

Abstract
The amnion is a specialized tissue in contact with the amniotic fluid, which is in a constantly changing state. To investigate the importance of epigenetic events in this tissue in the physiology and pathophysiology of pregnancy, we performed genome-wide DNA methylation profiling of human amnion from term (with and without labor) and preterm deliveries. Using the Illumina Infinium HumanMethylation27 BeadChip, we identified genes exhibiting differential methylation associated with normal labor and preterm birth. Functional analysis of the differentially methylated genes revealed biologically relevant enriched gene sets. Bisulfite sequencing analysis of the promoter region of the oxytocin receptor (OXTR) gene detected two CpG dinucleotides showing significant methylation differences among the three groups of samples. Hypermethylation of the CpG island of the solute carrier family 30 member 3 (SLC30A3) gene in preterm amnion was confirmed by methylation-specific PCR. This work provides preliminary evidence that DNA methylation changes in the amnion may be at least partially involved in the physiological process of labor and the etiology of preterm birth and suggests that DNA methylation profiles, in combination with other biological data, may provide valuable insight into the mechanisms underlying normal and pathological pregnancies.
1. Introduction
The human amnion is the inner layer of the fetal membranes composed of a monolayer of epithelial cells attached to a basement membrane overlying a collagen-rich stroma [1, 2]. This tissue, which encloses the amniotic fluid, protects the fetus from external mechanical forces and provides an environment that supports fetal movement and growth [3, 4]. The amnion is also a metabolically active tissue involved in the synthesis of various substances with important functions during pregnancy, including prostaglandins and cytokines [1, 5, 6]. It is particularly well known as a major source of prostaglandin E2, a potent molecule mediating cervical ripening and myometrial contraction [7–10], whose levels dramatically increase before and during labor [11, 12].
The amniotic membrane provides most of the tensile strength of the fetal membranes, and alterations in its integrity can lead to undesirable pregnancy outcomes such as preterm premature rupture of membranes (PPROMs) [1, 13], which complicates 3% of all pregnancies and is responsible for approximately one-third of all preterm births (PTBs) [14]. Given the important role of the amnion in the maintenance of pregnancy and parturition, investigation into molecular events occurring in this tissue may contribute to a better understanding of physiological and pathological processes involved in pregnancy.
Considering that the amniotic fluid is in a constantly changing state, it may be critical that the amnion properly responds to environmental cues from the amniotic fluid to accommodate the dynamic needs of the fetus, which could be mediated through epigenetic processes. A previous study by Wang et al. [15] has shown that matrix metalloproteinase 1 (MMP1), whose genetic variation is associated with susceptibility to PPROM [16], is regulated at the epigenetic level, specifically by DNA methylation, and that MMP1 promoter methylation status correlates with its expression in the amnion and association with PPROM. This finding suggests that the amnion represents an intriguing source of tissue for studying epigenetic events of potential physiological and pathological relevance.
In this study, we performed genome-wide methylation profiling of human term and preterm amnion in order to explore the possible importance of DNA methylation in physiologic labor as well as the etiology of PTB. In addition, independent of the genome-wide methylation study, we carried out methylation analysis of the promoter region of the oxytocin receptor (OXTR) gene whose role in human parturition is well established [17]. Given that OXTR expression in the amnion increases in association with the onset of labor [18] and that its aberrant methylation in other tissue types has been implicated in autism [19], a disorder that has been associated with PTB [20, 21], we sought to investigate if DNA methylation could represent one mechanism regulating OXTR gene function in the contexts of normal parturition and prematurity.
2. Materials and Methods
2.1. Placental Tissue Collection and Preparation
Fresh human placentas were collected in 2009 and 2010 at the University of Iowa Hospitals and Clinics in IA, USA and Instituto de Maternidad y Ginecología Nuestra Señora de las Mercedes in Tucumán, Argentina with signed informed consent and an institutional review board approval. We examined 121 placentas from three groups of patients undergoing: term cesarean delivery without labor (term no labor (TNL) group, n = 18), normal term vaginal delivery (term labor (TL) group, n = 40), and spontaneous preterm (<37 weeks of gestation) delivery (preterm labor (PTL) group, n = 63). Gestational age (GA) was determined using the first day of the last menstrual period as well as by ultrasound examination and was confirmed by assessment at birth. Each placenta was dissected into fetal (amnion, chorion) and maternal (decidua basalis) components within an hour of delivery. The amnion and chorion obtained from the extraplacental membranes (reflected membranes) were separated by blunt dissection under sterile conditions. Decidual tissue samples were macroscopically isolated from the surface of the basal plate of the placenta. After being cut into small pieces, the dissected tissues were placed in RNA later solution (Applied Biosystems, Carlsbad, CA, USA) and stored per manufacturer's recommendations until used. A subset of these samples was selected for genome-wide methylation analysis on the basis of their informativity in relation to our previous gene expression profiling study (unpublished). Additional samples used for validation experiments were selected primarily based on the quality of DNA or RNA extracted from the tissue samples.
2.2. DNA Preparation and Methylation Standards
Genomic DNA was extracted from placental tissue samples using the DNeasy Blood & Tissue Kit (QIAGEN, Valencia, CA, USA) following the manufacturer's protocol. The quality of the extracted DNA was evaluated by agarose gel electrophoresis. 500 ng of DNA was bisulfite-converted using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer's instructions, and used in subsequent experiments. Universal Methylated Human DNA Standard (Zymo Research), which is enzymatically methylated in vitro at all cytosines in CpG dinucleotides, was used as a positive control in the Illumina Infinium methylation assay. We also used Human Methylated and Non-methylated DNA Standards (Zymo Research) as positive and negative controls for methylation-specific PCR. Both of the standards are purified from DNMT1 and DNMT3b double-knockout HCT116 cells, but the methylated standard is enzymatically methylated at all cytosines in CpG dinucleotides.
2.3. Genome-Wide DNA Methylation Analysis
2.3.1. Illumina Infinium Methylation Assay
DNA methylation profiling was performed by the W.M. Keck Biotechnology Resource Laboratory at Yale University, using the Illumina Infinium HumanMethylation27 BeadChip (Illumina, San Diego, CA, USA). Details of the design and general properties of this platform have been previously described [22]. A total of 24 samples were assayed on two BeadChips (12 samples per chip) following the standard protocol provided by Illumina. The samples examined included 9 individual and 1 pooled amnion samples each from the TNL and TL groups, one pooled amnion sample from the PTL group obtained by combining 6 individual samples, and 3 controls (methylated DNA control treated with M.SssI methyltransferase (New England Biolabs, Ipswich, MA, USA), Universal Methylated Human DNA Standard (Zymo Research), and bisulfite-untreated control). These samples were selected from among patients who had participated in our previous gene expression profiling study (unpublished), performed independently of the current work. Based on this previous study, which showed heterogeneous global gene expression patterns among PTL samples, we only included one pooled PTL sample to assess a group DNA methylation average. The samples were arranged randomly on each chip and were processed in a blinded fashion. Table 1 summarizes the clinical characteristics of the three groups of samples studied.
Table 1.
Clinical characteristics of the three subject groups studied by genome-wide DNA methylation profiling.
| Parameter | TNL (n = 9)1 | TL (n = 9)1 | PTL (n = 6)2 |
|---|---|---|---|
| Gestational age (weeks)3 | 39.1 ± 0.8 | 38.8 ± 0.8 | 33.5 ± 2.6 |
| Race | |||
| White | 4 | 3 | 3 |
| Black | 0 | 0 | 1 |
| Other | 5 | 6 | 2 |
| Maternal age at delivery (years)3 | 29.7 ± 5.5 | 27 ± 4.4 | 28.7 ± 3.5 |
| (Range 22–38) | (Range 20–33) | (Range 25–33) | |
| Antibiotics during pregnancy or labor | |||
| Yes | 6 | 1 | 5 |
| No | 3 |
7 |
0 |
| Unknown | 0 | 1 | 1 |
| Birth weight (grams)3 | 3508.3 ± 267.2 | 3354.1 ± 348.6 | 2102.2 ± 724.6 |
| Infant gender | |||
| Female | 6 | 4 | 2 |
| Male | 3 | 5 | 4 |
1Examined both individually and as a pooled sample.
2Examined as a pooled sample.
3Data are presented as mean ± standard deviation (SD).
Abbreviations: TNL: term no labor; TL: term labor; PTL: preterm labor.
2.3.2. Quality Control and Statistical Analysis
Data analysis was conducted on a fee-for-service basis by the W.M. Keck Biostatistics Resource at Yale University with GenomeStudio Methylation Module v1.0 (Illumina). We evaluated the quality of the data based on the signals of assay built-in control probes (staining, hybridization, target removal, extension, bisulfite conversion, methylation signal specificity, background determination, and overall assay performance) and three experimental controls (two positive methylated controls and one non-bisulfite-converted control), and confirmed the reliability of our data. Principal component analysis (PCA) demonstrated that there is no significant batch effect among the three groups of samples examined. The methylation status of each interrogated CpG site was determined employing the β-value (defined as the fraction of methylation, calculation details described in a previous study [23]) method. An average β-value (AVG_Beta) for each CpG locus ranging from 0 (unmethylated) to 1 (completely methylated) was extracted utilizing the GenomeStudio software and used in further analyses. For the determination of differential methylation between two given groups, we used the Illumina custom error model. This model assumes a normal distribution of the methylation value (β) among replicates corresponding to a set of biological conditions (TNL, TL, and PTL). We prioritized differentially methylated CpG sites by difference score (DiffScore). DiffScore, which takes into account background noise and sample variability [24], was calculated using the following formula: DiffScore = 10sgn(β condition − β reference)log10 P, where β condition = β TL/β PTL, β reference = β TNL or β condition = β TNL/β PTL, β reference = β TL. The resulting differentially methylated CpG sites were annotated with respect to their nearest gene based on the information provided by Illumina. A more detailed description of the Illumina custom error model and the DiffScore has been provided previously [25].
2.3.3. Functional Enrichment Analysis
Differentially methylated genes (DMGs) with a DiffScore of >20 (equivalent to P-value of <0.01) were evaluated for functional enrichment using predefined gene sets from the Molecular Signatures Database (MSigDB) [26]. We searched for significantly enriched gene sets by computing overlaps between the lists of DMGs and the CP collection (canonical pathways, 880 gene sets) or the C5 collection (GO gene sets, 1454 gene sets) in the MSigDB. Gene sets with a P-value (based on the hypergeometric distribution) less than 0.05 were considered significant.
2.4. Bisulfite Sequencing (BS)
To validate methylation differences revealed by the genome-wide methylation assay, we performed bisulfite sequencing on urocortin (UCN), a gene identified as differentially methylated between the TL and PTL groups, and OXTR, a gene whose methylation status has recently been shown to be important in the pathogenesis of autism [19]. We investigated the methylation status of OXTR, given its significant role in parturition [17] and its labor-associated expression pattern in the amnion [18], which makes it a potential candidate gene for PTB. There are two OXTR CpG sites targeted by the Illumina Infinium BeadChip assay, both of which were not identified as being differentially methylated. However, because there is currently no evidence supporting the biological importance of the regions containing the two sites, we focused our BS analysis on CpG sites of known biological significance that are located in a different region of the OXTR gene. Primers for UCN were designed to cover the CpG site identified as being differentially methylated by genome-wide methylation profiling, using the default parameters of MethPrimer [27]. PCR amplification using the primer pair results in a 278 bp product that spans part of the promoter, exon 1, and part of intron 1 of UCN (-439 to -162 relative to translation start site (TSS)) containing 16 CpG sites. For OXTR, we used the same primers and PCR conditions as those used in the previous study [19]. PCR amplification using the primer set results in a 358 bp product that spans the OXTR promoter (-1195 to -838 relative to TSS) containing 22 CpG sites that has been associated with tissue-specific OXTR expression [28] and the development of autism [19]. The regions examined in both genes were located within CpG islands. We carried out our analysis using the same samples assayed on the BeadChips and eight additional independent PTL samples (TNL, n = 9; TL, n = 9; PTL, n = 14). Bisulfite-converted DNA was PCR-amplified using ZymoTaq DNA polymerase (Zymo Research). The resulting PCR products were run on an agarose gel and cloned into the pGEM-T Easy vector (Promega, Madison, WI, USA). Individual clones were isolated, amplified following standard protocols, and purified using the PureLink Quick Plasmid Miniprep Kit (Invitrogen, Carlsbad, CA, USA) per manufacturer's instructions. Ten clones per sample, on average, were isolated and sequenced at the University of Iowa DNA facility. Percentage methylation was determined for each CpG site similarly as done in previous work [19]. Statistical analysis was conducted using SigmaPlot 11.0 (Systat Software, San Jose, CA, USA). Significance of differential methylation (DM) was assessed using the t-test (two-tailed), Mann-Whitney (M-W) rank sum test (two-sided), one-way ANOVA, or Kruskal-Wallis (K-W) one-way ANOVA by ranks, as indicated in the text and/or figure legends. Post hoc analysis following ANOVA was performed using either the Holm-Sidak or Dunn's Method. A P < 0.05 was considered significant.
2.5. Encyclopedia of DNA Elements (ENCODE) ChIP-Seq Data
We examined the potential functional significance of the region of the OXTR gene containing CpG sites with statistically different DNA methylation status (CpGs-959 and -1084) using the ChIP-seq data from the ENCODE project available in the University of California Santa Cruz (UCSC) genome browser [29, 30]. We specifically used the suppressor of zeste 12 homolog (Drosophila) (SUZ12) and Pol2 ChIP-seq data generated by the laboratories of Michael Snyder at Stanford University and Vishy Iyer at the University of Texas Austin. The ChIP-Seq data were obtained using human cells (NT2-D1 for the SUZ12 data; GM18526, 18951, 19099, 19193, and ProgFib for the Pol2 data).
2.6. Methylation-Specific PCR (MSP)
Validation of DM was additionally carried out using methylation-specific PCR (MSP). Two pairs of primers (unmethylated and methylated) for each of the lysophosphatidic acid receptor 5 (LPAR5), paternally expressed 10 (PEG10), and solute carrier family 30 member 3 (SLC30A3) genes were designed using the MSP-specific default parameters of the MethPrimer program [27]. Bisulfite-converted DNA extracted from amnion tissues (TNL, n = 9; TL, n = 9; PTL, n = 14) was PCR-amplified using Biolase DNA polymerase (Bioline, Taunton, MA, USA). The resulting PCR products were visualized on a 2% agarose gel. Human Methylated and Non-methylated DNA Standards from Zymo Research were used as positive and negative controls.
2.7. RNA Extraction and Real-Time qRT-PCR
Total RNA was extracted from amnion (TNL, n = 14; TL, n = 34; PTL, n = 59) and decidua (TNL, n = 12; TL, n = 16; PTL, n = 31) tissues using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. The quality of extracted RNA was checked using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Reverse transcription was carried out with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems), using random hexamers as primers following the manufacturer's instructions. Real-time qRT-PCR was performed using synthesized cDNA as a template, gene-specific primers (UCN and OXTR) and Power SYBR Green PCR Master Mix (Applied Biosystems). The reactions (including no-template controls) were run in triplicate on the 7900HT Fast Real-Time PCR System (Applied Biosystems) using ACTB (beta actin) [31] as an endogenous reference. Data were analyzed with the SDS 2.4 software (Applied Biosystems), employing the comparative CT method [32]. Absence of nonspecific amplification was confirmed by dissociation curve analysis. Samples with a value that falls outside ±2 standard deviations of the group mean were defined as outliers and removed from the study. Statistical analysis was performed similarly as described above in the bisulfite sequencing section. Data were presented as mean ± standard error of the mean (SEM).
3. Results
3.1. Genome-Wide Patterns of DNA Methylation and Differentially Methylated CpG Loci between Term (Non-Labored and Labored) and Preterm Amnion Tissues
To investigate the possible involvement of epigenetic mechanisms in the physiology of normal labor and the pathogenesis of PTB, we examined the genome-wide methylation profiles of the amnion obtained following term (TNL and TL, n = 9 for each) and preterm (PTL, n = 6) deliveries using the Illumina Infinium BeadChip platform. The overall levels of DNA methylation in the experimental samples were low with third quartile AVG_Beta values between 0.4 and 0.55. Principal component analysis (PCA) placed the pooled TL and PTL samples close to each other and very distant from the pooled TNL sample (Figure 1), which indicates that the genome-wide methylation patterns in amnion tissues from the two spontaneous labor groups (regardless of gestational age (GA) at delivery) are more similar to each other than to those observed in non-labor tissues.
Figure 1.
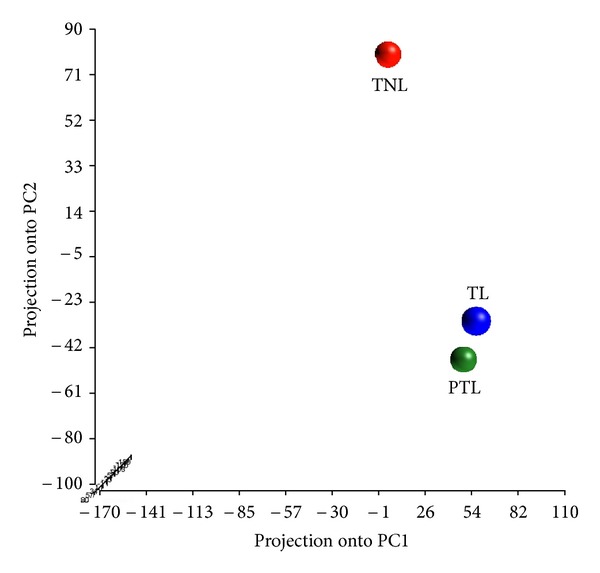
Principal component analysis (PCA) plot of DNA methylation profiles in term (non-labored and labored) and preterm amnion. Each colored dot represents a pooled DNA sample from term no labor (TNL), term labor (TL), or preterm labor (PTL) group. Note that the TNL sample is placed distantly from the TL or PTL samples, indicating that the TNL group displays distinctly different methylation patterns compared to the other two groups.
We also performed gene/locus level analysis of differential methylation (DM), searching for methylation changes associated with labor and/or PTB at specific CpG sites. Using the Illumina custom error model algorithm, we identified 65 CpG sites in 64 and 61 autosomal genes each that are differentially methylated between the TNL and TL groups and the TL and PTL groups, respectively with a DiffScore of >30 (equivalent to P-value of <0.001). Listed in Table 2 are the 15 most highly differentially methylated genes (DMGs). It was noted that among the genes with differentially methylated sites, although very few, were those belonging to special classes of genes, including noncoding RNAs and imprinted genes (such as Down syndrome critical region gene 10 (DSCR10), FBXL19 antisense RNA 1 (FBXL19-AS1), and paternally expressed 10 (PEG10) as shown in Table 2), many of which have regulatory functions in diverse biological processes.
Table 2.
List of top 15 differentially methylated autosomal genes in amnion tissues from term (TNL, TL) and preterm (PTL) deliveries ranked by statistical significance1.
| TNL versus TL2 | TL versus PTL3 | ||||
|---|---|---|---|---|---|
| Gene | Locus | CpG island4 | Gene | Locus | CpG island4 |
| IL32 | 16p13.3 | No | TOB1 | 17q21 | No |
| EDARADD | 1q42.3 | No | PNPLA3 | 22q13.31 | No |
| STK19 | 6p21.3 | No | ZNF671 | 19q13.43 | No |
| EXTL1 | 1p36.1 | No | DAB2IP 7 | 9q33.1–q33.3 | No |
| HLA-DQB2 | 6p21 | No | MFNG | 22q12 | No |
| MFSD3 | 8q24.3 | Yes | UCN | 2p23–p21 | Yes |
| RAB31 | 18p11.3 | Yes | EXOC3L2 | 19q13.32 | Yes |
| PNPLA3 | 22q13.31 | No | SLC44A2 | 19p13.1 | Yes |
| GRHPR | 9q12 | Yes | FBXL19-AS1 6 | 16p11.2 | Yes |
| MPHOSPH10 | 2p13.3 | No | DLGAP5 | 14q22.3 | Yes |
| PEG10 5 | 7q21 | No | SLC30A3 | 2p23.3 | Yes |
| DSCR10 6 | 21q22.13 | No | CHFR | 12q24.33 | No |
| SRRD | 22q12.1 | Yes | C11orf1 | 11q23.1 | No |
| POLI | 18q21.1 | Yes | SLC24A4 | 14q32.12 | No |
| OSTalpha | 3q29 | No | PI4KB | 1q21 | No |
1Statistical significance was determined based on P-values calculated from DiffScores. All genes listed here have a DiffScore >40 (corresponding to P-value of <0.0001).
2Genes most highly methylated in the TL group compared to the TNL group.
3Genes most highly methylated in the PTL group compared to the TL group.
4Defined by the CpG island track in the UCSC Genome Browser.
5An imprinted gene.
6Non-protein coding genes.
7A gene identified as having three non-island CpG sites with a DiffScore >40.
Abbreviations: TNL: term no labor; TL: term labor; PTL: preterm labor.
3.2. Functional Enrichment Analysis
To determine the biological significance of DMGs, functional annotation analysis was performed. Our approach involved examining the extent of overlap between our lists of DMGs and predefined annotated gene sets from the MSigDB [26] (see Section 2 for further details). For this analysis, we used gene lists with a less stringent P-value cutoff of <0.01 (corresponding to a DiffScore of >20), given the small number of DMGs (n = 65) with a P-value below 0.001. We found that 7 gene sets were significantly overrepresented (P < 0.05) in the list of 110 DMGs between the TNL and TL groups. The seven enriched gene sets included cation transport, ion channel activity, and those shown in Table 3, most of which are highly relevant to molecular processes involved in physiologic labor. Among the 186 DMGs between the TL and PTL groups, 17 gene sets were overrepresented. Many of the enriched gene sets were found to be associated with the regulation of cell behavior and extracellular matrix-cell interactions, including focal adhesion, cell junction, cell-substrate adherens junction, and integrin binding (Table 3).
Table 3.
Gene sets overrepresented among differentially methylated genes in amnion tissues from term (TNL, TL) and preterm (PTL) deliveries1.
| TNL versus TL | |
|---|---|
| Gene set2 | P-value3 |
| HEART_DEVELOPMENT | 0.012 |
| POSITIVE_REGULATION_OF_CYTOKINE_PRODUCTION | 0.017 |
| GATED_CHANNEL_ACTIVITY | 0.024 |
| REGULATION_OF_HEART_CONTRACTION | 0.041 |
| REGULATION_OF_CYTOKINE_PRODUCTION | 0.044 |
|
| |
| TL versus PTL | |
|
| |
| NEGATIVE_REGULATION_OF_TRANSFERASE_ACTIVITY | 0.007 |
| ADHERENS_JUNCTION4 | 0.011 |
| HEPARIN_BINDING | 0.014 |
| FOCAL_ADHESION_FORMATION | 0.02 |
| FOCAL_ADHESION | 0.024 |
1Presented are the top 5 most significantly enriched gene sets from C5 collection (GO gene sets).
2Defined in the Molecular Signatures Database (MSigDB).
3The cutoff for statistical significance was P = 0.05.
4Also identified as being enriched (P = 0.049) in the analysis performed with the CP collection.
Abbreviations: TNL: term no labor; TL: term labor; PTL: preterm labor.
3.3. Bisulfite Sequencing (BS) Analysis of Differential Methylation
To validate DM detected by genome-wide methylation profiling, we performed BS analysis on UCN, a gene identified as being overmethylated in the PTL group compared with the TL group with a DiffScore >50 (Table 2). We performed the same analysis on one additional gene named oxytocin receptor (OXTR) whose mRNA and protein expression has been shown to be markedly upregulated in association with labor in primary human amnion epithelial cells [18]. Previous studies have demonstrated that the methylation status of the promoter region of this gene is associated with tissue-specific OXTR expression [28] and the development of autism [19], a disorder linked to PTB [20, 21, 33, 34]. These findings intrigued us to investigate whether DNA methylation could represent one mechanism regulating the labor-associated activity of OXTR in the amnion. We selected the two genes (UCN and OXTR), given their crucial role in normal labor and parturition, which makes them potential candidate genes for PTB. Details on the regions amplified, samples used in the BS experiments, and statistical tests performed for the analysis of the sequencing results are given in Section 2 and Figure 2.
Figure 2.
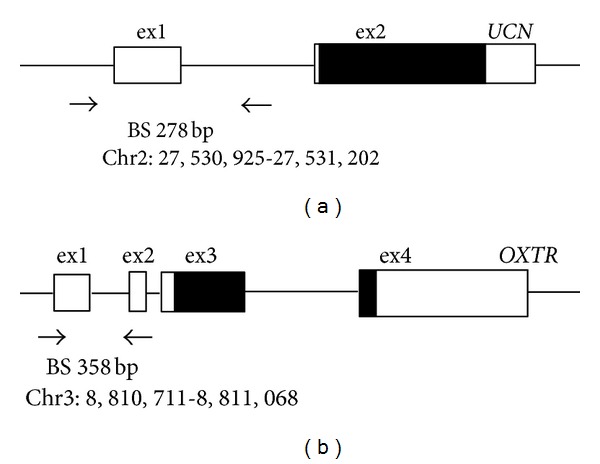
Schematic representation of CpG island regions of UCN (a) and OXTR (b) analyzed by bisulfite sequencing (BS). Black horizontal arrows denote BS PCR primer binding sites. Solid box: coding region; open box: untranslated region. The expected PCR product sizes and positions of the primer binding sites (chromosome and base count, NCBI Build GRCh37/hg19) are indicated. Further details on the PCR-amplified regions are provided in Section 2.
All 16 CpG dinucleotides interrogated in the UCN gene showed some degree of methylation with the ones at positions -361, -335, and -319, being more highly methylated (22.9–55.7%, Table 4) compared with those at other positions (1.1–17.1%). All except two CpG sites were overmethylated in the PTL samples compared to the TL samples, showing the expected direction of DM. However, the differences were not statistically significant.
Table 4.
UCN and OXTR promoter methylation status in the amnion and decidua from term (TNL, TL) and preterm (PTL) deliveries1.
| UCN | ||||||||
|---|---|---|---|---|---|---|---|---|
| Site2 | Amnion | Decidua | ||||||
| TL | PTL | P-value | TL | PTL | P-value | |||
| -1903 | 2.2% | 8.6% | 0.39 | 6.7% | 14.8% | 0.07 | ||
| -2793 | 2.2% | 11.4% | 0.07 | 15.6% | 12.5% | 0.97 | ||
| -3194 | 24.4% | 22.9% | 0.83 | 19.9% | 30.8% | 0.08 | ||
| -3354 | 23.3% | 27.1% | 0.56 | 23.2% | 25.8% | 0.75 | ||
| -3614 | 48.9% | 55.7% | 0.33 | 32.1% | 36.4% | 0.48 | ||
|
| ||||||||
| OXTR | ||||||||
| Site2 | Amnion | Decidua | ||||||
| TNL | TL | PTL | P-value | TNL | TL | PTL | P-value | |
|
| ||||||||
| -860 | 24.4% | 22.2% | 24.3% | 0.96 | 20% | 15.6% | 24.6% | 0.19 |
| -901 | 37.8% | 30% | 45.7% | 0.17 | 45.6% | 38.9% | 46.9% | 0.66 |
| -924 | 56.7% | 62.2% | 68.6% | 0.29 | 60% | 52.2% | 67.2% | 0.09 |
| -9345 | 50% | 41.1% | 59.3% | 0.22 | 46.7% | 42.2% | 57.1% | 0.02 |
| -9595 | 43.3% | 24.4% | 27.1% | 0.014 | 33.3% | 33.3% | 30.6% | 0.91 |
| -10845 | 4.4% | 4.4% | 5.7% | 0.97 | 10% | 0% | 8.7% | 0.008 |
1Presents average % methylation at each CpG site.
2Nucleotide positions relative to translation start site.
3CpG sites in UCN with the lowest P-value in each tissue type.
4CpG sites methylated at higher levels in both tissues than the average methylation level of all sites examined.
5CpG sites with statistically significant (P < 0.05) differential methylation in either tissue. Details of statistical tests used are described in Section 2.
Abbreviations: TNL: term no labor; TL: term labor; PTL: preterm labor.
For OXTR, since we had no priori data on the methylation status of the 22 CpG sites in amnion tissue, all three groups of samples (TNL, TL, and PTL) were examined. Consistent with the finding of Gregory et al. [19], 5 CpG sites at positions -959, -934, -924, -901, and -860 showed the highest levels (22.2–68.6%, Table 4) and variation in methylation, whereas very little or no methylation (0–5.7%) was observed at the other sites. We found that one (CpG-959) of the five sites was significantly differentially methylated among the three groups tested (one-way ANOVA, P = 0.014, Table 4). Pairwise comparisons (Holm-Sidak test) revealed significant differences between the TNL and TL groups (P = 0.017) and the TNL and PTL groups (P = 0.025) and borderline significant difference between the TL and PTL groups (P = 0.050), demonstrating more distinct differences in methylation at this site between non-labor and labor tissues than between term and preterm tissues.
To determine if the observed DM also occurs in other parts of the placenta where the genes are known to be expressed [17, 35], we extended our study to decidua tissues from the same groups of individuals. The decidua, which is of maternal origin, unlike the amnion of fetal origin [4], was selected, given that the function of OXTR in parturition has been well demonstrated in maternal tissue [17], and therefore, the examination of the decidua, along with the amnion, may allow us to compare the methylation state of the OXTR gene and possibly its importance in both fetal and maternal tissues.
The overall methylation patterns observed in the decidua were similar to those identified in the amnion. However, unlike in the amnion tissues, the methylation levels not at CpG-959, but at different sites (CpGs-934 and -1084), were found to be statistically significantly different (P = 0.02, 0.008, resp., K-W one-way ANOVA by ranks) among the three groups of the decidua tissues (Table 4). The CpG-1084 site, interestingly, was completely unmethylated in the TL group, whereas it was methylated to some small degree in the other two groups (TNL, 10%; PTL, 8.7%) (Table 4). Significant differences between the TL and TNL or PTL groups were confirmed by Dunn's post hoc test (P < 0.05). In the case of CpG-934, the difference was significant only between the TL and PTL groups. Taken together, it appears that there exist compartment-specific OXTR methylation patterns in the placenta.
3.4. Analysis of UCN and OXTR Gene Expression in the Amnion and Decidua
To evaluate the functional significance of the methylation status of the two genes, we performed gene expression analysis using qRT-PCR on an extended set of amnion and decidua tissues (n = 107, 59, resp.) from the three groups. Although the DM of UCN was not validated by BS, we observed a statistically significant 2.3-fold increase in its transcript levels in the PTL amnion samples compared to the TNL and TL samples (P < 0.001, K-W one-way ANOVA by ranks, Figure 3). There was also a statistically significant, but less than twofold increase in OXTR mRNA levels in the PTL amnion samples compared with the TL samples (P < 0.05, K-W one-way ANOVA by ranks, Dunn's post hoc test, Figure 3). The results were not replicated in the decidua samples for either gene. These findings suggest that the upregulation of UCN is specific to the amnion from spontaneous preterm deliveries, and that the DM observed in OXTR may not correlate with OXTR expression given that methylation generally plays a role in gene silencing.
Figure 3.
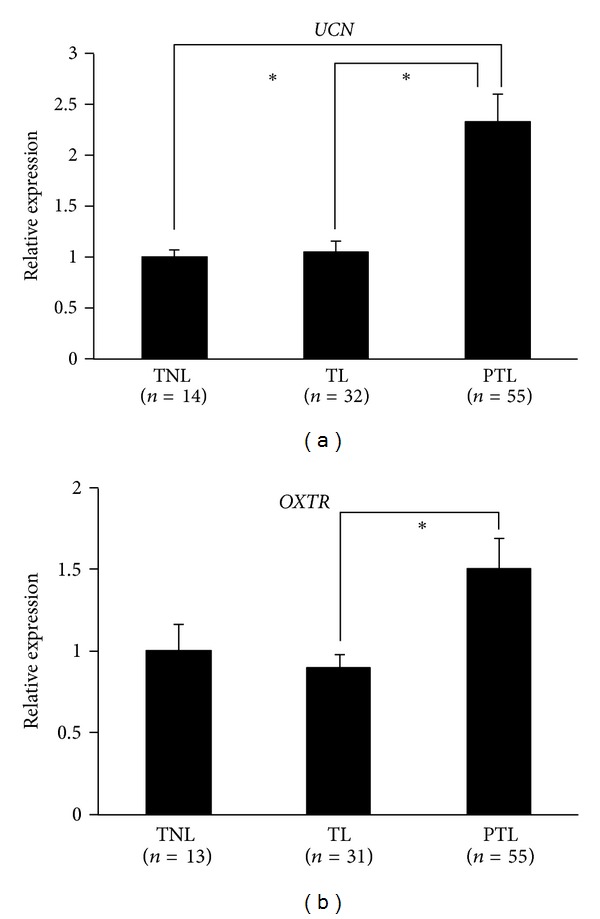
UCN and OXTR mRNA expression levels in term (non-labored and labored) and preterm amnion. Expression levels were normalized to that of beta-actin (ACTB). Experiments were performed in triplicate. Data presented are mean ± standard error of the mean (SEM). Asterisks represent statistically significant differences (P < 0.05, K-W one-way ANOVA by ranks followed by Dunn's post hoc test) between specified groups.
3.5. Methylation-Specific PCR (MSP) Analysis of Differential Methylation
As an alternative approach to validate DNA methylation differences captured by our genome-wide methylation study, we carried out methylation-specific PCR (MSP) for selected 3 DMGs between the TNL and TL groups (PEG10) and between the TL and PTL groups (LPAR5 and SLC30A3). Analysis of the same set of amnion samples used in BS revealed no intergroup differences in PEG10 and LPAR5 methylation (data not shown). However, the methylation status of SLC30A3 was in good agreement with our genome-wide methylation data with methylated MSP products present in 10 out of 14 (71%) PTL samples and none of the TL samples (Figure 4).
Figure 4.
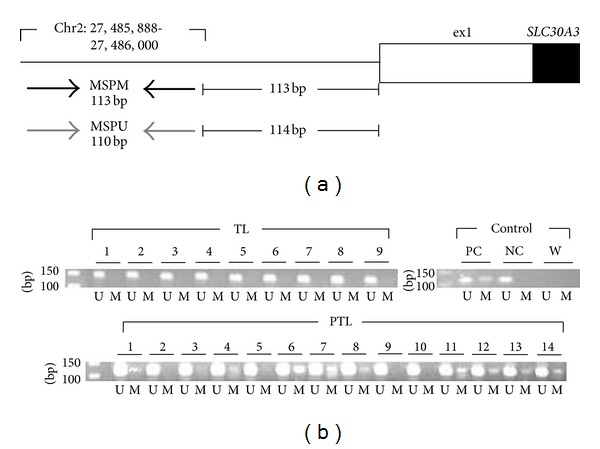
Methylation-specific PCR (MSP) analysis of SLC30A3. (a) Schematic representation of MSP primer binding sites. Black horizontal arrows: methylated-specific primer (MSPM) binding sites; gray horizontal arrows: unmethylated-specific primer (MSPU) binding sites. The expected PCR product sizes and positions of the primer annealing sites (chromosome and base count, NCBI Build GRCh37/hg19) are indicated. Solid box: coding region; open box: untranslated region. (b) Agarose gel electrophoresis of MSP products. M: product amplified with MSPM; U: product amplified with MSPU. PC: positive control, human methylated DNA standard; NC: negative control, human unmethylated DNA standard; W: water control (details can be found in Section 2). Note that the TL samples show only unmethylated PCR products, while many of the PTL samples show both methylated and unmethylated PCR products, indicative of partial methylation.
4. Discussion
The present study investigated if there exist unique genome-wide methylation signatures that distinguish among term (non-labored and labored) and preterm amnion tissues. Our methylation profiling revealed a higher degree of similarity between the methylation patterns in the TL and PTL pooled samples than those observed in the TNL pooled sample, suggesting the potential role of methylation in the regulation of labor, independent from GA. We identified a relatively small number of DMGs between the TNL and TL groups and the TL and PTL groups (65 genes each) at the P < 0.001 significance level. This observation may be attributed to the small sample size and the sample-to-sample variability related to GA. Gene set enrichment analysis of those genes revealed significant overrepresentation of pathways that appear to be functionally relevant (Table 3). The enrichment of pathways related to ion transport, ion channel activity, and cytokine production among the DMGs between the TNL and TL groups reflects biochemical and molecular events associated with the onset of labor, which, along with hormonal factors, help to initiate parturition. These results are at least partially in line with previous gene expression profiling studies reporting labor-associated cytokine-related gene signatures in human amniotic [36] and chorioamniotic [37] membranes. The overrepresentation of heart-(development and contraction) related gene sets may be explained by the presence of myofibroblasts in the connective tissue of the amnion [38], which have contractile ability [39], and hence are involved in heart rhythm regulation [40] and, possibly, prevention of excessive distension of the amniotic membrane [38]. The DMGs between the TL and PTL groups were enriched in gene sets involved in cell adhesion, cell-cell and cell-extracellular matrix interactions, which have crucial roles in the modulation of cellular behavior and tissue maintenance and organization [41]. This observation confirms the importance of intact fetal membranes as a critical factor in the maintenance of pregnancy. Another overrepresented gene set was the negative regulation of transferase activity. Given the versatile roles of transferases, differential methylation of this group of genes (including HEXIM1, SFN, CBLC, and DUSP2) may influence a wide range of cellular processes in a way that interferes with timely onset of labor and parturition. Among these genes, DUSP2 has previously been documented as being significantly upregulated following interleukin-1β (IL-1β) stimulation in myometrial cells [42], suggesting its potential role in the mediation of uterine contractions. It would be intriguing to examine how the activity of DUSP2 in the amnion may contribute to the process of parturition.
Our study at the individual gene level using BS revealed three CpG sites (CpGs-934, -959, and -1084) in OXTR that exhibit significant DM among the three groups of amnion and decidua tissues, which are of fetal and maternal origin, respectively [4]. Subsequent gene expression analysis demonstrated no correlation between gene expression and methylation and therefore, the functional significance of the observed DM remains undetermined. Previous work showed that site-specific methylation can result in transcriptional alterations through its effects on the interaction of transcription factors (TFs) with its cognate DNA sequence [43]. Currently, there are no known TF binding sites around CpG-959, which was previously identified as significantly hypermethylated in peripheral blood mononuclear cells from autistic patients compared with those from control patients [19]. However, Gregory et al. [19] have indicated that CpG-934, whose differential methylation has also been associated with autism, falls within predicted binding domains for v-rel reticuloendotheliosis viral oncogene homolog (avian) (c-Rel), zinc fingers and homeoboxes 2 (ZHX2), and lectin, galactoside-binding, soluble, 4 (LGALS4). Using ENCODE ChIP-seq data available in the UCSC genome browser, we also found that CpG-1084 falls within putative binding sites for SUZ12 and Pol2 (see Section 2 for more details), which warrants future studies to dissect the impact of the methylation status at this specific dinucleotide on the interactions between these TFs and their binding sites.
Despite the lack of any significant difference in UCN methylation levels between the TL and PTL groups, our observation of a significant, more than 2-fold increase in UCN mRNA levels in the PTL amnion tissues compared with the term tissues suggests a potential role of this gene in the etiology of PTB, which encodes an endogenous ligand for corticotropin releasing hormone receptor (CRHR) that mediates the action of CRH, one of the major endocrine factors in parturition [44]. Given that there are several putative binding sites for TFs (such as C7EBP, GATA, and MyoD) [45] upstream of the region examined in this study, it would be intriguing to investigate whether the methylation status of CpG dinucleotides encompassing those sites correlates with the observed gene expression patterns. It would also be worthwhile to examine if mechanisms other than methylation underlie the transcriptional regulation of UCN in the amnion.
Our MSP analysis identified another gene (SLC30A3) that might play a role in pathogenic processes of PTB. This gene, also known as ZNT3, encodes a zinc transporter responsible for zinc efflux from the cytoplasm to extracellular spaces or intracellular organelles [46]. Given the differential expression of SLC30A3 in relation to dietary zinc and/or glucose supply in mouse placenta [47] and beta cells [48], it is postulated that its dysregulated expression due to aberrant methylation in human amnion may influence nutritional homeostasis during pregnancy, ultimately, leading to PTB.
Our work was limited by the small sample size and the lack of control for gender-specific methylation differences [49, 50]. Another major limitation is that the PTL tissues were examined as a pooled sample, not individually. Previous studies have demonstrated that pooled DNA samples can be used to provide a reliable estimate of average group methylation when analyzed using high-throughput techniques such as MALDI-TOF mass spectrometry [51, 52]. Therefore, a DNA pooling approach using such systems could be employed in future studies for large-scale assessment of methylation variations in maternal and fetal tissues. Very recently, it has been shown that neonatal DNA exhibits a considerable degree of GA-associated variability in DNA methylation patterns [53]. Given this finding, a precisely stratified analysis based on GA may allow a more accurate characterization of DNA methylation profiles associated with term and preterm pregnancies.
5. Conclusion
This work provides preliminary evidence that DNA methylation changes may play at least a partial role in physiologic labor and the etiology of PTB, and suggests that DNA methylation profiles, together with other types of biological data, hold a promise for the identification of genes involved in normal parturition and preterm birth.
Conflict of Interests
The authors declare that there is no conflict of interests.
Acknowledgments
The authors thank all the families who participated in the study, and acknowledge the great efforts of Laura Knosp, Susan Berends, Mercedes López, Mirta Gladys Leguizamón, Silvina Argañaraz, Marta Padilla, and Azucena Singh who were critical in contacting and enrolling families and screening samples. They are grateful to the W.M. Keck Foundation Biotechnology Resource Laboratory and the Keck Biostatistics Resource at Yale University for help with conducting the genome-wide methylation experiment and analyzing the raw data. This research was supported by Grants from the National Institutes of Health (R01 HD052953 and HD57192) and the March of Dimes Foundation (FY2006-575 and FY2008-260).
References
- 1.Benirschke K. Pathology of the Human Placenta. New York, NY, USA: Springer; 2012. [Google Scholar]
- 2.Niknejad H, Peirovi H, Jorjani M, Ahmadiani A, Ghanavi J, Seifalian AM. Properties of the amniotic membrane for potential use in tissue engineering. European Cells and Materials. 2008;15:88–99. doi: 10.22203/ecm.v015a07. [DOI] [PubMed] [Google Scholar]
- 3.Toda A, Okabe M, Yoshida T, Nikaido T. The potential of amniotic membrane/amnion-derived cells for regeneration of various tissues. Journal of Pharmacological Sciences. 2007;105(3):215–228. doi: 10.1254/jphs.cr0070034. [DOI] [PubMed] [Google Scholar]
- 4.Carlson BM. Human Embryology and Developmental Biology. Philadelphia, PA, USA: Mosby/Elsevier; 2009. [Google Scholar]
- 5.Keelan JA, Sato T, Mitchell MD. Regulation of interleukin (IL)-6 and IL-8 production in an amnion-derived cell line by cytokines, growth factors, glucocorticoids, and phorbol esters. American Journal of Reproductive Immunology. 1997;38(4):272–278. doi: 10.1111/j.1600-0897.1997.tb00514.x. [DOI] [PubMed] [Google Scholar]
- 6.Keelan JA, Sato T, Mitchell MD. Interleukin (IL)-6 and IL-8 production by human amnion: regulation by cytokines, growth factors, glucocorticoids, phorbol esters, and bacterial lipopolysaccharide. Biology of Reproduction. 1997;57(6):1438–1444. doi: 10.1095/biolreprod57.6.1438. [DOI] [PubMed] [Google Scholar]
- 7.Ackerman WE, Summerfield TLS, Vandre DD, Robinson JM, Kniss DA. Nuclear factor-kappa B regulates inducible prostaglandin E synthase expression in human amnion mesenchymal cells. Biology of Reproduction. 2008;78(1):68–76. doi: 10.1095/biolreprod.107.061663. [DOI] [PubMed] [Google Scholar]
- 8.Bernstein P, Leyland N, Gurland P, Gare D. Cervical ripening and labor induction with prostaglandin E2 gel: a placebo-controlled study. American Journal of Obstetrics and Gynecology. 1987;156(2):336–340. doi: 10.1016/0002-9378(87)90279-1. [DOI] [PubMed] [Google Scholar]
- 9.Rayburn WF. Prostaglandin E2 gel for cervical ripening and induction of labor: a critical analysis. American Journal of Obstetrics and Gynecology. 1989;160(3):529–534. doi: 10.1016/s0002-9378(89)80020-1. [DOI] [PubMed] [Google Scholar]
- 10.Gibb W. The role of prostaglandins in human parturition. Annals of Medicine. 1998;30(3):235–241. doi: 10.3109/07853899809005850. [DOI] [PubMed] [Google Scholar]
- 11.Haluska GJ, Kaler CA, Cook MJ, Novy MJ. Prostaglandin production during spontaneous labor and after treatment with RU486 in pregnant rhesus macaques. Biology of Reproduction. 1994;51(4):760–765. doi: 10.1095/biolreprod51.4.760. [DOI] [PubMed] [Google Scholar]
- 12.Schellenberg JC, Kirkby W. Production of prostaglandin F(2α) and E2 in explants of intrauterine tissues of guinea pigs during late pregnancy and labor. Prostaglandins. 1997;54(3):625–638. doi: 10.1016/s0090-6980(97)00129-9. [DOI] [PubMed] [Google Scholar]
- 13.Oyen ML, Calvin SE, Landers DV. Premature rupture of the fetal membranes: is the amnion the major determinant? American Journal of Obstetrics and Gynecology. 2006;195(2):510–515. doi: 10.1016/j.ajog.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 14.Mercer BM. Preterm premature rupture of the membranes. Obstetrics and Gynecology. 2003;101(1):178–193. doi: 10.1016/s0029-7844(02)02366-9. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Ogawa M, Wood JR, et al. Genetic and epigenetic mechanisms combine to control MMP1 expression and its association with preterm premature rupture of membranes. Human Molecular Genetics. 2008;17(8):1087–1096. doi: 10.1093/hmg/ddm381. [DOI] [PubMed] [Google Scholar]
- 16.Fujimoto T, Parry S, Urbanek M, et al. A single nucleotide polymorphism in the matrix metalloproteinase-1 (MMP-1) promoter influences amnion cell MMP-1 expression and risk for preterm premature rupture of the fetal membranes. The Journal of Biological Chemistry. 2002;277(8):6296–6302. doi: 10.1074/jbc.M107865200. [DOI] [PubMed] [Google Scholar]
- 17.Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiological Reviews. 2001;81(2):629–683. doi: 10.1152/physrev.2001.81.2.629. [DOI] [PubMed] [Google Scholar]
- 18.Terzidou V, Blanks AM, Kim SH, Thornton S, Bennett PR. Labor and inflammation increase the expression of oxytocin receptor in human amnion. Biology of Reproduction. 2011;84(3):546–552. doi: 10.1095/biolreprod.110.086785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregory SG, Connelly JJ, Towers AJ, et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Medicine. 2009;7, article 62 doi: 10.1186/1741-7015-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larsson HJ, Eaton WW, Madsen KM, et al. Risk factors for autism: perinatal factors, parental psychiatric history, and socioeconomic status. American Journal of Epidemiology. 2005;161(10):916–925. doi: 10.1093/aje/kwi123. [DOI] [PubMed] [Google Scholar]
- 21.Limperopoulos C, Bassan H, Sullivan NR, et al. Positive screening for autism in ex-preterm infants: prevalence and risk factors. Pediatrics. 2008;121(4):758–765. doi: 10.1542/peds.2007-2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bibikova M, Le J, Barnes B, et al. Genome-wide DNA methylation profiling using Infinium assay. Epigenomics. 2009;1(1):177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- 23.Chowdhury S, Erickson SW, MacLeod SL, et al. Maternal genome-wide DNA methylation patterns and congenital heart defects. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0016506.e16506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chudin E, Kruglyak S, Baker SC, Oeser S, Barker D, McDaniel TK. A model of technical variation of microarray signals. Journal of Computational Biology. 2006;13(4):996–1003. doi: 10.1089/cmb.2006.13.996. [DOI] [PubMed] [Google Scholar]
- 25.Naumova OY, Lee M, Koposov R, Szyf M, Dozier M, Grigorenko EL. Differential patterns of whole-genome DNA methylation in institutionalized children and children raised by their biological parents. Development and Psychopathology. 2012;24(1):143–155. doi: 10.1017/S0954579411000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 28.Kusui C, Kimura T, Ogita K, et al. DNA methylation of the human oxytocin receptor gene promoter regulates tissue-specific gene suppression. Biochemical and Biophysical Research Communications. 2001;289(3):681–686. doi: 10.1006/bbrc.2001.6024. [DOI] [PubMed] [Google Scholar]
- 29.Birney E, Stamatoyannopoulos JA, Dutta A, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenbloom KR, Dreszer TR, Pheasant M, et al. ENCODE whole-genome data in the UCSC genome browser. Nucleic Acids Research. 2010;38(1):D620–D625. doi: 10.1093/nar/gkp961.gkp961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahn K, Huh JW, Park SJ, et al. Selection of internal reference genes for SYBR green qRT-PCR studies of rhesus monkey (Macaca mulatta) tissues. BMC Molecular Biology. 2008;9, article 78 doi: 10.1186/1471-2199-9-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Johnson S, Hollis C, Kochhar P, Hennessy E, Wolke D, Marlow N. Autism spectrum disorders in extremely preterm children. Journal of Pediatrics. 2010;156(4):525.e2–531.e2. doi: 10.1016/j.jpeds.2009.10.041. [DOI] [PubMed] [Google Scholar]
- 34.Johnson S, Marlow N. Preterm birth and childhood psychiatric disorders. Pediatric Research. 2011;69(5):11R–18R. doi: 10.1203/PDR.0b013e318212faa0. [DOI] [PubMed] [Google Scholar]
- 35.Gu Q, Clifton VL, Schwartz J, Madsen G, Sha JY, Smith R. Characterization of urocortin in human pregnancy. Chinese Medical Journal. 2001;114(6):618–622. [PubMed] [Google Scholar]
- 36.Han YM, Romero R, Kim JS, et al. Region-specific gene expression profiling: novel evidence for biological heterogeneity of the human amnion. Biology of Reproduction. 2008;79(5):954–961. doi: 10.1095/biolreprod.108.069260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haddad R, Tromp G, Kuivaniemi H, et al. Human spontaneous labor without histologic chorioamnionitis is characterized by an acute inflammation gene expression signature. American Journal of Obstetrics and Gynecology. 2006;195(2):394.e12–405.e12. doi: 10.1016/j.ajog.2005.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang T, Schneider J. Myofibroblasten im bindegewebe des menschlichen amnions. Zeitschrift für Geburtshilfe und Perinatologie. 1982;186:164–169. [PubMed] [Google Scholar]
- 39.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano: regulation of connective tissue remodelling. Nature Reviews Molecular Cell Biology. 2002;3(5):349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 40.Rohr S. Myofibroblasts in diseased hearts: new players in cardiac arrhythmias? Heart Rhythm. 2009;6(6):848–856. doi: 10.1016/j.hrthm.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 41.DuFort CC, Paszek MJ, Weaver VM. Balancing forces: architectural control of mechanotransduction. Nature Reviews Molecular Cell Biology. 2011;12(5):308–319. doi: 10.1038/nrm3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chevillard G, Derjuga A, Devost D, Zingg HH, Blank V. Identification of interleukin-1β regulated genes in uterine smooth muscle cells. Reproduction. 2007;134(6):811–822. doi: 10.1530/REP-07-0289. [DOI] [PubMed] [Google Scholar]
- 43.Bélanger AS, Tojcic J, Harvey M, Guillemette C. Regulation of UGT1A1 and HNF1 transcription factor gene expression by DNA methylation in colon cancer cells. BMC Molecular Biology. 2010;11, article 9 doi: 10.1186/1471-2199-11-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Florio P, Vale W, Petraglia F. Urocortins in human reproduction. Peptides. 2004;25(10):1751–1757. doi: 10.1016/j.peptides.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 45.Zhao L, Donaldson CJ, Smith GW, Vale WW. The structures of the mouse and human urocortin genes (Ucn and UCN) Genomics. 1998;50(1):23–33. doi: 10.1006/geno.1998.5292. [DOI] [PubMed] [Google Scholar]
- 46.Cousins RJ, Liuzzi JP, Lichten LA. Mammalian zinc transport, trafficking, and signals. The Journal of Biological Chemistry. 2006;281(34):24085–24089. doi: 10.1074/jbc.R600011200. [DOI] [PubMed] [Google Scholar]
- 47.Helston RM, Phillips SR, McKay JA, Jackson KA, Mathers JC, Ford D. Zinc transporters in the mouse placenta show a coordinated regulatory response to changes in dietary zinc intake. Placenta. 2007;28(5-6):437–444. doi: 10.1016/j.placenta.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 48.Smidt K, Jessen N, Petersen AB, et al. SLC30A3 responds to glucose- and zinc variations in β-cells and is critical for insulin production and in vivo glucose-metabolism during β-cell stress. PLoS ONE. 2009;4(5) doi: 10.1371/journal.pone.0005684.e5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuen RKC, Peñaherrera MS, von Dadelszen P, McFadden DE, Robinson WP. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. European Journal of Human Genetics. 2010;18(9):1006–1012. doi: 10.1038/ejhg.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cotton AM, Avila L, Penaherrera MS, Affleck JG, Robinson WP, Brown CJ. Inactive X chromosome-specific reduction in placental DNA methylation. Human Molecular Genetics. 2009;18(19):3544–3552. doi: 10.1093/hmg/ddp299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Docherty SJ, Davis OSP, Haworth CMA, Plomin R, Mill J. DNA methylation profiling using bisulfite-based epityping of pooled genomic DNA. Methods. 2010;52(3):255–258. doi: 10.1016/j.ymeth.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 52.Docherty SJ, Davis OS, Haworth CM, Plomin R, Mill J. Bisulfite-based epityping on pooled genomic DNA provides an accurate estimate of average group DNA methylation. Epigenetics Chromatin. 2009;2, article 3 doi: 10.1186/1756-8935-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schroeder JW, Conneely KN, Cubells JC, et al. Neonatal DNA methylation patterns associate with gestational age. Epigenetics. 2011;6(12):1498–1504. doi: 10.4161/epi.6.12.18296. [DOI] [PMC free article] [PubMed] [Google Scholar]