

Abstract
This paper describes the effects of a switch to velaglucerase alfa in a group of adult patients with type 1 Gaucher disease, all of whom had previously had their dose reduced as a consequence of the worldwide imiglucerase shortage. Thirty-two patients from two large European Gaucher centers switched to treatment with velaglucerase alfa after 1-8.5 months of dose reduction. The course of important Gaucher disease parameters was studied at four time points: one year before the shortage, just before the shortage, before a switch to velaglucerase and after up to one year of treatment with velaglucerase. These parameters included hemoglobin concentration, platelet count, plasma chitotriosidase activity in all patients, and spleen and liver volumes (as well as bone marrow fat fraction images) in 10 patients. Decreases in platelet counts as a result of reduced treatment with imiglucerase were quickly restored on treatment with velaglucerase alfa. Chitotriosidase activity declined overall after switching. Five out of 10 patients had an increase in liver volume of at least 10% after six months of velaglucerase treatment, which was reversible in 3. Most patients received infusions at home and no important side effects were observed. Velaglucerase alfa appears to be a safe and effective alternative for imiglucerase.
Key words: velaglucerase alfa, type 1 Gaucher disease, platelet count
Introduction
Gaucher disease (GD; OMIM #230800) is one of the most common lysosomal storage disorders. Clinical features are the result of a deficiency of the lysosomal enzyme glucocerebrosidase (EC 3.2.1.45). The disorder is classically divided in three subtypes of which type 1 is the most common. Type 1 can be distinguished from the other two types by the absence of typical neurological involvement as seen in the neuronopathic forms.1,2 The main manifestations of GD type 1 are cytopenia, hepatosplenomegaly and bone complications.1 Since the early 90s, therapy has become available in the form of infusions with macrophage-targeted enzyme: placental-derived alglucerase, later replaced by the recombinant form, imiglucerase (Genzyme Corp., MA, USA). Enzyme therapy is highly effective in restoring blood counts to normal, decreasing visceral disease and enlargement, and improving bone symptoms and quality of life.3,4
In June 2009, Genzyme identified a virus (vesivirus 2117) in one of their bioreactors and the subsequent shutdown of their production facility resulted in a severe shortage of imiglucerase worldwide.5 Due to the shortage, most patients were obliged to continue their infusions at a reduced dose or even had to interrupt infusions. Because GD is a heterogeneous disorder, the implications of these variable dose reductions were not easy to predict. To address issues concerning the shortage, a meeting of European stakeholders was organized in September 2009.5 The possible applications were discussed of alternative and emerging therapies such as the registered substrate inhibitor miglustat (Actelion Therapeutics) and two recombinant enzyme preparations: velaglucerase (h-GCB, Shire Human Genetic Therapies, MA, USA) and taliglucerase (pr-GCD, Protalix Biotherapeutics, Carmiel, Israel). Both enzyme preparations had completed phase III clinical trials but had yet to be registered at the time the shortage emerged.5
On the 26th of February 2010, the US Food and Drug Administration approved velaglucerase alfa for long-term enzyme therapy for patients with type 1 GD. This was followed by approval from the European Medicines Agency on the 26th of August 2010. Authorization was granted on the results of a Phase I/II clinical trial with a 5-year extension study and three recently completed Phase II trials. Results show that velaglucerase is safe and that its effectiveness is comparable to imiglucerase.6-10 As a consequence of the shortage, several centers switched patients to velaglucerase. This paper describes the clinical and biochemical effects of a switch to velaglucerase in a subgroup of adult patients with type 1 GD followed at two large European centers.
Design and Methods
Referral centers
In two large European referral centers, the Academic Medical Centre in Amsterdam (The Netherlands) and Addenbrooke's Hospital in Cambridge (UK), all adult patients who switched from treatment with imiglucerase to treatment with velaglucerase during the imiglucerase shortage were included in this retrospective analysis. Dutch patients received their first infusion with velaglucerase in the hospital and then switched to home treatment, while UK patients received at least two infusions in the hospital. Home infusions were either administered by a visiting nurse or by the patients themselves. There was a substantial variation in original enzyme replacement therapy (ERT) doses between patients as a result of individualized dosing regimens in which a patient's dose was based on disease severity and individual response. Patients started velaglucerase alfa essentially at dosages equal to their original imiglucerase dose, with the exception of patient n. 9 in whom the dose was doubled.
Information concerning data collection, haemoglobin level and platelet count, chitotriosidase activity, imaging and stastical analysis is available in the Online Supplementary Appendix.
Results and Discussion
Thirty-two adult patients switched to treatment with velaglucerase alfa: 12 patients from The Netherlands and 20 from the UK. Mean age was 52 years (range 28-72), Twenty patients were women and 17 of the 32 patients had been splenectomized.
Two patients in whom the period in which the dose was reduced was less than one month are not discussed further. Both were stable during this short period of dose reduction and remained stable after switching to velaglucerase.
Four patients were excluded from the analysis. In one patient, a laboratory error at the start of treatment with velaglucerase led to missing data. One patient on subjective grounds of lack of effectiveness asked to stop velaglucerase after six months of therapy; this patient returned to treatment with imiglucerase when this became available after a further six months without treatment.,
Finally, 2 patients suffered from complications unrelated to treatment with velaglucerase and this influenced the laboratory outcomes in such a way that effects of treatment could not be assessed. The first patient, a 60-year old woman was quadraplegic before treatment and developed severe pneumonia requiring ICU admission. The second patient, a 62-year old man, had a longstanding history of severe GD that had required a splenectomy and included multiple bone complications. A monoclonal gammopathy of undetermined significance (MGUS) had been identified at the age of 42 years and a few weeks after switching to velaglucerase, he progressed to multiple myeloma.
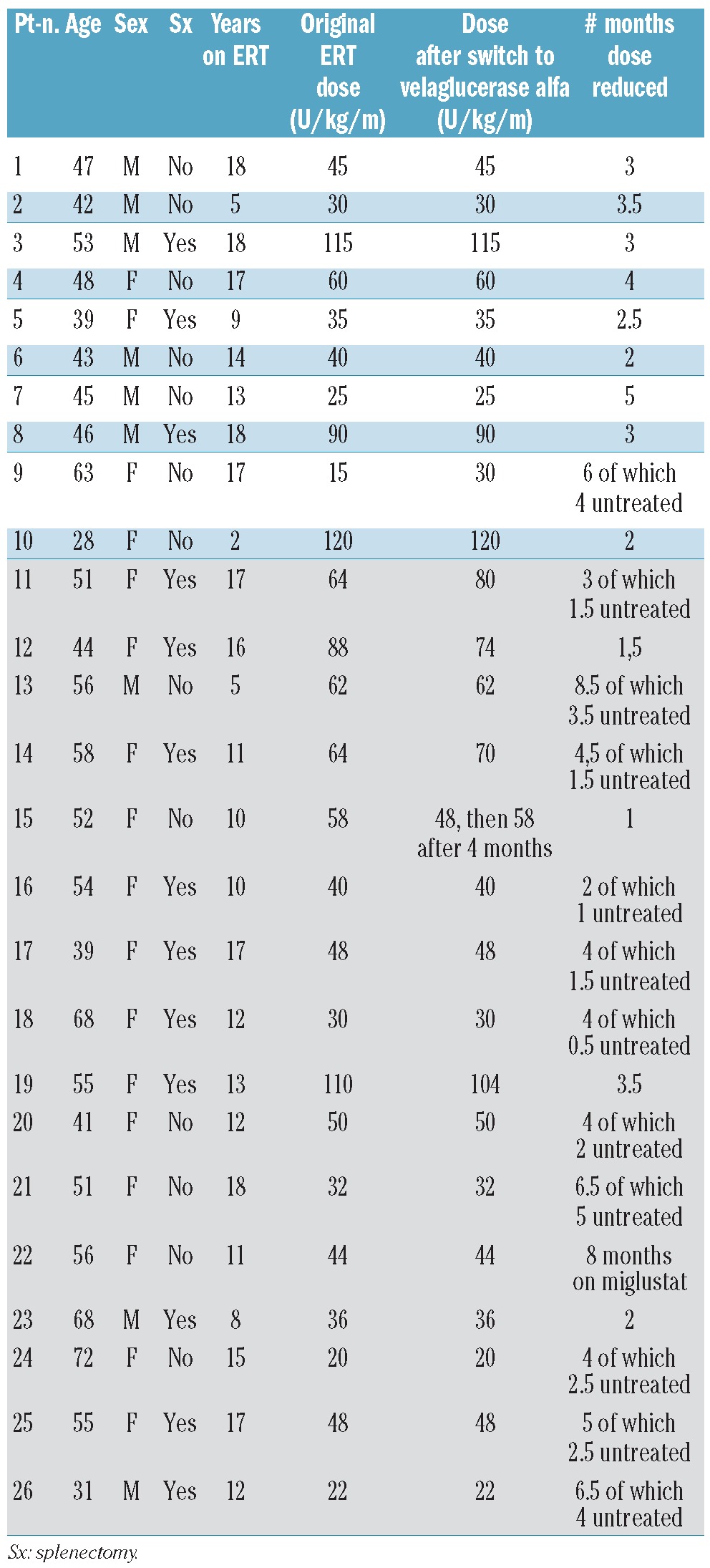
Table 1 provides an overview of the baseline characteristics in the 26 patients who underwent analysis. Median dose prior to reduction was 47 u/kg/month (range 15-120 u/kg/month). Patients had been dose reduced ranging from 1-8.5 months (median 3.7 months). Patients experienced multiple-staged dose reductions during shortage. One patient (Pt. n. 22) switched to miglustat. Median dose after switching was 47 u/kg/month (range 20-120 u/kg/month).
Table 1.
Baseline characteristics of the 26 patients who switched to treatment with velaglucerase. UK patients are depicted in the gray cells (nr. 11-26).
Hemoglobin concentration and platelet count
One patient (Pt. n. 21 in Table 1) showed an increase in hemoglobin concentration in the year before the shortage. This patient was treated for uterine fibroids causing iron deficiency anemia. Hemoglobin concentrations were stable during the shortage and also after switching to treatment with velaglucerase in all patients.
A deterioration in platelet counts was seen in 2 patients before the shortage (Pt. ns. 20, 22) and in 3 patients during the shortage (Pt. ns. 4, 13, 20). Treatment with velaglucerase resulted in stabilization (Pt. n. 20) or improvements (Pt. n. 4, 13, 22) in these patients. A favorable response was also observed in patients 6 and 10 who had shown no foregoing decline. In patient n. 9, who received a double dose of velaglucerase compared to the original imiglucerase dose, platelet count and hemoglobin levels were stable.
Four patients (Pt. ns. 4, 6, 15, 22) show a platelet count of less than 100×109/L at the moment of switching to treatment with velaglucerase, despite long-term treatment with ERT (ranging from 10-17 years). These patients did not differ from non-thrombocytopenic patients with regard to their original dose or years on ERT, but had generally larger spleens.
Chitotriosidase activity
One patient (Pt. n. 25) was deficient for chitotriosidase activity and in 2 patients follow up was incomplete (Pt. ns. 9, 10).
Chitotriosidase activity decreased in 3 patients in the year before the shortage (Pt. n. 13, 16, 23). Four patients showed an increase in chitotriosidase activity during the shortage (Pt. nd. 16, 20, 22, 26), while one patient demonstrated a decline (Pt. n. 7). Two patients exhibited a decrease in chitotriosidase activity of over 30% after switching to treatment with velaglucerase compared with values at the start of treatment with velaglucerase (Pt. ns. 22, 26).
Imaging
In 9 out of 10 Dutch patients, magnetic resonance imaging (MRI) was performed at the start of treatment with velaglucerase, and after six and 12 months. Five patients showed an increase in liver volume of 10% or more after six months of treatment with velaglucerase (Pt. ns. 1, 3, 6, 7, 10). In one patient (Pt. n. 7) this was accompanied by an increase in spleen volume of 11 %. In 3 patients, liver volume returned to baseline values after twelve months of treatment (Pt. ns. 1, 6, 10). In one of these patients (Pt. n. 6), spleen volume showed a decline of 10% or more after 12 months of treatment.
Quantitative chemical shift imaging (QCSI) was performed in 7 patients. No patient showed a relative change of 20% or more in QCSI values compared with baseline.
Adverse events
One patient had a suspected rib fracture while on a reduced dose (Pt. n. 11).
As mentioned previously, 2 patients were excluded from the analysis in this paper due to adverse events that were considered to be unrelated to their switch in treatment. A third patient (Pt. n. 24) suffered a right hemisphere cerebral infarct judged as ‘probably unrelated’ to the velaglucerase infusions. Finally, another patient (Pt. n. 22) suffered from back pain during the initial infusions with velaglucerase that was thought to be related. Back pain was present during the first 3 infusions, but resolved without sequelae at the fourth infusion.
Patient-related factors associated with outcome
There was no statistical difference between patients who did, compared with those who did not show progression during the shortage in age, treatment history, original ERT dose or duration of the shortage. Non-splenectomized patients had significantly lower platelet counts at each time point and showed a significantly greater increase in platelet count in response to treatment with velaglucerase.
Discussion
This study shows that switching to velaglucerase alfa at a dose that is equivalent to the imiglucerase dose before the shortage, is effective in most adult patients with Gaucher disease. Reductions in platelet count were generally quickly restored and chitotriosidase activity declined, after increasing during the shortage period. In addition to its effectiveness, velaglucerase was also safe and can be administered at home, as previously reported by Zimran et al.9,10 The first (two) infusions in the hospital were all uneventful, after which patients immediately switched to home treatment. No adverse events were reported during a total of approximately 600 infusions at home.
The reason for the observed temporary increases in liver volume at six months in 5 out of 10 patients is unclear. It may have been the result of a variation in imaging technique, although this rarely exceeds 10% when measurements are performed with the same technique. An increase in spleen volume was seen in only one patient, indicating a temporary worsening of the disease. For the other 4 patients, a more specific effect on the liver cannot be excluded, but the fact that during follow up no laboratory abnormalities or clinical symptoms occurred and liver volume had reduced to baseline values in 3 patients at 12 months makes a toxic effect unlikely. Interestingly, a recent paper by Elstein et al.10 reported positive effects on liver volume as measured by ultrasound of a switch to velaglucerase alfa in most patients with a mean decrease of 14%, but increases of 10% or more in 6 out of 19 patients after 12 months of treatment with velaglucerase alfa.
This study also clearly illustrates the rapid response of chitotriosidase to the effects of dose reduction during the shortage period, as well as its quick restoration after switching to velaglucerase at a dose equivalent to the original imiglucerase dose. This study combines the data of two different treatment centers and consequently different assays were used to measure chitotriosidase activity. As this may have influenced the variation in this marker, we have used the relative changes in chitotriosidase activity and established identical response patterns. The observations are in line with other studies showing that plasma chitotriosidase activity may increase in response to a treatment interruption11-15 or a switch from treatment with ERT to substrate reduction therapy (SRT).16
Previous studies have shown velaglucerase alfa to be effective and safe in treating therapy naïve patients6-10 or after a switch from stable imiglucerase dose to an equal velaglucerase dose6,7,10 Recently, Elstein et al.10 reported a socalled ‘booster-effect’ with velaglucerase alfa in patients who switched from long-term imiglucerase therapy. Their study concerned a different patient population as their patients who switched had either experienced an off-treatment period or an immediate switch at equal dose from imiglucerase to velaglucerase treatment. Our results agree with their findings that velaglucerase alfa is safe and effective. This study was unable to evaluate the possibility of a ‘booster-effect’ because all patients experienced a period in which they were treated with reduced dosages.
The current report is a retrospective analysis of available data and not the result of a research protocol. The decision to switch patients to treatment with velaglucerase was made on the basis of the disease history of individual patients (e.g. splenectomized patients, patients with persisting bone disease), as well as more practical considerations, such as patient's willingness to switch to a new form of treatment and receive their first infusion in a hospital setting. While the possibility of ascertainment bias can, therefore, not be excluded, the pattern of deterioration during the shortage for this cohort is comparable to papers by Zimran and Goldblatt in dose-reduced patients. More particularly, the experiences with velaglucerase reported here represent the real world experience of a substantial number of Gaucher patients who were regarded at risk for deterioration during the shortage and as such provide important information on the potential consequences of any future crises in enzyme supply.
Figure 1.

Hemoglobin concentration, platelet count and chitotriosidase activity, results one year prior to the last measurement before the shortage, results at the last measurement before the shortage, results at the last measurement before the switch to treatment with velaglucerase, results after one year of treatment with velaglucerase. Open circles are UK patients, cubes are Dutch patients. Hemoglobin concentration is shown separately for men and women. Platelet counts are shown for splenectomized patients and non-splenectomized patients separately because splenectomized patients usually have higher platelet counts. Splenectomized patients are represented by dotted lines.
It would be interesting to collect information on quality of life (QoL) as deterioration of disease manifestations as a consequence of the shortage might occur. However, neither QoL measures nor overall severity scores, such as the severity score index (SSI) or disease severity scoring system (DS3), were available for analysis.17,18
The prolonged restrictions in the supply of imiglucerase over more than two years highlight the critical importance of non-exclusivity in the licensing and approval of biological agents for the treatment of serious diseases. The advantage of having alternative therapies with the same mode of action cannot be overemphasized. A third enzyme preparation, taliglucerase alfa, has received marketing approval by the FDA and a Phase III clinical trial studying the efficacy of a new oral compound for substrate reduction therapy, eliglustat tartrate, is being conducted. To determine the optimal form of therapy for individual patients, it is necessary to be able to compare the safety and efficacy of these treatments to one another. In the aftermath of the imiglucerase shortage, several reports have been published describing the consequences of this shortage and each of these has contributed to our knowledge and understanding of its effects.12-14 However, with the relatively small numbers of patients hitherto described, current reports provide incomplete information about therapeutic outcomes. While this criticism may also apply to our experience so far, it is clear that by pooling such information we can generate an opportunity to learn which patients will benefit most from a specific treatment regimen. The ‘booster-effect’ reported by Elstein10 in some, but not all patients after a switch to velaglucerase alfa can only underscore this.
Since velaglucerase alfa appears to be as safe and effective as imiglucerase, the cost-effectiveness ratio may become a factor in the choice of treatment. It is hoped that with more competitive products on the market, the price will be reduced, without any differences between regions. This can improve access to treatment in non-Western countries, where numerous Gaucher disease patients currently remain untreated.
Supplementary Material
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures: The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Beutler E, Grabowski GA. Gaucher Disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D. eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001. p. 3636 [Google Scholar]
- 2.Biegstraaten M, van Schaik I, Aerts JM, Hollak CE. ‘Non-neuronopathic’ Gaucher disease reconsidered. Prevalence of neurological manifestations in a Dutch cohort of type I Gaucher disease patients and a systematic review of the literature. J Inherit Metab Dis. 2008;31(3):337-49 [DOI] [PubMed] [Google Scholar]
- 3.Barton NW, Furbish FS, Murray GJ, Garfield M, Brady RO. Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc Natl Acad Sci USA. 1990;87(5):1913-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Doppelt SH, Hill SC, et al. Replacement therapy for inherited enzyme deficiency--macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991;324(21):1464-70 [DOI] [PubMed] [Google Scholar]
- 5.Hollak CE, vom Dahl S, Aerts JM, Belmatoug N, Bembi B, Cohen Y, et al. Force Majeure: therapeutic measures in response to restricted supply of imiglucerase (Cerezyme) for patients with Gaucher disease. Blood Cells Mol Dis. 2010;44(1):41-7 [DOI] [PubMed] [Google Scholar]
- 6.US Food and Drug Administration (FDA) FDA labelling information-Vpriv (velaglucerase alfa). 2010
- 7.European Medicines Agency (EMA) European Public Assessment Report-Vpriv. 2010
- 8.Elstein D, Cohn GM, Wang N, Djordjevic M, Brutaru C, Zimran A. Early achievement and maintenance of the therapeutic goals using velaglucerase alfa in type 1 Gaucher disease. Blood Cells Mol Dis. 2011;46(1):119-23 [DOI] [PubMed] [Google Scholar]
- 9.Zimran A, Altarescu G, Philips M, Attias D, Jmoudiak M, Deeb M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood. 2010;115(23):4651-6 [DOI] [PubMed] [Google Scholar]
- 10.Elstein D, Altarescu G, Maayan H, Phillips M, Abrahamov A, Hadas-Halpern I, et al. Booster-effect with velaglucerase alfa in patients with Gaucher disease switched from long-term imiglucerase therapy: Early Access Program results from Jerusalem. Blood Cells Mol Dis. 2012;48(1):45-50 [DOI] [PubMed] [Google Scholar]
- 11.Czartoryska B, Tylki-Szymanska A, Lugowska A. Changes in serum chitotriosidase activity with cessation of replacement enzyme (cerebrosidase) administration in Gaucher disease. Clin Biochem. 2000;33(2):147-9 [DOI] [PubMed] [Google Scholar]
- 12.Giraldo P, Irun P, Alfonso P, Dalmau J, Fernandez-Galan MA, Figueredo A, et al. Evaluation of Spanish Gaucher disease patients after a 6-month imiglucerase shortage. Blood Cells Mol Dis. 2011;46(1):115-8 [DOI] [PubMed] [Google Scholar]
- 13.Goldblatt J, Fletcher JM, McGill J, Szer J, Wilson M. Enzyme replacement therapy “drug holiday”: results from an unexpected shortage of an orphan drug supply in Australia. Blood Cells Mol Dis. 2011;46(1):107-10 [DOI] [PubMed] [Google Scholar]
- 14.Zimran A, Altarescu G, Elstein D. Nonprecipitous changes upon withdrawal from imiglucerase for Gaucher disease because of a shortage in supply. Blood Cells Mol Dis. 2011;46(1):111-4 [DOI] [PubMed] [Google Scholar]
- 15.vom Dahl S, Poll LW, Haussinger D. Clinical monitoring after cessation of enzyme replacement therapy in M. Gaucher. Br J Haematol. 2001;113(4):1084-7 [DOI] [PubMed] [Google Scholar]
- 16.Giraldo P, Alfonso P, Atutxa K, Fernandez-Galan MA, Barez A, Franco R, et al. Realworld clinical experience with long-term miglustat maintenance therapy in type 1 Gaucher disease: the ZAGAL project. Haematologica. 2009;94(12):1771-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinreb NJ, Cappellini MD, Cox TM, Giannini EH, Grabowski GA, Hwu WL, et al. A validated disease severity scoring system for adults with type 1 Gaucher disease. Genet Med. 2010;12(1):44-51 [DOI] [PubMed] [Google Scholar]
- 18.Zimran A, Kay A, Gelbart T, Garver P, Thurston D, Saven A, et al. Gaucher disease. Clinical, laboratory, radiologic, and genetic features of 53 patients. Medicine (Baltimore) 1992;71(6):337-53 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.