

Abstract
Diamond-Blackfan anemia is an autosomal dominant disease due to mutations in nine ribosomal protein encoding genes. Because most mutations are loss of function and detected by direct sequencing of coding exons, we reasoned that part of the approximately 50% mutation negative patients may have carried a copy number variant of ribosomal protein genes. As a proof of concept, we designed a multiplex ligation-dependent probe amplification assay targeted to screen the six genes that are most frequently mutated in Diamond-Blackfan anemia patients: RPS17, RPS19, RPS26, RPL5, RPL11, and RPL35A. Using this assay we showed that deletions represent approximately 20% of all mutations. The combination of sequencing and multiplex ligation-dependent probe amplification analysis of these six genes allows the genetic characterization of approximately 65% of patients, showing that Diamond-Blackfan anemia is indisputably a ribosomopathy.
Key words: Diamond-Blackfan anemia, multiplex ligation-dependent probe amplification, ribosomal protein gene deletion
Introduction
Diamond-Blackfan anemia (DBA, #MIM105650) is a rare congenital pure red cell aplasia characterized by normochromic macrocytic anemia, reticulocytopenia, and normocellular bone marrow with a selective deficiency of erythroid precursors. Forty percent of patients show physical malformations involving head, thumb, heart and urogenital system.1,2
Most cases are apparently sporadic, but the disease can be inherited with an autosomal dominant pattern. Penetrance is incomplete and expressivity widely variable, even in patients from the same family.2,3
The major DBA gene, ribosomal protein (RP) S194 is mutated in approximately 25% of patients.5 Heterozygous mutations in an increasing number of other genes encoding RPs of the small (RPS7, RPS10, RPS17, RPS24, RPS26) or the large (RPL5, RPL11, RPL35A) ribosomal subunits have been reported in DBA patients.5-11 However, point mutations in the coding regions of these genes are present in only 50% of DBA patients, as shown by a series of sequencing studies that also included a screening of almost all RP genes.5,8,10
This suggests that DBA has a further genetic heterogeneity due to non-RP genes or that there are also other mutations besides those detected by traditional sequencing. Polymerase chain reaction (PCR)-based methods used for conventional mutation detection fail to identify heterozygous deletions because the normal allele masks these mutations. In sequence analysis, apparent LOH (loss of heterozygosity) of one or more intragenic single nucleotide polymorphisms (SNPs) may be the only sign of a deletion in family studies.
Strategies that detect copy number variations are, therefore, necessary to complement sequencing. We previously used the multiplex ligation-dependent probe amplification (MLPA) technique to detect RPS19 gene deletions or duplications.12
The finding of partial or complete RP deletions/duplications in DBA patients using CGH array, SNP array or quantitative PCR has been recently reported.13-15
In order to better characterize the molecular defect in our cohort of Italian DBA patients who were negative for RP gene mutations by standard sequencing, we developed an MLPA synthetic probe set that allows the concurrent analysis of the six principal RP genes: RPS19, RPL5, RPL11, RPL35A, RPS17 and RPS26.
Design and Methods
Patients
Patients were selected from the Italian DBA Registry that includes 173 Italian subjects from 162 families. Diagnosis was reached following the criteria suggested by the DBA International Clinical Consensus (ICC) Consortium.2 All the patients were screened for mutations in ten genes.
RPS19 mutations were found in 44 of 162 (27%) unrelated DBA patients using a strategy that included sequencing and the MLPA technique. The sequencing analysis of RPS24, RPS26, RPL5, and RPL11, revealed mutations in 46 of 162 (28%) unrelated patients. No mutations were found in RPS10, RPS14, RPS16, RPS17, or RPL35A.
Seventy-two (45%) unrelated DBA patients showed no mutations in these genes and were included in this study. Informed consent was obtained from all patients and/or their legal guardians.
MLPA design and method
MLPA oligonucleotide probes were designed according to the “Designing synthetic MLPA probes” protocol v.8 (MRC-Holland, Amsterdam, The Netherlands) that uses 12 probes per assay. Each synthetic probe was composed of two 5′ and 3′ half-probes containing unique target-specific sequence and universal primer sequences 5′-GGGTTCCCTAAGGGTTGGA and 5′-TCTA-GATTGGATCTTGCTGGCAC on their 5′ and 3′ ends, respectively. Primers were designed in regions that did not include single nucleotide polymorphisms (SNPs) (http://genome.ucsc.edu/).
Because of the constraint of including 12 probes, we were forced to choose a strategy aimed at the screening of whole gene deletions or duplications. The probemix contained probes interrogating 1-3 exons per each RP gene (Online Supplementary Table S1, Figure 1A and B).
Figure 1.

(A) Schematic drawing showing the location of the MLPA (1-3 probes per gene) and realtime PCR probes (1-4 per gene). NCBI Genomic reference sequences: RPS17 NC_000015.9 (82821161..82824865), RPS19 NC_000019.9 (42363988..42375484), RPS26 NC_000012.11 (56435686..56438007), RPL5 NC_000001.10 (93297594..93307481), RPL11 NC_000001.10 (24018269..24022915), RPL35A NC_000003.11 (197677052..197682722). (B) MLPA peak corresponding to RP exons. (C) MLPA chromatogram of patients carrying RP deletion/duplication. Arrows indicate the MLPA peak corresponding to the deleted/duplicated exon. MLPA peaks: 1=RPL35A exon 1, 2=RPL5 exon 1, 3=RPS19 exon 6, 4=RPS19 exon 2, 5=RPS17 exon 4, 6=RPL5 exon 6, 7=RPS26 exon 4, 8=RPL35A exon 5, 9=RPS26 exon 1, 10=RPL11 exon 4, 11=RPS17 exon 1, 12=RPS19 exon 1. Pt: patient.
MLPA was performed on genomic DNA according to the manufacturer's instructions. We used the P200-A1 Human DNA reference kit that includes reference probes and MLPA control fragments (http://www.mrc-holland.com).
The PCR products were diluted 1:15 in water, 1 mL of this solution was mixed with 10 mL of HiDi formamide (ABI) and 0.2 μL of ROX500 size standard. The amplification products were separated by capillary electrophoresis on an ABI 3100 Avant Genetic Analyzer (Applied Biosystems, Warrington, UK). Genescan v3.7 software was used to analyze the runs and to retrieve peak intensities corresponding to each probe in the different samples and the integrated peak areas were exported to an Excel 2003 spreadsheet (Microsoft, CA, USA).
Normalization was obtained by dividing the peak area of each probe's amplification product by the combined peak areas of all probes (intra-normalization). We then performed an inter-sample normalization dividing these intra-normalized probe ratios of a sample by the average intra-normalized probe ratio of all reference samples (a mixture of DNA samples from healthy individuals).
Threshold values of loss and gain of a genetic material were set at 0.7 and 1.30, respectively. All samples showing evidence for deletions or duplications were replicated at least twice.
Characterization of large deletions and duplications
RPS17, RPS19, RPS26, RPL5, RPL11, RPL35A gene copy number was confirmed by quantitative duplex polymerase chain reaction (PCR) using primers designed with the Universal Probe Library (UPL) Technology (http://www.roche-applied-science.com/) (Figure 1A).
Reactions were carried out in triplicate in a total volume of 20 μL using 10 μL of 2X TaqMan Universal PCR Master mix (P/N4324018), 1 μL of 20X RNaseP assay as endogenous control (VIC dye, P/N 4316844), 0.2 μM of forward and reverse specific primers, 0.1 mM of specific UPL probe (6FAM, labeled, Roche Diagnostics) and 20 ng of genomic DNA (Online Supplementary Table S2).
PCR was performed using thermal cycling conditions suggested by the manufacturers in a 96-well clear optical reaction plate on an ABI-Prism7500 Fast instrument (Applied Biosystems).
Gene copy number was calculated using the comparative delta-delta Ct method: we normalized the ΔCt of the sample to the ΔCt of DNA of a healthy control pool.
Results and Discussion
MLPA analysis led to the identification of deletions in 14 of 72 patients (19.4%): 4 were in RPS17, 3 in RPS26 and RPL35A, 2 in RPL11 and one in RPS19 and RPL5. All probes showed a half-dosage suggesting whole gene deletion in heterozygosity. Patient 5 showed a partial RPS19 gene deletion, involving the last exon only (exon 6). All the deletions are expected to suppress the protein expression. In one patient, a duplication involving the whole RPL35A gene was identified (Figure 1C). All gene copy number variations were confirmed by real-time PCR analysis (Figure 2).
Figure 2.

Real-time PCR results.
Using MLPA, we verified the presence of copy number changes in the available parents, and found that in 3 cases they were inherited: one RPS17 deletion (patient 3), one RPL11 deletion (patient 10) and the RPL35A duplication (patient 15). In the remaining cases, deletions are likely to be de novo or at very low grade of somatic mosaicism (Table 1).
Table 1.
Clinical features of DBA RP deleted/duplicated patients.
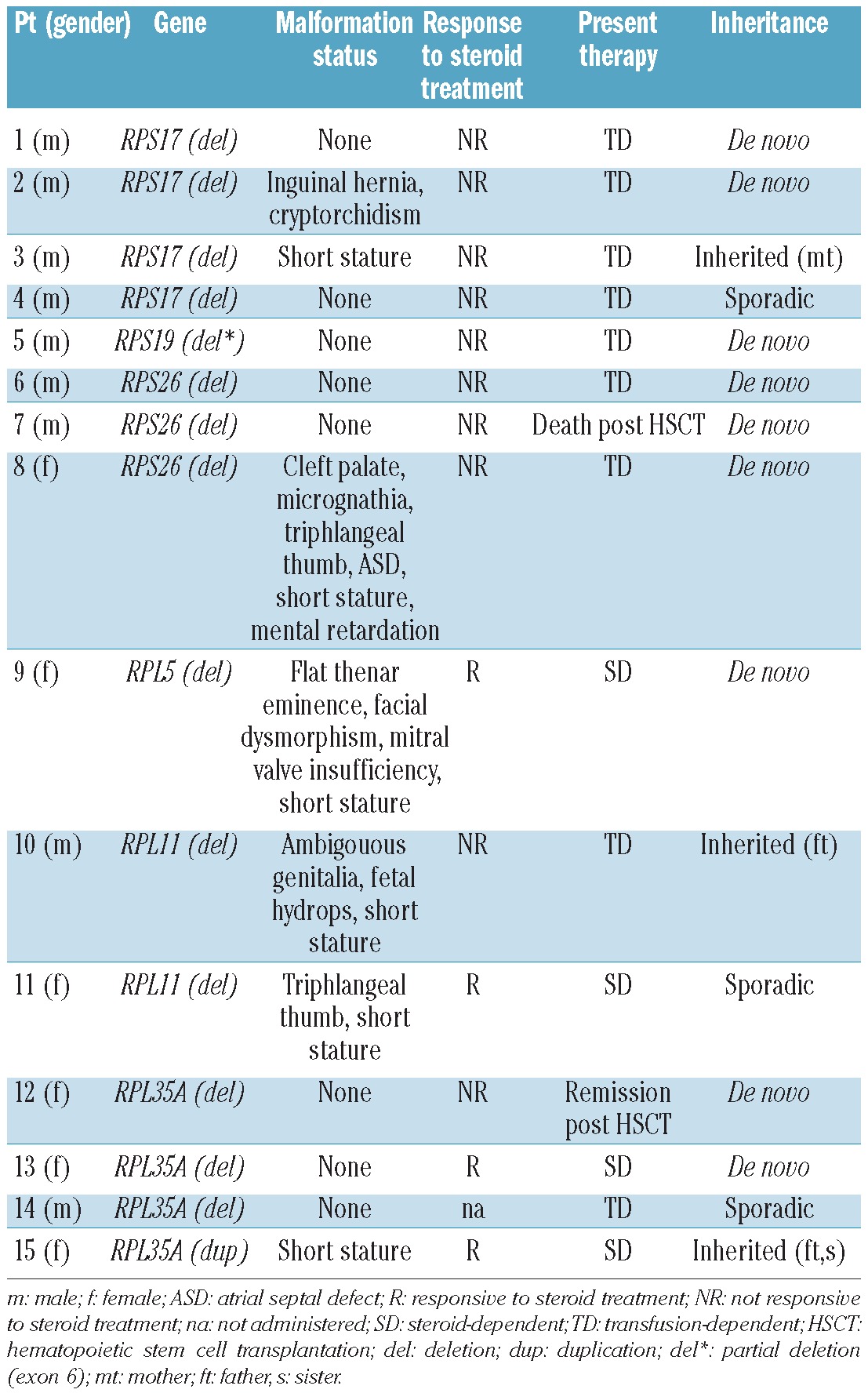
All patients with copy number changes (9 males, 6 females) displayed early-onset anemia (age at diagnosis 0-12 months). All 3 patients with RPL5 or RPL11 deletions, one patient with RPS17 and one with RPS26 deletion showed somatic malformations. We did not consider short stature as a somatic malformation because it was difficult to evaluate in the context of severe anemia, iron overload and chronic corticosteroid use. Specifically, the RPS26 deleted patient had multiple malformations and she was also mentally retarded. As suspected on the basis of the severe clinical features, this deletion involved also two genes localized upstream and downstream RPS26 (CDK2 and RNF41 genes) for at least an estimated deletion extension of ~240 kb.
A steroid dependence was present in 4 of 15 patients, and 9 of 15 never responded to steroid treatment and were, therefore, transfusion dependent. Two patients underwent hematopoietic stem cell transplantation: one achieved clinical remission and the other died due to transplant-related toxicity (Table 1).
As also shown in our previous work, we support the use of MLPA as an efficient and rapid assay for the analysis of gene dosage alterations.12 The main result of this study is the identification of a relatively high frequency of large rearrangements in the DBA genes analyzed.
Interestingly, we found for the first time whole-gene deletions of RPL11 and a complete RPL35A duplication. Although RP gene deletions are certainly causal, because all DBA mutations so far are LOF mutations, further studies are needed to assess whether the RPL35A duplication is a low-penetrance pathogenetic mutation or a silent variation (Table 1).
Mutation screening of the RP genes with a combination of sequencing and MLPA reached an overall rate of 65% (105 of 162). In our cohort, genomic deletions in six genes represent 18% of all mutations. Therefore, deletion screening should be taken into account in planning diagnostic strategies for mutations in DBA genes. Since we cannot exclude the presence of small exonic deletions or duplications, our future goal is to produce a multiplex assay able to screen all exons of the six main genes. In conclusion, we developed a reliable and robust MLPA analytical method for RP DBA genes that represents a useful complement to DNA sequencing analysis in patients affected by DBA.
Acknowledgments
part of the work mentioned in this article was supported by grants from Regione Piemonte (to UR), from Istituto Piemontese per la Ricerca sulla DBA (to ID and UR), from Banca del Piemonte (to UR), from Telethon (to ID) from ENERCA (to ID). We also thank the Daniella Maria Arturi Foundation for supporting communication among DBA researchers, the AIEOP centers (Italian Association for Pediatric Hematology and Oncology) and all the DBA families.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures: The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Lipton JM, Atsidaftos E, Zyskind I, Vlachos A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: an update from the Diamond Blackfan Anemia Registry. Pediatr Blood Cancer. 2006;46(5):558-64 [DOI] [PubMed] [Google Scholar]
- 2.Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, Ramenghi U, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142(6):859-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campagnoli MF, Garelli E, Quarello P, Carando A, Varotto S, Nobili B, et al. Molecular basis of Diamond-Blackfan anemia: new findings from the Italian registry and a review of the literature. Haematologica. 2004;89(4):480-9 [PubMed] [Google Scholar]
- 4.Draptchinskaia N, Gustavsson P, Andersson B, Pettersson M, Willig TN, Dianzani I, et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat Genet. 1999;21(2):169-75 [DOI] [PubMed] [Google Scholar]
- 5.Boria I, Garelli E, Gazda HT, Aspesi A, Quarello P, Pavesi E, et al. The ribosomal basis of Diamond-Blackfan Anemia: mutation and database update. Hum Mutat. 2010;31(12):1269-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gazda HT, Grabowska A, Merida-Long LB, Latawiec E, Schneider HE, Lipton JM, et al. Ribosomal protein S24 gene is mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2006;79(6):1110-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond-Blackfan anemia. Hum Mutat. 2007;28(12):1178-82 [DOI] [PubMed] [Google Scholar]
- 8.Doherty L, Sheen MR, Vlachos A, Choesmel V, O'Donohue MF, Clinton C, et al. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia. Am J Hum Genet. 2010;86(2):222-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farrar JE, Nater M, Caywood E, McDevitt MA, Kowalski J, Takemoto CM, et al. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood. 2008;112(5):1582-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gazda HT, Sheen MR, Vlachos A, Choesmel V, O'Donohue MF, Schneider H, et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet. 2008; 83(6):769-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quarello P, Garelli E, Carando A, Brusco A, Calabrese R, Dufour C, et al. Diamond-Blackfan anemia: genotype-phenotype correlations in Italian patients with RPL5 and RPL11 mutations. Haematologica. 2010;95(2):206-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quarello P, Garelli E, Brusco A, Carando A, Pappi P, Barberis M, et al. Multiplex ligation-dependent probe amplification enhances molecular diagnosis of Diamond-Blackfan anemia due to RPS19 deficiency. Haematologica. 2008;93(11):1748-50 [DOI] [PubMed] [Google Scholar]
- 13.Farrar JE, Vlachos A, Atsidaftos E, Carlson-Donohoe H, Markello TC, Arceci RJ, et al. Ribosomal protein gene deletions in Diamond-Blackfan anemia. Blood. 2011; 118(26):6943-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuramitsu M, Sato-Otsubo A, Morio T, Takagi M, Toki T, Terui K, et al. Extensive gene deletions in Japanese patients with Diamond-Blackfan anemia. Blood. 2012; 119(10):2376-84 [DOI] [PubMed] [Google Scholar]
- 15.Gazda H, Landowski M, Buros C, Vlachos A, Sieff CA, Newburger PE, Niewiadomska E, et al. Array Comparative Genomic Hybridization of Ribosomal Protein Genes In Diamond-Blackfan Anemia Patients; Evidence for Three New DBA Genes, RPS8, RPS14 and RPL15, with Large Deletion or Duplication. Blood.2010;116(Abstract 1007). [Google Scholar]