

Abstract
Background
The RUNX1 (AML1) gene is a frequent mutational target in myelodysplastic syndromes and acute myeloid leukemia. Previous studies suggested that RUNX1 mutations may have pathological and prognostic implications.
Design and Methods
We screened 93 patients with cytogenetically normal acute myeloid leukemia for RUNX1 mutations by capillary sequencing of genomic DNA. Mutation status was then correlated with clinical data and gene expression profiles.
Results
We found that 15 out of 93 (16.1%) patients with cytogenetically normal acute myeloid leukemia had RUNX1 mutations. Seventy-three patients were enrolled in the AMLCG-99 trial and carried ten RUNX1 mutations (13.7%). Among these 73 patients RUNX1 mutations were significantly associated with older age, male sex, absence of NPM1 mutations and presence of MLL-partial tandem duplications. Moreover, RUNX1-mutated patients had a lower complete remission rate (30% versus 73% P=0.01), lower relapse-free survival rate (3-year relapse-free survival 0% versus 30.4%; P=0.002) and lower overall survival rate (3-year overall survival 0% versus 34.4%; P<0.001) than patients with wild-type RUNX1. RUNX1 mutations remained associated with shorter overall survival in a multivariate model including age and the European LeukemiaNet acute myeloid leukemia genetic classification as covariates. Patients with RUNX1 mutations showed a unique gene expression pattern with differential expression of 85 genes. The most prominently up-regulated genes in patients with RUNX1-mutated cytogenetically normal acute myeloid leukemia include lymphoid regulators such as HOP homeobox (HOPX), deoxynucleotidyltransferase (DNTT, terminal) and B-cell linker (BLNK), indicating lineage infidelity.
Conclusions
Our findings firmly establish that RUNX1 mutations are a marker of poor prognosis and provide insights into the pathogenesis of RUNX1 mutation-positive acute myeloid leukemia.
Key words: RUNX1, mutations, prognosis, acute myeloid leukemia
Introduction
The transcription factor RUNX1 is the fusion partner of RUNX1T1 (ETO) in the recurring t(8;21)(q22;q22) translocation present in 8-13% of adult patients with de novo acute myeloid leukemia (AML).1RUNX1 is a key regulator of hematopoiesis and is involved in hematopoietic stem cell emergence and regulation.2 The structure of the RUNX1 protein is characterized by an N-terminal RUNT domain, which mediates DNA-binding as well as an interaction with core-binding-factor beta (CBFB), and a C-terminal transactivation domain.3 Point mutations in RUNX1 were initially described in AML secondary to myelodysplastic syndromes, radiation exposure or chemotherapy, with the frequency in these settings being 8 to 10%.4 Subsequently, analyses of cytogenetically heterogeneous AML cohorts found RUNX1 mutations in 6-33% of patients.5-7 The mutational spectrum includes N-terminal missense mutations, affecting mostly the RUNT domain, and C-terminal truncating mutations, deleting the transactivation domain. Both missense and truncating mutations were reported not only to cause a loss of normal RUNX1 function, but also to act in a dominant negative fashion on the transactivation capacity of wild-type RUNX1.3 In minimally differentiated AML (AML M0) with RUNX1 mutations, deregulation of lymphoid genes was observed, indicating linage infidelity.8 Mutations in RUNX1 are associated with a poor prognosis in cohorts of patients with cytogenetically heterogeneous AML.5-7 We, therefore, studied the prognostic implications of RUNX1 mutations in a cohort homogeneous with regards to both cytogenetics (only cytogenetically normal AML; CN-AML) and treatment (all patients treated on the AMLCG-1999 trial). To learn more about the biology of RUNX1-mutated AML, we analyzed differential gene expression in cases of CN-AML with RUNX1 mutations versus cases with wild-type RUNX1.
Design and Methods
Patients
Ninety-three adult patients with CN-AML with available material and gene expression data were analyzed for RUNX1 mutations. Seventy-three were enrolled in the multicenter AMLCG-1999 trial of the German AML Cooperative Group, and an additional 20 CN-AML patients were not treated in the trial and could not, therefore, be evaluated for outcome, but were studied for RUNX1 mutation status and gene expression profiles. Diagnostics were performed centrally at the Laboratory for Leukemia Diagnostics, University of Munich (Germany), and included standard cytomorphology, cytogenetics, fluorescence in situ hybridization and testing for FLT3-internal tandem duplications (ITD), MLL-partial tandem duplications (PTD), and NPM1, CEBPA, NRAS, KIT, IDH1 (R132) and IDH2 (R140 and R172) mutations. The diagnosis of CN-AML was based on the analysis of at least 20 metaphases in more than 90% of patients, and on the analysis of at least ten metaphases in the remaining patients. All patients received intensive cytarabine-based double-induction and consolidation chemotherapy.9 The AMLCG-1999 trial is registered at ClinicalTrials.gov (NCT00266136) and was approved by the local institutional review boards of all participating centers. Informed consent was obtained from all patients in accordance with the Declaration of Helsinki.
Mutation screening
The entire open reading frame of RUNX1 (NM_001754.4) was analyzed from genomic DNA using polymerase chain reaction amplification with exon-spanning primers and bidirectional DNA sequencing on an ABI 3100 Avant instrument. Primer sequences are listed in Online Supplementary Table S1.
Microarray analyses
Bone marrow samples taken before treatment was commenced were analyzed using Affymetrix HG-U133 A/B oligonucleotide microarrays (Affymetrix, Santa Clara, CA, USA). Details regarding sample preparation, hybridization and image acquisition have been described previously.10,11 In order to combine individual oligonucleotide probes to probe sets and to annotate these probe sets to genes, we used custom chip definition files based on the GeneAnnot database (available online at http://www.xlab.unimo.it/GA_CDF/).12 In contrast to standard Affymetrix annotations, in these custom chip definition files each gene is represented by one single probe set comprising only probes that exclusively match the gene of interest. This approach reduces the multiple testing burden by decreasing the total number of probe sets, and potentially increases the specificity of the analyses by eliminating cross-hybridizing probes. Data were normalized using the variance stabilizing normalization algorithm13 and expression values were calculated by the median polish method.
Differentially expressed probe sets were identified by comparing RUNX1-mutated and RUNX1-wild type patients, using a permutation-based algorithm to adjust for multiple testing.14 Genes were called significant if their adjusted q value was <0.05 and the fold change between the two groups was >1.5 or <0.66.15 Microarray analyses were performed using the R software package, version 2.13.0.16
To identify functionally related sets of genes which are deregulated in RUNX1-mutated CN-AML, we performed gene set enrichment analysis. Gene sets were obtained from the curated ‘canonical pathways’ (c2:cp) collection of the Molecular Signatures Database (MSigDB version 3.0.; http://www.broadinstitute.org/gsea/msigdb/).17
Only gene sets containing between 15 and 200 individual genes (654 of the 880 total gene sets) were included in the analysis. Gene sets were considered significant at a false discovery rate (adjusted for gene set size and multiple testing) of q<0.10
Statistical analysis
Fisher's exact test was used to compare categorical clinical variables of the RUNX1-mutated and RUNX1-wild-type cohorts. For the continuous variables we used the Mann-Whitney U-test. The clinical endpoints of complete remission, non-responsive AML, relapse-free survival and overall survival were defined as reported previously.11,18 In brief, patients with more than 5% residual bone marrow blasts after induction treatment were judged to be nonresponders. Relapse-free survival was defined as time from the date of complete remission until relapse or death, regardless of cause. Overall survival was defined as time from study entry until death from any cause. Patients alive without an event were censored at the time of their last follow-up.
The prognostic impact of RUNX1 mutations was first evaluated according to the Kaplan-Meier method and the log-rank test. To adjust for important clinical and molecular prognostic variables, we derived a multivariate Cox model for overall survival with age as a continuous parameter (10-year difference), European LeukemiaNet (ELN) genetic group and RUNX1 mutational status as covariates.
Results
Patient's characteristics and clinical outcome
In a cohort of 93 adult CN-AML patients, 15 (16.1%) were found to carry RUNX1 mutations (Figure 1A, Table 1). Four patients carried several RUNX1 mutations. Among the 73 patients enrolled in the AMLCG-1999 trial, ten (13.7%) had RUNX1 mutations. The clinical and molecular characteristics of these 73 patients are listed in Table 2. Compared to wild-type RUNX1, RUNX1 mutations were associated with older age (P=0.001), male sex (P=0.005), higher lactate dehydrogenase levels (P=0.003) and a trend towards a lower white blood cell count (P=0.08).
Figure 1.

(A) Overview of mutations in RUNX1. Linear structure of the RUNX1 protein (NP_001745.2) includes the N-terminal RUNT domain and the C-terminal transcriptional activation domain (TAD). Amino acid (aa) changes resulting from mutations found in our cohort of CN-AML patients are detailed. The graph was generated using the software DOG 2.0.25 (B) Distribution of mutations in RUNX1 and eight additional genes in 93 CN-AML patients. Additional mutations are shown for patients with RUNX1 mutations (n=15) or wild-type RUNX1 (n=78). Seventy-three CN-AML patients were enrolled in the AMLCG-99 clinical trial (left panel). Another 20 CN-AML patients were not homogenously treated (right panel). Genes analyzed for mutations are indicated on the left side.
Table 1.
Molecular details of RUNX1 mutations in 94 CN-AML patients. Fifteen CN-AML patients carried RUNX1 mutations. Four out of these 15 patients had several RUNX1 mutations. Sequence variations in the cDNA and protein are indicated with reference to the longest isoform of RUNX1 (NM_001754.4). UPN: Unique Patient Number.
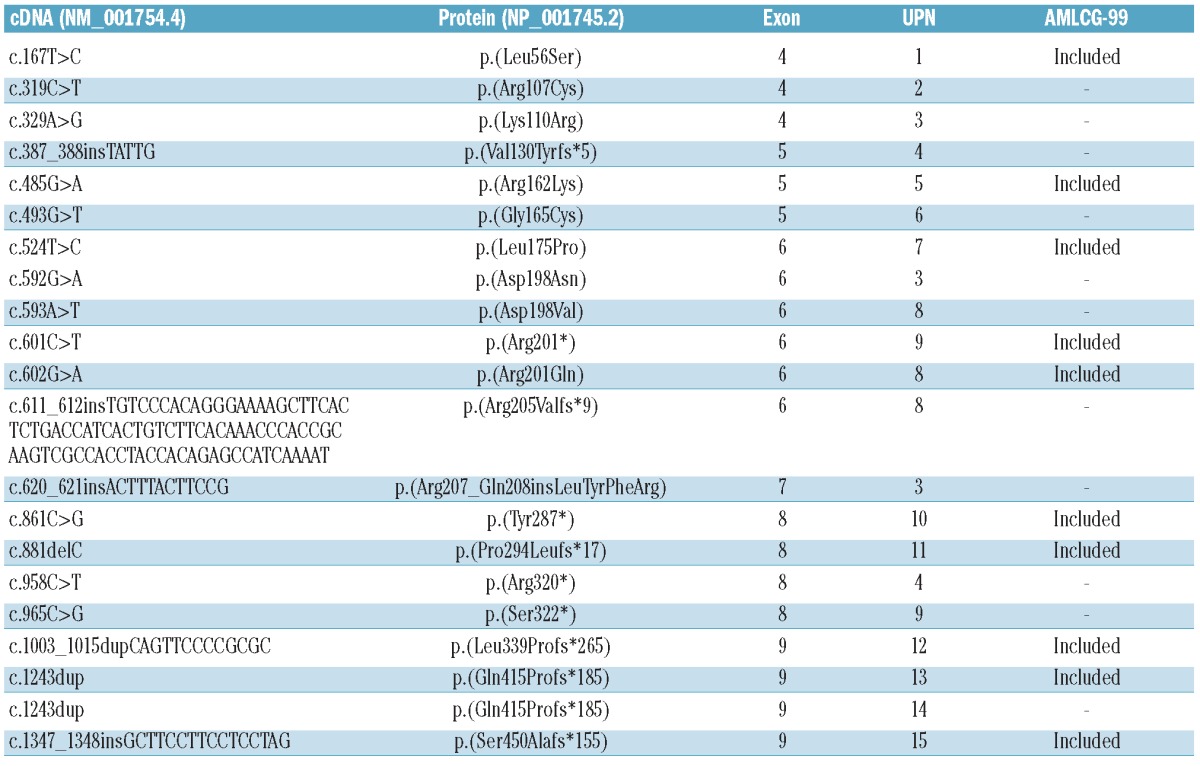
Table 2.
Patient's characteristics. Correlation of clinical characteristics and RUNX1 mutation status is indicated for 73 patients enrolled in the AMLCG-99.
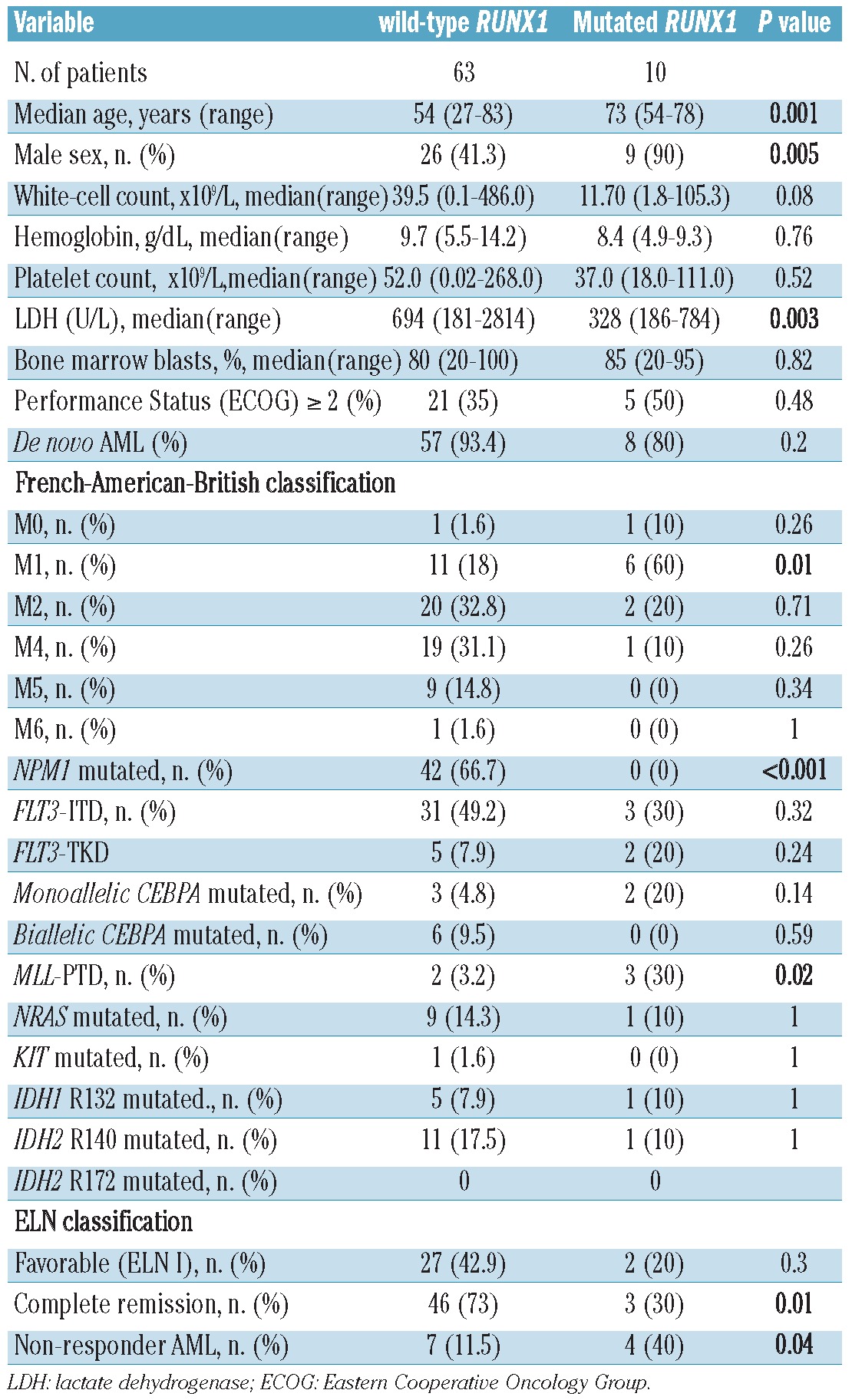
No patient with mutated RUNX1 carried a concurrent NPM1 mutation, while the frequency of NPM1 mutations in the RUNX1-wild-type group was 66.7% (P<0.001). MLL-PTD were more frequent among patients with RUNX1-mutations than among patients with wild-type RUNX1 (P=0.02). There was no significant association of RUNX1 mutations with FLT3-ITD, CEBPA, NRAS, KIT, IDH1 (R132), or IDH2 (R140/R172) (Figure 1B).
Only three of the ten (30%) RUNX1-mutated patients achieved a complete remission after intensive induction treatment, whereas the complete remission rate in the control group was 46/63 (73%; P=0.01). Four of ten (40%) RUNX1-mutated patients were primarily refractory to induction treatment, whereas this rate was only 11.5% in the control group (P=0.04).
The log-rank test identified RUNX1 mutations as a significant strong negative predictor of relapse-free survival (P=0.002) and overall survival (P<0.001). The 3-year relapse-free and overall survival rates for RUNX1 mutated patients were 0%, whereas they were 30.4% (relapse-free survival) and 34.4% (overall survival) for patients with wild-type RUNX1.
Kaplan-Meier estimates were calculated to display the negative prognostic influence of RUNX1 mutations on overall survival in all 73 study patients and the ELN intermediate I and elderly subgroups of patients with AML (Figure 2).
Figure 2.
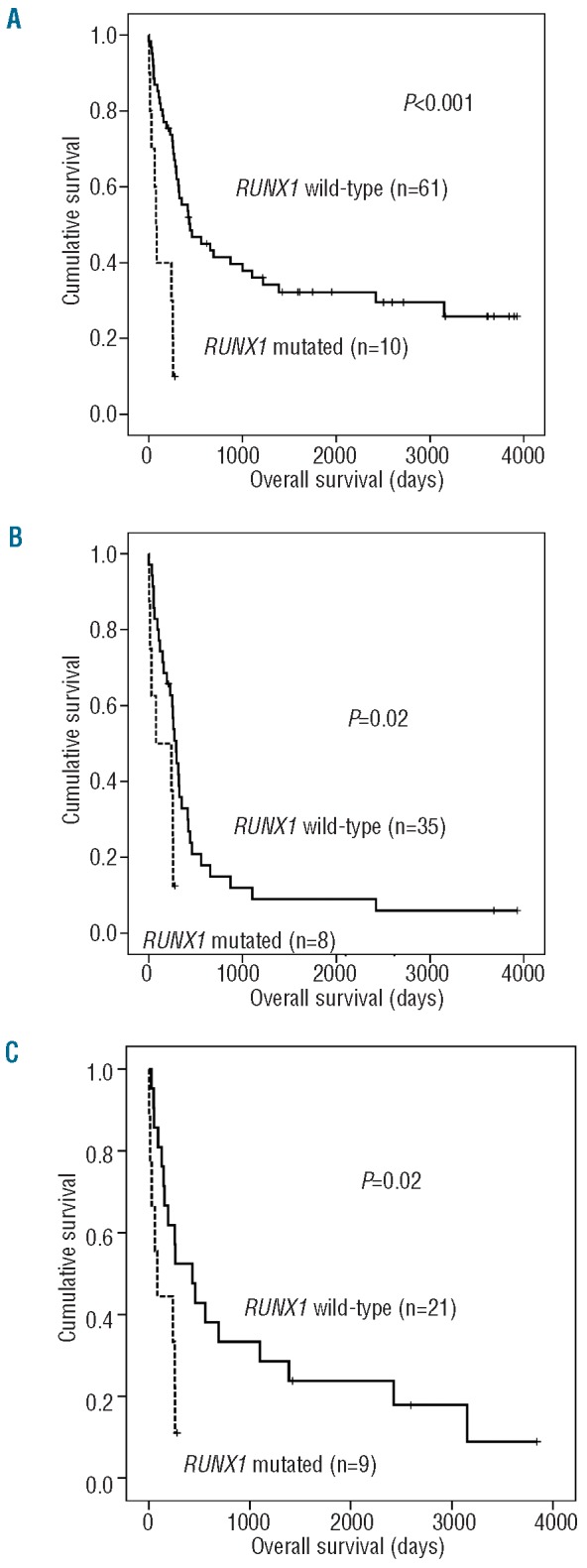
Influence of RUNX1 mutations on clinical outcome. Kaplan-Meier estimates for intensively treated CN-AML patients with or without RUNX1 mutations. The censored patient in the RUNX1 mutated group experienced a relapse and was then lost to follow up. (A) The median overall survival in RUNX1 mutated patients was 75 days compared to 442 days for patients with RUNX1 wild-type status. (B) For ELN Intermediate I patients (CN-AML with wild-type CEBPA and wild-type NPM1 and/or FLT3-ITD) median overall survival in RUNX1 mutated patients was 75 days compared to 293 days for patients without this mutation. (C) In elderly AML patients (≥ 60 years) the median overall survival for RUNX1-mutated patients was 86 days and 432 days for patients with wild-type RUNX1.
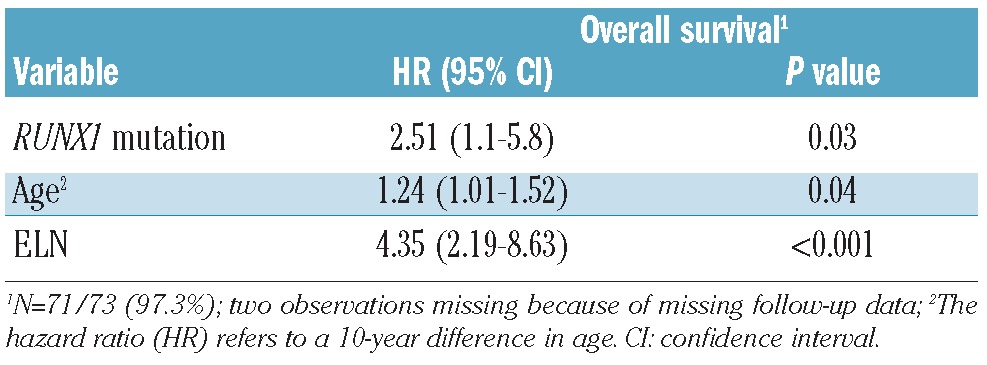
In a multivariate model for overall survival, including age (10-year differences), the ELN genetic groups and RUNX1 mutational status, all covariates were statistically significant parameters (Table 3).
Table 3.
Multivariate Cox regression model with covariates RUNX1 mutational status, age (10-year difference) and the European LeukemiaNet AML risk classification (ELN).
Identification of genes differentially expressed between RUNX1-mutated and RUNX1-wild type cases
To gain insights into the biology of RUNX1-mutated CN-AML, we derived RUNX1 mutation-associated gene expression signatures. Of note, RUNX1 mutations were found exclusively in CN-AML patients with wild-type NPM1, while over 60% of RUNX1-wild type patients carried NPM1 mutations which themselves are associated with a strong gene expression signature.19 To avoid confounding our analyses through the impact of NPM1 mutations, we only analyzed patients with wild-type NPM1. Comparing 15 RUNX1-mutated/NPM1-wild-type patients and 26 RUNX1-wild-type/NPM1-wild-type ones, we identified a set of 85 differentially expressed genes (Figure 3 and Online Supplementary Table S2). Sixty-nine genes showed higher expression in the RUNX1-mutated cases, while 16 genes were down-regulated. The most prominently up-regulated genes in RUNX1-mutated CN-AML include lymphoid regulators such as HOP homeobox (HOPX), deoxynucleotidyltransferase, terminal (DNTT) and B-cell linker (BLNK), indicating lineage infidelity.
Figure 3.
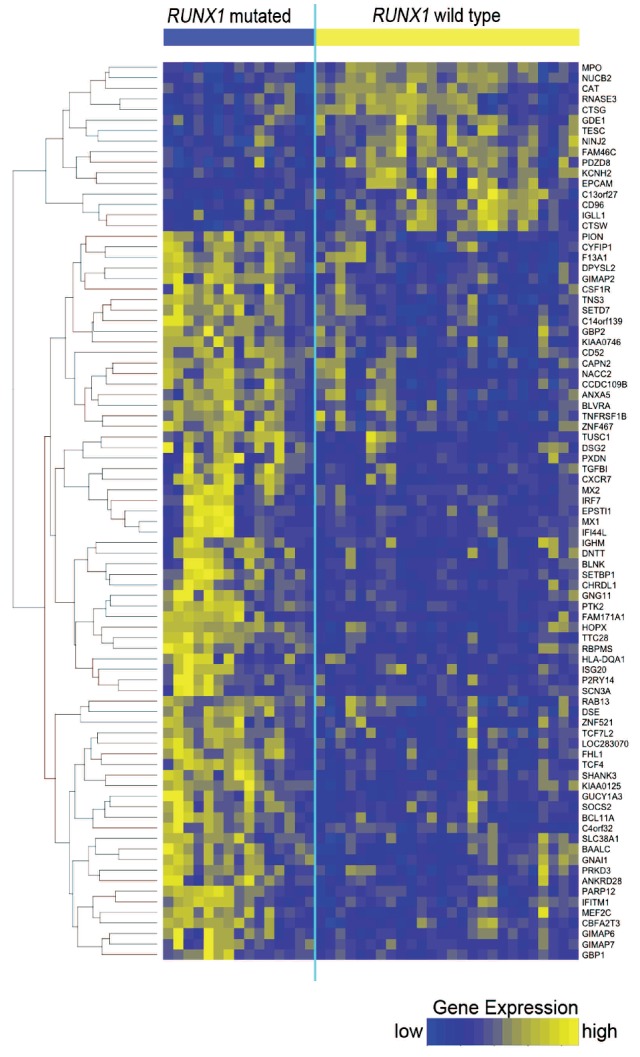
Heatmap of genes differentially expressed between RUNX1-mutated and RUNX1-wild-type patients. Each column represents one of 41 CN-AML patients, grouped according to RUNX1 mutation status, and each row represents one of 85 genes that were differentially expressed. Yellow indicates high and blue indicates low gene expression.
To investigate whether specific functional pathways are over-represented among the genes deregulated in RUNX1-mutated CN-AML, we performed gene set enrichment analysis. We found that 71 gene sets were significantly enriched in the RUNX1-mutated patients, while 51 gene sets were enriched in the RUNX1-wild-type patients (Online Supplementary Table S3). Gene sets up-regulated in RUNX1-mutated patients included signaling pathways highly expressed in lymphoid cells, such as the B-cell receptor (BCR) signaling pathway and the toll-like receptor 4 (TLR4) and NOTCH1 pathways (Online Supplementary Figure S1 A-C). Conversely, pathways related to DNA synthesis, DNA repair and DNA damage response pathways were down-regulated in RUNX1-mutated AML (Online Supplementary Figure S1 D-F).
Discussion
In our analysis of a homogeneous and uniformly treated cohort of CN-AML patients enrolled in the AMLCG-99 trial, we found RUNX1 mutations in 13.7% of patients. This frequency is similar to that reported by Tang et al.6 who found mutations in 13.9% of CN-AML, but higher than the frequency reported by Gaidzik et al. (3.9%)5 who only studied patients below the age of 60 years. We confirmed that RUNX1 mutations are more frequent in elderly, male patients and that these mutations are associated with some established genetic markers such as MLL-PTD (positively) and NPM1 mutations (negatively).5-7 In our cohort, NPM1 and RUNX1 mutations were mutually exclusive.
Our analyses revealed that RUNX1 mutations are a highly significant predictor of inferior outcomes, including a lower complete remission rate, shorter relapse-free survival and shorter overall survival. A high proportion of patients with RUNX1 mutations did not respond to intensive induction treatment, and only three out of ten (30%) achieved a complete remission. Even these three responders all died within 9.5 months (two in relapse; one in complete remission). RUNX1 mutations were a significant covariate in a multivariate model for overall survival including age (≥60 years), the ELN genetic classification and RUNX1 mutational status. These findings are consistent with those in a study by Tang et al.,6 who reported that RUNX1 mutations are associated with shorter relapse-free survival and overall survival in homogeneously treated CN-AML patients. In contrast, Gaidzik et al. previously reported a negative prognostic impact of RUNX1 mutations in a cytogenetically heterogeneous cohort, but found no significant impact on relapse-free or overall survival within the CN-AML subset.5 Of note, their study only included younger patients, suggesting that the negative impact of mutated RUNX1 might be age-related. Our findings strongly suggest that CN-AML patients with RUNX1 mutations do not benefit from standard treatment. Screening for RUNX1 mutations might, therefore, identify candidates for alternative treatment approaches. In summary, RUNX1 mutational status might be considered for inclusion in a revised version of the ELN AML risk classification, particularly for older patients.
In addition, we demonstrated that patients with RUNX1-mutated CN-AML have a distinct gene expression pattern characterized by differential expression of 85 genes. Twenty-six out of these 85 differentially expressed genes were previously reported to be deregulated in RUNX1 mutated AML M0 (minimally differentiated AML according to the French-American-British classification), indicating that the expression of these genes is very likely to be influenced by RUNX1 mutations.8 These 26 genes include the T-cell markers DNTT and BLNK, suggesting that mutations in the early hematopoietic stem cell regulator RUNX1 may disturb differentiation resulting in lineage infidelity in early progenitor cells. Since AML M0 is cytogenetically diverse, the study by Silva et al. is limited by the influence of multiple cytogenetic aberrations including complex karyotype and trisomy 13.8,20 Furthermore, Silva et al. limited their mutation screening to the RUNT-domain of RUNX1, which likely resulted in an underestimation of the RUNX1 mutation burden. In contrast, our study of RUNX1 mutations in CN-AML is not biased by the impact of cytogenetic aberrations on gene expression and an underestimation of RUNX1 mutations (the complete coding sequence of RUNX1 gene was sequenced). Gaidzik et al. also studied the association between RUNX1 mutational status and gene expression in a large cohort of AML patients including various cytogenetic subgroups.5 However, that cohort included only seven CN-AML patients with RUNX1 mutations and several different microarray platforms were used in their study, thus limiting the comparability with the study presented here.
High levels of expression of several genes that we found up-regulated in RUNX1-mutated CN-AML, namely DNTT, SETBP1, BAALC and PTK2, had previously been shown to be associated with adverse prognosis in AML.21-24
In summary, we provide further evidence for the unfavorable impact of RUNX1 mutations on clinical outcomes in a cytogenetically homogeneous and uniformly treated cohort of AML patients. Compared to previous studies on RUNX1 mutation-related gene expression signatures which were based on cytogenetically diverse cohorts of patients,5,8 our study specifically focused on CN-AML. Our findings reveal the unique biology of RUNX1-mutation-positive AML and may provide the basis for the development of novel diagnostic tools and therapies. Importantly, our findings that RUNX1 mutations in elderly CN-AML patients are associated with a dismal prognosis should aid in defining these patients as a group that could potentially benefit from alternative treatment strategies.
Supplementary Material
Acknowledgments:
the authors thank the participating centers of the AMLCG clinical trial.
Funding: this work was funded by a Deutsche Krebshilfe grant (109031) to PAG and SKB, a grant from the BMBF to SKB (NGFN Plus, PKL-01-GS0876-6) and a grant from the DFG to SKB (SFB 684, A6).
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures: The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Peterson LF, Zhang DE. The 8;21 translocation in leukemogenesis. Oncogene. 2004;23(24):4255-62 [DOI] [PubMed] [Google Scholar]
- 2.Swiers G, de Bruijn M, Speck NA. Hematopoietic stem cell emergence in the conceptus and the role of Runx1. Int J Dev Biol. 2010;54(6-7):1151-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harada Y, Harada H. Molecular pathways mediating MDS/AML with focus on AML1/RUNX1 point mutations. J Cell Physiol. 2009;220(1):16-20 [DOI] [PubMed] [Google Scholar]
- 4.Osato M. Point mutations in the RUNX1/AML1 gene: another actor in RUNX leukemia. Oncogene. 2004;23(24):4284-96 [DOI] [PubMed] [Google Scholar]
- 5.Gaidzik VI, Bullinger L, Schlenk RF, Zimmermann AS, Rock J, Paschka P, et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J Clin Oncol. 2011;29(10):1364-72 [DOI] [PubMed] [Google Scholar]
- 6.Tang JL, Hou HA, Chen CY, Liu CY, Chou WC, Tseng MH, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114(26):5352-61 [DOI] [PubMed] [Google Scholar]
- 7.Schnittger S, Dicker F, Kern W, Wendland N, Sundermann J, Alpermann T, et al. RUNX1 mutations are frequent in de novo AML with noncomplex karyotype and confer an unfavorable prognosis. Blood. 2011;117(8):2348-57 [DOI] [PubMed] [Google Scholar]
- 8.Silva FP, Swagemakers SM, Erpelinck-Verschueren C, Wouters BJ, Delwel R, Vrieling H, et al. Gene expression profiling of minimally differentiated acute myeloid leukemia: M0 is a distinct entity subdivided by RUNX1 mutation status. Blood. 2009;114(14):3001-7 [DOI] [PubMed] [Google Scholar]
- 9.Buchner T, Berdel WE, Schoch C, Haferlach T, Serve HL, Kienast J, et al. Double induction containing either two courses or one course of high-dose cytarabine plus mitoxantrone and postremission therapy by either autologous stem-cell transplantation or by prolonged maintenance for acute myeloid leukemia. J Clin Oncol. 2006;24(16):2480-9 [DOI] [PubMed] [Google Scholar]
- 10.Haferlach T, Kohlmann A, Schnittger S, Dugas M, Hiddemann W, Kern W, et al. Global approach to the diagnosis of leukemia using gene expression profiling. Blood. 2005;106(4):1189-98 [DOI] [PubMed] [Google Scholar]
- 11.Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112(10):4193-201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrari F, Bortoluzzi S, Coppe A, Sirota A, Safran M, Shmoish M, et al. Novel definition files for human GeneChips based on GeneAnnot. BMC Bioinformatics. 2007;8:446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huber W, von Heydebreck A, Sultmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics. 2002;18(Suppl 1):S96-104 [DOI] [PubMed] [Google Scholar]
- 14.Scheid S, Spang R. A stochastic downhill search algorithm for estimating the local false discovery rate. IEEE/ACM Trans Comput Biol Bioinform. 2004;1(3):98-108 [DOI] [PubMed] [Google Scholar]
- 15.Shi L, Jones WD, Jensen RV, Harris SC, Perkins RG, Goodsaid FM, et al. The balance of reproducibility, sensitivity, and specificity of lists of differentially expressed genes in microarray studies. BMC Bioinformatics. 2008;9(Suppl 9):S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.R_Development_Core_Team R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: 2006:{ISBN} 3-900051-07-0 [Google Scholar]
- 17.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krug U, Rollig C, Koschmieder A, Heinecke A, Sauerland MC, Schaich M, et al. Complete remission and early death after intensive chemotherapy in patients aged 60 years or older with acute myeloid leukaemia: a web-based application for prediction of outcomes. Lancet. 2010;376(9757):2000-8 [DOI] [PubMed] [Google Scholar]
- 19.Becker H, Marcucci G, Maharry K, Radmacher MD, Mrozek K, Margeson D, et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: a Cancer and Leukemia Group B study. J Clin Oncol. 2010;28(4):596-604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silva FP, Lind A, Brouwer-Mandema G, Valk PJ, Giphart-Gassler M. Trisomy 13 correlates with RUNX1 mutation and increased FLT3 expression in AML-M0 patients. Haematologica. 2007;92(8):1123-6 [DOI] [PubMed] [Google Scholar]
- 21.Venditti A, Del Poeta G, Buccisano F, Tamburini A, Cox-Froncillo MC, Aronica G, et al. Prognostic relevance of the expression of Tdt and CD7 in 335 cases of acute myeloid leukemia. Leukemia. 1998;12(7):1056-63 [DOI] [PubMed] [Google Scholar]
- 22.Cristobal I, Blanco FJ, Garcia-Orti L, Marcotegui N, Vicente C, Rifon J, et al. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood. 2010;115(3):615-25 [DOI] [PubMed] [Google Scholar]
- 23.Recher C, Ysebaert L, Beyne-Rauzy O, Mansat-De Mas V, Ruidavets JB, Cariven P, et al. Expression of focal adhesion kinase in acute myeloid leukemia is associated with enhanced blast migration, increased cellularity, and poor prognosis. Cancer Res. 2004;64(9):3191-7 [DOI] [PubMed] [Google Scholar]
- 24.Baldus CD, Tanner SM, Ruppert AS, Whitman SP, Archer KJ, Marcucci G, et al. BAALC expression predicts clinical outcome of de novo acute myeloid leukemia patients with normal cytogenetics: a Cancer and Leukemia Group B study. Blood. 2003;102(5):1613-8 [DOI] [PubMed] [Google Scholar]
- 25.Ren J, Wen L, Gao X, Jin C, Xue Y, Yao X. DOG 1.0: illustrator of protein domain structures. Cell Res. 2009;19(2):271-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.