

Abstract
Macroautophagy (autophagy herein) is a cellular catabolic mechanism activated in response to stress conditions including starvation, hypoxia and misfolded protein accumulation. Abnormalities in autophagy were associated with pathologies including cancer and neurodegenerative diseases. Hence, elucidation of the signaling pathways controlling autophagy is of utmost importance. Recently we and others described microRNAs (miRNAs) as novel and potent modulators of the autophagic activity. Here, we describe MIR181A (hsa-miR-181a-1) as a new autophagy-regulating miRNA. We showed that overexpression of MIR181A resulted in the attenuation of starvation- and rapamycin-induced autophagy in MCF-7, Huh-7 and K562 cells. Moreover, antagomir-mediated inactivation of endogenous miRNA activity stimulated autophagy. We identified ATG5 as an MIR181A target. Indeed, ATG5 cellular levels were decreased in cells upon MIR181A overexpression and increased following the introduction of antagomirs. More importantly, overexpression of ATG5 from a miRNA-insensitive cDNA construct rescued autophagic activity in the presence of MIR181A. We also showed that the ATG5 3′ UTR contained functional MIR181A responsive sequences sensitive to point mutations. Therefore, MIR181A is a novel and important regulator of autophagy and ATG5 is a rate-limiting miRNA target in this effect.
Keywords: macroautophagy, mammalian autophagy regulation, microRNA, hsa-miR-181a, ATG5, starvation, rapamycin, MTOR
Introduction
Autophagy is a highly conserved catabolic pathway of the cell, degrading long-lived proteins and damaged organelles.1 Autophagy is active at a basal level under normal conditions, but is rapidly upregulated following exposure to stress factors including nutrient/hormone deprivation, hypoxic stress, accumulation of misfolded proteins or bacterial invasion.2,3 Autophagy plays important roles in various organismal processes such as development and aging and abnormalities in autophagy lead to pathologies including neurodegenerative diseases and cancer.4-7
The hallmark of autophagy is the formation of double- or multimembrane vesicles in the cytosol called autophagosomes. These vesicles eventually fuse with lysosomes to form autolysosomes where cargo degradation occurs using lytic enzymes.1,8 There are several important protein complexes acting in concert during autophagosome formation including the ATG12–ATG5-ATG16L1 and ATG8 (or MAP1LC3, abbreviated in the text as LC3, in mammals) conjugation systems.9 In the first system, following a ubiquitination-like reaction involving E1- and E2-like enzymes ATG7 and ATG10, respectively, the ATG12 ubiquitin-like protein is covalently conjugated to the intermolecular lysine 130 of the ATG5 protein. Then, ATG12–ATG5 forms a larger complex with, and oligomerizes through, ATG16L1.9 In the second ubiquitination-like reaction, LC3 protein is covalently conjugated to the lipid PE (phosphatidylethanolamine) and this event is necessary for autophagic membrane elongation and vesicle completion.10 In addition to the activity of ATG7 (E1-like) and ATG3 (E2-like) enzymes, LC3-lipid conjugation requires the E3-like activity of ATG12–ATG5-ATG16L1 complex.11 To be primed for conjugation, a C-terminal glycine residue of LC3 has to be exposed following its cleavage by ATG4 proteins.12,13
While free LC3 (also called LC3-I) is soluble, its lipid-conjugated form (LC3-II) is associated with growing and complete autophagosomes. Hence, LC3 conjugation is used as a molecular marker of autophagosome generation and accumulation.12 A second autophagy marker is degradation of the autophagy receptor SQSTM1/p62, reflecting autolysosomal lytic activity and autophagic flux.14
Mechanisms regulating mammalian autophagy still need further investigation. Accumulating data indicate that, in addition to intracellular signaling mechanisms, miRNAs play an important and central role in autophagy regulation.15,16 miRNAs are ~22 nucleotide-long noncoding RNAs that are expressed endogenously in various organisms from plants to mammals.17 They control biological events by either triggering degradation of their target mRNAs, and/or through inhibition of their translation. They do so selectively, by recognizing specific sequences called miRNA-response elements (MREs), generally present in the 3′ UTR of target mRNAs.18
Recent reports provided evidence that under stress conditions, a number of miRNAs including MIR30A, MIR101, MIR130A, and MIR196 are capable of modulating autophagic activity by changing intracellular levels of key autophagy proteins.19-22 Indeed, as a result of an unbiased miRNA screen, we discovered that MIR376B regulated autophagy by directly targeting autophagy genes BECN1 (Beclin 1 gene) and ATG4C.23 Another miRNA that we discovered during this screen was MIR181A1/hsa-miR-181a-1/hsa-miR-213 (abbreviated as MIR181A herein). Here, we describe the role of this miRNA in the control of autophagy. We demonstrated that MIR181A blocked starvation- and rapamycin-induced autophagy in cancer cell lines. In light of our results, we provide evidence for a key autophagy protein, ATG5, as a rate-limiting and direct autophagy-related target of MIR181A.
Results
MIR181A blocked starvation-induced autophagy in MCF-7 cells
An unbiased screen using GFP-LC3 as a read-out and starvation as a stimulus in MCF-7 cells that we described recently,23 led us to identify MIR181A (miRbase accession number: MI0000289) as a novel microRNA potentially regulating autophagy. The MIR181 family consists of at least four members, namely MIR181A-D, and, they were shown to play important roles in various biological events including development, differentiation, hematopoiesis, immune modulation and muscle adaptation to exercise.24 MIR181A is widely expressed in several tissues including brain, thymus, and bone marrow and its levels were shown to change in response to stress.25-27
We first confirmed that MIR181A had an effect on autophagy. As shown in Figure 1A and B, overexpression of MIR181A significantly blocked starvation-induced GFP-LC3 dot accumulation. In line with these results, starvation-activated lipid conjugation of free LC3-I to the autophagic membrane-associated LC3-II was attenuated in the extracts of cells following MIR181A transfection (Fig. 1C) and degradation of the autophagy receptor SQSTM1 following starvation was less in miRNA-transfected cell extracts (Fig. 1D). These results introduced MIR181A as a new miRNA controlling autophagy.

Figure 1. Overexpression of MIR181A resulted in decreased autophagic activity in MCF-7 cells. (A) MIR181A blocked starvation-induced GFP-LC3 dot formation in MCF-7 cells. Cells were cotransfected with MIR181A or control construct (MIR-CNT) together with GFP-LC3 plasmid and autophagy was assessed under no starvation or starvation (2 h) conditions. White arrows indicate clusters of the GFP-LC3 dots in cells. (B) Quantitative analysis of the experiments in (A). MIR181A overexpression, but not control (MIR-CNT) overexpression, blocked starvation-induced autophagy (mean ± SD of independent experiments, n = 3, **p < 0.01. N.S., not significant). (C) MIR181A decreased starvation-induced conversion of LC3-I to LC3-II in MCF-7 cells. Immunoblot results of extracts from nonstarved (STV-) or starved (STV+) cells (n = 3). LC3-II/LC3-I densitometric ratios are marked. ACTB was used as a loading control. (D) MIR181A blocked starvation induced SQSTM1 degradation in MCF-7 cells (n = 3). ACTB was used as a loading control. SQSTM1/ACTB densitometric ratios were marked. (E) MIR181A blocked rapamycin-induced GFP-LC3 dot formation. Cells were cotransfected with GFP-LC3 plasmid and MIR181A or MIR-CNT, treated with DMSO (carrier) or rapamycin (2.5 µM, 24 h). (F) Quantitative analysis of the experiments in (E) (mean ± SD of independent experiments, n = 4, *p < 0.05. N.S., not significant). (G) MIR181A decreased LC3-I to LC3-II conversion stimulated by rapamycin (RAP) in MCF-7 cells (n = 2). LC3-II/LC3-I ratios are marked. (H) MIR181A blocked rapamycin-induced SQSTM1 degradation in MCF-7 cells (n = 2).
It should be noted that LC3-I migrated as a double band in some immunoblots. Since two unrelated antibodies recognized both bands, in this paper we considered both bands as LC3-I (Fig. S1 and see Materials and Methods for details).
MIR181A blocked rapamycin-induced autophagy in MCF-7 cells
We then checked whether MIR181A overexpression also had an effect on rapamycin-induced autophagy. Rapamycin stimulates autophagy through inhibition of MTOR (mammalian target of rapamycin).28 In line with starvation-related results, rapamycin-induced GFP-LC3 dot formation (Fig. 1E and F), LC3 lipidation (Fig. 1G) and SQSTM1 degradation (Fig. 1H) were decreased following overexpression of MIR181A. Therefore, we concluded that, in addition to its effects on starvation-induced autophagy, MIR181A also inhibited rapamycin-induced autophagic activity in MCF-7 cells.
Effect of MIR181A on autophagy was observed in two more cell lines
In order to check whether the miRNA effects that we observed in the MCF-7 breast cancer cell line were cell type-specific or not, we performed autophagy tests in two other cell types, Huh-7 cells (a human hepatocarcinoma cell line) and K562 cells (a human chronic myelocytic leukemia cell line). MIR181A overexpression in Huh-7 cells significantly attenuated starvation- and rapamycin-induced GFP-LC3 dot formation (Fig. 2A and B; Fig. 2E and F, respectively). Similar to the results in MCF-7 cells, overexpression of MIR181A resulted in a decrease in stimulus-activated LC3-I/LC3-II conversion (Fig. 2C and G) and SQSTM1 degradation (Fig. 2D and H) in Huh-7 cells. In line with these data, both LC3-I/LC3-II conversion and SQSTM1 degradation were attenuated in K562 cells overexpressing MIR181A but not the control miRNA (Fig. S2).

Figure 2. Effect of MI181A on autophagy is not cell-type dependent. (A) MIR181A blocked starvation-induced GFP-LC3 dot formation in Huh-7 cells. Cells were cotransfected with GFP-LC3 plasmid and MIR181A or MIR-CNT and autophagy was assessed under no starvation or starvation (4 h) conditions. (B) Quantitative analysis of the experiments in (A) (mean ± SD of independent experiments, n = 3, *p < 0.05. N.S., not significant). (C) MIR181A expression decreased starvation-induced LC3-I to LC3-II conversion in Huh-7 cells (n = 3). LC3-II/LC3-I ratios are marked. (D) MIR181A blocked starvation-induced SQSTM1 degradation in Huh-7 cells (n = 2). SQSTM1/ACTB ratios are marked. (E) MIR181A blocked rapamycin-induced GFP-LC3 dot formation. Autophagy was assessed following DMSO or rapamycin treatment (2.5 µM, 24 h). (F) Quantitative analysis of the experiments in (E) (mean ± SD of independent experiments, n = 3, **p < 0.01. N.S., not significant). (G) MIR181A resulted in decreased rapamycin-induced conversion of LC3-I to LC3-II in Huh-7 cells (n = 2). RAP, rapamycin. LC3-II/LC3-I ratios are marked. (H) MIR181A blocked rapamycin-induced SQSTM1 degradation in Huh-7 cells (n = 2). SQSTM1/ACTB ratios are marked.
These results showed that MIR181A blocked autophagy in at least three cell lines originating from different tissues. Therefore, the effect of MIR181A on autophagy might be cell-type independent.
Tests with lysosomal protease inhibitors
To confirm that MIR181A inhibited autophagy-induced LC3 turnover and SQSTM1 degradation, and therefore autophagic vesicle flux, we performed similar autophagy tests in the presence or absence of lysosomal protease inhibitors E64d and pepstatin A. Indeed, addition of the lysosomal inhibitors led to a prominent accumulation of LC3-II and SQSTM1 in MIR-CNT transfected cells, pointing to the presence of a normal autophagic flux under these conditions. Yet, since MIR181A blocked autophagic vesicle generation, inhibitor-related accumulation of LC3-II and SQSTM1 during starvation was relatively less in MCF-7 cells overexpressing MIR181A compared with controls (Fig. S3A and S3B). Similar results were obtained in Huh-7 cells following autophagy stimulation by starvation (Fig. S3E and S3F). Rapamycin stimulus also led to similar results in both cell types (Fig. S3C and S3D; Fig. S3G and S3H).
To further confirm these results, we also followed the appearance of free GFP in GFP-LC3 transfected cells in the presence or absence of lysosomal hydrolase inhibitors. It was previously reported that free GFP fragments generated from the lysosomal cleavage of GFP-LC3 accumulated best during rapamycin treatment, and under these conditions for stimulating autophagy, addition of late-stage autophagy inhibitors such as E64d/pepstatin A further increased free GFP levels.29,30 As shown in Figure S4, following rapamycin treatment, a free GFP band appeared on the blots. Addition of E64d/pepstatin A resulted in a further increase in free GFP band intensity in controls but not in MIR181A-overexpressing cells. Hence, attenuation of autophagic activity by MIR181A led to a lower amount of GFP-LC3 degradation and free GFP accumulation in cell expressing the miRNA.
Effect of antagomir-mediated inhibition of endogenous MIR181A on autophagy
Antagomirs, or anti-miRNAs, counteract miRNA effects by specifically inhibiting endogenous mature miRNAs. To test the effects of endogenous MIR181A inhibition on autophagy, we transfected cells with MIR181A-specific antagomirs (Ant-181a) or control antagomirs (CNT-Ant) and analyzed autophagy under nonstarved or starved conditions. We observed that starvation-dependent GFP-LC3 dot formation was modestly but significantly increased (Fig. 3A and B) and LC3-I to LC3-II conversion was stimulated (Fig. 3C), hence autophagy was accelerated in cells transfected with Ant-181a but not with control antagomirs. Moreover, SQSTM1 degradation following starvation was more prominent after Ant-181a transfection compared with controls (Fig. 3D). Therefore, inhibition of endogenous MIR181A using antagomirs led to a further stimulation of the autophagic activity during starvation, indicating that endogenous MIR181A contributes to the limitation of stress-activated autophagic responses in cells.

Figure 3. Inhibition of endogenous MIR181A using antagomirs (Ant-181a) stimulated autophagic activity. (A) Ant-181a enhanced starvation-induced GFP-LC3 dot formation. CNT-Ant, control antagomirs. (B) Quantitative analysis of the experiments in (A). NO STV, No starvation. STV, starvation for 20 min. (mean ± SD of independent experiments, n = 3, *p < 0.05, **p < 0.01). (C) Ant-181a, but not CNT-Ant stimulated starvation (STV, 4 h)-stimulated LC3-I to LC3-II conversion in MCF-7 cells (n = 4). LC3-II/LC3-I densitometric ratios are marked. (D) Ant-181a, but not CNT-Ant resulted in the stimulation of SQSTM1 protein degradation following starvation (4 h) in MCF-7 cells (n = 3). SQSTM1/ACTB ratios are marked.
Autophagy-related target of MIR181A
To unravel the mechanism of autophagy inhibition by MIR181A, we searched for autophagy genes containing potential MIR181A MREs in their 3′ UTRs using publicly available bioinformatics tools Microcosm Targets and TargetScan. ATG5 (GenBank accession number: NM_004849.2, 1177–1200) was identified as a MIR181A target by both bioinformatics tools. The predicted interaction between MIR181A and the ATG5 3′ UTR was shown in Figure 4A. To confirm the bioinformatics-based predictions, we performed immunoblot analysis in control or MIR181A transfected cell extracts using an ATG5-specific antibody. Indeed, ATG5 protein levels were decreased in MCF-7 cells that were overexpressing MIR181A under both fed and starved conditions (Fig. 4B). Similarly, ATG5 protein levels were decreased in MIR181A transfected Huh-7 (Fig. S5A) and K562 cells (Fig. S5B). miRNA expression also affected target transcript levels in cells. A decrease in ATG5 mRNA levels was observed by qPCR in cells following transfection with MIR181A but not with MIR-CNT (Fig. 4C). Conversely, introduction of the antagomir Ant-181a (blocking endogenous MIR181A), but not control antagomirs, resulted in an increase in ATG5 protein levels in cells (Fig. 4D).
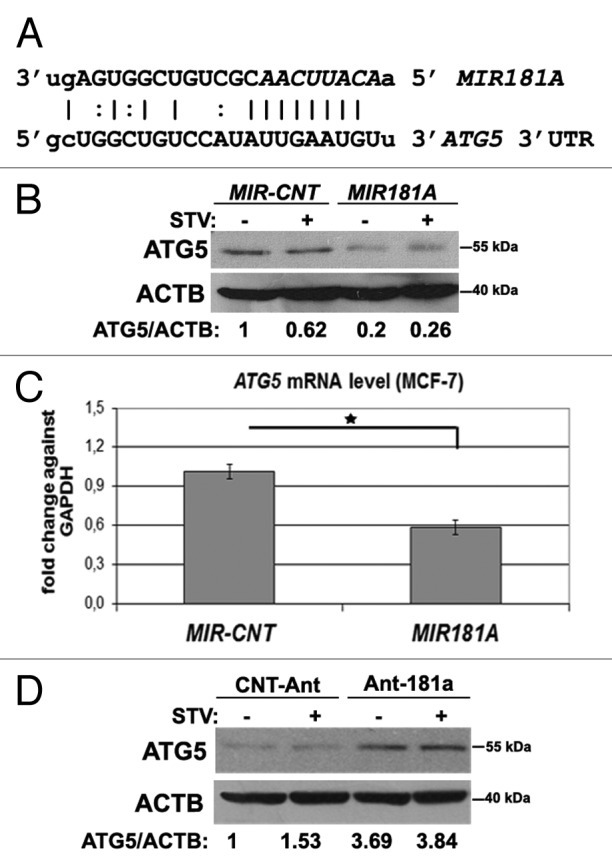
Figure 4.MIR181A affected ATG5 levels in MCF-7 cells. (A) MIR181A target sequence in the 3′ UTR of the ATG5 mRNA. The MIR181A seed sequence is marked in italics. (B) ATG5 protein levels were decreased following MIR181A overexpression in MCF-7 cells. Immunoblots of MIR-CNT or MIR181A transfected cells that were nonstarved (STV-) or starved (STV+) (n = 3). ACTB was used as a loading control. ATG5/ACTB band densitometric ratios are shown. (C) Quantitative PCR (qPCR) analysis of ATG5 mRNA levels in control (MIR-CNT) or MIR181A transfected MCF-7 cells (mean ± SD of independent experiments, n = 3, *p < 0.05). Data were normalized using GAPDH mRNA. (D) ATG5 protein levels were increased following antagomir-181a (Ant-181a) transfection. CNT-Ant, control antagomirs (n = 2). ATG5/ACTB ratios are shown.
To further validate that ATG5 downregulation was responsible for the autophagy-related effects of MIR181A, we performed “rescue experiments.” In these experiments, ATG5 protein was overexpressed from a plasmid lacking the MIR181A response element; it was therefore resistant to miRNA-mediated downregulation. Hence, although MIR181A overexpression decreased endogenous ATG5 protein levels significantly, cells cotransfected with the miRNA and the ATG5 plasmid possessed near physiological (approx. 1.36x) levels of total ATG5 protein (Fig. 5A). Under these conditions, the MIR181A-mediated suppression of autophagy observed during starvation-induced autophagy was reversed upon co-expression of the ATG5 protein (Fig. 5B and C). In other words, introduction of the ATG5 protein was sufficient to bring autophagy back to normal levels even in the presence of the miRNA. These results demonstrated that ATG5 was the rate-limiting target of MIR181A for autophagy inhibition.
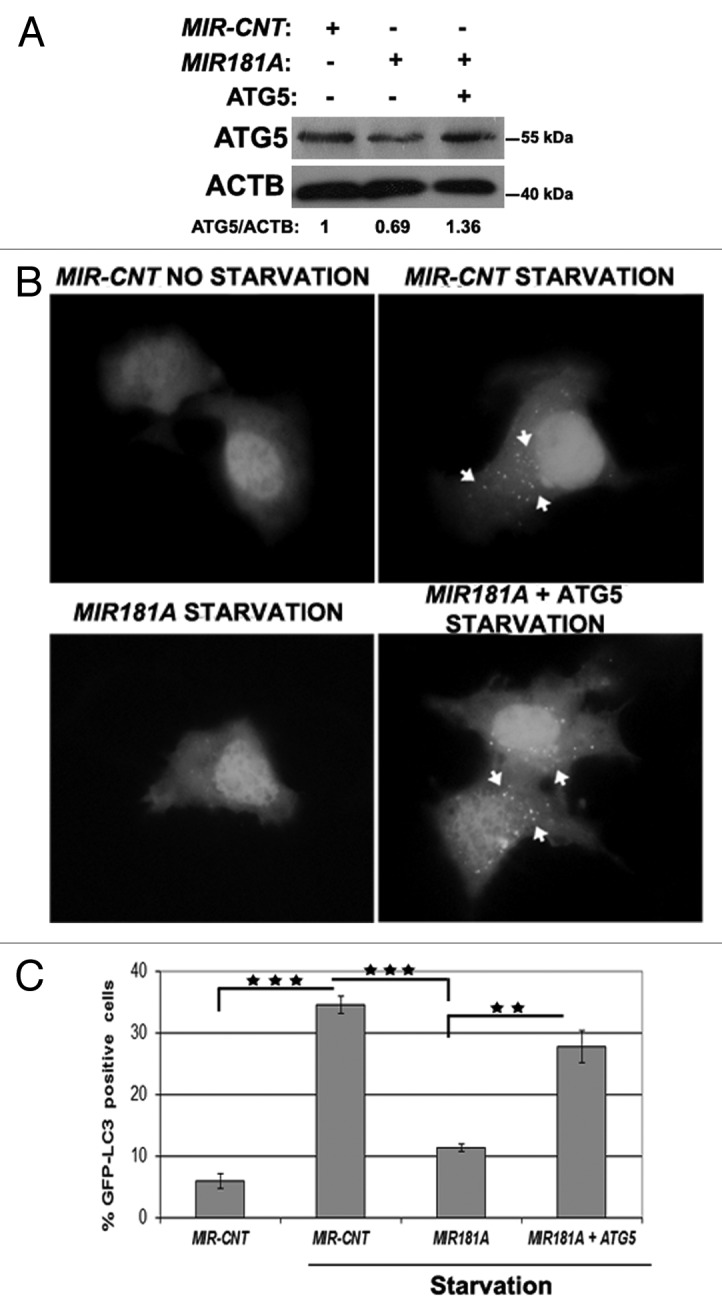
Figure 5. ATG5 overexpression rescued cells from MIR181A-mediated autophagy inhibition. MCF-7 cells were cotransfected with MIR181A or MIR-CNT and an ATG5 expression plasmid lacking the MIR181A target region. Autophagy was evaluated. (A) Immunoblot analysis of showing ATG5 and ACTB protein levels following indicated transfections (n = 2). ATG5/ACTB ratios are shown. (B) GFP-LC3 dot formation before or after starvation (2 h). ATG5, MIR181A-insensitive ATG5 expression plasmid. (C) Quantitative analysis of GFP-LC3 dot formation (mean ± SD of independent experiments, n = 3, **p < 0.01 and ***p < 0.001).
Other potential autophagy-related targets
BCL2, an antiapoptotic member of the BCL2 family that was involved in autophagy regulation through sequestration of the autophagy protein BECN1, is among the known targets of MIR181A.31-34 In our system, overexpression of the miRNA did not significantly affect the levels of BCL2 protein under nonstarved or starved conditions in MCF-7 cells (Fig. S6A), whereas, in line with other reports,35,36 there was no detectable BCL2 protein expression in Huh-7 cells (Fig. S6B). Therefore, we think that BCL2 regulation by MIR181A did not contribute to the autophagy modulatory effects described here.
We also checked whether there are other autophagy-related targets of MIR181A. Since MIR181A could block MTOR inhibitor rapamycin-induced autophagy, and ATG5 is downstream of MTOR in the autophagy pathway, we wondered whether the upstream MTOR pathway would be affected by the overexpression of this miRNA. To check the effect of MIR181A on MTOR activity, we analyzed the phosphorylation status of an MTOR target protein, RPS6KB/p70S6K. Although under basal/fed condition overexpression of MIR181A led to an unexpected increase in RPS6KB phosphorylation, this difference between MIR181A and control transfected cells vanished following autophagy activation by starvation (Fig. S7). Thus, although it is possible that MIR181A-induced MTOR-activation could contribute to its autophagy-blocking effects, miRNA overexpression could not overcome starvation-induced MTOR inhibition in our hands.
We also checked the effect of MIR181A overexpression on previously identified autophagy-related miRNA targets.23 We found that MIR181A overexpression had no significant effect on the levels of the MIR30A and 376B target BECN1 protein, or the MIR376B target ATG4C protein (Fig. S8A and S8B).
Using the bioinformatics tools mentioned above, we next analyzed ATG5 and ATG12 mRNA 3′ UTRs for MIR376B-specific MREs. The ATG12 3′ UTR but not that of ATG5 contained a potential MIR376B response element (Fig. S8C). In qPCR tests, we observed no change in ATG12 transcript levels in MIR376B overexpressing cells (Fig. S8D). On the other hand, MIR376B overexpression could result in a decrease in ATG12–ATG5 protein complex levels (Fig. S8E). Therefore, MIR376B might potentiate the inhibitory effect of MIR181A on autophagy, possibly through regulation of ATG12 protein levels. Further studies are needed to resolve this issue.
All together we concluded that, although it is possible that some other targets of MIR181A could contribute to its biological effects, ATG5 seems to be the major and rate-limiting autophagy-related target of this microRNA.
ATG5 as direct target of MIR181A
To further validate the effect of MIR181A on ATG5, the region in the 3′ UTR of the ATG5 mRNA containing a potential MIR181A response element was cloned into the 3′ UTR part of a luciferase vector. In parallel, we also created a mutant version of this construct by introducing base changes to the crucial binding residues, mainly in the miRNA seed sequence-binding region (Fig. 6A). Cotransfection of MIR181A together with the wild-type luciferase vector in 293T cells resulted in a significant decrease in the luciferase activity compared with control levels (Fig. 6B, wild-type). In contrast, MIR181A had no significant effect on the levels of luciferase expressed from the mutant construct; here the luciferase activity was similar to control levels (Fig. 6B, mutant). These results showed that, MIR181A controlled the levels of ATG5 by directly targeting the above described MRE present in the 3′ UTR region of the ATG5 gene.

Figure 6. ATG5 as a direct target of MIR181A and changes in endogenous MIR181A levels during cellular stress. (A) A scheme representing luciferase constructs with wild-type (wt) or mutant 3′ UTR MIR181A MRE sequences of ATG5. Mutations are marked in lower case letters and underlined. (B) Normalized luciferase activity in lysates from 293T cells co-transfected with wild-type or mutant ATG5-luciferase constructs and MIR181A or MIR-CNT (mean ± SD of independent experiments, n = 3, **p < 0.01. N.S., not significant). (C) TaqMan quantitative PCR (qPCR) analysis of endogenous MIR181A levels under control (CNT, no starvation) or starvation (STV, 2 h and 4 h) conditions. Endogenous MIR181A levels were not responsive to starvation treatment (mean ± SD of independent experiments, n = 3, N.S., not significant). TaqMan qPCR data were normalized using U6 small nuclear 1 (RNU6-1) (U6) mRNA levels. (D) TaqMan qPCR analysis of endogenous MIR181A levels in cells treated with DMSO (carrier) or rapamycin. Endogenous MIR181A levels were increased following rapamycin treatment (mean ± SD of independent experiments, n = 4, *p < 0.05).
Effect of autophagy inducers on endogenous MIR181A levels
We wondered whether MIR181A endogenous miRNA levels were responsive to the stress-inducing stimuli used in this study. We starved cells for 2 or 4 h, or treated them with the MTOR-inhibitor rapamycin. Interestingly, starvation did not lead to a significant change in the miRNA levels (Fig. 6C). In contrast, rapamycin treatment induced a prominent increase in endogenous MIR181A levels (Fig. 6D). Since MIR181A-specific antagomir introduction could further lead to an increase in the target ATG5 protein levels (see above), we believe that even under starvation conditions, steady-state endogenous levels of the miRNA were still functional to counteract autophagy. Yet, we showed that MIR181A levels might be upregulated in response to certain stress-stimuli such as rapamycin.
Discussion
In this study, we introduced MIR181A as a novel autophagy-related microRNA. MIR181A overexpression inhibited GFP-LC3 dot formation, LC3-I to LC3-II conversion and SQSTM1 degradation. These results were valid for two commonly used autophagy stimuli, namely starvation and rapamycin and in at least three cell types originating from different tissues (MCF-7 breast cancer, Huh-7 liver cancer and K562 chronic myelocytic leukemia cell lines). Additionally, antagomir-mediated suppression of cellular endogenous MIR181A was shown to accelerate the autophagic response. All together, these data established that MIR181A is one of the key miRNAs in autophagy regulation.
We identified ATG5 as a target of MIR181A. We showed that MIR181A overexpression attenuated ATG5 protein and mRNA levels and, more importantly, reintroduction of ATG5 protein in the presence of MIR181A reversed autophagy blockage by the miRNA. The effect of MIR181A on ATG5 was direct since the MRE found in the 3′ UTR region of the ATG5 gene was responsive to MIR181A, and introduction of mutations to this sequence abolished the miRNA effect. So, our results established ATG5 as a rate-limiting and important target of MIR181A during autophagy regulation.
Results presented in this work may be summarized in a model as follows (Fig. 7). Cellular ATG5 amount above a certain threshold is required to form functional ATG12–ATG5-ATG16L1 complexes, subsequent LC3 lipidation and autophagy activation. The presence of miRNAs including MIR181A under stress conditions leads to a fall in ATG5 levels below the threshold, and therefore results in a decrease in LC3 lipidation and inhibition of autophagic activity. So, stress signals stimulating autophagy seem to be counteracted by inhibitor miRNAs such as MIR181A, limiting uncontrolled and potentially harmful autophagic activity in cells.
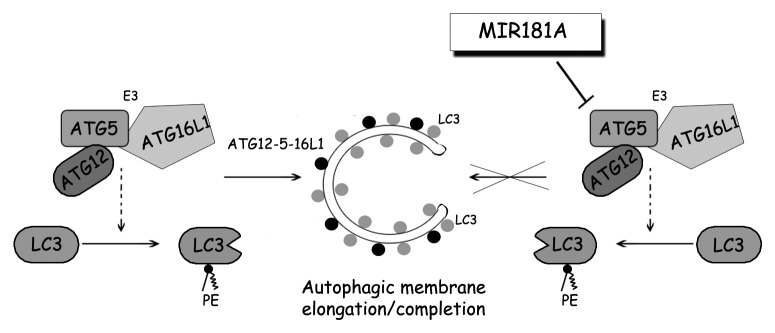
Figure 7. A model depicting the effect of MIR181A on autophagy pathways. MIR181A regulates stress-induced autophagy by targeting the key autophagy protein ATG5. The resulting decrease in ATG12–ATG5-ATG16L1 activity leads to attenuation of LC3 lipidation and inhibits the membrane elongation and completion stage of autophagy.
While this manuscript was in preparation, Huang et al. published a report suggesting a connection between ATG5 and MIR181A.37 Working on the mechanisms of cisplatin resistance in squamous cell carcinoma cells JHU-029, they discovered that a TP63 isoform α, ΔNp63α, downregulated MIR181A.37,38 In this context, they showed that MIR181A inhibitors led to an increase in ATG5 protein levels and, mimics and inhibitors of the miRNA modulated expression from a luciferase construct fused to the full length 3′ UTR region of ATG5.37 Yet, the importance of MIR181A or ATG5 downregulation for cisplatin-induced autophagy is still obscure, and the above-mentioned study did not experimentally refine a MIR181A response element in ATG5 mRNA. Nevertheless, possible involvement of MIR181A in the regulation of autophagy activated by stimuli other than starvation or rapamycin is intriguing.
Another autophagy-related miRNA, MIR30A, was recently shown to regulate ATG5 levels in chronic myeloid leukemia (CML) cells including K562.39 In this report, MIR30A overexpression decreased and antagomirs against this miRNA increased BECN1 and ATG5 levels. Interestingly, MIR30A-mediated changes in BECN1 and ATG5 levels and modulation of cytoprotective autophagy influenced the efficacy of the BCR-ABL tyrosine kinase inhibitor drug imatinib on CML cells. Likewise, MIR181A was able to affect ATG5 levels and autophagy in K562 CML cells. Yet, the influence of MIR181A on imatinib responses and its possible crosstalk with MIR30A needs to be studied. Of note, MIR30A overexpression did not influence endogenous MIR181A levels in cells (Fig. S9).
MIR181A was previously shown to be involved in various events including development, differentiation, hematopoiesis, immune modulation and muscle adaptation to exercise.27,40-42,43,44 In fact, autophagy was described as an important regulator of similar physiological events.45-47 Study of the contribution of autophagy-related effects of MIR181A in these and other overlapping scenarios could provide valuable information about the importance of this miRNA under physiological conditions.
A complex connection exists between autophagy and cancer. Autophagy deficiency was associated with increased tumor formation in animal models and, mutations or epigenetic changes in autophagy genes and proteins were documented in some human cancer types.48-50 Therefore, autophagy was proposed as a mechanism protecting cells from cancerous transformation.3,51 On the other hand, some tumor types become addicted to autophagy and under these conditions autophagy seems to control tumor necrosis and inflammation in response to metabolic stress.52 Hence, autophagy seems to serve as a cancer cell survival mechanism in established tumors. Consequently, the combination of autophagy inhibition with cancer therapy was proposed as a novel approach to potentiate tumor cell death.7,53 Interestingly, MIR181A is downregulated in human squamous cell and non-small-cell lung cancers and, upregulation of the same miRNA is observed in multiple myeloma and head and neck cancers.54-57 In cancers with a high MIR181A expression, autophagy inhibition through MIR181A-mediated targeting of ATG5 might modulate survival responses and contribute to cancer cell sensitization to chemotherapy. In line with this, MIR181A overexpression could potentiate the effects of radiation therapy or chemotherapy agents Ara-C, vincristine and cisplatin in glioma, leukemia, gastric cancer or lung cancer cell lines.33,34,58,59 Similarly, we could observe that MIR181A overexpression potentiated the toxicity of the cancer therapy agent cisplatin in MCF-7 breast cancer cells (Fig. S10). So, at least in cancer types harboring MIR181A level changes, autophagy modulation by this miRNA might have an impact on the survival of the transformed cells and affect their responses to chemotherapy. Further studies are required to explore the contribution of MIR181A expression to chemotherapy sensitivity of different types of cancers.
Materials and Methods
Plasmid constructs
Details of the microRNA screening procedure and miRNA vectors were previously described.23,60 As a control, human telomerase RNA (hTR) was used. For the dual luciferase assay, the MRE found in the 3′ UTR region of the ATG5 mRNA and its mutated version were cloned into the pGL3-control vector (Promega E1741) at the 3′ region of the luciferase gene using following linker primers: ATG5 wildtype primers: 5′- CTAGAGCTGGCTGTCCATATTGAATGTTGAATTCT- 3′, 5′- CTAGAGAATTCAACATTCAATATGGACAGCCAGCT-3′. ATG5 mutant primers: 5′-CTAGAGGTGGGTCTCCATATTCTAACATGAATTCT-3′, 5′- CTAGAGAATTCATGTTAGAATATGGAGACCCACCT-3′. XbaI restriction sites between the stop codon and polyadenylation signal were used.
Cell culture and transfections
MCF-7 human breast carcinoma cells, Huh-7 human hepatocellular carcinoma cells and 293T human embryonic kidney cells were cultured in DMEM (Sigma, 05671) supplemented with 10% (v/v) fetal bovine serum (FBS; Biochrom KG, S0115) and antibiotics (Penicillin/Streptomycin; Biological Industries, 03-031-1B) in a 5% CO2-humidified incubator at 37°C. K562 cells were cultured in RPMI medium (Invitrogen, 31870) supplemented with 10% FBS and antibiotics. In order to induce autophagy, starvation in Earle’s Balanced Salt solution (EBSS; Biological Industries, BI02-010-1A) or rapamycin (2.5 µM, 24 h) (Sigma-Aldrich, R0395) were used.
Transient transfection of MCF-7, Huh-7 and K562 cells was performed using either polyethyleneimine (PEI; Polysciences Inc., 23966) transfection method as previously described23 or AMAXA nucleofector (Lonza Nucleofector II AAD-1001S) with program E-014 and the Nucleofector kit V (Lonza, VCA-1003) according to the manufacturer’s instructions. 293T cells were transfected using calcium phosphate coprecipitation method according to standard protocols.61
GFP-LC3 analyses
48 h after cotransfection of miRNAs and GFP-LC3, MCF-7 cells and Huh-7 cells were incubated for 2 h or 4 h in EBSS medium or in DMEM medium/FBS containing 2.5 µM rapamycin for 24 h. Cells were fixed in 3.7% formaldehyde for 20 min, washed with PBS, mounted and inspected under 60× magnification using a BX60 fluorescence microscope (Olympus, BX60). Basal autophagy threshold was determined as 10 GFP-LC3 dots per MCF-7 cell and 15 GFP-LC3 dots per Huh-7 cell. At least 150 GFP positive cells per condition were counted and the graphs were plotted as percentage of GFP-LC3 dot positive cells over total number of transfected cells.
GFP-LC3 analyses of antagomir-transfected cells were done as described above but to better observe differences in the exponential phase of autophagic vesicle formation, tests were performed in the presence of lysosomal protease inhibitors E64d (10 μg/ml) (Santa Cruz, SC201280A) and pepstatin A (10 μg/ml) (Sigma, P5318) and after 20 min of starvation.
Target prediction for miRNA
miRNA targets were identified using publicly available bioinformatics tools Microcosm Targets (www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/) and TargetScan Human (www.targetscan.org/).
Immunoblot analysis and antibodies
Protein extraction was performed with RIPA buffer (50 mM TRIS-HCl pH 7.4, 150 mM NaCl, 1% NP40, 0.25% Na-deoxycholate) supplemented with complete protease inhibitor cocktail (Roche, 04-693-131-001) and 1mM phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich, P7626). Cell extracts (30 µg) were separated in 15% SDS-polyacrylamide gels and transferred to PVDF membranes (Roche, 03010040001). Following blockage in 5% nonfat milk in PBST (3.2 mM Na2HPO4, 0.5 mM KH2PO4, 1.3 mM KCl, 135 mM NaCl and 0.05% Tween 20, pH 7.4) for 1 h at RT, membranes were incubated in 3% BSA-PBST solutions containing primary antibodies (ab): anti-ATG5 ab (Sigma-Aldrich, A0856, dilution 1:2000), anti-LC3B ab (Sigma-Aldrich, L7543, dilution 1:2000), anti-SQSTM1/p62 ab (BD Transduct. Lab, 610832, 1:1000), anti-ACTB ab (Sigma-Aldrich, A5441, dilution 1:7500) and anti-BCL2 ab (Santa Cruz, sc-7382, 1:1000), anti-RPS6KB/p70S6 kinase ab (Cell Signaling, 9202, 1:1000), anti-Phospho-RPS6KB/p70S6 kinase Thr389 ab (Cell Signaling, 9206, 1:1000), anti-GFP ab (Roche, 11-814-460-001, 1:1000). Then, secondary mouse or rabbit antibodies coupled to horseradish peroxidase (anti-mouse: Jackson Immunoresearch Laboratories, 115035003; anti-rabbit: Jackson Immunoresearch laboratories, 111035144, dilutions 1:10.000) were applied in 5% milk/PBST for 1 h at RT and protein bands were revealed with chemiluminescence. Band intensities were quantified using ImageJ software.62
LC3-I was migrating as a double band in some experiments with the Sigma antibody used in this study. To understand whether one of the bands was a nonspecific band, we migrated cell extracts in parallel, blotted and incubated them with two different polyclonal LC3 antibodies, namely MBL antibody (PM036, raised against aa 1 to 120 of MAP1LC3B) and Sigma antibody (used in this study, L7543, raised against aa 2 to 15 of LC3B). Albeit the antibodies showed different affinities for LC3-I and LC3-II, both polyclonal antibodies revealed two LC3-I bands of similar sizes (Fig. S1). It is very unlikely that two different polyclonal antibodies raised against two different polypeptides would reveal nonspecific bands of the same size. Therefore, we were convinced that both bands in LC3 blots corresponded to LC3-I forms and they were not nonspecific bands. Further studies are required to understand modification(s) leading to LC3-I double-band formation.
RNA isolation and RT-PCR analysis
Total RNA was extracted using TRIzol reagent (Sigma-Aldrich, T9424) according to the manufacturer’s instructions. cDNA was reverse transcribed from total RNA (DNase treated) using M-MuLV reverse transcriptase (Fermentas, EP0351) and random hexamers (Invitrogen, 48190-011).
Real-time RT-PCR for ATG5 and ATG12 mRNA quantification
SYBR Green Quantitative RT-PCR kit (Roche, 04-913-914-001) and an iCycler iQ thermal cycler (BioRad) were used for single step qRT-PCR reactions. To activate the SYBR green, an initial cycle of 95°C, 10 min was performed followed by, PCR reactions: 40 cycles of 95°C for 15 sec and 60°C for 1 min. Then a thermal denaturation protocol was used to generate the dissociation curves for the verification of amplification specificity (a single cycle of 95°C for 60 sec, 55°C for 60 sec and 80 cycles of 55°C for 10 sec). Changes in mRNA levels were quantified using the 2−ΔΔCT method using GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA as control. Primers used during the study were: ATG5 primers 5′-TCAGCCACTGCAGAGGTGTTT-3′; 5′-GGCTGCAGATGGACAGTTGCA-3′; GAPDH primers 5′-AGCCACATCGCTCAGACAC-3′; 5′-GCCCAATACGACCAAATCC-3′; and ATG12 primers 5′- CAGTTTACCATCACTGCCAAAA-3′; 5′- ACAAAGAAGTGGGCAGTAGAGC-3′. Reactions were performed in duplicates and number of independent experiments (n) was marked.
TaqMan RT-PCR for endogenous MIR181A quantification
TaqMan qRT-PCR reactions were performed using FastStart Universal Probe Master kit (ROCHE, 04913957001) and an iCycler iQ thermal cycler (BioRad) according to the protocols described previously.23 Primers and the probe used during the study were: Stem-loop primer, 5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAACTCACCGACAGCGTTGAATGTT-3′. Forward primer, 5′-AACATTCAACGCTGTCGGTGAGT-3′. Reverse primer, 5′-GTGCAGGGTCCGAGGT-3′. Probe, 5′(6-FAM)-CTGGATACGACAACTCAC-(TAMRA-sp)3′.
Dual luciferase-reporter assay
Vectors containing wild-type or mutated MIR181A MREs from ATG5 3′ UTR were cotransfected with MIR181A into 293T cells. 48 h later, cells were lysed. Firefly and renilla luciferase activities were measured using a dual luciferase-reporter assay system (Promega, E1910). Results were evaluated through normalization of the firefly luciferase activity with renilla luciferase activity.
Antagomir tests
miRIDIAN microRNA Hairpin Inhibitors (antagomirs) against hsa-mir181a (IH-30055307) and control antagomir miRIDIAN microRNA hairpin inhibitor negative control (IN001005-01-05) were purchased from Dharmacon. Negative control was based on cel-miR-67 (mature sequence: UCACAACCUCCUAGAAAGAGUAGA and Accession Number: MIMAT0000039), which has minimal sequence similarity with human miRNAs.
Transfection of antagomirs (200 nM) was performed using either the PEI transfection method23 or AMAXA nucleofector (Lonza Nucleofector II AAD-1001S) with program E-014 and the Nucleofector kit V (Lonza, VCA-1003) according to the manufacturer’s instructions.
Statistical analyses
Statistical analyses were performed using Student’s two-tailed t-test. Data were represented as means of ± SD of n independent experiments. Values of p < 0.05 were considered as significant.
Supplementary Material
Acknowledgments
pCI-hApg5 and GFP-LC3 plasmids were kindly provided by Noboru Mizushima and Tomatsu Yoshimori, respectively. We would also like to thank Carlos le Sage for technical assistance, and Gozuacik Lab members and Stuart James Lucas for fruitful discussions and critical reading of the manuscript. K562 cells were a kind gift of Ayhan Bilir. This work was supported by the Scientific and Technological Research Council of Turkey (TUBITAK) 1001 Grant and Sabanci University. D.G. is a recipient of EMBO Strategical Development and Installation Grant (EMBO-SDIG) and Turkish Academy of Sciences (TUBA) GEBIP Award. G.K. and K.A.T. are recipients of Yousef Jameel and TUBITAK-BIDEB Scholarships, respectively. The authors have no financial conflict of interests.
Glossary
Abbreviations:
- ACTB
actin beta
- ATG5
autophagy-related 5
- ATG12
autophagy-related 12
- ATG16L1
autophagy-related 16-like 1
- BECN1
Beclin 1
- miRNA
microRNA
- LC3
microtubule-associated protein 1 light chain 3
- MIR181A
human microRNA-181A and its gene
- PE
phosphatidylethanolamine
- MTOR
mechanistic target of rapamycin
- MRE
miRNA-response element
- BCL2
B-cell lymphoma2
- RPS6KB/P70S6K
ribosomal protein S6 kinase
- SQSTM1/P62
sequestosome 1
- RAP
rapamycin
- STV
starvation
- E+P
E64d/pepstatin A
- RT-PCR
reverse transcriptase-polymerase chain reaction
- MTT
thiazolyl blue tetrazolium blue
- Ant-181a
MIR181A-specific antagomir
- CNT-Ant
control antagomir
- TRIzol
trizol Reagent
- PEI
polyethylenimine
- DMSO
dimethyl sulfoxide
- U6
U6 small nuclear 1 (RNU6-1)
- GFP
green fluorescent protein
- MIR376B
human microRNA-376B and its gene
- EBSS
Earle’s Balanced Salt Solution
- MIR30A
human microRNA-30A and its gene
- QPCR
quantitative PCR
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/autophagy/article/23117
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/23117
References
- 1.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 2.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 3.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 4.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–30. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–95. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 6.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–61. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 7.Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25:1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20:748–62. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 9.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 10.Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151:263–76. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 12.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem. 2004;279:47704–10. doi: 10.1074/jbc.M407016200. [DOI] [PubMed] [Google Scholar]
- 14.Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–14. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu LL, Wen X, Bao JK, Liu B. MicroRNA-modulated autophagic signaling networks in cancer. Int J Biochem Cell Biol. 2012;44:733–6. doi: 10.1016/j.biocel.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Xu J, Wang Y, Tan X, Jing H. MicroRNAs in autophagy and their emerging roles in crosstalk with apoptosis. Autophagy. 2012;8:873–82. doi: 10.4161/auto.19629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 18.Voorhoeve PM, Agami R. Classifying microRNAs in cancer: the good, the bad and the ugly. Biochim Biophys Acta. 2007;1775:274–82. doi: 10.1016/j.bbcan.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 19.Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy. 2009;5:816–23. doi: 10.4161/auto.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frankel LB, Wen J, Lees M, Høyer-Hansen M, Farkas T, Krogh A, et al. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011;30:4628–41. doi: 10.1038/emboj.2011.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kovaleva V, Mora R, Park YJ, Plass C, Chiramel AI, Bartenschlager R, et al. miRNA-130a targets ATG2B and DICER1 to inhibit autophagy and trigger killing of chronic lymphocytic leukemia cells. Cancer Res. 2012;72:1763–72. doi: 10.1158/0008-5472.CAN-11-3671. [DOI] [PubMed] [Google Scholar]
- 22.Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43:242–5. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 23.Korkmaz G, le Sage C, Tekirdag KA, Agami R, Gozuacik D. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy. 2012;8:165–76. doi: 10.4161/auto.8.2.18351. [DOI] [PubMed] [Google Scholar]
- 24.Seoudi AM, Lashine YA, Abdelaziz AI. MicroRNA-181a - a tale of discrepancies. Expert Rev Mol Med. 2012;14:e5. doi: 10.1017/S1462399411002122. [DOI] [PubMed] [Google Scholar]
- 25.Shi L, Cheng Z, Zhang J, Li R, Zhao P, Fu Z, et al. hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res. 2008;1236:185–93. doi: 10.1016/j.brainres.2008.07.085. [DOI] [PubMed] [Google Scholar]
- 26.Belkaya S, Silge RL, Hoover AR, Medeiros JJ, Eitson JL, Becker AM, et al. Dynamic modulation of thymic microRNAs in response to stress. PLoS One. 2011;6:e27580. doi: 10.1371/journal.pone.0027580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–6. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 28.Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int J Biochem Cell Biol. 2004;36:2445–62. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, et al. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy. 2011;7:188–204. doi: 10.4161/auto.7.2.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Hui L, Xu W. miR-181a sensitizes a multidrug-resistant leukemia cell line K562/A02 to daunorubicin by targeting BCL-2. Acta Biochim Biophys Sin (Shanghai) 2012;44:269–77. doi: 10.1093/abbs/gmr128. [DOI] [PubMed] [Google Scholar]
- 32.Ouyang YB, Lu Y, Yue S, Giffard RG. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion. 2012;12:213–9. doi: 10.1016/j.mito.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen G, Zhu W, Shi D, Lv L, Zhang C, Liu P, et al. MicroRNA-181a sensitizes human malignant glioma U87MG cells to radiation by targeting Bcl-2. Oncol Rep. 2010;23:997–1003. doi: 10.3892/or_00000725. [DOI] [PubMed] [Google Scholar]
- 34.Bai H, Cao Z, Deng C, Zhou L, Wang C. miR-181a sensitizes resistant leukaemia HL-60/Ara-C cells to Ara-C by inducing apoptosis. J Cancer Res Clin Oncol. 2012;138:595–602. doi: 10.1007/s00432-011-1137-3. [DOI] [PubMed] [Google Scholar]
- 35.Huang YL, Chou CK. Bcl-2 blocks apoptotic signal of transforming growth factor-beta in human hepatoma cells. J Biomed Sci. 1998;5:185–91. doi: 10.1007/BF02253468. [DOI] [PubMed] [Google Scholar]
- 36.Okamoto K, Muraguchi T, Shidoji Y. Enhanced Glucose Requirement in Human Hepatoma-derived HuH-7 Cells by Forced Expression of the bcl-2 Gene. J Clin Biochem Nutr. 2008;43:101–8. doi: 10.3164/jcbn.2008053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Y, Guerrero-Preston R, Ratovitski EA. Phospho-ΔNp63α-dependent regulation of autophagic signaling through transcription and micro-RNA modulation. Cell Cycle. 2012;11:1247–59. doi: 10.4161/cc.11.6.19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang Y, Chuang A, Hao H, Talbot C, Sen T, Trink B, et al. Phospho-ΔNp63α is a key regulator of the cisplatin-induced microRNAome in cancer cells. Cell Death Differ. 2011;18:1220–30. doi: 10.1038/cdd.2010.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu Y, Yang L, Zhao M, Zhu S, Kang R, Vernon P, et al. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia. 2012;26:1752–60. doi: 10.1038/leu.2012.65. [DOI] [PubMed] [Google Scholar]
- 40.Ryan DG, Oliveira-Fernandes M, Lavker RM. MicroRNAs of the mammalian eye display distinct and overlapping tissue specificity. Mol Vis. 2006;12:1175–84. [PubMed] [Google Scholar]
- 41.Ji JF, Yamashita T, Budhu A, Forgues M, Jia HL, Li CL, et al. Identification of microRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells. Hepatology. 2009;50:472–80. doi: 10.1002/hep.22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Gocek E, Liu CG, Studzinski GP. MicroRNAs181 regulate the expression of p27Kip1 in human myeloid leukemia cells induced to differentiate by 1,25-dihydroxyvitamin D3. Cell Cycle. 2009;8:736–41. doi: 10.4161/cc.8.5.7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsitsiou E, Lindsay MA. microRNAs and the immune response. Curr Opin Pharmacol. 2009;9:514–20. doi: 10.1016/j.coph.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Safdar A, Abadi A, Akhtar M, Hettinga BP, Tarnopolsky MA. miRNA in the regulation of skeletal muscle adaptation to acute endurance exercise in C57Bl/6J male mice. PLoS One. 2009;4:e5610. doi: 10.1371/journal.pone.0005610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 46.Kuballa P, Nolte WM, Castoreno AB, Xavier RJ. Autophagy and the immune system. Annu Rev Immunol. 2012;30:611–46. doi: 10.1146/annurev-immunol-020711-074948. [DOI] [PubMed] [Google Scholar]
- 47.Masiero E, Sandri M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy. 2010;6:307–9. doi: 10.4161/auto.6.2.11137. [DOI] [PubMed] [Google Scholar]
- 48.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–82. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaza N, Kohli L, Roth KA. Autophagy in brain tumors: a new target for therapeutic intervention. Brain Pathol. 2012;22:89–98. doi: 10.1111/j.1750-3639.2011.00544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol. 2009;3:366–75. doi: 10.1016/j.molonc.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mathew R, White E. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev. 2011;21:113–9. doi: 10.1016/j.gde.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen N, Karantza V. Autophagy as a therapeutic target in cancer. Cancer Biol Ther. 2011;11:157–68. doi: 10.4161/cbt.11.2.14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao W, Shen H, Liu L, Xu J, Xu J, Shu Y. MiR-21 overexpression in human primary squamous cell lung carcinoma is associated with poor patient prognosis. J Cancer Res Clin Oncol. 2011;137:557–66. doi: 10.1007/s00432-010-0918-4. [DOI] [PubMed] [Google Scholar]
- 55.Gao W, Yu Y, Cao H, Shen H, Li X, Pan S, et al. Deregulated expression of miR-21, miR-143 and miR-181a in non small cell lung cancer is related to clinicopathologic characteristics or patient prognosis. Biomed Pharmacother. 2010;64:399–408. doi: 10.1016/j.biopha.2010.01.018. [DOI] [PubMed] [Google Scholar]
- 56.Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci U S A. 2008;105:12885–90. doi: 10.1073/pnas.0806202105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nurul-Syakima AM, Yoke-Kqueen C, Sabariah AR, Shiran MS, Singh A, Learn-Han L. Differential microRNA expression and identification of putative miRNA targets and pathways in head and neck cancers. Int J Mol Med. 2011;28:327–36. doi: 10.3892/ijmm.2011.714. [DOI] [PubMed] [Google Scholar]
- 58.Galluzzi L, Morselli E, Vitale I, Kepp O, Senovilla L, Criollo A, et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res. 2010;70:1793–803. doi: 10.1158/0008-5472.CAN-09-3112. [DOI] [PubMed] [Google Scholar]
- 59.Zhu W, Shan X, Wang T, Shu Y, Liu P. miR-181b modulates multidrug resistance by targeting BCL2 in human cancer cell lines. Int J Cancer. 2010;127:2520–9. doi: 10.1002/ijc.25260. [DOI] [PubMed] [Google Scholar]
- 60.Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006;124:1169–81. doi: 10.1016/j.cell.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 61.Oral O, Oz-Arslan D, Itah Z, Naghavi A, Deveci R, Karacali S, et al. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis. 2012;17:810–20. doi: 10.1007/s10495-012-0735-0. [DOI] [PubMed] [Google Scholar]
- 62.Abramoff MD, Magalhães PJ, Ram SJ. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.