

Abstract
We recently reported that transgenic mice with cardiac-restricted overexpression of CCN2/CTGF have substantially increased tolerance towards ischemia/reperfusion injury. The purpose of this study was to investigate to what extent fully differentiated cardiac myocytes are direct targets of CCN2, and to resolve the signaling mechanisms that convey the cardioprotective actions of CCN2. Akt and GSK-3β were identified as putative intermediaries of intracellular signaling stimulated by recombinant human CCN2 (rhCCN2). Concentration-effect experiments revealed CCN2-stimulated phosphorylation of Akt (Ser473) and downstream GSK-3β (Ser9) with EC50 ~250 nmol/L. CCN2-stimulated phosphorylation of Akt and GSK-3β was sensitive to inhibition of PI3-kinase (LY294002). Phosphorylation of GSK-3β was also sensitive to Akt-inhibition (API-2), demonstrating CCN2-engendered activation of a PI3-kinase/Akt/GSK-3β-signaling pathway. A C-terminal peptide fragment of CCN2 (11.2 kD) displayed partial agonist activity, while two short peptides derived from the Thrombospondin- and the IGFBP- homology domains of CCN2, respectively, additively inhibited rhCCN2-stimulated Akt-phosphorylation. The viability of cardiac myocytes subjected to hypoxia/reoxygenation injury or doxorubicin-induced oxidative stress was assessed by assays of adenylate kinase and lactate dehydrogenase released from dying cells. Cardiac myocytes exposed to CCN2 displayed increased tolerance towards hypoxia/reoxygenation and doxorubicin-induced oxidative stress, an effect that was abrogated by inhibition of PI3-kinase. The cytoprotective actions of CCN2 reflected in the transcriptome of CCN2-stimulated cardiac myocytes (anti-apoptosis, stress, and wound-response gene programs). In conclusion, this study discloses the novel findings that cardiac myocytes are CCN2 target cells in which CCN2 increases tolerance towards hypoxia and oxidative stress via PI3-kinase-dependent Akt/GSK-3β signaling.
Keywords: CCN2/CTGF, Cardiac myocyte, Cytoprotection, Cardioprotection, Signaling, Akt/GSK-3β
Introduction
CCN2/CTGF, connective tissue growth factor, is a secreted, matricellular protein that belongs to the CCN family (acronym for the first three recognized members of this family; Cyr61/CTGF/Nov). The CCN proteins are involved in a wide array of cellular and more complex biologic functions depending on cellular context (for review, see Brigstock et al. (Rachfal and Brigstock 2005)). As alluded to by its name, increased tissue contents of CCN2 is often found in conditions associated with fibrosis as in wound healing with scar formation or inflammatory disorders with increased production of collagen (Chuva de Sousa Lopes et al. 2004). However, it has not yet been demonstrated that CCN2 is a bona fide growth factor sufficient to elicit fibrosis by itself. Indeed, genetically-engineered mice with increased tissue expression of CCN2, as well as other studies, indicate that CCN2 by itself is not sufficient to elicit fibrosis (Ahmed et al. 2011; Chujo et al. 2009; Nakanishi et al. 2001). Rather, additional factors appear to be required for fibrosis (Chujo et al. 2009). CCN2 is highly expressed in the developing heart, and can be identified in endothelial cells, cardiac fibroblasts, as well as cardiac myocytes (Chuva de Sousa Lopes et al. 2004). However, in the postnatal heart CCN2 is more or less completely repressed. Several studies have reported substantial induction of myocardial CCN2 in heart failure of various etiologies (Chen et al. 2000; Ahmed et al. 2005). Yet, the role of CCN2 in the pathophysiologic mechanisms of heart failure remains largely unknown. Data indicating that CCN2 may act as a cell survival factor have prompted studies attempting to resolve the role of CCN2 in ischemia/reperfusion injury. In two reports of transgenic mice with cardiac-restricted overexpression of CCN2, the first report did not find altered tolerance of hearts overexpressing CCN2 towards ischemia/reperfusion injury, whereas the later study reported robust increase of tolerance towards ischemia/reperfusion injury of CCN2-expressing hearts (Panek et al. 2009; Ahmed et al. 2011). Apparently, overexpression of CCN2 was substantially higher in the hearts displaying increased tolerance towards ischemia/reperfusion injury. Corroborating the evidence of a cardioprotective function of CCN2 was also the observation that rhCCN2 reduced infarct size of ex vivo-perfused hearts subjected to ischemia/reperfusion injury (Ahmed et al. 2011). However, to what extent the cardioprotective properties of CCN2 could be elicited by direct action on cardiac myocytes was not investigated. The CCN proteins are assumed to function by interaction with extracellular matrix proteins. Although a cognate receptor for CCN2, or any of the other CCN proteins, has not yet been convincingly identified, several reports indicate that CCN proteins may also interact with plasma membrane proteins, e.g. integrin receptors or LRP5/6 (LDL receptor-associated protein-5/6) (Rachfal and Brigstock 2005). Furthermore, the intracellular signaling pathways of CCN2 have not yet been properly characterized, although intracellular signaling mechanism may vary with cell type or cellular context (Rachfal and Brigstock 2005). The major objectives of this study were 1) to investigate to what extent the cardioprotective properties reported for CCN2 could be based on direct cytoprotective actions of CCN2 on adult cardiac myocytes exposed to hypoxia/reoxygenation, i.e. simulated ischemia/reperfusion injury, 2) to identify the intracellular signaling pathways of CCN2 in the terminally differentiated, adult cardiac myocyte, and 3) to resolve to what extent the identified signaling mechanisms and resultant transcriptome were involved in the cytoprotective actions of CCN2.
Materials and methods
Reagents
Recombinant full-length, human CCN2 (rhCCN2) was contract-purified from the cell culture medium of transfected HEK 293 cells by EMP Genetech (Germany), essentially as previously described (Ahmed et al. 2004). The purified protein was resolved by sodium dodecylsulphate polyacrylamide gel electrophoresis (SDS-PAGE), and the homogeneity and integrity of the protein was assessed by Coomassie brilliant blue staining of the gel and by Western blot analysis. rhCCN2 was subjected to deglycosylation under non-denaturing conditions using Protein Deglycosylation Mix (Endo-α-N-Acetylgalactosaminidase, PNGase F, Neuraminidase, β1-4 Galactosidase, β-N-Acetylglucosaminidase) provided by New England Biolabs Inc. (MA, USA). rhCCN2, Reaction Buffer, and enzyme cocktail were mixed, and the reaction was subsequently incubated at 37 °C for four hours. rhCCN2 was also subjected to heat-treatment, as an eppendorf tube containing rhCCN2 was placed in boiling water for 15 min. The recombinant 11.2 kD C-terminal fragment of human CCN2 was from Peprotech EC Ltd. (UK). Synthetic peptides derived from the primary sequence of human CCN2 (GenBank ID: CAG46559) were custom-made from GenScript Inc. (NJ, USA). The peptides were P1, amino acids 200–224 (LVQTTEWSACSKTCGMGISTRVTND), from the Thrombospondin homology domain and P2, amino acids 47–66 (VSLVLDGCGCCRVCAKQLGE), from the Insulin-like growth factor binding protein-like (IGFBP) homology domain of human CCN2. All antibodies were purchased from Cell Signaling Technology, Inc. (MA, USA). The selective phosphatidylinositol 3-kinase (PI3-kinase) inhibitor LY294002, its corresponding negative control compound LY303511, and the selective Akt inhibitor API-2 were from Tocris Bioscience Inc. (MO, USA). Ad-GRK2ct, recombinant adenovirus encoding carboxyl terminal fragment of GRK2/βARK1, was kindly provided by Dr. Walter J. Koch, Jefferson Medical College, Philadelphia, USA (Drazner et al. 1997).
Isolation of adult cardiac myocytes and maintenance of primary cultures
Ca2+-tolerant cardiac myocytes were isolated from mouse hearts (C57/BL6 mice, 10–16 weeks of age) by coronary perfusion with collagenase type 2 (Worthington Biochemical Corp., NJ, USA) as described by O’Connell et al. (O’Connell et al. 2007). The cells were plated in laminin-coated tissue culture wells (Invitrogen Inc., CA, USA) in Minimum Essential Medium (MEM) with Hanks’ Balanced Salt Solution (HBSS), supplemented with 10 % serum, 100 U/mL penicillin-G, 2 mmol/L ATP, and 10 mmol/L 2,3-butanedione monoxime (BDM). After two hours the plating medium was replaced with culture medium (MEM with HBSS, supplemented with 0.1 % bovine serum albumin (BSA), 100 U/mL penicillin-G, and 10 mmol/L BDM). The cardiac myocytes were maintained in primary culture for up to 72 h in humidified atmosphere containing 2 % CO2. All animal studies were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Pub. No. 85–23, revised 1996) and were approved by the Chief Veterinarian of the Vivarium at Oslo University Hospital- Rikshospitalet and the national board for animal research, the Norwegian Animal Research Authority.
Analysis of protein- and phosphoprotein levels
For Western blot analysis, extracts of cardiac myocytes were prepared in solubilization buffer containing 10 mmol/L Tris–HCl (pH 7.4), 1 % SDS, 10 mmol/L NaF, and 2 mmol/L Na3VO4. The samples were denatured in Laemmli buffer, and separated by 12 % SDS-PAGE. The PVDF membrane was blocked in Tris-buffered saline (TBS) with 0.1 % Tween-20 and 5 % nonfat dry milk, and subsequently incubated with primary antibody in TBS with 0.1 % Tween-20 and 5 % BSA. Horseradish peroxidase-conjugated anti-rabbit IgG was used as secondary antibody, and the protein bands were visualized with the chemiluminescent substrate LumiGLO Reserve (KPL Inc., MD, USA). Protein/phosphoprotein levels in cell lysates from Rat-2-Fibroblasts, COS-7 cells, CHO cells, or HEK 293 cells stimulated with rhCCN2, deglycosylated rhCCN2, heat-treated rhCCN2, or C-peptide were determined by ELISA using the Luminex technology (Bio-Rad Laboratories Inc., CA, USA). Briefly, antibody coupled beads were added to wells of a 96-well plate, the wells were washed, and thereafter incubated with cell lysates for 30 min. The wells were washed again, and biotin-labeled detection antibodies were added. After another wash, the wells were incubated with fluorophore-labeled streptavidin reporter before recording in the Bio-Plex array reader.
Doxorubicin induced oxidative stress
Cardiac myocytes were preincubated in the absence or presence of 200 nmol/L rhCCN2 for 20 h. Subsequently, the cells were exposed to 15 μmol/L doxorubicin (Tocris) for additional 20 h before analysis of markers of cell damage.
Hypoxia/Reoxygenation-induced cell death
Cardiac myocytes were preincubated in the absence or presence of rhCCN2 (200 nmol/L) for 20 h and subsequently subjected to consecutive cycle of hypoxia and reoxygenation in order to simulate the ischemia/reperfusion injury of tissues following transient occlusion of blood supply. After the preincubation, the cell culture medium was replaced with glucose-free cell culture medium pre-equilibrated with 1 % O2 and then, the cells were incubated in a hypoxia chamber (1 % O2, 2 % CO2, and 97 % N2) for four hours followed by reoxygenation (21 % O2, 2 % CO2, 77 % N2) for 1 h.
Assays of cardiac myocyte cell death and viability
Cell death was assessed by analysis of adenylate kinase (AK) and lactate dehydrogenase activities (LDH) released into the cell culture medium from dying cells. Both AK and LDH release assays are established methods for assessment of cell viability based on the principle that cell death leads to increase of membrane permeability and effusion of cytoplasmic enzymes into the extracellular medium (Aras et al. 2008; Crouch et al. 1993). AK activities in the cell culture medium were determined by the ToxiLight assay (Lonza Group Ltd., Switzerland) according to the manufacturer’s instructions. Samples of cell culture medium were transferred into a 96-well plate (20 μL/well), and 100 μL/well of AK Detection Reagent was added. Luminescence was recorded by a microplate reader (F12900 GENios, Tecan Group Ltd., Switzerland). Release of LDH activities from dying cardiac myocytes were quantified using the Cytotoxicity Detection Kit (Roche Ltd., Switzerland). Cell culture medium was sampled and centrifuged, and the supernatant (100 μL) was mixed with 100 μL of the reaction mixture according to the manufacturer’s instructions. Absorbance was measured at 492 nm (reference wavelength 630 nm) with a microplate reader. Serial dilutions of cell culture medium disclosed linear correlations between sample amount and AK and LDH activities assayed. Assay of cell viability was performed by assessing release of either of the enzymes in cells treated with rhCCN2 versus cells exposed to vehicle relative to total enzyme release in complete-kill wells and calculated according to previously published methodology as follows (Aras et al. 2008):
 |
In the above equation Ei refers to enzyme release in individually treated wells, i.e. the CCN2-treated wells. Complete lysis of cardiac myocytes was achieved in 1 % Triton X-100.
TUNEL assay of apoptosis
Apoptosis of cardiac myocytes was determined by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). Cells were seeded in 6-well plates and subjected to hypoxia/reoxygenation as described above. TUNEL staining was performed using the In Situ Cell Death Detection Kit (Roche) according to the manufacturer’s instructions, and analyzed by fluorescence microscope (Axiovert S100, Carl Zeiss, Germany). TUNEL positive cells and total number of cells (Hoechst-stained) were counted. A minimum of ten visual fields per well were randomly selected and analyzed.
RNA purification
Total RNA was isolated using the RNeasy system (Qiagen, Germany). The amount and quality of the extracted RNA were measured by both a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, DE, USA) and a Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA).
Analysis of gene expression
RNA was reverse transcribed with TaqMan Reverse Transcription Reagents Kit. Real-time quantitative PCR was run in triplicates for each sample, using TaqMan Pre-Developed Assay Reagents and The ABI Prism 7900 Sequence Detection System and software (Applied Biosystems, CA, USA), according to the manufacturer’s instructions.
DNA microarray analysis
Cardiac myocytes were cultured in the absence (n = 6) or presence (n = 6) of 200 nmol/L rhCCN2 for 48 h. Total RNA was subjected to genome-wide expression analysis using Illumina gene expression bead arrays. MouseWG-6 v2 bead chips (Illumina Inc., CA, USA) with 48,000 transcripts were used. RNA was amplified using the Illumina TotalPrep-96 RNA Amplification Kit (Ambion Inc., TX, USA) with 500 ng of total RNA as input material. cRNA was synthesized overnight (14 h), labeled, and hybridized to the chips at 58 °C overnight, according to the manufacturer’s protocol. After hybridization and stringent wash, the chips were stained with streptavidin-Cy3 (GE Healthcare, UK) and scanned with an Illumina bead array reader. The scanned images were imported into BeadStudio 3.1.3.0 (Illumina Inc.) for extraction, quality control, and quantile normalization. Data were log2 transformed, and a moderated t-statistic was used for significance analysis. A list of differentially expressed genes was generated using topTable (Illumina), and the false discovery rate was controlled by Benjamini and Hochberg’s method. The annotation file MouseWG-6_V2_0_R2_11278593_A.bgx was used. Gene Ontology analysis was performed to look for terms that were overrepresented in the list of differentially expressed genes. A statistical comparison between the differentially expressed genes (q-value <0.01) and the whole data set was conducted by Fisher’s exact test, and p-values were Bonferroni corrected for multiple comparisons. Microarray data were deposited at GEO-NCBI with the following accession number: GSE36073. (The microarray service was provided by Norwegian Microarray Consortium, supported by the functional genomics program of the Research Council of Norway.)
Statistical analysis
The experimental groups were assessed by two-tailed un-paired Student’s t test. P values <0.05 were considered statistically significant. For multiple comparisons one-way ANOVA was performed, and post-hoc analysis with Bonferroni’s test was employed.
Results
CCN2 activates the PI3-kinase/Akt/GSK-3β salvage kinase pathway
Stimulation of adult cardiac myocytes with rhCCN2 (100 and 250 nmol/L) revealed robust increase in phospho-Akt (Ser473) and phospho-GSK-3β (Ser9) activities. On the other hand, stimulation of cardiac myocytes with rhCCN2 did not alter phospho-JNK (Thr183/Tyr185), phospho-P38 (Thr180/Tyr182), phospho-ERK1/2 (Thr202/204), phospho-ERK5 (Thr218/Tyr220), phospho-TrkA (Tyr490), phospho-Smad1/2/3 (Ser463/465/467/423/425), or phospho-Stat3 (Tyr705) levels in cardiac myocytes (Fig. 1a shows western blots and Fig. 1b densitometric analysis). Western blot analysis with site-specific phospho-serine antibodies revealed concentration-dependent increase of phospho-Akt (Ser473) (Fig. 2a) and phospho-GSK-3β (Ser9) (Fig. 2b) in cardiac myocytes stimulated with rhCCN2. Consistent with the phosphorylation of GSK-3β at serine-9 and ensuing inhibition of GSK-3β activities, rhCCN2 led to reduced levels of phospho-glycogen synthase (GS) (Ser641) in cardiac myocytes (Fig. 2c). CCN2-stimulated phosphorylation of Akt and GSK-3β was sensitive to inhibition of PI3-kinase by LY294002 (Fig. 3a and b), and complete inhibition of CCN2-stimulated phosphorylation of Akt and GSK-3β was observed with LY294002 at 50 μmol/L. Phosphorylation of GSK-3β (Ser9) was also sensitive to inhibition of Akt with the Akt-selective inhibitor API-2 (1–10 μmol/L) (Fig. 3c). Inhibition of CCN2-stimulated phosphorylation of GSK-3β by both LY294002 and API-2 provides strong evidence of a CCN2-stimulated intracellular PI3-kinase/Akt/GSK-3β-signaling pathway in cardiac myocytes. p70s6K, a kinase downstream of Akt, was also phosphorylated by CCN2 and its activation was sensitive to PI3-kinase inhibition with LY294002 (Fig. 3d). To further investigate the mechanisms of CCN2-induced activation of Akt, competitive inhibition of the pleckstrin homology (PH) domain of Akt was tested by means of recombinant adenovirus-mediated transduction of the carboxyl-terminal region of G protein-coupled receptor kinase-2 (GRK2ct) in cardiac myocytes. Expression of GRK2ct, containing a prototypical PH-domain, substantially attenuated CCN2-stimulated phospho-Akt (Ser9) levels in cardiac myocytes, providing evidence of a PH domain-mediated plasma membrane targeting mechanism in activation of Akt by CCN2 (Fig. 3e and f).
Fig. 1.


a. Western blots of indicated phospho-protein levels and total protein levels in cardiac myocytes stimulated with 100 nmol/L or 250 nmol/L rhCCN2 for 30 min. Figure demonstrates three replicate lanes of each treatment (control/stimulation). The Western blot images are all representative of three independent experiments. b. Densitometric analysis of Western blots in a. Histograms show phospho-protein levels relative to total protein contents. Data are mean ± SEM of n = 3 in each group, *p < 0.05 vs control
Fig. 2.
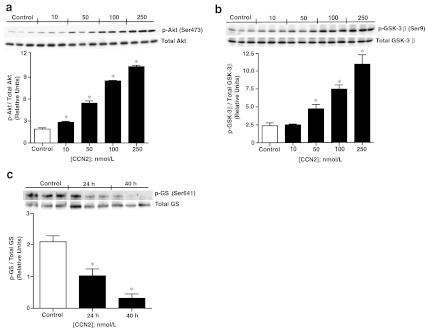
Activation of Akt/GSK-3β signaling pathway by CCN2. a Western blot analysis of phospho-Akt (Ser473) and b phospho-GSK-3β (Ser9) contents in cardiac myocytes stimulated with increasing concentrations of rhCCN2 for 30 min. c Western blot analysis of phospho-glycogen synthase (Ser641) levels in cardiac myocytes stimulated with 250 nmol/L rhCCN2 for 24 and 40 h. The histograms demonstrate densitometric analysis of phospho-protein levels relative to respective total protein contents. Data are mean ± SEM of n = 3 in each group, *p < 0.05 vs. control. The figure is representative of three independent experiments
Fig. 3.

CCN2-mediated activation of Akt/GSK-3β signaling pathway is sensitive to PI3-kinase- and Akt-inhibiton. a Western blot analyses of phosphoprotein and total protein levels of Akt, b-c GSK-3β, and d p70S6K in cardiac myocytes incubated for 15 min without (Control) or with 100 nmol/L rhCCN2 (CCN2), or with 100 nmol/L rhCCN2 in the presence of the PI3-kinase inhibitor LY294002 (CCN2 + LY) or in presence of the Akt inhibitor API-2 (CCN2 + API-2). e Inhibition of phospho-Akt (Ser473) activities by competitive inhibition of pleckstrin homology (PH) domain of Akt by expression of GRK2ct. Western blot demonstrating phospho-Akt (Ser473) and total Akt levels in cardiac myocytes expressing GRK2ct or LacZ in the absence or presence of rhCCN2. Cells were incubated for 40 h after infection with Ad-GRK2ct or Ad-LacZ at 1,000× multiplicity of infection (MOI), then stimulated with rhCCN2 (100 nmol/L) for 30 min. f Western blot analysis of GRK2ct expression in cardiac myocytes 40 h after infection with 1,000 MOI Ad-GRK2ct or Ad-LacZ. The histograms demonstrate densitometric analysis of indicated phospho-protein activities relative to total contents of the respective proteins. Data are mean ± SEM, n = 3 in each group, *p < 0.05. The figure is representative of three independent experiments
For investigation of the efficacy and potency of rhCCN2 in activation of Akt/GSK-3 signaling, Rat-2 fibroblasts were employed and stimulated in the presence of an extended range of concentrations of CCN2. ELISA (Bio-Plex/Luminex) disclosed concentration-dependent increase of phospho-Akt (Ser473) and phospho-GSK-3α/β (Ser21/Ser9) levels in stimulated cells, with half-maximal effects (EC50) of rhCCN2 at approximately 200 nmol/L. Maximal responses of rhCCN2 were obtained at concentrations between 500 nmol/L and 1 μmol/L. (Fig. 4a and b). rhCCN2 also stimulated increase of phospho-Akt (Ser473) and phospho-GSK-3α/β (Ser21/9) levels in COS-7 cells, CHO cells, and HEK 293 cells (data not shown).
Fig. 4.

Comparison of efficacy of deglycosylated rhCCN2, heat-treated rhCCN2, and C-peptide with native rhCCN2. Concentration-effect curves of deglycosylated rhCCN2 (a-b), heat-treated (c-d), and C-peptide (e-f) compared with rhCCN2 in Rat-2–fibroblasts. The phosphoprotein levels were examined by Bio-Plex/Luminex. Each point is mean ± SEM (n = 3)
Deglycosylated and heat-treated rhCCN2 activates Akt and GSK-3α/β
The purified rhCCN2 was essentially homogenous as assessed by SDS-PAGE and subsequent staining of the gel with Coomassie brilliant blue (Fig. 5a, lane 1). Western blot analysis revealed that purified, rhCCN2 migrated as two bands with molecular mass around 38–40 kD, both of which displayed strong immunoreactivity against anti-CCN2 antibody (Fig. 5a, lane 2). rhCCN2 has previously been reported to migrate as two discrete bands of approximate molecular mass 38–40 kD, with alteration of electrophoretic mobility after deglycosylation (Bohr et al. 2010). rhCCN2 was deglycosylated under non-denaturing conditions by enzymatic reactions, and deglycosylation was confirmed by mobility shift on SDS-PAGE (Fig. 5b). As shown in Fig. 4a and b, the potency and efficacy of deglycosylated rhCCN2 were similar to those of native, glycosylated rhCCN2 in phosphorylation of Akt (Ser473) and GSK-3α/β (Ser21/Ser9) in Rat-2-fibroblasts, indicating that glycosylation of CCN2 is not essential for stimulation of the intracellular Akt/GSK-3β signaling pathway. Likewise, heat-treated rhCCN2 stimulated phosphorylation of Akt and GSK-3α/β with similar efficacy and potency as native rhCCN2 (Fig. 4c and d). In contrast, rhCCN2 pretreated with the strong reducing agent dithiothretiol (DTT, 10 mmol/L) was not able to phosphorylate Akt (data not shown), suggesting that the activity of CCN2 is dependent on intact disulfide bonds.
Fig. 5.

Assessment of integrity and homogeneity of rhCCN2. a Purified recombinant human CCN2 resolved by SDS-PAGE and stained with Coomassie brilliant blue (lane 1) or subjected to Western blot analysis (lane 2). b Coomassie staining of native rhCCN2 (Control) and deglycosylated rhCCN2 (Deglycos.)
Peptides derived from the primary sequence of CCN2 with agonist or antagonist activity
The 11.2 kD C-terminal peptide fragment of human CCN2 (C-peptide) displayed partial agonist activity compared with full-length rhCCN2 at phosphorylation of Akt (Ser473) (Fig. 4e) and GSK-3α/β (Ser21/Ser9) (Fig. 4f) in Rat-2-fibroblasts. The C-peptide was also substantially less potent than the native protein. The peptides derived from the Thrombospondin-homology (P1) and the IGFBP-homology (P2) domains of CCN2 (Fig. 6b) did not display agonist activity in terms of capacity to stimulate phosphorylation of Akt (Ser473) (data not shown). However, both peptides inhibited CCN2-stimulated phosphorylation of Akt (Ser473), and the inhibitory actions of P1 and P2 were additive (Fig. 6a).
Fig. 6.

a Phospho-Akt levels in Rat-2-fibroblasts stimulated in the absence or presence of rhCCN2 (50 nmol/L) for 30 min, or co-incubated with P1 and/or P2 in two different concentrations (12.5 μmol/L (μM) and 50 μmol/L) together with rhCCN2 (50 nmol/L). Phosphoprotein activities were analyzed by Bio-Plex/Luminex. Each point is mean ± SEM (n = 3). *p < 0.05 vs. CCN2, †p < 0.05 vs. CCN2 +12.5 μmol/L P1 and vs. CCN2 +12.5 μmol/L P2, #p < 0.05 vs. CCN2 +50 μmol/L P1. b Diagram of the modules of CCN2, with the peptides derived from the Thrombospondin-homology (P1) and the IGFBP-homology (P2) domains marked
rhCCN2 increases tolerance towards doxorubicin-induced oxidative stress and hypoxia/reoxygenation injury in cardiac myocytes
The effects of rhCCN2 on viability of adult cardiac myocytes following hypoxia/reoxygenation and oxidative stress were investigated. Incubation of cardiac myocytes in the presence of rhCCN2 (200 nmol/L) protected cardiac myocytes against doxorubicin-induced oxidative stress and cell death as demonstrated by reduced release of LDH and AK to the cell culture medium (Fig. 7a). Similarly, rhCCN2 increased survival (viability) of cardiac myocytes exposed to hypoxia/reoxygenation (Fig. 7b).
Fig. 7.

Cardiac myocyte cell death quantified by analysis of release of lactate dehydrogenase (LDH) and adenylate kinase activities into the cell culture medium. a Viability of cardiac myocytes pretreated in the absence (Dox) or presence (Dox + CCN2) of 200 nmol/L rhCCN2 for 20 h, and then incubated with 15 μmol/L doxorubicin for 20 h. b Viability of cardiac myocytes preincubated in the absence (Control hypoxia) or presence (CCN2) of 200 nmol/L rhCCN2 for 20 h, then exposed to hypoxia (1 % O2) for four hours, before one hour reoxygenation with ambient O2 (simulated ischemia/reperfusion). Cell viability is expressed as the mean ± SEM percentage of the survival of untreated control cells (Control/Control normoxia), n = 6 in each group, *p < 0.05. The figure is representative of three independent experiments
PI3-kinase inhibition abolishes the cytoprotective effect of rhCCN2
To investigate to what extent PI3-kinase/Akt signaling pathways were involved in the CCN2-mediated protective mechanisms, cardiac myocytes were stimulated with rhCCN2 in the absence or presence of the PI3-kinase inhibitor LY294002, and then subjected to hypoxia/reoxygenation injury. As shown in Fig. 8a and b, inhibition of PI3-kinase abolished the cytoprotective effects of rhCCN2, indicating that the protective effect of rhCCN2 in cardiac myocytes is mediated by PI3-kinase/Akt. The negative control compound, LY303511, with respect to the PI3-kinase inhibitory activity of LY294002 did not influence cell viability (Fig. 8a and b). TUNEL-staining of cardiac myocytes exposed to hypoxia/reoxygenation showed decreased number of apoptotic cells in samples preincubated in the presence of rhCCN2 (200 nmol/L) (Fig. 8c). Figure 8d and e show examples of TUNEL positive and negative cardiac myocytes, respectively.
Fig. 8.

Analysis of cell death quantified by LDH and adenylate kinase released into the cell culture medium of cardiac myocytes exposed to hypoxia (1 % O2) for four hours and subsequently one hour reoxygenation to ambient O2 (simulated ischemia/reperfusion). a-b Viability of cells preincubated for 20 h in the absence (Control hypoxia) or presence of 200 nmol/L rhCCN2 (CCN2), or with 200 nmol/L rhCCN2 and 10 μmol/L of PI3-kinase inhibitor LY294002 (CCN2 + LY294002), or 200 nmol/L rhCCN2 and 10 μmol/L LY303511 (Negative control with respect to PI3-kinase inhibitory activity of LY294002) (CCN2 + LY303511), before exposure to hypoxia and reoxygenation. Control cells were not exposed to hypoxia or rhCCN2 (Control normoxia). Cell viability is expressed as the mean ± SEM percentage of the survival of untreated control cells, n = 6 in each group, *p < 0.05 vs. control. c TUNEL of cells preincubated in the absence or presence of 200 nmol/L rhCCN2 for 20 h, then exposed to hypoxia and subsequently reoxygenation. Histogram demonstrates mean of three independent assays (number of % apoptotic cells from a minimum of 10 visual fields per assay). Data are mean ± SEM. *p < 0.05 vs. control. d Example of TUNEL positive cardiac myocytes. e Example of TUNEL negative cardiac myocytes
Analysis of the transcriptome of cardiac myocytes stimulated with rhCCN2
Illumina gene expression bead arrays (MouseWG-6 v2 bead chips) were used to analyze the transcriptome of cardiac myocytes incubated in the absence or presence of rhCCN2 (200 nmol/L) for 48 h. The total number of genes analyzed was 38,805. Based on statistical analysis with a moderated t-statistic (Benjamini and Hochberg’s method to control the false discovery rate), 6,476 genes were found to be significantly up- or down-regulated in cells stimulated with CCN2. Gene ontology analysis revealed that genes included under the terms “Anti-apoptosis” (P = 0.01; a subordinate of “Apoptosis-related genes” (P = 1.6*10−5)), “Response to wounding” (P = 2.6*10−8), and “Response to stress” (P = 3.8*10−5) were significantly overrepresented in cardiac myocytes exposed to rhCCN2. Graphical presentation of the genes included under these terms is provided by heat maps illustrating the relative expression levels of these genes (Fig. 9 and 10). A listing of the top 15 genes displaying the greatest fold up- or down-regulation is presented in Table 1. Several of the most highly up-regulated genes in this list have previously been reported to exert cardioprotective actions and increase tolerance towards ischemia/reperfusion injury, i.e. WISP1 (CCN4), GDF15, and lipocalin-2 (Venkatesan et al. 2010; Xu et al. 2006; Roudkenar et al. 2009), or activate cellular prosurvival mechanisms (the dual specificity tyrosine phosphorylation-regulated kinase DYRK3) (Guo et al. 2010). Up-regulation of these genes in cardiac myocytes stimulated with rhCCN2 for 48 h were confirmed by real-time qPCR using sequence specific Taqman probes (Fig. 11). CCN2-stimulated induction of WISP1 and GDF15, among others on the top-15 list, was also found to be sensitive to inhibition of Akt with API-2, demonstrating that the transcriptome of CCN2-stimulated cardiac myocytes at least in part reflected activation of Akt signaling pathways (Fig. 11). However, CCN2-stimulated increase of lipocalin-2 and DYRK3 mRNA expression did not appear to be sensitive to Akt-inhibition (Fig. 11).
Fig. 9.

Heat maps illustrating relative expression levels of differentially expressed genes relating to the Gene Ontology terms “response to wounding” and “anti-apoptosis” in cardiac myocytes stimulated in the absence or presence of rhCCN2 (200 nmol/L) for 48 h. Yellow/red and blue colors indicate expression levels above and below average, respectively
Fig. 10.

Heat maps illustrating relative expression levels of differentially expressed genes relating to the Gene Ontology terms “response to stress” and “apoptosis” (apoptosis-related genes) in cardiac myocytes stimulated in the absence or presence of rhCCN2 (200 nmol/L) for 48 h. Yellow/red and blue colors indicate expression levels above and below average, respectively
Table 1.
The most up- and down-regulated genes from the DNA microarray analysis of cardiac myocytes cultured in the absence (n = 6) or presence (n = 6) of 200 nmol/L CCN2 for 48 h
| Description | Symbol | Fold change |
|---|---|---|
| WNT1 inducible signaling pathway protein 1 | Wisp1 | 5,9 |
| Growth differentiation factor 15 | Gdf15 | 4,8 |
| Lipocalin 2 | Lcn2 | 4,3 |
| Solute carrier family 40 (iron-regulated transporter), member 1 | Slc40a1 | 4,1 |
| Adenosine monophosphate deaminase 3 | Ampd3 | 3,7 |
| Myosin, light polypeptide kinase 2, skeletal muscle | Mylk2 | 3,4 |
| Breakpoint cluster region | Bcr | 3,3 |
| Chemokine (C-X-C motif) ligand 1 | Cxcl1 | 3,3 |
| Dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 3 | Dyrk3 | 3,3 |
| Ankyrin repeat domain 10 | Ankrd10 | 3,1 |
| Cytokine inducible SH2-containing protein | Cish | 3,1 |
| Feminization 1 homolog b (C. elegans) | Fem1b | 3,1 |
| Sulfiredoxin 1 homolog (S. cerevisiae) | Srxn1 | 3,0 |
| Teashirt zinc finger family member 1 | Tshz1 | 3,0 |
| Solute carrier family 6 (neurotransmitter transporter, taurine), member 6 | Slc6a6 | 3,0 |
| Lymphocyte antigen 6 complex, locus A | Ly6a | −8,3 |
| Radical S-adenosyl methionine domain containing 2 | Rsad2 | −6,3 |
| Aquaporin 1 | Aqp1 | −5,6 |
| Interferon-induced protein with tetratricopeptide repeats 3 | Ifit3 | −4,8 |
| Lymphocyte antigen 6 complex, locus C1 | Ly6c1 | −4,7 |
| Lymphocyte antigen 6 complex, locus E | Ly6e | −4,7 |
| Interferon, alpha-inducible protein 27 like 2A | Ifi27l2a | −4,6 |
| Claudin 5 | Cldn5 | −4,6 |
| Adenylate cyclase 4 | Adcy4 | −4,0 |
| Thymic stromal lymphopoietin | Tslp | −3,8 |
| Serine (or cysteine) peptidase inhibitor, clade A, member 3N | Serpina3n | −3,8 |
| Caveolin-1, caveolae protein | Cav1 | −3,8 |
| Histocompatibility 2, T region locus 23 | H2-T23 | −3,8 |
| Multimerin 2 | Mmrn2 | −3,7 |
| Lysozyme 1 | Lyz1 | −3,6 |
Fig. 11.

Histograms of quantitative real-time RT-PCR analyses of mRNA levels of WISP1, GDF15, Lipocalin-2, and DYRK3, relative to 18S rRNA levels in cardiac myocytes incubated for 48 h without rhCCN2, with 200 nmol/L rhCCN2, or with 200 nmol/L rhCCN2 in combination with 10 μmol/L API-2. Data are mean ± SEM, n = 3 in each group, *p < 0.05
Discussion
The present study discloses the novel findings that CCN2 increases the tolerance of adult, fully differentiated cardiac myocytes towards hypoxia/reoxygenation-induced oxidative stress and cell death. The cytoprotective effect of CCN2 was engendered by direct action on cardiac myocytes and was also supported by activation of stress response- and anti-apoptotic gene programs. The principal signaling pathway that conferred the cytoprotective actions of CCN2, was a PI3-kinase sensitive Akt/GSK-3β signaling pathway.
In a recent report we demonstrated that CCN2 increases the tolerance of the heart towards ischemia/reperfusion injury both in transgenic mice with cardiac-restricted overexpression of CCN2, as well as in ex vivo perfused mouse hearts exposed to rhCCN2 (Ahmed et al. 2011). However, an issue not yet resolved, was to what extent CCN2 acted through modulation of extracellular matrix protein functions, or rather exerted its cardioprotective effects by direct action on cardiac myocytes. Despite the fact that a receptor for CCN2 has not yet been properly characterized, the current study reveals that cardiac myocytes are direct targets of rhCCN2. The conclusion that the Akt/GSK-3β phosphokinase cascade was the relevant signaling pathway in the CCN2-mediated cytoprotection was supported by several observations. First, phosphorylation of Akt at Ser473 and GSK-3β at Ser9 were the only robustly regulated phosphokinases detected in cardiac myocytes stimulated with rhCCN2. Second, Akt-mediated phosphorylation of GSK-3β at Ser9 with subsequent inhibition of GSK-3β, reflected in reduced phosphorylation of glycogen synthase, a canonical substrate of GSK-3β. Inhibition of GSK-3β activities has previously been reported to be a nodal point in protection of the heart towards ischemia-reperfusion injury through inhibition of the mitochondrial transition pore (Juhaszova et al. 2004; Juhaszova et al. 2009). Third, the transcriptome of CCN2-stimulated cardiac myocytes revealed induction of gene expression programs with recognized cytoprotective functions in hypoxia/reoxygenation injury and oxidative stress. In this respect, several of the most substantially up-regulated genes in cardiac myocytes treated with rhCCN2 have previously been reported to confer cardioprotection towards ischemia/reperfusion injury, such as for example WISP1/CCN4, GDF-15, and lipocalin-2 (Venkatesan et al. 2010; Xu et al. 2006; Roudkenar et al. 2009;). In congruence with the ultimate role of Akt/GSK-3β signaling in CCN2-mediated cytoprotection, as demonstrated in this study, CCN2-stimulated induction of WISP1/CCN4 and GDF15 expression were also sensitive to inhibition of Akt. Thus, the gene expression signature caused by prolonged exposure to CCN2 apparently corroborates the immediate protective effects of Akt/GSK-3β phosphokinase signaling. Yet, the observation that inhibition of PI3-kinase/Akt signaling did not prevent all CCN2-mediated gene regulation may suggest that CCN2 also activates other signaling pathways. The latter contention would be consistent with the more robust cardioprotection observed in Langendorff-perfused hearts from transgenic mice with cardiac-restricted overexpression of CCN2 versus that of hearts exposed to rhCCN2 immediately before ischemia (Ahmed et al. 2011). However, activation of the Akt/GSK-3β phosphokinase cascade was the only confidently activated signaling pathway observed upon exposure of adult cardiac myocytes to rhCCN2. In other cell types CCN2 has been reported to activate TrkA receptor signaling, p38 or ERK1/2 MAP kinases, or SMAD2/3 signaling depending on cellular context (Rachfal and Brigstock 2005). Neither of these pathways was stimulated by CCN2 in the adult cardiac myocytes. Thus, CCN2-engendered signaling appears to be dependent on both cell type, and extent of cell differentiation. Although a receptor for CCN2 has not yet been convincingly characterized, the competitive inhibition of CCN2-stimulated activation of Akt by overexpression of the PH domain of GRK2ct, as shown in this study, demonstrates that direct action of CCN2 on cardiac myocytes elicits mechanisms that activate Akt at the plasma membrane interface.
Gene ontology analysis of the CCN2-stimulated transcriptome of cardiac myocytes revealed activation of cellular and biological processes that help increasing the tolerance of cardiac myocytes towards hypoxia/reoxygenation-induced injury, i.e. anti-apototic genes, stress-responsive genes, and wound-response genes. A recent study reported that recombinant hCCN2 induces pro-fibrotic and pro-inflammatory factors in the H9c2 cardiac myocyte cell line (Wang et al. 2010). The gene ontology analysis of the CCN2-mediated transcriptome of fully differentiated, adult cardiac myocytes did not disclose statistically significant regulation of pro-inflammatory or pro-fibrotic gene programs. Yet, specific proinflammatory cytokines, for example interleukin-6 and interleukin-1β, were found to be slightly upregulated (fold change 1.2 and 1.3, respectively), while TNF-α and interleukin-8 mRNA levels were not regulated in the DNA microarray of CCN2-stimulated cardiac myocytes in our study. Such proinflammatory factors may also be survival factors in ischemia/reperfusion injury despite their more dubious role upon prolonged presence. It should be noted, however, that transgenic mice with cardiac-restricted overexpression of CCN2 did not reveal an inflammatory morphologic substrate in the heart (Ahmed et al. 2011).
An emerging issue related to the use of purified, recombinant human CCN2 is to what extent CCN2 was the active principal or substance in the purified preparation that stimulated phosphorylation of Akt and GSK-3β. Concentration effect curves of CCN2-stimulated phosphorylation of Akt and GSK-3β in Rat-2-fibroblasts revealed half-maximum stimulation at approximately 200 nmol/L of CCN2, with maximal effective concentrations between 500 nmol/L and 1 μmol/L. CCN2 stimulated phosphorylation of Akt and exerted cytoprotective actions in adult cardiac myocytes within the same range of concentrations. Furthermore, a peptide from the c-terminal region of CCN2 (11.2 kDa C-terminal fragment of human CCN2), previously reported to exert agonist effects, was a partial agonist with respect to phosphorylation of Akt and GSK-3β, albeit substantially less potent than the full-length protein. Indeed, this study is first to provide data of efficacy and potency relationships of the the C-terminal CCN2 fragment versus full-length CCN2. In addition, peptides derived from the Thrombospondin and IGFBP homology domains of CCN2 inhibited CCN2-stimulated phosphorylation of Akt and GSK-3β. The two peptides exerted additive effects, suggesting that competition at several epitopes of CCN2 were involved in inhibition of CCN2-stimulated phosphorylation of Akt and GSK-3β. Thus, the data consistently indicate that recombinant CCN2 was the active principal in the purified preparation. The similar potencies and efficacies of glycoylated versus deglycosylated rhCCN2 indicate that glycosylation of CCN2 does appear to affect signaling per se, but may rather affect other properties of CCN2, for example binding of extracellular matrix proteins and distribution among tissues.
CCN2-stimulated increase of phospho-Akt (Ser473) levels has previously been reported in neonatal rat cardiac myocytes (Hayata et al. 2008). In the same study CCN2 also stimulated phosphorylation of several members of the MAP kinase family, that is ERK1/2, JNK, and P38. However, it is well known that neonatal cardiac myocytes respond very differently compared with fully differentiated adult cardiac myocytes. Interestingly, CCN1, another member of the CCN-family, has also been reported to protect neonatal rat cardiac myocytes from oxidative stress via an Akt-sensitive signaling pathway (Yoshida et al. 2007). Furthermore, WISP1/CCN4 ameliorated doxorubicin-induced death of adult mouse cardiac myocytes by several mechanisms, including inhibition of p38 and JNK MAP kinases, as well as activation of Akt (Venkatesan et al. 2010). However, to what extent phosphorylation and activation of Akt is a class specific effect of CCN-proteins is currently not known. These issues await recombinant expression and purification of the different members of the CCN family.
In conclusion, this study demonstrates that adult cardiac myocytes are direct targets of CCN2, and that CCN2 stimulates the Akt/GSK-3β signaling pathway, with resulting induction of a transcriptome that increases the tolerance of cardiac myocytes towards hypoxia/reoxygenation injury and oxidative stress. Thus, recombinant CCN2, or a compound with its agonist properties, may be pursued as a putative novel pharmacologic intervention in acute coronary syndromes in order to reduce infarct size.
Acknowledgments
This study was supported by grants from the Norwegian Council for Cardiovascular Research under the Norwegian Health Association, The Research Council of Norway, and the South Eastern Norway Regional Health Authority.
Disclosures
The authors Ingvild Tronstad Moe, M. Shakil Ahmed, and Håvard Attramadal are inventors in a patent application related to the potential application of CCN2/CTGF as a cardioprotectant (PCT/GB2009/002218;WO/2010/032007).
Abbreviations
- rhCCN2
Recombinant full-length, human CCN2
- GSK-3β
Glycogen synthase kinase 3 beta
- AK
Adenylate kinase
- LDH
Lactate dehydrogenase
References
- Ahmed MS, Gravning J, Martinov VN, von Lueder TG, Edvardsen T, Czibik G, Moe IT, Vinge LE, Øie E, Valen G, Attramadal H. Mechanisms of novel cardioprotective functions of CCN2/CTGF in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2011;300:H1291–H1302. doi: 10.1152/ajpheart.00604.2010. [DOI] [PubMed] [Google Scholar]
- Ahmed MS, Øie E, Vinge LE, Yndestad A, Andersen GØ, Andersson Y, Attramadal T, Attramadal H. Connective tissue growth factor-a novel mediator of angiotensin II-stimulated cardiac fibroblast activation in heart failure in rats. J Mol Cell Cardiol. 2004;36:393–404. doi: 10.1016/j.yjmcc.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Ahmed MS, von Lueder TG, Øie E, Kjekshus H, Attramadal H. Induction of myocardial connective tissue growth factor in pacing-induced heart failure in pigs. Acta Physiol Scand. 2005;184:27–36. doi: 10.1111/j.1365-201X.2005.01416.x. [DOI] [PubMed] [Google Scholar]
- Aras MA, Hartnett KA, Aizenman E (2008) Assessment of cell viability in primary neuronal cultures. Curr Protoc Neurosci Chapter 7:Unit [DOI] [PubMed]
- Bohr W, Kupper M, Hoffmann K, Weiskirchen R. Recombinant expression, purification, and functional characterisation of connective tissue growth factor and nephroblastoma-overexpressed protein. PLoS One. 2010;5:e16000. doi: 10.1371/journal.pone.0016000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. 2000;32:1805–1819. doi: 10.1006/jmcc.2000.1215. [DOI] [PubMed] [Google Scholar]
- Chujo S, Shirasaki F, Kondo-Miyazaki M, Ikawa Y, Takehara K. Role of connective tissue growth factor and its interaction with basic fibroblast growth factor and macrophage chemoattractant protein-1 in skin fibrosis. J Cell Physiol. 2009;220:189–195. doi: 10.1002/jcp.21750. [DOI] [PubMed] [Google Scholar]
- Chuva de Sousa Lopes SM, Feijen A, Korving J, Korchynskyi O, Larsson J, Karlsson S, Ten DP, Lyons KM, Goldschmeding R, Doevendans P, Mummery CL. Connective tissue growth factor expression and Smad signaling during mouse heart development and myocardial infarction. Dev Dyn. 2004;231:542–550. doi: 10.1002/dvdy.20162. [DOI] [PubMed] [Google Scholar]
- Crouch SP, Kozlowski R, Slater KJ, Fletcher J. The use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J Immunol Methods. 1993;160:81–88. doi: 10.1016/0022-1759(93)90011-U. [DOI] [PubMed] [Google Scholar]
- Drazner MH, Peppel KC, Dyer S, Grant AO, Koch WJ, Lefkowitz RJ. Potentiation of beta-adrenergic signaling by adenoviral-mediated gene transfer in adult rabbit ventricular myocytes. J Clin Invest. 1997;99:288–296. doi: 10.1172/JCI119157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem. 2010;285:13223–13232. doi: 10.1074/jbc.M110.102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayata N, Fujio Y, Yamamoto Y, Iwakura T, Obana M, Takai M, Mohri T, Nonen S, Maeda M, Azuma J. Connective tissue growth factor induces cardiac hypertrophy through Akt signaling. Biochem Biophys Res Commun. 2008;370:274–278. doi: 10.1016/j.bbrc.2008.03.100. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD, Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004;113:1535–1549. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3beta in cardioprotection. Circ Res. 2009;104:1240–1252. doi: 10.1161/CIRCRESAHA.109.197996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi T, Yamaai T, Asano M, Nawachi K, Suzuki M, Sugimoto T, Takigawa M. Overexpression of connective tissue growth factor/hypertrophic chondrocyte-specific gene product 24 decreases bone density in adult mice and induces dwarfism. Biochem Biophys Res Commun. 2001;281:678–681. doi: 10.1006/bbrc.2001.4379. [DOI] [PubMed] [Google Scholar]
- O’Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007;357:271–296. doi: 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
- Panek AN, Posch MG, Alenina N, Ghadge SK, Erdmann B, Popova E, Perrot A, Geier C, Dietz R, Morano I, Bader M, Ozcelik C. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS One. 2009;4:e6743. doi: 10.1371/journal.pone.0006743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachfal AW, Brigstock DR. Structural and functional properties of CCN proteins. Vitam Horm. 2005;70:69–103. doi: 10.1016/S0083-6729(05)70003-0. [DOI] [PubMed] [Google Scholar]
- Roudkenar MH, Halabian R, Roushandeh AM, Nourani MR, Masroori N, Ebrahimi M, Nikogoftar M, Rouhbakhsh M, Bahmani P, Najafabadi AJ, Shokrgozar MA. Lipocalin 2 regulation by thermal stresses: protective role of Lcn2/NGAL against cold and heat stresses. Exp Cell Res. 2009;315:3140–3151. doi: 10.1016/j.yexcr.2009.08.019. [DOI] [PubMed] [Google Scholar]
- Venkatesan B, Prabhu SD, Venkatachalam K, Mummidi S, Valente AJ, Clark RA, Delafontaine P, Chandrasekar B. WNT1-inducible signaling pathway protein-1 activates diverse cell survival pathways and blocks doxorubicin-induced cardiomyocyte death. Cell Signal. 2010;22:809–820. doi: 10.1016/j.cellsig.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, McLennan SV, Allen TJ, Twigg SM. Regulation of pro-inflammatory and pro-fibrotic factors by CCN2/CTGF in H9c2 cardiomyocytes. J Cell Commun Signal. 2010;4:15–23. doi: 10.1007/s12079-009-0083-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kimball TR, Lorenz JN, Brown DA, Bauskin AR, Klevitsky R, Hewett TE, Breit SN, Molkentin JD. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res. 2006;98:342–350. doi: 10.1161/01.RES.0000202804.84885.d0. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Togi K, Matsumae H, Nakashima Y, Kojima Y, Yamamoto H, Ono K, Nakamura T, Kita T, Tanaka M. CCN1 protects cardiac myocytes from oxidative stress via beta1 integrin-Akt pathway. Biochem Biophys Res Commun. 2007;355:611–618. doi: 10.1016/j.bbrc.2007.01.195. [DOI] [PubMed] [Google Scholar]