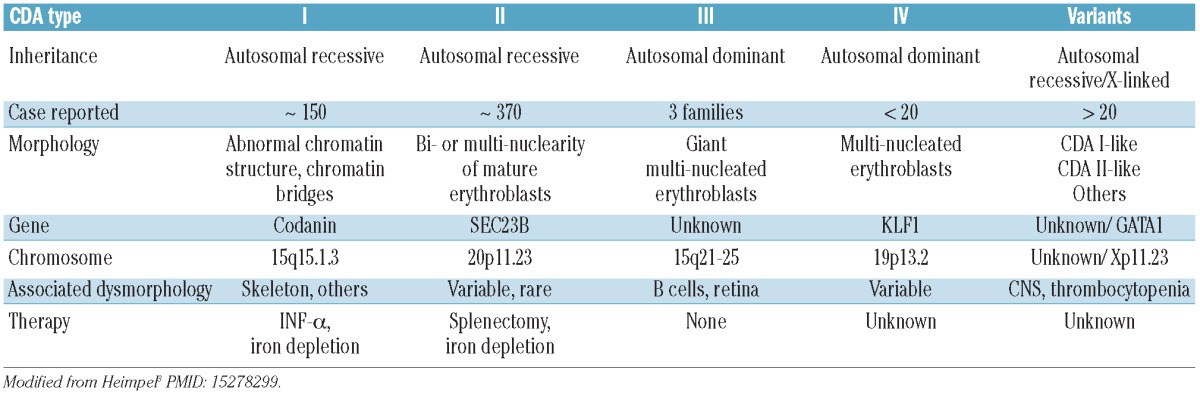
Hematologic parameters in CDA type I and II. The histograms show hemoglobin values, mean cell volume (MCV) and absolute reticulocyte count in CDA type I and II patients. The number of patients enrolled in each comparison is indicated in brackets. Student's t-test was used to compare differences between two groups.
Complications and therapeutic approaches
Cholelithiasis, splenomegaly and iron overload are the most prevalent complications of CDA types I and II. The co-inheritance of UGT1A (TA)7/(TA)7 genotype, i.e. Gilbert syndrome, could account for an increased rate of gallstones.42 In both CDA I and in CDA II, as well as in all ineffective erythropoiesis conditions, secondary hemochromatosis is the most important long-term complication. Hemochromatosis can lead to organ damage if not recognized and appropriately treated. Molecular mechanisms of iron overload have been recently characterized. 43,44 In both conditions, iron loading is not dependent on transfusions (but could be increased by them). Iron loading may be alleviated by ongoing iron loss, such as menstrual bleeding or pregnancies. The study by Tamary on iron overload pathogenesis in CDA I patients supports the notion that high levels of GDF15 contribute to this pathology that,44 because of its overexpression, occurs together with ineffective erythropoiesis and positively correlates with ferritin levels in adults. In 2011, Casanovas demonstrated that serum hepcidin to ferritin ratio is strongly decreased in CDA patients compared to control subjects; thus showing that serum hepcidin concentrations are inappropriately low. Taken together, these data support a role for GDF15 in limiting hepcidin expression in response to iron overload, and increasing iron absorption under erythropoietic stress conditions. However, GDF15 concentrations are significantly lower in CDA II compared to CDA I patients, despite a similar degree of iron overload in both patient groups. We can speculate that additional signals may determine hepatic hepcidin expression and the degree of iron overload in CDA II (i.e. TWSG1, SMAD7).43
Decision-making depends on age, CDA type, severity of expression and comorbidity. Most CDA patients have only mild or moderate anemia and do not require medical intervention. About 50% and 10% of neonates with CDA I45 and CDA II,46 respectively, need at least one erythrocyte transfusion, and some remain transfusion-dependent in the following years. In most adolescents and adults, the need for transfusions is limited to aplastic crises, pregnancy, severe infections or major operations. Furthermore, the co-inheritance of other red cell defects, such as alpha or beta thalassemia and G6PD deficiency, worsens the clinical phenotype to a severe and/or transfusion-dependent condition.47,48 Transfusions contribute to iron overload, and this risk has to be individually weighed against the failure to thrive in infants and children with severe anemia.
With regard to the distinct erythroid hyperplasia, vitamin B12 and folic acid supplements are frequently used, although without any evidence of efficacy. Use of erythropoietin formulations also appear to be ineffective. Interferon (IFN)-α seems to be effective in improving the chronic anemia and splenomegaly in CDA I, but whether this shows efficacy in other types of CDAs remains uncertain. As in other types of IFN-α therapy, it is possible to hypothesize that several polymorphisms could modulate the IFN-α response.49 The pathophysiological basis of the beneficial effect of interferon in CDA I is still not understood. A study on cell lines treated with IFN-α has established which genes are up- or down-regulated by this drug.50
Splenectomy leads to a moderate but sustained increase in Hb concentration and decrease in serum bilirubin levels, but it does not prevent further iron loading. This may be explained by the observation that iron loading is more closely correlated to the expansion of the erythroid marrow than to the anemia itself. The main benefit of splenectomy is abrogation of transfusion requirements and increase in the Hb concentration in severe cases. However, the Hb levels post-splenectomy did not reach normal values. 7,8 In other patients, it is advisable to follow the guidelines for mild cases of HS.51 Splenectomy is not recommended in CDA I and II and individual decisions have to be made in CDA variants with transfusion dependency and enlarged spleen. Cholecystectomy is often indicated in all types of CDA, and decision making should follow the common guidelines for cholelithiasis.51
Since the main problem encountered by CDA patients after the first years of life is iron overload, ferritin levels should be controlled at least annually, even in patients with light or moderate anemia. Adequate treatment with regular phlebotomies leads to normal ferritin concentrations, indicating reversal of iron overload in patients with CDA. Very often, CDA patients do not support phlebotomy treatment and in these cases oral chelating agents could be proposed. Since data correlating serum ferritin levels to tissue iron are inadequate, prospective studies using non-invasive techniques for liver iron determination are required. For the moment. management of iron overload should follow the guidelines for thalassemia.
Allogeneic bone marrow transplantation (BMT) from an HLA-identical sibling was successful in 6 transfusiondependent children with very severe CDA and in one adult with CDA II and beta-thalassemia trait. This treatment can abolish transfusion dependency, thus preventing progression of tissue damage related to iron overload. The result should be both longer life expectancy and better quality of life.46
Molecular genetics and pathogenesis
CDA type I
The gene responsible for CDA I (CDAN1 gene) was mapped to the long arm of chromosome 15 between 15q15.1q15.3 by homozygosity mapping performed in 25 CDA I patients from four large consanguineous Israeli Bedouin families with a high degree of consanguinity.52 The CDAN1 gene was successively cloned with 28 exons spanning 15 Kb and encoding a protein named codanin-1. In unrelated patients of European, Bedouin and Asian origin, different point mutations were detected. Approximately 90% of patients with a bone marrow suggesting CDA I have codanin-1 gene mutations (Figure 3).53-54 The existence of families with definite phenotype of CDA I in which no mutation of the CDAN1 gene has been found, suggested either a promoter defect or the presence of a second disease locus (genetic heterogeneity). The latter observation has been suggested by the exclusion of linkage with chromosome 15 in several families.17 The vast majority of patients with confirmed diagnosis of CDA I showed mutations of at least one allele from exons 6 to 28 within CDAN1 and more than 30 unique mutations have been identified so far (see Online Supplementary Table S1 for complete mutational spectrum of CDAN1). All these seem to be independent events and, up to now, no particularly frequent mutations have been reported in the literature. Interestingly, no homozygotes or compound heterozygotes for null-type mutations have been identified, supporting an earlier view that codanin-1 may have a unique function and may be essential during development.
Figure 3.
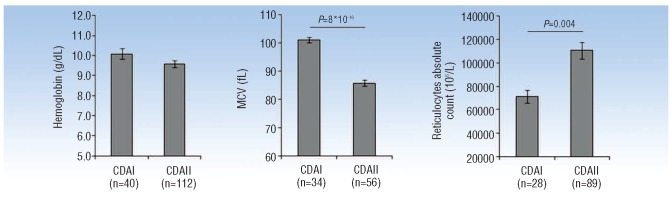
Distribution of CDAN1 and SEC23B mutations. The pie chart shows the distribution of the different types of mutations found to date in CDAN1 and SEC23B genes (between 2002 and 2012).
Codanin-1 is a ubiquitously expressed protein that has still not been well characterized. It seems to be related to chromosome structure and it must be involved in mitotic process. In 2009, Tamary et al. showed that codanin-1 is a direct transcriptional target of the E2F1 transcription factor and that its levels increase during S-phase and decrease during mitosis.55 In another study, Tamary and colleagues found that codanin-1 binds to Asf1a (histone H3/H4 chaperone anti-silencing 1),56 involved in the chromatin structure dynamics by its role in nucleosome assembly and disassembly. In particular, Asf1a binds histone H3/H4 in the cytoplasm and in complex with importin-4 accompanies histone dimers into the nucleus where they are transferred to downstream chromatin assembly factors.57-60 Very recently, Ask and colleagues confirmed that this histone chaperone is a direct binding partner of codanin-1.61 They suggested that binding of codanin-1 inhibits dissociation of histones from Asf1a that cannot, therefore, be deposited onto DNA. Furthermore, they demonstrated that codanin-1 exerts a dominant-negative effect on S-phase progression chiefly by interfering with Asf1 function. Indeed, codanin-1 mutants found in CDA I patients are defective in Asf1 regulation and this defect might underline at least some of the phenotype associated with CDA I.
Recently, another contribution attempted to define the role of codanin-1 in pathophysiology of CDA I. Renella et al. investigated localization, distribution and interactions of codanin-1 in CDA I patients and generated a murine knock-out model for CDAN1. No gross differences between normal and patient samples both in the amounts of histone proteins or various epigenetic marks of histone tails were found, suggesting that histone signatures involved in maintenance of chromatin structure and epigenetic regulation are globally maintained in CDA I.62 The authors demonstrated codanin-1 distribution in both nucleus and cytoplasm of normal primary human erythroblasts. This localization pattern was unchanged in CDA I erythroblasts. Although no differences in the localization patterns of various nuclear proteins were observed between patients and control erythroblasts, the localization of HP1α, a key component of heterochromatin, was found to be markedly perturbed. HP1α accumulates in the Golgi apparatus of CDA I but not in normal erythroblasts. The authors confirmed that the abnormal localization of HP1α in CDA I patients is confined to the intermediate erythroblast maturation stage, where the characteristic ultrastructural chromatin pattern of CDA I is observed. Furthermore, they suggested that an abnormality in codanin-1 could be responsible for the aberrant localization of HP1α. Interestingly, by confocal immunofluorescence, they also found codanin-1 co-localizes partially with SEC23B, the protein mutated in CDA II, suggesting a molecular link between the two major types of CDAs. As expected by the molecular studies in human patients, the total absence of codanin-1 is lethal. Renella et al. generated the first murine Cdan1 gene-trap model demonstrating its widespread expression during embryonic development. Cdan1gt/gt homozygotes die in utero before the onset of primitive erythropoiesis, suggesting that Cdan1 has other critical roles during embryogenesis.62
CDA type II
For a long time, the pathognomonic hypoglycosylation of the band 3 was indicated as the cause of CDA II. Three genes (MANII, MANA and GnTII), encoding for the enzymes involved in this abnormal glycosylation, were first established as putative causative genes of this condition. However, they were excluded by linkage analysis.63 In the same year, genome-wide linkage analysis localized the disease gene to a 5-cM region of chromosome 20q11.2.64 The identification of a CDA II-linked region led to the sequencing and subsequent exclusion of numerous genes along the pericentromeric segment of the long arm of chromosome 20.65 In a refined contig build (build 36.3), the markers with the highest CDA II lod scores overlap the minimal homozygosity region on the short arm of chromosome 20.66 So, a joint approach of functional mapping and homozygosity mapping by genome-wide SNP analysis finally allowed the gene to be re-mapped, right across the centromere. With the assumption that the cis, median and trans N-glycan Golgi processing of erythroblast glycoproteins was impaired, the SEC23B (CDAN2) gene became a likely candidate for CDA II.67 A proteomic approach led to the same result.68 Sequencing analysis in 33 patients from 28 unrelated families from the main European Registries showed a wide spectrum of different mutations in the SEC23B gene in either the compound heterozygous or homozygous state.67 In the same study, an in vitro model of gene silencing demonstrated that suppression of SEC23B expression recapitulates the cytokinesis defect, with a significant increase in the percentage of binuclearity and an increased size of nuclei in SEC23B silenced cells. Knockdown of zebrafish sec23b also leads to aberrant erythrocyte development. Indeed, sec23b zebrafish morphants show a significant increase in immature, binucleated erythrocytes.67 The disease gene encodes the cytoplasmic coat protein (COP)II component SEC23B, involved in the secretory pathway of eukaryotic cells. COPII is a multi-subunit complex which mediates accumulation of secretory cargo, deformation of the membrane and generation of subsequent anterograde transport of correctly folded cargo that bud from the ER towards the Golgi apparatus.69 This pathway is critical for membrane homeostasis, localization of proteins within cells and secretion of extracellular factors.69,70 To date, mutations in other COPII components have been assigned to human genetic disorders. Alterations in SAR1B are identified as the etiology of the chylomicron retention disease, Anderson disease and Marinesco-Sjogren syndrome,71 while mutations in the SEC23A gene cause craniolenticulosutural dysplasia (CLSD or Boyadjiev-Jabs syndrome).72 Interestingly, the latter gene is a paralog of SEC23B. The specificity of the CDA II phenotype seems to be determined by tissue-specific expression of SEC23B versus SEC23A during erythroid differentiation.67 Alternatively, this specificity could be explained by the presence of tissue-specific cargoes (such as band 3 in red blood cells), which might require high levels and full function of a specific COPII component to be correctly transported.73
A recent study on 42 CDA II patients from the Italian and the French Registries showed a relationship between the mutations and various biological parameters. In this study, patients were divided into two groups according to their genotype: 1) patients with two missense mutations and 2) patients with one nonsense and one missense mutation. Compound heterozygosity for a missense and nonsense mutation tended to produce more severe clinical presentations than homozygosity or compound heterozygosity for two missense mutations. Homozygosity or compound heterozygosity for two nonsense mutations was not found; this was considered to be lethal.74 Until now, 53 different causative mutations have been described in the SEC23B gene (Figure 3; see Online Supplementary Table S1 for complete mutational spectrum of CDAN2).8,14,75-77 Very recently, Sec23b deficient mice (Sec23b gt/gt) from ES cells with a genetrap cassette inserted into the last intron of Sec23b were generated. Sec23b gt/gt mice are born with no apparent anemia phenotype, but die shortly after birth, with degeneration of professional secretory tissues, pancreas, as well as salivary glands. However, no significant differences in red blood cell count, hemoglobin or hematocrit levels were observed in blood collected from wild-type (WT) and Sec23b gt/gt neonates.78 The disparate phenotypes in mouse and human could result from residual SEC23B function associated with the hypomorphic mutations observed in humans or, alternatively, might be explained by a speciesspecific shift in function between the closely related SEC23 paralogs.78 These data demonstrate that Sec23b deficient humans and mice exhibit disparate phenotypes, apparently restricted to CDA II in humans and a prominent neonatal pancreatic insufficiency in mice.
Other congenital dyserythropoietic anemias
Some genes have been already associated with CDA variants. One of these is the gene encoding for the transcriptional factor GATA-1. Mutations in GATA-1 are directly linked to deregulated formation of certain blood cell lineages (Figure 1). Several rare syndromes are caused by defects in GATA-1 gene expression or a malformed protein product.79 They are: X-linked thrombocytopenia (XLT), X-linked thrombocytopenia with thalassemia (XLTT), congenital erythropoietic porphyria (CEP), Xlinked dyserythropoietic anemia and thrombocytopenia (XDAT), transient myeloproliferative disorder (TMD), acute megakaryoblastic leukaemia (AMKL) associated with trisomy 21, and dyserythropoietic anemia associated with the production of a short isoform, GATA-1s.80 The latter is due to a mutation occurring at the last nucleotide of the exon 2 donor splice site and affecting GATA-1 splicing. 80 Very recently, this mutation has been found in 2 siblings affected by Diamond-Blackfan anemia (DBA), a dominant disorder characterized by reduced proliferation and survival of erythroid progenitors leading to hypoproliferative anemia.81 Although the bone marrow examination from these patients confirmed the clinical diagnosis of DBA, the absence of a dominant inheritance, as well as the mildly low platelet count in one of 2 patients, make it more appropriate to consider these cases as DBA-like conditions. This further underlined the variability of the GATA-1-related phenotypes. GATA-1 has two zinc finger domains essential for normal function. The C-terminal finger is necessary for DNA binding. The N-terminal finger mediates interaction with FOG-1 (for friend of GATA-1), a cofactor of GATA-1. In 2000, Nichols described a family with XDAT due to Val205Met substitution.82 This highly conserved residue is necessary for GATA-1:FOG-1 interaction, fundamental in megakaryocyte and erythroid development. Another base mutation that results in Gly208Arg substitution within the highly conserved portion of the Nterminal finger domain has been associated to dyserythropoietic anemia and macrothrombocytopenia.83
Recently, alteration in the erythroid transcriptional factor KLF1 has been associated to CDA type IV84 in the case of 2 patients with a hitherto unclassified CDA in whom the Glu325Lys missense mutation in KLF1 was identified (Figure 1). Patients showed severe anemia at birth and required repeated transfusion during childhood, persistent expression of ɛ and ζ embryonic globin, an HbF level of 40%, novel intra-erythroblastic and intra-erythrocytic inclusions, and deficiency of erythroid proteins CD44 and aquaporin 1. The marrow aspirate studies revealed active erythropoiesis with some dyserythropoietic features. KLF1 is an erythroid transcription factor, and extensive studies in mouse models have shown that it plays a critical role in the expression of globin genes, but also in the expression of a wide spectrum of genes potentially essential for erythropoiesis.85-87 The unique features of this type of CDA confirm the key role of KLF1 during human erythroid differentiation.
Finally, the case of a patient with mevalonate kinase deficiency (MKD) and congenital dyserythropoietic anemia has been described. The clinical phenotype was variable, ranging from the hyperimmunoglobulinemia D and periodic fever syndrome (HIDS), to mevalonic aciduria (MA), a severe metabolic disease. Genomic sequencing of the mevalonate kinase gene revealed compound heterozygosity for a missense mutation previously described in MA (Val310Met) and a novel missense mutation (Tyr116Hys). In contrast, sequencing of the SEC23B gene revealed no mutations, suggesting that the bone marrow abnormalities were causally related to the MKD.88
Conclusions
Congenital dyserythropoietic anemias belong to a heterogeneous group of hereditary disorders, both at clinical and genetic levels. The identification of their causative genes has shown that these conditions have different molecular mechanisms that induce disturbances of cell maturation and cell division during erythropoiesis. Some altered proteins seem to be involved in the chromatin structure dynamics, as well as in histone metabolism, such as codanin-1 in CDA I. Others are transcription factors, involved in the synthesis of many proteins that are important for erythroid differentiation: e.g. KLF1 and GATA1 in CDA IV and XDAT, respectively. CDA II seems to belong to a group of diseases that involve the transport of newly synthesized proteins from ER to the Golgi.
Recent advances partially clarify the pathogenesis of the maturation arrest in the most common CDAs. Mutations in codanin-1 seem to lead to high proliferation rates in early erythroid progenitors that sensitize these cells to chromatin assembly defects; this in turn can jeopardize chromatin organization and chromosome segregation during mitosis.
The pathogenesis of CDA II seems to be due to abnormal vesicular transport ER-Golgi. We could hypothesize that this transport involves a specific group of proteins implicated in cytokinesis, and this could explain the basis of morphological abnormality. However, the effective relationship between mutations in the SEC23B gene and ineffective erythropoiesis is still not understood.
Despite the recent advances in our understanding of the molecular pathogenesis of CDAs, several issues still remain unsolved. In particular, it remains unclear why erythropoiesis is principally sensitive to codanin-1 and SEC23B mutations, since both are ubiquitously expressed proteins. Also, why does the abnormal morphology of erythroid cells during maturation involve only a small percentage of affected cells? Future studies may prove useful in defining the pathogenetic mechanisms involved in these conditions, and possibly this will lead to new drugs becoming available.
Supplementary Material
Acknowledgments
Funding: this work was supported by Ministry of University and Research of Italy (MIUR), by Telethon (Italy) (GGP09044), by contract grant number: MURPS 35-126/Ind, by contract grant sponsor: Regione Campania, by contract grant number: DGRC 1901/2009.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures: The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Crookston JH, Crookston MC, Burnie KL, Francombe WH, Dacie JV, Davis JA, Lewis SM. Hereditary erythroblastic multinuclearity associated with a positive acidifiedserum test: a type of congenital dyserythropoietic anaemia. Br J Haematol. 1969;17(1):11-26 [DOI] [PubMed] [Google Scholar]
- 2.Heimpel H, Wendt F. Congenital dyserythropoietic anemia with karyorrhexis and multinuclearity of erythroblasts. Helv Med Acta. 1968;34(2):103-15 [PubMed] [Google Scholar]
- 3.Heimpel H. Congenital dyserythropoietic anemias: epidemiology, clinical significance, and progress in understanding their pathogenesis. Ann Hematol. 2004;83(10):613-21 [DOI] [PubMed] [Google Scholar]
- 4.Sandstrom H, Wahlin A. Congenital dyserythropoietic anemia type III. Haematologica. 2000;85(7):753-7 [PubMed] [Google Scholar]
- 5.Lind L, Sandström H, Wahlin A, Eriksson M, Nilsson-Sojka B, Sikström C, Holmgren G. Localization of the gene for congenital dyserythropoietic anemia type III, CDAN3, to chromosome 15q21-q25. Hum Mol Genet. 1995;4(1):109-12 [DOI] [PubMed] [Google Scholar]
- 6.Wickramasinghe SN. Congenital dyserythropoietic anaemias: clinical features, haematological morphology and new biochemical data. Blood Rev. 1998;12(3):178-200 [DOI] [PubMed] [Google Scholar]
- 7.Renella R, Wood WG. The congenital dyserythropoietic anemias. Hematol Oncol Clin North Am. 2009;23(2):283-306 [DOI] [PubMed] [Google Scholar]
- 8.Iolascon A, Russo R, Delaunay J. Congenital dyserythropoietic anemias. Curr Opin Hematol. 2011;18(3):146-51 [DOI] [PubMed] [Google Scholar]
- 9.Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745-55 [DOI] [PubMed] [Google Scholar]
- 10.Heimpel H, Matuschek A, Ahmed M, Bader-Meunier B, Colita A, Delaunay J, et al. Frequency of congenital dyserythropoietic anemias in Europe. Eur J Haematol. 2010;85(1):20-5 [PubMed] [Google Scholar]
- 11.Gulbis B, Eleftheriou A, Angastiniotis M, Ball S, Surrallés J, Castella M, et al. Epidemiology of rare anaemias in Europe. Adv Exp Med Biol. 2010;686:375-96 [DOI] [PubMed] [Google Scholar]
- 12.Kawabata H, Doisaki S, Okamoto A, Uchiyama T, Sakamoto S, Hama A, et al. A case of congenital dyserythropoietic anemia type 1 in a Japanese adult with a CDAN1 gene mutation and an inappropriately low serum hepcidin-25 level. Intern Med. 2012;51(8):917-20 [DOI] [PubMed] [Google Scholar]
- 13.Russo R, Gambale A, Esposito MR, Serra ML, Troiano A, De Maggio I, et al. Two founder mutations in the SEC23B gene account for the relatively high frequency of CDA II in the Italian population. Am J Hematol. 2011;86(9):727-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu G, Niu S, Dong A, Cai H, Anderson GJ, Han B, Nie G. A Chinese family carrying novel mutations in SEC23B and HFE2, the genes responsible for congenital dyserythropoietic anaemia II (CDA II) and primary iron overload, respectively. Br J Haematol. 2012;158(1):143-5 [DOI] [PubMed] [Google Scholar]
- 15.Iolascon A, Servedio V, Carbone R, Totaro A, Carella M, Perrotta S, et al. Geographic distribution of CDA-II: did a founder effect operate in Southern Italy? Haematologica. 2000;85(5):470-4 [PubMed] [Google Scholar]
- 16.Heimpel H, Schwarz K, Ebnöther M, Goede JS, Heydrich D, Kamp T, et al. Congenital dyserythropoietic anemia type I (CDA I): molecular genetics, clinical appearance, and prognosis based on longterm observation. Blood. 2006;107(1):334-40 [DOI] [PubMed] [Google Scholar]
- 17.Tamary H, Offret H, Dgany O, Foliguet B, Wickramasinghe SN, Krasnov T, et al. Congenital dyserythropoietic anaemia, type I, in a Caucasian patient with retinal angioid streaks (homozygous Arg1042Trp mutation in codanin-1). Eur J Haematol. 2008;80(3):271-4 [DOI] [PubMed] [Google Scholar]
- 18.Parez N, Dommergues M, Zupan V, Chambost H, Fieschi JB, Delaunay J, et al. Severe congenital dyserythropoietic anaemia type I: prenatal management, transfusion support and alpha-interferon therapy. Br J Haematol. 2000;110(2):420-3 [DOI] [PubMed] [Google Scholar]
- 19.Shalev H, Kapleushnik Y, Haeskelzon L, Degani O, Kransnov T, Sphilberg O, et al. Clinical and laboratory manifestations of congenital dyserythropoietic anemia type I in young adults. Eur J Haematol. 2002;68(3):170-4 [DOI] [PubMed] [Google Scholar]
- 20.Tamary H, Dgany O, Proust A, Krasnov T, Avidan N, Eidelitz-Markus T, et al. Clinical and molecular variability in congenital dyserythropoietic anaemia type I. Br J Haematol. 2005;130(4):628-34 [DOI] [PubMed] [Google Scholar]
- 21.Heimpel H, Kellermann K, Neuschwander N, Hogel J, Schwarz K. The morphological diagnosis of congenital dyserythropoietic anemia: results of a quantitative analysis of peripheral blood and bone marrow cells. Haematologica. 2010;95(6):1034-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iolascon A, D'Agostaro G, Perrotta S, Izzo P, Tavano R, Miraglia del Giudice B. Congenital dyserythropoietic anemia type II: molecular basis and clinical aspects. Haematologica. 1996;81(6):543-59 [PubMed] [Google Scholar]
- 23.Fermo E, Bianchi P, Notarangelo LD, Binda S, Vercellati C, Marcello AP, et al. CDAII presenting as hydrops foetalis: molecular characterization of two cases. Blood Cells Mol Dis. 2010;45(1):20-2 [DOI] [PubMed] [Google Scholar]
- 24.Heimpel H, Anselstetter V, Chrobak L, Denecke J, Einsiedler B, Gallmeier K, et al. Congenital dyserythropoietic anemia type II: epidemiology, clinical appearance, and prognosis based on long-term observation. Blood. 2003;102(13):4576-81 [DOI] [PubMed] [Google Scholar]
- 25.Bird AR, Jacobs P, Moores P. Congenital dyserythropoietic anaemia (type II) presenting with haemosiderosis. Acta Haematol. 1987;78(1):33-6 [DOI] [PubMed] [Google Scholar]
- 26.Halpern Z, Rahmani R, Levo Y. Severe hemochromatosis: the predominant clinical manifestation of congenital dyserythropoietic anemia type 2. Acta Haematol. 1985;74(3):178-80 [DOI] [PubMed] [Google Scholar]
- 27.Hines GL. Paravertebral extramedullary hematopoiesis (as a posterior mediastinal tumor) associated with congenital dyserythropoietic anemia. J Thorac Cardiovasc Surg. 1993;106(4):760-1 [PubMed] [Google Scholar]
- 28.Imran A, Mawhinney R, Swirsky D, Hall C. Paravertebral extramedullary haemopoiesis occurring in a case of congenital dyserythropoietic anaemia type II. Br J Haematol. 2008;140(1):1. [DOI] [PubMed] [Google Scholar]
- 29.Lugassy G, Michaeli J, Harats N, Libson E, Rachmilewitz EA. Paravertebral extramedullary hematopoiesis associated with improvement of anemia in congenital dyserythropoietic anemia type II. Am J Hematol. 1986;22(3):295-300 [DOI] [PubMed] [Google Scholar]
- 30.Steiner W, Denzlinger C, Weiss M. [Paravertebral extramedullary hematopoiesis in congenital dyserythropoietic anemia type II (CDA II)]. Rontgenpraxis. 1995;48(4):110-2 [PubMed] [Google Scholar]
- 31.Tamura H, Matsumoto G, Itakura Y, Terai H, Ikebuchi K, Mitarai T, Isoda K. A case of congenital dyserythropoietic anemia type II associated with hemochromatosis. Intern Med. 1992;31(3):380-4 [DOI] [PubMed] [Google Scholar]
- 32.Verwilghen RL, Lewis SM, Dacie JV, Crookston JH, Crookston MC. Hempas: congenital dyserythropoietic anaemia (type II). Q J Med. 1973;42(166):257-78 [PubMed] [Google Scholar]
- 33.Alloisio N, Texier P, Denoroy L, Berger C, Miraglia del Giudice E, Perrotta S, et al. The cisternae decorating the red blood cell membrane in congenital dyserythropoietic anemia (type II) originate from the endoplasmic reticulum. Blood. 1996;87(10):4433-9 [PubMed] [Google Scholar]
- 34.Breton-Gorius J, Daniel MT, Clauvel JP, Dreyfus B. [Ultrastructural abnormalities of erythroblasts and erythrocytes in 6 cases of congenital dyserythropoietic anemia]. Nouv Rev Fr Hematol. 1973;13(1):23-49 [PubMed] [Google Scholar]
- 35.Verwilghen RL, Tan P, De Wolf-Peeters C, Broeckaert-van O, Louwagie AC. Cell membrane anomaly impeding cell division. Experientia. 1971;27(12):1467-8 [DOI] [PubMed] [Google Scholar]
- 36.Wong KY, Hug G, Lampkin BC. Congenital dyserythropoietic anemia type II: ultrastructural and radioautographic studies of blood and bone marrow. Blood. 1972;39(1):23-30 [PubMed] [Google Scholar]
- 37.Baines AJ, Banga JP, Gratzer WB, Linch DC, Huehns ER. Red cell membrane protein anomalies in congenital dyserythropoietic anaemia, type II (HEMP AS). Br J Haematol. 1982;50(4):563-74 [DOI] [PubMed] [Google Scholar]
- 38.Mawby WJ, Tanner MJ, Anstee DJ, Clamp JR. Incomplete glycosylation of erythrocyte membrane proteins in congenital dyserythropoietic anaemia type II (CDA II). Br J Haematol. 1983;55(2):357-68 [DOI] [PubMed] [Google Scholar]
- 39.Scartezzini P, Forni GL, Baldi M, Izzo C, Sansone G. Decreased glycosylation of band 3 and band 4.5 glycoproteins of erythrocyte membrane in congenital dyserythropoietic anaemia type II. Br J Haematol. 1982;51(4):569-76 [DOI] [PubMed] [Google Scholar]
- 40.Tomita A, Parker CJ. Aberrant regulation of complement by the erythrocytes of hereditary erythroblastic multinuclearity with a positive acidified serum lysis test (HEMPAS). Blood. 1994;83(1):250-9 [PubMed] [Google Scholar]
- 41.De Franceschi L, Turrini F, del Giudice EM, Perrotta S, Olivieri O, Corrocher R, et al. Decreased band 3 anion transport activity and band 3 clusterization in congenital dyserythropoietic anemia type II. Exp Hematol. 1998;26(9):869-73 [PubMed] [Google Scholar]
- 42.Perrotta S, del Giudice EM, Carbone R, Servedio V, Schettini F, Jr, Nobili B, Iolascon A. Gilbert's syndrome accounts for the phenotypic variability of congenital dyserythropoietic anemia type II (CDA-II). J Pediatr. 2000;136(4):556-9 [DOI] [PubMed] [Google Scholar]
- 43.Casanovas G, Swinkels DW, Altamura S, Schwarz K, Laarakkers CM, Gross HJ, et al. Growth differentiation factor 15 in patients with congenital dyserythropoietic anaemia (CDA) type II. J Mol Med (Berl). 2011;89(8):811-6 [DOI] [PubMed] [Google Scholar]
- 44.Tamary H, Shalev H, Perez-Avraham G, Zoldan M, Levi I, Swinkels DW, et al. Elevated growth differentiation factor 15 expression in patients with congenital dyserythropoietic anemia type I. Blood. 2008;112(13):5241-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shalev H, Kapelushnik J, Moser A, Dgany O, Krasnov T, Tamary H. A comprehensive study of the neonatal manifestations of congenital dyserythropoietic anemia type I. J Pediatr Hematol Oncol. 2004;26(11):746-8 [DOI] [PubMed] [Google Scholar]
- 46.Iolascon A, Sabato V, de Mattia D, Locatelli F. Bone marrow transplantation in a case of severe, type II congenital dyserythropoietic anaemia (CDA II). Bone Marrow Transplant. 2001;27(2):213-5 [DOI] [PubMed] [Google Scholar]
- 47.Menike D, Wickramasinghe SN. Effects of four species of interferon-alpha on cultured erythroid progenitors from congenital dyserythropoietic anaemia type I. Br J Haematol. 1998;103(3):825-30 [DOI] [PubMed] [Google Scholar]
- 48.Shamseddine A, Taher A, Jaafar H, Haidar JH, Nasr R, Arzoumanian V, et al. Interferon alpha is an effective therapy for congenital dyserythropoietic anaemia type I. Eur J Haematol. 2000;65(3):207-9 [DOI] [PubMed] [Google Scholar]
- 49.Persico M, Capasso M, Russo R, Persico E, Crocè L, Tiribelli C, Iolascon A. Elevated expression and polymorphisms of SOCS3 influence patient response to antiviral therapy in chronic hepatitis C. Gut. 2008;57(4):507-15 [DOI] [PubMed] [Google Scholar]
- 50.Iolascon A, Volinia S, Borriello A, Giordani L, Moretti A, Servedio V, et al. Genes transcriptionally modulated by interferon alpha2a correlate with the cytokine activity. Haematologica. 2004;89(9):1046-53 [PubMed] [Google Scholar]
- 51.Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ. Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update. Br J Haematol. 2012;156(1):37-49 [DOI] [PubMed] [Google Scholar]
- 52.Tamary H, Shalmon L, Shalev H, Halil A, Dobrushin D, Ashkenazi N, et al. Localization of the gene for congenital dyserythropoietic anemia type I to a <1-cM interval on chromosome 15q15.1-15.3. Am J Hum Genet. 1998;62(5):1062-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dgany O, Avidan N, Delaunay J, Krasnov T, Shalmon L, Shalev H, et al. Congenital dyserythropoietic anemia type I is caused by mutations in codanin-1. Am J Hum Genet. 2002;71(6):1467-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ru YX, Zhu XF, Yan WW, Gao JT, Schwarz K, Heimpel H. Congenital dyserythropoietic anemia in a Chinese family with a mutation of the CDAN1-gene. Ann Hematol. 2008;87(9):751-4 [DOI] [PubMed] [Google Scholar]
- 55.Noy-Lotan S, Dgany O, Lahmi R, Marcoux N, Krasnov T, Yissachar N, et al. Codanin-1, the protein encoded by the gene mutated in congenital dyserythropoietic anemia type I (CDAN1), is cell cycle-regulated. Haematologica. 2009;94(5):629-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tamary H, Marcoux N, Noy-Lotan S, Yaniv I, Dgany O. Codanin-1, the Product of the Gene Mutated In Congenital Dyserythropoietic Anemia Type I (CDA I), Binds to Histone Chaperone Asf1a and Inhibits Its Nucleosome Assembly Activity. Blood. 2010;116(21):442 [Google Scholar]
- 57.Alvarez F, Munoz F, Schilcher P, Imhof A, Almouzni G, Loyola A. Sequential establishment of marks on soluble histones H3 and H4. J Biol Chem. 2011;286(20):17714-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Campos EI, Fillingham J, Li G, Zheng H, Voigt P, Kuo WH, et al. The program for processing newly synthesized histones H3.1 and H4. Nat Struct Mol Biol. 2010;17(11):1343-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Koning L, Corpet A, Haber JE, Almouzni G. Histone chaperones: an escort network regulating histone traffic. Nat Struct Mol Biol. 2007;14(11):997-1007 [DOI] [PubMed] [Google Scholar]
- 60.Jasencakova Z, Scharf AN, Ask K, Corpet A, Imhof A, Almouzni G, Groth A. Replication stress interferes with histone recycling and predeposition marking of new histones. Mol Cell. 2010;37(5):736-43 [DOI] [PubMed] [Google Scholar]
- 61.Ask K, Jasencakova Z, Menard P, Feng Y, Almouzni G, Groth A. Codanin-1, mutated in the anaemic disease CDAI, regulates Asf1 function in S-phase histone supply. EMBO J. 2012;31(8):2013-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Renella R, Roberts NA, Brown JM, De Gobbi M, Bird LE, Hassanali T, et al. Codanin-1 mutations in congenital dyserythropoietic anemia type 1 affect HP1{alpha} localization in erythroblasts. Blood. 2011;117(25):6928-38 [DOI] [PubMed] [Google Scholar]
- 63.Iolascon A, Miraglia del Giudice E, Perrotta S, Granatiero M, Zelante L, Gasparini P. Exclusion of three candidate genes as determinants of congenital dyserythropoietic anemia type II (CDA-II). Blood. 1997;90(10):4197-200 [PubMed] [Google Scholar]
- 64.Gasparini P, Miraglia del Giudice E, Delaunay J, Totaro A, Granatiero M, Melchionda S, et al. Localization of the congenital dyserythropoietic anemia II locus to chromosome 20q11.2 by genomewide search. Am J Hum Genet. 1997;61(5):1112-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lanzara C, Ficarella R, Totaro A, Chen X, Roberto R, Perrotta S, et al. Congenital dyserythropoietic anemia type II: exclusion of seven candidate genes. Blood Cells Mol Dis. 2003;30(1):22-9 [DOI] [PubMed] [Google Scholar]
- 66.Denecke J, Marquardt T. Congenital dyserythropoietic anemia type II (CDAII/HEMPAS): where are we now? Biochim Biophys Acta. 2009;1792(9):915-20 [DOI] [PubMed] [Google Scholar]
- 67.Schwarz K, Iolascon A, Verissimo F, Trede NS, Horsley W, Chen W, et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet. 2009;41(8):936-40 [DOI] [PubMed] [Google Scholar]
- 68.Bianchi P, Fermo E, Vercellati C, Boschetti C, Barcellini W, Iurlo A, et al. Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum Mutat. 2009;30(9):1292-8 [DOI] [PubMed] [Google Scholar]
- 69.Fromme JC, Orci L, Schekman R. Coordination of COPII vesicle trafficking by Sec23. Trends Cell Biol. 2008;18(7):330-6 [DOI] [PubMed] [Google Scholar]
- 70.Lee MC, Miller EA, Goldberg J, Orci L, Schekman R. Bi-directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol. 2004;20:87-123 [DOI] [PubMed] [Google Scholar]
- 71.Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, et al. Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet. 2003;34(1):29-31 [DOI] [PubMed] [Google Scholar]
- 72.Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, Hur DJ, et al. Cranio-lenticulosutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet. 2006;38(10):1192-7 [DOI] [PubMed] [Google Scholar]
- 73.De Matteis MA, Luini A. Mendelian disorders of membrane trafficking. N Engl J Med. 2011;365(10):927-38 [DOI] [PubMed] [Google Scholar]
- 74.Iolascon A, Russo R, Esposito MR, Asci R, Piscopo C, Perrotta S, et al. Molecular analysis of 42 patients with congenital dyserythropoietic anemia type II: new mutations in the SEC23B gene and a search for a genotype-phenotype relationship. Haematologica. 2010;95(5):708-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Punzo F, Bertoli-Avella AM, Scianguetta S, Della Ragione F, Casale M, Ronzoni L, et al. Congenital dyserythropoietic anemia type II: molecular analysis and expression of the SEC23B gene. Orphanet J Rare Dis. 2011;6:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Russo R, Esposito MR, Asci R, Gambale A, Perrotta S, Ramenghi U, et al. Mutational spectrum in congenital dyserythropoietic anemia type II: identification of 19 novel variants in SEC23B gene. Am J Hematol. 2010;85(12):915-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Amir A, Dgany O, Krasnov T, Resnitzky P, Mor-Cohen R, Bennett M, et al. E109K is a SEC23B founder mutation among Israeli Moroccan Jewish patients with congenital dyserythropoietic anemia type II. Acta Haematol. 2011;125(4):202-7 [DOI] [PubMed] [Google Scholar]
- 78.Tao J, Zhu M, Wang H, Afelik S, Vasievich MP, Chen XW, et al. SEC23B is required for the maintenance of murine professional secretory tissues. Proc Natl Acad Sci USA. 2012;109(29):E2001-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ciovacco WA, Raskind WH, Kacena MA. Human phenotypes associated with GATA-1 mutations. Gene. 2008;427(1-2):1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hollanda LM, Lima CS, Cunha AF, Albuquerque DM, Vassallo J, Ozelo MC, et al. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet. 2006;38(7):807-12 [DOI] [PubMed] [Google Scholar]
- 81.Sankaran VG, Ghazvinian R, Do R, et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012;122(7):2439-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nichols KE, Crispino JD, Poncz M, White JG, Orkin SH, Maris JM, Weiss MJ. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet. 2000;24(3):266-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Del Vecchio GC, Giordani L, De Santis A, De Mattia D. Dyserythropoietic anemia and thrombocytopenia due to a novel mutation in GATA-1. Acta Haematol. 2005;114(2):113-6 [DOI] [PubMed] [Google Scholar]
- 84.Arnaud L, Saison C, Helias V, Lucien N, Steschenko D, Giarratana MC, et al. A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am J Hum Genet. 2010;87(5):721-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Drissen R, von Lindern M, Kolbus A, Driegen S, Steinlein P, Beug H, et al. The erythroid phenotype of EKLF-null mice: defects in hemoglobin metabolism and membrane stability. Mol Cell Biol. 2005; 25(12):5205-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nuez B, Michalovich D, Bygrave A, Ploemacher R, Grosveld F. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature. 1995;375(6529):316-8 [DOI] [PubMed] [Google Scholar]
- 87.Perkins AC, Sharpe AH, Orkin SH. Lethal beta-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. 1995;375(6529):318-22 [DOI] [PubMed] [Google Scholar]
- 88.Samkari A, Borzutzky A, Fermo E, Treaba DO, Dedeoglu F, Altura RA. A novel missense mutation in MVK associated with MK deficiency and dyserythropoietic anemia. Pediatrics. 2010;125(4):e964-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.