

Abstract
The most bacteria-like mitochondrial genome known is that of the jakobid flagellate Reclinomonas americana NZ. This genome also encodes the largest known gene set among mitochondrial DNAs (mtDNAs), including the RNA subunit of RNase P (transfer RNA processing), a reduced form of transfer–messenger RNA (translational control), and a four-subunit bacteria-like RNA polymerase, which in other eukaryotes is substituted by a nucleus-encoded, single-subunit, phage-like enzyme. Further, protein-coding genes are preceded by potential Shine–Dalgarno translation initiation motifs. Whether similarly ancestral mitochondrial characters also exist in relatives of R. americana NZ is unknown. Here, we report a comparative analysis of nine mtDNAs from five distant jakobid genera: Andalucia, Histiona, Jakoba, Reclinomonas, and Seculamonas. We find that Andalucia godoyi has an even larger mtDNA gene complement than R. americana NZ. The extra genes are rpl35 (a large subunit mitoribosomal protein) and cox15 (involved in cytochrome oxidase assembly), which are nucleus encoded throughout other eukaryotes. Andalucia cox15 is strikingly similar to its homolog in the free-living α-proteobacterium Tistrella mobilis. Similarly, a long, highly conserved gene cluster in jakobid mtDNAs, which is a clear vestige of prokaryotic operons, displays a gene order more closely resembling that in free-living α-proteobacteria than in Rickettsiales species. Although jakobid mtDNAs, overall, are characterized by bacteria-like features, they also display a few remarkably divergent characters, such as 3′-tRNA editing in Seculamonas ecuadoriensis and genome linearization in Jakoba libera. Phylogenetic analysis with mtDNA-encoded proteins strongly supports monophyly of jakobids with Andalucia as the deepest divergence. However, it remains unclear which α-proteobacterial group is the closest mitochondrial relative.
Keywords: complete mtDNA sequences, genome evolution, gene migration to nucleus, excavates
Introduction
Mitochondria are organelles of α-proteobacterial origin that contribute to ATP and metabolite production in the eukaryotic cell and typically contain their own mitochondrial DNA (mtDNA). The evolutionary transformation of the endosymbiont to an organelle was accompanied by drastic genome reduction. Of the initial approximately 1,000–8,000 genes (estimated from the gene content of contemporary bacteria; National Center for Biotechnology Information (NCBI) Genome Database), only 0.5–1.2% are retained in mtDNAs. "Domestication" of the endosymbiont rendered many of its biological functions (e.g., biotin synthesis) unnecessary, leading to the elimination of redundant genes. A further drastic reduction of coding capacity resulted from massive gene migration from mtDNA to the nucleus. Nuclear genes acquired from the endosymbiont generally encode components of the organelle itself, such as transporters, building blocks of the inner membrane, metabolic enzymes, and proteins of the oxidative phosphorylation and protein synthesis machinery. However, the exact the number of nuclear genes of α-proteobacterial origin is still a matter of debate (reviewed in Gray et al. 2001).
The most gene-rich mtDNA reported to date is that of Reclinomonas americana NZ (Lang et al. 1997), which is a member of the jakobids. (Note that the term "jakobids" has been used previously to circumscribe "core-jakobids" plus malawimonads [e.g., Lang et al. 1999; O'Kelly and Nerad 1999].) However, the inclusion of malawimonads in this assemblage is not supported from a phylogenetic point of view (e.g., Rodríguez-Ezpeleta et al. 2007; Hampl et al. 2009; Derelle and Lang 2011). Jakobids are bacterivorous unicellular eukaryotes characterized by two flagella, one of which is directed posteriorly, and a feeding groove along the body used for capture and ingestion of small particles and bacteria (Flavin and Nerad 1993; O'Kelly 1993). The mitochondrial genome of R. americana (designated below as Reclinomonas-94) specifies as many as 96 assigned genes including 65 proteins and 31 structural RNAs, plus two open reading frames (ORFs) longer than 100 residues (Lang et al. 1997). Reclinomonas-94 genes otherwise rarely found in mtDNA encode NADH dehydrogenase subunits 7–11 (nad7–nad11), succinate dehydrogenase subunits, mitoribosomal proteins, ABC and twin arginine transporters, and RNase P-RNA. Genes identified in at most one other mtDNA code for the Tu elongation factor A, ATP synthase subunit 3, and cox11 involved in cytochrome oxidase assembly. Exclusively present in Reclinomonas-94 mtDNA are genes for six additional large subunit (LSU) mitoribosomal proteins, four subunits of bacteria-type RNA-polymerase, secY specifying a protein transporter, and ssrA, which encodes (a reduced form of) transfer–messenger RNA (tmRNA) (Jacob et al. 2004); four of the above listed genes (atp4, ssrA, tatA, and tatC) were not reported in the initial publication (Lang et al. 1997) but rather detected later. In addition to the unusually large gene complement and genes encoding a bacteria-like RNA-polymerase, this mtDNA exhibits remarkably primitive features such as putative Shine–Dalgarno (SD) motifs and gene clusters closely resembling prokaryotic operons. These combined features prompted the apt description of the mitochondrion of Reclinomonas-94 as "the mitochondrion that time forgot" (Palmer 1997).
The above findings raised a number of intriguing questions. Is Reclinomonas-94 a rare exception or do other jakobids have similarly ancestral mtDNAs? Can we recognize evolutionary trends by comparing various jakobid mitochondrial genomes? To address these questions, we sequenced eight mtDNAs from jakobids belonging to the genera Andalucia (Lara et al. 2006), Histiona (Flavin and Nerad 1993), Jakoba (Patterson 1990), Reclinomonas (Flavin and Nerad 1993), and Seculamonas (Edgcomb et al. 2001; O'Kelly CJ, unpublished data). We find that mtDNAs from all jakobids are considerably more eubacteria-like than mtDNA from any other eukaryote. Moreover, one of the jakobids that branches basally to all other jakobids in the mitochondrial protein-based phylogenetic tree has retained even more mtDNA-encoded genes than Reclinomonas-94.
Materials and Methods
Strains and Culture
The jakobid strains used in this study (listed in table 1) were obtained from the American Type Culture Collection (ATCC), except for Andalucia godoyi, which was kindly provided by A. Simpson (Lara et al. 2006). The large variety of (food) bacteria that are present in the original strain isolates was reduced by repeated dilution in growth medium so as to retain only a few jakobid cells, and by adding to these isolates precultured live Enterobacter aerogenes (ATCC 13048) bacteria as food. We used WCL culture medium for R. americana species (ATCC 50394, 50283, 50284, 50633), Histiona aroides (ATCC 50634), Seculamonas ecuadoriensis (ATCC 50688), and A. godoyi, and F/2 medium for the two jakobids that were isolated from marine environments, Jakoba libera (ATCC 50422) and J. bahamiensis (ATCC 50695; currently not listed at ATCC’s website). Detailed recipes for the media are described at http://megasun.bch.umontreal.ca/People/lang/FMGP/methods.html. Cultures (500 ml) in 2.5 l Erlenmeyer flasks were gently shaken at 22 °C and daily supplemented with live bacteria. Cells were harvested by centrifugation in the early stationary growth phase (after 2–5 days), when most food bacteria were consumed.
Table 1.
General Features of Jakobid mtDNAsa
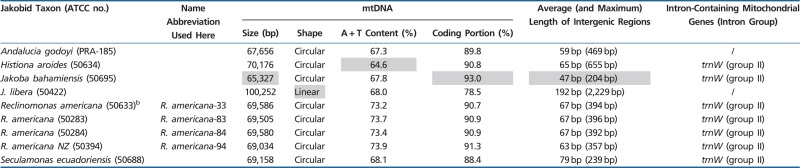 |
aShading highlights notable characteristics.
bGenBank Accession no. NC_001823.
Purification and Sequencing of mtDNA
Jakobid cells were broken mechanically to extract total DNA, and mtDNA was isolated by CsCl-bisbenzimide equilibrium gradient centrifugation, based on a higher A + T content than nuclear DNA (Lang and Burger 2007). Random libraries were constructed from mtDNA that was fragmented by nebulization and then cloned and sequenced (Sanger; Licor sequencer) by a whole-genome shotgun approach (Lang and Burger 2007). Because the mtDNA preparation of H. aroides was considerably contaminated with nuclear DNA (which is nearly as A + T-rich as mtDNA), random sequencing was combined with sequencing of DNA amplified by long polymerase chain reaction.
Genome Annotation
Gene annotation was performed with the automated tool MFannot (http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl) developed in house. In brief, MFannot predicts group I and group II introns, tRNAs, RNase P-RNA, and 5S ribosomal RNA (rRNA) with Erpin as a search engine (Gautheret and Lambert 2001), based on RNA structural profiles established by us. Exons of protein-coding genes are inferred in a first round with Exonerate (Slater and Birney 2005) and then for less well-conserved genes with HMMER (Eddy S; http://hmmer.janelia.org), based on models for all known mtDNA-encoded proteins. Only sequence positions that are aligned with confidence are retained for model construction. Mini-exons (as short as 3 nt) that are not resolved by Exonerate but inferred by the presence of orphan introns are detected as missing protein regions in multiple protein alignments. The precise placement of small exons is based on the best fit of Hidden Markov Model (HMM) protein profiles and on the fit with conserved nucleotide sequence profiles of group I or group II exon–intron boundaries. Genes encoding the small subunit (SSU) and LSU rRNAs are predicted with HMM profiles covering the most highly conserved domains, allowing precise placement of the SSU rRNA termini but only approximate positioning of LSU rRNA ends. The latter termini, as well as the precise exon–intron boundaries of rRNA genes, are predicted manually using comparative structure modeling. In any case, automated annotations are complemented by manual analyses to account for MFannot warnings (e.g., potential trans-spliced genes, gene fusions, frame shifts, alternative translation initiation sites, and failure to identify mini-introns), and find features that are not (yet) recognized by automated procedures (e.g., tmRNA genes). Unidentified ORFs located adjacent to genes that in bacteria are arranged in operons were examined individually using Position-Specific Iterated-Basic Local Alignment Search Tool (PSI-BLAST; Altschul et al. 1997). In addition, we built HMM profiles of the positional counterpart in bacteria and searched all ORFs against this profile. ORFs with significant or close-to-significant sequence similarity were further validated or rejected by inspecting multiple protein alignments.
Mitochondrial tmRNAs were searched for with a covariance model that was built from an alignment of previously identified jakobid tmRNAs (Jacob et al. 2004), using the cmbuild and cmcalibrate tools that are included in the most recent implementation of Infernal (version 1.1rc1; Eddy S; http://hmmer.janelia.org). Only confidently aligned nucleotides were used for model building and searching, by applying the—hand option. The model identifies the circularly permuted gene sequences with high confidence (E value <1.9e−12) and even recognizes the 3′-part of the continuous J. libera gene (E value: 5.8e−3).
In Silico Search for Sequence Motifs and Genome Signatures
Basic sequence manipulations (formatting, reverse complementation, and translation) were conducted with the in-house tool FLIP. Genic and intergenic regions of jakobid mtDNA were extracted using PEPPER, also developed in-house. These software tools are described at http://megasun.bch.umontreal.ca/ogmp/ogmpid.html and are available on request. Sequence identities of intergenic regions were evaluated using FASTA (Pearson 2000).
To locate potential SD motifs, that is, sequence motifs complementary to the inferred 3′-end of mitochondrial SSU rRNA (5′-CUCCUUUOH), we used RNA hybrid available on the bibiserver at http://bibiserv.techfak.uni-bielefeld.de/rnahybrid (Rehmsmeier et al. 2004). Hairpin elements in intergenic regions were identified with RNAalifold (Bernhart et al. 2008).
For the prediction of the probable origins and termini of replication in jakobid mtDNAs, we applied the cumulative GC skew technique that measures the asymmetric strand distribution of G and C for individual fixed-length windows along the sequence, that is (G − C) / (G + C), and then sums the scores values. Cumulative skew plotted along the sequence indicate probable origin (local minimum) and terminus (local maximum) of replication. For this analysis, we used the gc_skew implementation at the webserver http://gcat.davidson.edu/DGPB/gc_skew/gc_skew.html (Grigoriev 1999).
Destabilized helical DNA regions, which are often associated with replication origins, promoters, and protein-binding sites were predicted with the SIDD tool (Bi and Benham 2004; Zhabinskaya and Benham 2011) (webserver at http://benham.genomecenter.ucdavis.edu/sibz/). Overlapping 10-kbp genome portions were analyzed with the parameters DNA_Type = linear, temperature = 310 K, and Superhelical_Density = −0.055. Superhelically induced duplex destabilization, which facilitates or creates local sites of strand separation, are inferred from particular dinucleotide repeats and the equilibrium probability of transition from right-handed B to left-handed Z form of DNA.
Potential bacteria-like promoters were predicted with BPROM (http://linux1.softberry.com), which was chosen among various public web-accessible predictors, because it yields for jakobid mtDNAs a reasonable number (in the order of 200) of potential sites per genome, instead of 0 or >1,000, obtained with other tools tested. BPROM searches for sequence motifs derived from validated bacterial functional sites collected in the DPInteract database (Robison et al. 1998). The algorithm of promoter identification is based on linear discriminant function that accounts for sequence characteristics of promoter regions. Because the score is a logarithmic value with the neutral score being 0, any value >0 is more likely than not to be a promoter. For bacterial sequences, the threshold is usually set to 1 or more, but because jakobid mtDNA sequences are A + T-rich and probably produce more false positives as a result, we calculated a specific threshold for each genome. To do so, mtDNA sequences were randomly shuffled with SHUFFLE DNA (webserver at http://bioinformatics.org/sms/), and these sequences were analyzed again with BPROM. The highest score in the random-shuffled genome sequences was 9.24 for A. godoyi and 15.51 for Reclinomonas-94 mtDNA. Hits with scores below these values are therefore considered false positives.
Recent acquisition of foreign genome portions via horizontal transfer in J. libera mtDNA was tested with IGIPT (Jain et al. 2011) (webserver at http://bioinf.iiit.ac.in/IGIPT), by comparing the codon bias and amino acid bias of the ORFs at the termini of the linear chromosome (dpo, orf98, orf339, orf436, and orf686) with that of typical mitochondrial genes, as well as by scanning for changes in dinucleotide composition along the mtDNA. For calculation of codon bias, the average frequencies for codons specifying a particular amino acid are normalized, following which differences in codon usage of one gene set relative to another are determined. The amino acid bias is based on residue frequencies of a particular gene set compared with the average frequencies for the genome. The dinucleotide bias calculation assesses differences between the observed dinucleotide frequencies and those expected from random associations of mononucleotide frequencies. We set the search parameters as follows: standard deviation = 1.5 for filtering results to be reported; window size = 10,000 kb.
Phylogenetic Analysis
The data set contains 19 mitochondrion-encoded proteins (Cox1, 2, 3, Cob, Atp1, 6, 9, and Nad1, 2, 3, 4, 4L, 5, 6, 7, 9, 10, 11, TufA) from representative (preferably slowly evolving) taxa for which complete mtDNA sequences are available and from the jakobids studied here (the very closely related Reclinomonas species are represented by Reclinomonas-94). Protein collections were managed and automatically aligned, trimmed, and concatenated with Mams (developed in house; Lang BF and Rioux P, unpublished). Mams uses MUSCLE (Edgar 2004) for an initial alignment, followed by a refinement step with HMMalign (Eddy S; http://hmmer.janelia.org) and the elimination of all sequence positions with posterior probabilities lower than 1. The final data set contained 52 taxa and 5,791 amino acid positions (alignment is available from the authors upon request).
For phylogenetic analyses by Bayesian inference (PhyloBayes [Lartillot and Philippe 2004]), we used the default CAT model, six discrete categories, four independent chains, 14,000 cycles (corresponding to ∼770,000 generations), and the –dc parameter to remove constant sites. The first 10,000 cycles were discarded as burn-in. The robustness of internal branches was evaluated based on jackknife replicates (100 at 65%) rather than by bootstrap analysis, because the latter method generates duplicated sequence sites whose modeling is problematic with the Bayesian approach. Maximum likelihood analysis was performed with RaxML-HPC (Stamatakis 2006) v7.2.2, using the LG model (PROTGAMMALGF), and the fast bootstrapping option (100 replicates).
Sequence Deposition in Public Repositories
The annotated mtDNA sequences of jakobids (accession numbers KC353352–59) have been deposited in GenBank.
Results
General Features of Mitochondrial Genomes
Among characterized mitochondrial genomes, those in jakobids are of intermediate size, ranging from 65 to 100 kbp (table 1). Jakobid mtDNAs are circular mapping except for J. libera whose mtDNA is a linear monomer. The A + T content of these mitochondrial genomes is moderate (64–74%), and the proportion of coding regions (including introns) versus intergenic sequence is high (80–93%). To our knowledge, only mtDNA from the red alga Chondrus crispus is more compact, with coding sequences in that case amounting to approximately 96%.
Gene Content of Mitochondrial Genomes
Generally, mitochondrial genomes encode a small set of proteins and RNAs that are involved in oxidative phosphorylation (“OXPHOS”) and protein synthesis. In a few eukaryotes, the products of mtDNA-encoded genes also take part in protein import and maturation, respiratory complex assembly, and tRNA processing. Only in jakobids are mtDNA-specified genes implicated in transcription (RNA polymerase) and translational quality control (tmRNAs). Table 2 provides an overview of jakobid mitochondrial genes and the biological processes in which they participate. The mitochondrial gene content of individual jakobids is compiled in tables 3 and 4, compared with that of two other gene-rich protists.
Table 2.
Genes in Jakobid mtDNA and Their Functions
 |
aBlack, common mitochondrial genes; blue, expanded gene set, present in mtDNAs of various protists and plants; red, predominantly found in jakobids (rpl32 is otherwise only known from Vitis vinifera and Populus alba mt DNA [GenBank accession no. YP_002608375; BAG80685]; tufA in Hartmannella vermiformis mtDNA [Burger et al., GenBank accession no. GU828005]; rpl19 in Hartmannella, Malawimonas californiana, and M. jakobiformis [Gray et al. 2004]; ssrA in oomycete mtDNA [Lang et al., in preparation]; and cox11 in Naegleria [Burger et al., GenBank accession no.AF288092]); red underline, exclusively found in jakobid mtDNAs. For a previous description of "non-standard" mitochondrial genes, see Gray et al. (2004).
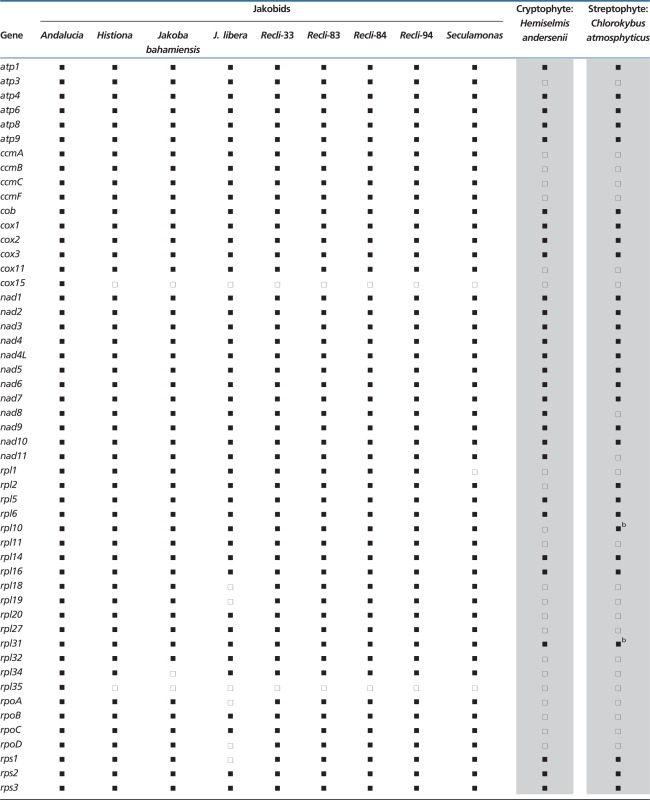
Table 3.
Comparison of Protein-Coding Gene Sets in Jakobid mtDNAsa
 |
 |
aTaxon abbreviations, Recli-33, Reclinomonas americana-33; Recli-83, R. americana-83; Recli-84, R. americana-84; Recli-94, R. americana-94. For complete taxon names, see table 1.
bGene not annotated in GenBank record.
Table 4.
Comparison of Gene Sets Coding for Structural RNAs in Jakobid mtDNAsa
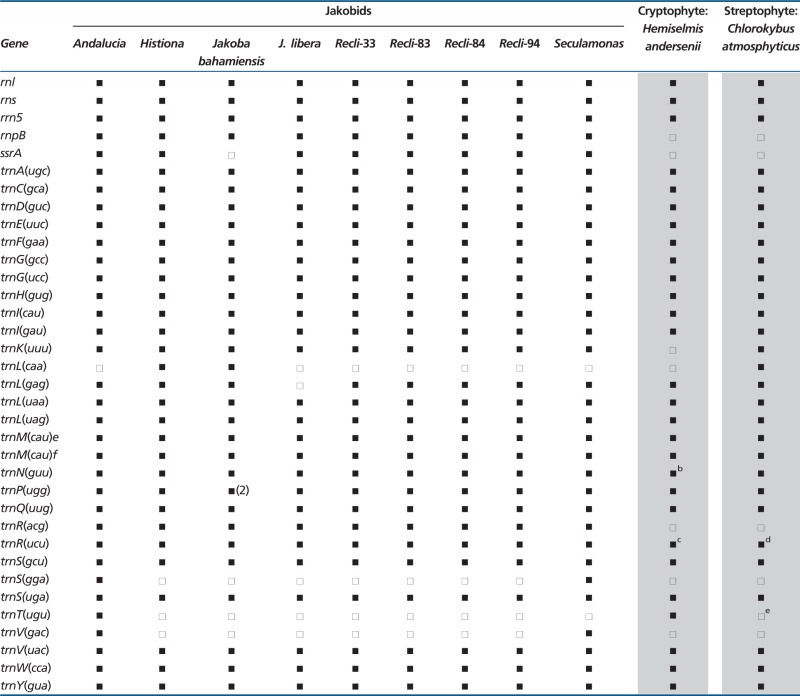 |
aTaxa as in table 3.
bPlus trnN(auu).
cPlus trnR(cgc) and trnR(gcg).
dPlus trnR(ucg).
eInstead trnT(ggu).
Tables 3 and 4 show that although Andalucia mtDNA does not have a secY gene, it is clearly the eukaryote with the largest number of identified mitochondrial genes (exactly 100), superseding Reclinomonas in that regard. Only Andalucia mtDNA carries rpl35 (specifying a LSU ribosomal protein) and cox15 (whose protein product is involved in cytochrome oxidase assembly), genes that have relocated to the nucleus in all other eukaryotes. Andalucia cox15 particularly resembles its homolog in Tistrella mobilis, a free-living α-proteobacterium, sharing 38% sequence identity over the entire protein and three specific indel signatures; sequence identities with other bacterial and eukaryotic homologs are below 28% (a multiple protein sequence alignment is shown in supplementary fig. S1, Supplementary Material online). Another remarkable feature is the presence of a mtDNA-encoded trnT in Andalucia, which is lacking in all other jakobids where it is most likely imported from the cytosol, because it is essential for translation of mtDNA-encoded genes.
The jakobid with the fewest mitochondrial genes is J. libera, with gene losses among protein-coding and tRNA genes (tables 3 and 4). Interestingly, this mtDNA is also the only one among jakobids to possess a gene (dpo) clearly related to family B DNA polymerases. This gene (or pseudogene, see Discussion) was most likely acquired secondarily, together with several ORFs and tandem repeat arrays located at both ends of the linear mtDNA, via integration of a mobile plasmid into the mitochondrial genome. Horizontal transfer is corroborated by significantly different sequence signatures (dinucleotide composition, codon usage, amino acid frequency, and G + C bias at different codon positions) in the terminal regions compared with those in the central portion of the molecule (supplementary tables S1 and S2, Supplementary Material online). Plasmid-mediated gene gain in mtDNA is frequently observed in fungal lineages (Fricova et al. 2010 and references therein) but has been also reported in other organismal groups (e.g., Takano et al. 1997).
In addition to the above genes of known function, jakobid mitochondrial genomes contain several hypothetical protein-coding genes (ORFs; table 5). Some of these ORFs may represent functional genes, considering that a SD-like sequence motif is located 7–15 nt upstream of the reading frame (see later). Two groups of ORFs are conserved across the four Reclinomonas mtDNAs, notably an ORF in the range of 169–181 amino acids in length and another 717–746 residues long; we refer to these two ORF families as recli-orf169–181 and recli-orf717–746. Members of these families not only share significant sequence similarity (26.4–84.1% identity over >97% of the protein’s length) but are also located in the same synteny block (fig. 1). Interestingly, Histiona mtDNA contains ORFs (orf163 and orf753, referred to as hist-orf163 and hist-orf753) of similar size and located in the same positional context as their counterparts in Reclinomonas taxa. However, sequence similarity is borderline; only hist-orf163 (E value: 3.6e−5) but not hist-orf753 is detected when searching with the Reclinomonas-specific HMM profiles against Histiona ORFs. Similarly, the search of Reclinomonas ORF-HMM profiles against assigned mitochondrial and α-proteobacterial proteins does not return hits above the reporting threshold.
Table 5.
Hypothetical Protein-Coding Genes in Jakobid mtDNAsa
 |
aNumber after "orf" indicates the number of amino acids contained in that ORF. ORFs of identical length but different sequence residing in the same mtDNA are distinguished by −1, −2, etc. ORFs occurring in the same synteny context in different mtDNAs are highlighted by shared color shading, and those with a SD-like sequence motif 7–15 nt upstream of the ATG codon (free energy < −8.5 kcal for pairing with anti-SD sequence in SSU rRNA) are marked by an asterisk.
bSD-like motifs absent from genome.
cTwo copies of identical sequence present at both ends of the linear chromosome.
Fig. 1.—

Gene maps of jakobid mtDNAs. Black boxes represent genes. Gene-name colors depict taxonomic gene distribution: black, common genes found in most animal, fungal, and protist mtDNAs; blue, expanded gene set, mostly present in protist and plant mtDNAs; red, genes predominantly found in jakobid mtDNAs (for details, see table 2); green, hypothetical protein-coding genes (ORFs). ORFs of identical length but different sequence are discriminated by the suffixes -1, -2, etc. Genes on the outer circle are transcribed in a clockwise direction, and those on the inner circle are transcribed counterclockwise. Transfer RNAs are indicated by the one-letter code of their cognate amino acid. Clockwise map order of tRNA genes is represented by increasing distance of the gene label from the circle’s center. The group II intron in trnW is shown as a yellow box. The innermost circle serves as size marker. Arcs indicate the largest synteny block (fig. 2). Unfilled arrows indicate the positions of predicted promoters. Black, filled arrowheads and the encircled "T" specify the predicted positions of replication origins and terminators, respectively. (A) Andalucia godoyi, (B) Histiona aroides, (C) Jakoba bahamiensis, and (D) J. libera. The linear J. libera mitochondrial genome is presented as an open circle, with red dots symbolizing the two extremities of the mtDNA molecule. Red arrows denote inverted repeats. The J. libera map shows only ORFs ≥ 98 residues. For the full listing of ORFs, see table 5. (E) Reclinomonas americana-94 (GenBank Accession no. NC_001823). The map of the other three Reclinomonas strains is identical except for the size of equipositional ORFs (see list in boxes). (F) Seculamonas ecuadoriensis.
Intergenic Regions, Gene Order, and Orientation
Figure 1 depicts the gene maps of the nine jakobid mtDNAs described here. In the four Reclinomonas strains, mitochondrial gene order is identical. These genomes have significant sequence similarity even in intergenic regions. The highest values are observed between Reclinomonas-33 and Reclinomonas-84, with an average sequence identity in intergenic regions of 80%. The least resemblance among Reclinomonas strains is between Reclinomonas-33 and Reclinomonas-94, where the identity average is 61%. In contrast, mtDNAs of the two Jakoba taxa differ considerably. Gene order varies due to numerous transpositions and inversions, and equivalent intergenic sequences have diverged to an extent that has erased any detectable similarity.
Despite considerable differences in gene order between the non-Reclinomonas jakobids, eight synteny blocks of more than four genes are found across jakobids (fig. 2 and supplementary fig. S2, Supplementary Material online). Foremost in these clusters are genes specifying ribosomal proteins (rps and rpl), but genes encoding NADH dehydrogenase subunits (nad), cytochrome c maturation proteins (ccm), and succinate dehydrogenase subunits (sdh) are present as well. Most synteny blocks are relicts of bacterial operons, and because the jakobid gene clusters are densely packed (with genes sometimes overlapping), it is likely that they represent polycistronic transcription units, as in bacteria.
Fig. 2.—

Gene order comparison of jakobid mtDNA and α-proteobacteria. Conservation of ribosomal gene organization in the mitochondrial genome of jakobids, compared with the generally contiguous L11, L10, Beta, streptomycin (Str), S10, spectinomycin (Spc), and Alpha operons of α-proteobacteria. Species abbreviations and GenBank Accession nos. are: Azos. spec., Azospirillum sp., NC_013854; Magn. magn., Magnetospirillum magneticum, NC_007626; Midi. mito., Candidatus Midichloria mitochondrii, NC_015722; Orie. tsut., Orientia tsutsugamushi str. Ideda, NC_010793; Rhod. cent., Rhodospirillum centenum, NC_011420; Rhod. rubr., Rhodospirillum rubrum ATCC 11170, NC_007643; Rick. prow., Rickettsia prowazekii str. Madrid, NC_000963; Tist. mobi., Tistrella mobilis KA081020-065, NC_017966; and Wolb-Droso., Wolbachia endosymbiont of Drosophila melanogaster, NC_002978.
We compared the gene order of jakobid mtDNAs with that of nine diverse α-proteobacterial genomes. Figure 2 aligns the longest common jakobid synteny block with the corresponding, usually contiguous, α-proteobacterial operons (L11, L10, Beta, Str, S10, Spc, and Alpha). Jakobids collectively display not only specific deletions (e.g., rpl12) but also an insertion (nad11-nad1-cox11-cox3 inserted in the Str cluster). In α-proteobacteria, nad and cox genes are typically part of separate, larger nad and cox operons. Interestingly, the gene arrangement in jakobid mitochondria is overall more similar to that in free-living α-proteobacteria, compared with Rickettsia-like intracellular pathogens that share a cluster discontinuity (fig. 2).
Ribosomal RNAs
The mitochondrial SSU and LSU rRNAs of the nine jakobids are strikingly bacteria-like, conforming closely to the standard models for Escherichia coli 16S and 23S rRNA, respectively. Sequences share a high degree of nucleotide identity with each other and with their E. coli counterparts, particularly within the "universal core" that defines the functionally most critical portions of the rRNAs. In all jakobid mitochondrial LSU rRNAs, a 5′-5.8S rRNA-like domain and a 3′-4.5S rRNA-like region are readily apparent at the level of both primary and secondary structure. Mitochondrial SSU rRNAs of jakobids contain a 3′-terminal pyrimidine-rich motif that is complementary to purine-rich SD-like elements upstream of the start of many protein-coding genes in the corresponding mitochondrial genomes (see later).
Overall, sequence identity ranges from approximately 70% to >95%, with the SSU rRNAs of the 33, 83, and 84 strains of R. americana being colinear and virtually identical in sequence, as are the LSU rRNAs. At the other extreme, for both the SSU and LSU mitochondrial rRNAs, the homologous Jakoba rRNAs are as divergent from one another in both primary and secondary structure as they are from their homologs in the other jakobids. As anticipated, length variation among the jakobid sequences and with the corresponding E. coli sequence is almost exclusively confined to highly divergent variable regions of secondary structure, previously identified from comparisons of SSU and LSU rRNA homologs. An alignment of jakobid mitochondrial and E. coli SSU rRNAs is shown in supplementary figure S3, Supplementary Material online.
Jakobid mtDNAs encode a recognizable 5S rRNA, which also conforms closely to the corresponding bacterial 5S rRNA in primary and secondary structure, as previously reported for R. americana-94 mitochondrial 5S rRNA (Lang et al. 1996). The four Reclinomonas mitochondrial 5S rRNA sequences are colinear and virtually identical in sequence, whereas those of the two Jakoba species exhibit substantial variation. The most divergent of jakobid mitochondrial 5S rRNAs is that of J. bahamiensis.
Transfer RNAs
Not all mitochondrial genomes contain a full complement of tRNA genes, with the missing tRNAs being imported into the organelle (Gray et al. 2004; Lang et al. 2011). Among the jakobid mitochondrial genomes analyzed here, only that of Andalucia encodes a complete set of tRNAs capable of reading all codons, assuming a mechanism whereby U in the first position of the anticodon permits recognition of all codons in a four-codon family (e.g., tRNAAla with anticodon UGC). The 29 different tRNAs specified by Andalucia mtDNA include separate initiator and elongator tRNAMet isoacceptors, as well as an apparent tRNAIle having a CAU anticodon. In this case, as in bacteria, the first position (C) of the anticodon presumably undergoes modification to lysidine (L), thereby enabling the LAU anticodon to read the AUA codon as isoleucine.
Of the 29 tRNAs encoded by Andalucia mtDNA, 25 are shared with all the other jakobids (an additional mitochondrial tRNALeu with GAG anticodon is present in all nine species except J. libera). Two further tRNAs, tRNASer and tRNAVal with GGA and UAC anticodons, respectively, are selectively shared between Andalucia and Seculamonas. Only Andalucia contains a native (mtDNA-encoded) tRNAThr, whereas only Histiona and J. bahamiensis mtDNAs encode a tRNALeu with CAA anticodon.
All jakobid mitochondrial tRNA sequences are able to assume the canonical cloverleaf secondary structure of a conventional tRNA. Occasional deviations from the typical structure are mostly supported by their occurrence in more than one jakobid. Examples include A rather than U at position 8 in tRNAAla(ugc) in all four Reclinomonas strains; C rather than A or G at position 9 in tRNAGlu(uuc) in all the jakobids; and a purine–purine mismatch in the first position of the anticodon stem of tRNAHis(gug) and a UxU mismatch in tRNAIle(cau) at the fourth anticodon stem position (both of the latter cases in Histiona and the four Reclinomonas strains). A particularly notable feature involves the first position of the anticodon loop of the elongator tRNAMet, which almost universally in tRNA is a pyrimidine. Atypically, this position is A in the elongator tRNAMet of Andalucia, J. libera, and the four Reclinomonas strains (but G in Seculamonas). A is also found in this position in the mitochondrial elongator tRNAMet of many (although not all) other protists.
In two Seculamonas mitochondrial tRNAs, tRNAGlu(uuc) and tRNASer(gga), mismatches in the first three acceptor stem positions suggested the possibility of tRNA editing, an inference verified experimentally (Leigh and Lang 2004). In these two instances, mismatches are converted to standard base pairs via removal and replacement of nucleotides at the 3′-end of the transcript, rather than at the 5′-end, as in a number of other tRNA editing systems (Lonergan and Gray 1993a, 1993b; Laforest et al. 1997). Aside from these two tRNAs, there is no compelling evidence that other jakobid mitochondrial tRNAs undergo 5′- or 3′-editing. The homologous Andalucia tRNASer(gga), for example, has a fully base-paired acceptor stem encoded in the mtDNA, and in other cases, nonstandard base pairs in the acceptor stem are overwhelmingly GċU or UċG, which in Seculamonas have been shown not to be edited (nor is an acceptor stem U × U mismatch in the mitochondrial tRNAHis of this organism) (Leigh and Lang 2004). With respect to the possibility of tRNA editing, the only case that perhaps warrants further investigation is the elongator tRNAMet of J. bahamiensis, in which the first three acceptor stem positions are GċT, A × C, and TċG.
Finally, we note that the position immediately upstream of the beginning of the mitochondrial trnH(gug) gene—the −1 position—is G in all the jakobids. Because the G−1 position is an almost universal feature of tRNAHis, constituting a required identity element for histidylation, the jakobid mitochondrial tRNAHis potentially acquires G−1 via an abnormal RNase P cleavage during pre-tRNA processing, as occurs in bacteria (Jackman et al. 2012). If so, such a pathway would presumably obviate a requirement for a mitochondrial tRNAHis guanylyltransferase, the enzyme that adds G−1 to the cytoplasmic tRNAHis in eukaryotes.
Examination of codon usage indicates that all jakobids employ the standard genetic code for mitochondrial translation (i.e., TGA does not specify Trp as in mitochondria of many other eukaryotes). Unlike in non-jakobid mitochondria that employ the standard genetic code, TGA stop codons occur relatively frequently in Andalucia (9 of 65 assigned genes), followed by 4/61 in J. libera; the other jakobids use this termination codon only once or twice, or not at all. TAG termination codons are also used relatively frequently in jakobid mitochondria, the ratio TAG:TAA ranging from 0.4 in J. bahamiensis to 0.1 in Reclinomonas-83. Mitochondrial TAG stop codons are reasonably abundant in land plants (Arabidopsis thaliana with a ratio of 0.2) and some fungi (e.g., in the Gigaspora rosea and Glomus irregulare [Nadimi et al. 2012]), whereas absent in the large majority of the other eukaryotes. In bacteria, TAA and TAG codons are served by the peptide release factor RF1, whereas a second factor, RF2, recognizes TAA and TGA (Scolnick et al. 1968). In mitochondria, TGA seems to co-occur with a nucleus-encoded RF2-like factor (Duarte et al. 2012), which we expect to be present also in Andalucia and J. libera mitochondria.
RNase P-RNA
P-RNA is the catalytic subunit of a ribonucleoprotein particle (RNP) that processes tRNA 5′-ends via endonucleolytic cleavage of precursor transcripts (Peck-Miller and Altman 1991). Both the cytoplasm and mitochondria have their own RNase P complex. The structural RNA associated with the mitochondrial RNP is rarely encoded by mtDNA (only present in a few fungi and protists); when it is, its secondary structure is often highly derived, rendering identification difficult (e.g., Martin and Lang 1997; Seif et al. 2003, 2005).
We discovered RNase P-RNA (rnpB) genes in all nine jakobid mtDNAs (Erpin search results are listed in supplementary table S3, Supplementary Material online), and the inferred RNA secondary structure of three representatives is depicted in figure 3. The Andalucia two-dimensional (2D) structure stands out as the smallest and most derived among jakobids, lacking P3, P12, and P19 pairings that are otherwise present in all jakobids (note that P19 is a hallmark of α-proteobacterial P-RNAs). In contrast to their bacterial counterparts (Stark et al. 1978; Altman 1989) and despite their bacteria-like RNA 2D-structure, P-RNAs of the four jakobids investigated biochemically (Reclinomonas-94, J. bahamiensis, J. libera, and S. ecuadoriensis) are unable to catalyze pre-tRNA cleavage in the absence of protein factors (Seif et al. 2006). Therefore, the structurally reduced RNA molecule of Andalucia mitochondria is most likely also inactive by itself. Interestingly, the predicted 3′-end of the Andalucia rnpB directly abuts the downstream trnG(ucc) gene. Thus, 5′-tRNA processing by RNase P would simultaneously generate the mature 3′-terminus of its own P-RNA subunit.
Fig. 3.—

Comparison of Andalucia and Reclinomonas mtP-RNA secondary structures. The Andalucia structure stands out as the shortest and most derived among jakobids, lacking P3, P12, and P19 pairings that are otherwise present in all jakobids (additionally, P19 is a hallmark feature of α-proteobacterial P-RNAs). The predicted 3′-end of the Andalucia mtP-RNA is adjacent to the downstream tRNAGly(UCC) gene, that is, 5′-tRNA processing by RNase P is responsible for 3′-maturation of its own RNA subunit. In most other jakobid mtDNAs, the tRNAGly(UCC) gene is located directly upstream of the rnpB gene and transcribed from the opposite strand.
Transfer–Messenger RNA
Bacteria and some plastids use tmRNAs to recognize and liberate translation complexes that have stalled on mRNAs lacking a stop codon. tmRNAs also earmark incomplete proteins for proteolysis by appending a short, conserved peptide (for a review, see Karzai et al. 2000). In the first reaction step, the tRNAAla-like domain of this RNA molecule triggers addition of a nonencoded Ala at the end of incomplete peptide chains. This step is followed by translation of the short mRNA-like region, which adds a signal peptide to the incomplete protein, marking the latter for degradation by C-terminal-specific proteases (Williams et al. 1999; Keiler et al. 2000; Zvereva et al. 2001). In α-proteobacteria and certain cyanobacteria, tmRNAs consist of two separate pieces that are held together by RNA-RNA interactions. In the corresponding gene (designated ssrA), the tRNA- and mRNA-like domains are circularly permutated with respect to the gene product (Keiler et al. 2000); consequently, the gene remained unrecognized for many years.
With the exception of J. bahamiensis mtDNA, jakobid mitochondrial genomes encode tmRNA but of a reduced form that lacks the mRNA-like domain (Jacob et al. 2004), which likely restricts tmRNA function to liberating stalled ribosomes. Similar to their α-proteobacterial ancestors, all mtDNA-encoded tmRNAs except that of J. libera have the two-piece configuration (Jacob et al. 2004) and contain the known tRNAAla identity elements, notably a GċU base pair at the third position of the acceptor stem and an A as the discriminator nucleotide preceding the CCA tail (Komine et al. 1994). All these features are also present in the inferred Andalucia tmRNA (fig. 4); however, compared with mitochondrial tmRNAs of the other jakobids, Andalucia’s is notable for its condensed secondary structure and shortened T-loop with an uncommon sequence. Similarly divergent is ssrA of J. libera mtDNA, which exists in a contiguous, nonpermutated form. Finally, no ssrA was detected in the mitochondrial genome of J. bahamiensis, despite a highly sensitive covariance search that even recognizes the structurally distinct J. libera sequence (supplementary table S4, Supplementary Material online).
Fig. 4.—

Comparison of tmRNA secondary structures from Andalucia, Reclinomonas-94, and Jakoba libera. Nucleotides marked blue are conserved in bacterial and jakobid tmRNAs. Nucleotides in red are identical among jakobid homologs. Yellow highlighting indicates the typical tRNAAla identity element that is common to tRNAAla and tmRNAs. This element is characterized by a GċU base pair (at the third position of the acceptor stem in the tRNA) and an A (the discriminator nucleotide in the tRNA) preceding the CCA tail (Komine et al. 1994). The blue line shows the processing sites corresponding to tRNA 5′- and 3′-termini, and the site of post-transcriptional CCA addition is indicated. The Andalucia tmRNA is characterized by a short, compact structure and a shortened T-loop with an unusual sequence compared with the other counterparts.
Replication of mtDNA
Circular bacterial chromosomes typically replicate by the theta mode starting at a single bidirectional origin and ending at the replication terminus, roughly opposite of the origin. The leading strand generally has an excess of guanines (G) relative to cytosines (C), a bias most likely due to different regimes of mutations or DNA repair in the leading versus the lagging strand during theta replication (Lobry 1996). Analysis of the GC skew ((G − C/G + C)) along one strand of a given bacterial chromosome provides a good indication of the position of the origin and terminus of replication (Grigoriev 1998; Salzberg et al. 1998; for illustration, see supplementary fig. S4A, Supplementary Material online). However, to a minor degree, strand-specific GC skew may also arise as a result of codon bias in protein-coding regions (Tillier and Collins 2000 and references therein).
We generated cumulative GC skew plots of jakobid mtDNAs (supplementary fig. S4B–F, Supplementary Material online; for predicted sites see fig. 1). Only the mtDNA of R. americana strains exhibits a prominent bimodal GC skew curve, characteristic of the classical bidirectional theta mode. The relatively close spacing of the curves’ minimum and maximum suggests that the replication origin and termination sites are not situated opposite to one another on the circular-mapping chromosome but rather close to each other, so that replication proceeds asymmetrically, with the clockwise replication covering as much as approximately 80% of the genome. mtDNA in Histiona exhibits a similar, but much less pronounced, curve as that of Reclinomonas. In contrast, the GC skew of J. bahamiensis mtDNA is strikingly homogenous along the sequence, resembling the situation in the yeast Candida glabrata (supplementary fig. S4G, Supplementary Material online), where experimental evidence suggests rolling circle replication of mtDNA. The GC skew graphs of the remaining three circular jakobid mtDNAs do not allow inferences about the replication mechanism.
The linear J. libera mtDNA displays a GC skew curve with a major central maximum (supplementary fig. S4E, Supplementary Material online), suggesting unidirectional replication that starts at both ends and terminates in the middle of the genome. This situation is reminiscent of linear-plasmid (invertron) replication in Neurospora (Chan et al. 1991) and is consistent with a plasmid insertion event as postulated above, transforming the mitochondrial genome architecture from circular to linear. Genome linearization mediated by plasmid insertion has been previously demonstrated in maize mitochondria, where the distal sequences of the linearized mtDNA originate from a plasmid (Schardl et al. 1984). As in autonomously replicating linear mitochondrial plasmids, mtDNA termini in maize carry a covalently attached terminal protein, which is thought to prime DNA synthesis during replication (Sakaguchi 1990).
The linear mitochondrial genome of Candida subhashii also appears to replicate similar to invertrons (Fricova et al. 2010) (supplementary fig. S4I, Supplementary Material online). It was postulated that replication of this mtDNA relies on the mitochondrion-encoded plasmid-derived B-family DNA polymerase (dpo), but biochemical or genetic evidence for this activity is lacking. The complete and seemingly functional dpo gene in C. subhashii mtDNA may reflect a gene acquisition that took place recently, leaving insufficient time to accumulate mutations. For J. libera, it is even less likely that the mitochondrial dpo gene plays a role in replication, because the deduced Dpo protein lacks a 21-residue-long stretch in the C-terminal region that is otherwise conserved in C. subhashii mitochondrial Dpo and other members of B-family DNA polymerases. Note that dpo genes also reside in select circular-mapping mtDNAs; to our knowledge, however, in all cases, these sequences undergo rapid mutational decay (e.g., Burger et al. 1999; Barroso et al. 2001; Nadimi et al. 2012).
Potential Promoters and SD Motifs
Because most jakobid mitochondrial genomes encode a multisubunit RNA polymerase, we attempted to predict bacteria-like promoters in two of the minimally derived jakobid mtDNAs, those of Andalucia and Reclinomonas-94. Combining predicted DNA-duplex destabilized regions (potential promoter motifs) and using a promoter score threshold inferred from shuffled sequence (see Materials and Methods), we detect four and seven promoter candidates in Andalucia and Reclinomonas-94 mtDNA, respectively (supplementary table S5, Supplementary Material online; arrows in fig. 1). Obviously, experimental transcript data will be needed to confirm the functionality of bacteria-like promoters proposed here in jakobid mtDNAs.
In bacteria, SD-like sequence motifs assure selection of the proper translation initiation codon by pairing with the 3′-end of SSU rRNA. Up to this point, the only organism with convincing putative mitochondrial SD-like sequence motifs had been Reclinomonas-94 (Lang et al. 1997). Here we show that, with the exception of J. libera, such motifs are also present in the other jakobid mtDNAs (supplementary table S6, Supplementary Material online). These motifs are complementary to a pyrimidine-rich sequence stretch at the inferred 3′-end of mitochondrial SSU rRNA (5′-CUCCUUUOH, compared with 5′-CUCCUUAOH in E.oli 16S rRNA) and located 7–15 nt upstream of the initiator ATG of protein-coding genes. The largest number of 6-nt long SD-like motifs (5′-AAAGGA-3′) is present in Andalucia mtDNA, upstream of 28 of 64 assigned protein-coding genes. In this genome, another 22 genes are preceded by a 5-residue-long motif (5′-AAAGG-3′) that is probably also functional. In the other jakobids, the number of 6- and 5-nt-long SD-like motifs decreases from 31 to 15 in the order Reclinomonas-94 > Reclinomonas-84 > Reclinomonas-83 > Seculamonas > Histiona > J. bahamiensis. In J. libera mtDNA, where SD-like motifs are absent, genes are preceded by stretches of A- or T-rich sequence.
Other Regulatory Sequence Elements
Other potential regulatory cis-elements in jakobid mtDNAs include conspicuous palindromic sequences in intergenic regions that have a propensity to form hairpin secondary structures. Although mitochondrial genomes of most jakobids exhibit up to 10 hairpin elements with a stem of ≥10 bp, that of Seculamonas is particularly palindrome-rich (>40 elements). Interestingly, six jakobid mtDNAs share a hairpin element located immediately downstream of rnl. Such hairpins may play a number of biological roles including the control of RNA processing and translation. For example, hairpin sequence elements in 3′-untranslated regions of mitochondrial mRNAs have been suggested to be recognition sites for RNases (Schuster et al. 1986) and thus may mediate processing of polycistronic precursor transcripts as well as end processing of single-gene transcripts. Alternatively, stem-loop structures at the 3′-terminus of mRNAs have been found to stabilize transcripts in both bacteria and chloroplasts (Stern and Gruissem 1987; Manley and Proudfoot 1994; Rochaix 1996; Leigh and Lang 2004). Finally, hairpin sequence elements seen in jakobid mtDNAs could play a part in transcription termination. In bacteria, for instance, one of the mechanisms is ρ-independent termination, which involves the formation of a stem-loop structure in the transcript upstream of a U-rich sequence stretch (Richardson 2002).
Phylogeny and Evolutionary Inferences Based on Mitochondrial Proteins
We conducted a phylogenetic analysis employing PhyloBayes and the CAT model, and using concatenated mtDNA-encoded protein sequences from jakobids (with Reclinomonas-94 representing all strains of this genus), representatives of other major eukaryotic groups, and α-proteobacteria as an outgroup (a total of 59 species and 5,805 aligned amino acid positions). The resulting tree demonstrates monophyly of jakobids and unambiguous positioning of A. godoyi as the deepest divergence within this group (fig. 5). Most phylogenetic relationships in the tree are consistent with inferences based on nuclear gene data; most inconsistencies are not well supported in the mitochondrial phylogeny and are probably artifactual due to either the relatively small data matrix and/or known phylogenetic artifacts such as long-branch attraction (LBA; Felsenstein 1978). As in other analyses published previously (e.g., Andersson et al. 1998; Derelle and Lang 2011), the tree shown here specifically groups mitochondria with Rickettsiales. However, this alliance may result from LBA, further exacerbated by A + T sequence bias (Foster and Hickey 1999), as suggested by the fast evolutionary rates and the short common branch uniting these two lineages (fig. 5).
Fig. 5.—

Phylogeny based on mtDNA-encoded proteins. The tree was constructed based on concatenated mtDNA-encoded protein sequences from jakobids, slowly evolving representatives of other, major eukaryotic groups, 16 α-proteobacteria (a total of 52 species and 5,791 aligned amino acid positions), and Magnetococcus as a close, non α-proteobacterial outgroup species. Reclinomas-94 represents all four, very similar Reclinomonas species studied here. The tree shown was inferred with PhyloBayes, the CAT + Gamma model, and six discrete categories (Lartillot and Philippe 2004), based on 19 mtDNA-encoded, concatenated proteins. Numbers indicate jackknife support values; branches without numbers have 100% support values. Short, basal branches with less than 60% support were collapsed. Posterior probability values (PhyloBayes analysis with four independent chains) other than 1.0 are indicated in brackets, following the corresponding jackknife value. Maximum likelihood inference predicts a similar tree (not shown), except for a strong (100% bootstrap value) regrouping of the fast-evolving Amoebozoa with haptophytes, which is probably an LBA artifact. Full taxon names and corresponding GenBank accession numbers for bacterial sequences: Agrobacterium radiobacter, NC_011985, NC_011983; Alphaproteobacterium BAL199, NZ_ABHC01000000; Azospirillum brasilense, NC_016617; Bradyrhizobium japonicum, NC_004463; Ehrlichia canis, NC_007354; Magnetococcus sp., NC_008576; Micavibrio aeruginosavorus, NC_016026; Magnetospirillum magnetotacticum, NZ_AAAP00000000; Mezorhizobium loti, NC_002682, NC_002679, NC_002678; Midichloria mitochondrii, NC_015722; Neorickettsia sennetsu, NC_007798; Orientia tsutsugamushi str. Ideda, NC_010793; Novosphingobium aromaticivorans, NC_007794; Rhodospirillum centenum, NC_011420; Rhodospirillum rubrum ATCC 11170, NC_007643; Rickettsia prowazekii str. Madrid, NC_000963; Tistrella mobilis, NC_017966; and Wolbachia endosymbiont of Drosophila melanogaster, NC_002978. Full taxon names and corresponding GenBank accession numbers for mitochondrial sequences: Acanthamoeba castellanii, NC_001637; Andalucia godoyi, this report, Bigelowiella natans, HQ840955; Capsaspora owczarzaki, (unpublished, available upon request); Chaetosphaeridium globosum, NC_004118; Chondrus crispus, NC_001677; Chattonella marina, NC_013837; Cyanidioschyzon merolae, NC_000887; Cyanophora paradoxa, NC_017836; Desmarestia viridis, NC_007684; Emiliania huxleyi, NC_005332; Geodia neptuni, NC_006990; Glaucocystis nostochinearum, NC015117; Hartmannella vermiformis, NC_013986; Hemiselmis andersenii, NC_010637; Histiona aroides, this report; Jakoba bahamiensis, this report; J. libera, this report; Malawimonas californiana (unpublished, available upon request); Marchantia polymorpha, NC_001660; Mesostigma viride, NC_008240; Nephroselmis olivacea, NC_008239; Malawimonas jakobiformis, NC_002553; Monosiga brevicollis, NC_004309; Pavlova lutheri, HQ908424; Phytophthora infestans, NC_002387; Porphyra purpurea, NC_002007; Prototheca wickerhamii, NC_001613; Reclinomonas americana, NC_001823; Rhodomonas salina, NC_002572; Seculamonas ecuadoriensis, this report; and Thalassiosira pseudonana, NC_007405.
Discussion
Differential Gene Migration as a Factor Driving mtDNA Diversity
Evolutionary gene loss from mtDNA is mostly a consequence of gene migration to the nucleus. For example, atp1, atp3, and atp4, present in mtDNA of jakobids and a few other eukaryotes, reside in the nucleus of animals and fungi. Gene loss can also be due to functional substitution by a nuclear gene. This scenario likely applies to tRNAThr, because an mtDNA-specified gene is missing in all jakobids except Andalucia (table 4). Import of tRNAs into mitochondria has been demonstrated experimentally in several eukaryotes (reviewed in Alfonzo and Söll 2009).
A more complicated case is the mitochondrial RNA polymerase. In eight of the nine jakobids studied here, mtDNA carries all four genes (rpoA-D) specifying a typical multisubunit bacteria-like RNA polymerase, and our in silico analyses predict the presence of bacteria-like promoter motifs. Jakoba libera is the only jakobid having an incomplete set of mitochondrion-encoded bacteria-like RNA polymerase genes, with rpoA and rpoD genes apparently missing. Again, the latter two genes may have emigrated to the nucleus, with the result that the J. libera mitochondrial α2ββ′σ RNA polymerase is assembled from mitochondrion- and cytosol-synthesized proteins. Alternatively, the remaining rpo genes in J. libera mtDNA may be vestigial, and mitochondrial transcription may be performed either by only these two core polymerase subunits or, as in the large majority of eukaryotes, by a T3/T7-phage-like RNA polymerase (Cermakian et al. 1996, 1997). Finally, it is also conceivable that in J. libera mitochondria, bacteria-like and phage-like transcription machineries operate simultaneously and mediate expression of distinct subsets of genes, as seen in chloroplasts of certain plants (Gray and Lang 1998). To gain insight into the biological role of the mitochondrion-encoded rpoB and rpoC in J. libera, sequence information from the nuclear genome will be required as well as biochemical experiments.
What Is the Origin of cox15?
The mtDNA-encoded cox15 gene in Andalucia is most intriguing, as its protein sequence closely resembles Cox15 of the free-living α-proteobacterium Tistrella, both in terms of sequence similarity and indel signatures (supplementary fig. S1, Supplementary Material online). The latter are not shared with either members of Rickettsiales, or other α-proteobacteria, or most surprisingly, with the nucleus-encoded versions throughout eukaryotes. It is conceivable that the mitochondrial endosymbiont was closely related to Tistrella and that cox15 was introduced into the nuclear genome via mitochondrion-to-nucleus gene migration followed by a rapid change of indel signatures in the nuclear gene. Other more complex scenarios are also plausible, such as horizontal gene transfer from bacteria—transiently associated with eukaryotes—to either the mitochondrion or the nucleus. However, these scenarios are not testable with the currently available data, because the Cox15 protein is relatively short and insufficiently conserved. Meaningful phylogenetic analyses would require genome data from Tistrella relatives, plus sequences from nucleus-encoded cox15 versions in jakobids and other protist lineages. Note that Tistrella is listed under Rhodospirillaceae in the NCBI taxonomy, but according to our analysis shown here, it diverges basally to α-proteobacteria and without apparent affinity to any of its subgroups.
Evolution of Jakobids from a Mitochondrial Perspective
Although the depicted mitochondrial phylogeny is well resolved and comprehensive in terms of spanning the entire range of known jakobid diversity, the data set of mtDNA-encoded protein sequences is still too small to resolve the relationship of jakobids to other eukaryotes and in particular to the jakobid-like (in an ultrastructural sense; O'Kelly and Nerad 1999) malawimonads. However, even the large nuclear data sets are unable to reproducibly position jakobids relative to malawimonads, and analyses with different data sets do not concur (Rodríguez-Ezpeleta et al. 2007; Hampl et al. 2009; Derelle and Lang 2011). Still, the tree based on mtDNA-encoded proteins provides convincing support for jakobid monophyly; it is also the first to place Andalucia godoyi as the deepest divergence within jakobids, to cluster Histiona as a sister taxon of Reclinomonas, and to unite the two Jakoba species (fig. 5).
As a result, key events in mitochondrial genome evolution in jakobids can be "dated" by mapping their occurrence onto the tree (fig. 6). For example, the loss of the tRNAThr gene and the group II intron insertion in the tRNATrp gene likely took place very early in jakobid evolution, and the tRNA intron was probably lost secondarily in the lineage leading to J. libera. Editing of mitochondrial tRNAs at their 3′-ends, which occurs exclusively in S. ecuadoriensis, probably evolved recently in this particular branch. The most dramatic evolutionary events occurred in the J. libera lineage, notably loss of mtDNA-encoded rpoA and rpoD genes (table 3), acquisition of long intergenic regions and numerous ORFs, divergent RNA secondary structures (e.g., fig. 4), loss of SD-like motifs, reversion of tmRNA to a nonpermutated continuous molecule, and genome linearization. This evolutionary acceleration may have been triggered by a plasmid insertion into J. libera mtDNA, as discussed earlier.
Fig. 6.—
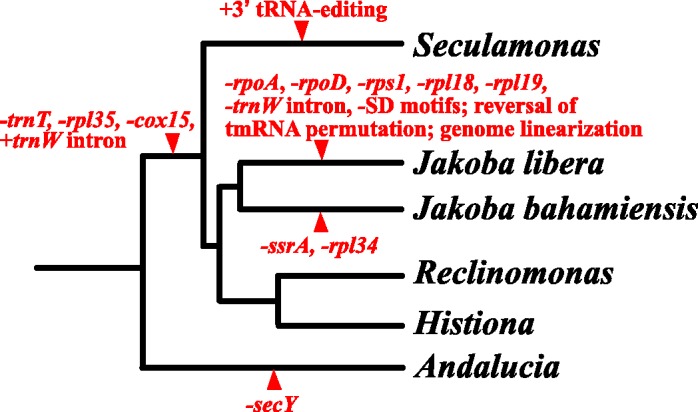
Evolutionary changes in jakobid mitochondrial genomes. The events are mapped onto the phylogenetic tree of jakobids assuming the smallest number of steps. −, loss; +, gain. For alternative evolutionary histories of cox15, see Discussion. The tree topology is taken from figure 5. For full species names, see the legend of figure 5.
Conclusions and Outlook
Jakobids stand out from other eukaryotes by reason of the elevated gene complement of their mitochondrial genomes, which specify molecular functions not encoded by any other mtDNA. Jakobids are also exceptional as to the bacteria-like features of mtDNA-encoded structural RNAs and bacteria-like regulatory elements presumably used in mitochondrial gene expression. Although Reclinomonas-94 was initially characterized as a unique organism whose mtDNA is extraordinarily ancestral, we show here that this protist belongs to a sizeable eukaryotic group that had remained unrecognized for decades.
The analysis of jakobid mitochondrial genomes reported here raises numerous new research questions. For example, it would be interesting to validate predictions of translation initiation, operons, transcription terminators, and replication origins. In addition, a biochemical characterization of jakobid mitochondrial RNA polymerases in J. libera would be in order. However, such studies will not be trivial, as jakobids are difficult to culture, and no protocols are yet available for isolating pure and intact mitochondria from these organisms (the main hurdle being that the organelle is firmly integrated with other subcellular structures [O'Kelly 1993]). Similarly, genetic manipulations that would allow the exploration of gene function have not been established for any jakobid.
Because the gene complement indicates that the mitochondrial genome of Andalucia is the most slowly evolving of all mtDNAs, we expect as well that its nuclear genome has preserved primitive features. Therefore, we recently initiated a collaborative Andalucia nuclear genome project. With a complete nuclear gene complement becoming available, it will be worthwhile to establish Andalucia as a eukaryotic model organism for biochemical studies, because it holds the promise of opening a window on the early evolution of cellular components, biological processes, and molecular functions of the eukaryotic cell.
Supplementary Material
Supplementary figures S1–S4 and tables S1–S6 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
G.B., B.F.L., and M.W.G. designed the study in the context of the former Organelle Genome Mega Sequencing Program (OGMP), a Canadian collaboration aimed at sequencing mtDNAs of poorly studied protist taxa, in an effort to gauge eukaryotic diversity. B.F.L. purified and cultured all protists used in this study and prepared mtDNA. G.B. (director of the OGMP Laboratory) supervised library construction and sequencing of mtDNA from J. bahamiensis, J. libera, and Reclinomonas-84, Reclinomonas-94, and conducted sequence assembly and annotation of these genomes. L.F. performed library construction and sequencing of mtDNAs from A. godoyi, H. aroides, Reclinomonas-33, Reclinomonas-83, and S. ecuadoriensis, and B.F.L. assembled and annotated these genomes. G.B., B.F.L., and M.W.G. analyzed the genome data in detail. G.B. coordinated the preparation of the manuscript and drafted a first version, and B.F.L. and M.W.G. participated in writing. All authors approved the final manuscript. The authors thank Dr Charley O’Kelly (Friday Harbor Laboratory) for continuous advice on any practical and theoretical aspect related to jakobids, purification of strains, and optimization of culture conditions. Further, they thank Dr Tom Nerad (previously ATCC) and Dr C. O’Kelly for providing jakobid strains. Andalucia godoyi was isolated by Dr E. Lara and kindly provided to us by Dr Alastair Simpson (Dalhousie University). The authors also thank Isabelle Plante, Dimitri Vzdornov, and Yun Zhu (previously OGMP Laboratory, Université de Montréal) for excellent technical assistance and Dr Matus Valach (Université de Montréal) for comments on the manuscript. This work was supported by the Medical Research Council of Canada (grant number SP-34), the Canadian Institutes of Health Research (grant number MSP-14226), the Canadian Institute for Advanced Research (fellowships for M.W.G. and B.F.L. and salary support for G.B.), and the Canada Research Chairs program (B.F.L. and M.W.G.).
Literature Cited
- Alfonzo JD, Söll D. Mitochondrial tRNA import—the challenge to understand has just begun. Biol Chem. 2009;390:717–722. doi: 10.1515/BC.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman S. Ribonuclease P: an enzyme with a catalytic RNA subunit. Adv Enzymol Relat Areas Mol Biol. 1989;62:1–36. doi: 10.1002/9780470123089.ch1. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson SGE, et al. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature. 1998;396:133–140. doi: 10.1038/24094. [DOI] [PubMed] [Google Scholar]
- Barroso G, Bois F, Labarère J. Duplication of a truncated paralog of the family B DNA polymerase gene Aa-polB in the Agrocybe aegerita mitochondrial genome. Appl Environ Microbiol. 2001;67:1739–1743. doi: 10.1128/AEM.67.4.1739-1743.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhart SH, Hofacker IL, Will S, Gruber AR, Stadler PF. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics. 2008;9:474. doi: 10.1186/1471-2105-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi C, Benham CJ. WebSIDD: server for predicting stress-induced duplex destabilized (SIDD) sites in superhelical DNA. Bioinformatics. 2004;20:1477–1479. doi: 10.1093/bioinformatics/bth304. [DOI] [PubMed] [Google Scholar]
- Burger G, Saint-Louis D, Gray MW, Lang BF. Complete sequence of the mitochondrial DNA of the red alga Porphyra purpurea. Cyanobacterial introns and shared ancestry of red and green algae. Plant Cell. 1999;11:1675–1694. doi: 10.1105/tpc.11.9.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermakian N, et al. On the evolution of the single-subunit RNA polymerases. J Mol Evol. 1997;45:671–681. doi: 10.1007/pl00006271. [DOI] [PubMed] [Google Scholar]
- Cermakian N, Ikeda TM, Cedergren R, Gray MW. Sequences homologous to yeast mitochondrial and bacteriophage T3 and T7 RNA polymerases are widespread throughout the eukaryotic lineage. Nucleic Acids Res. 1996;24:648–654. doi: 10.1093/nar/24.4.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan BS-S, Court DA, Vierula PJ, Bertrand H. The kalilo linear senescence-inducing plasmid of Neurospora is an invertron and encodes DNA and RNA polymerases. Curr Genet. 1991;20:225–237. doi: 10.1007/BF00326237. [DOI] [PubMed] [Google Scholar]
- Derelle R, Lang BF. Rooting the eukaryotic tree with mitochondrial and bacterial proteins. Mol Biol Evol. 2011;29:1277–1289. doi: 10.1093/molbev/msr295. [DOI] [PubMed] [Google Scholar]
- Duarte I, Nabuurs SB, Magno R, Huynen M. Evolution and diversification of the organellar release factor family. Mol Biol Evol. 2012;29:3497–3512. doi: 10.1093/molbev/mss157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgcomb VP, Roger AJ, Simpson AG, Kysela DT, Sogin ML. Evolutionary relationships among “jakobid” flagellates as indicated by alpha- and beta-tubulin phylogenies. Mol Biol Evol. 2001;18:514–522. doi: 10.1093/oxfordjournals.molbev.a003830. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Cases in which parsimony and compatibility methods will be positively misleading. Syst Biol. 1978;27:401–410. [Google Scholar]
- Flavin M, Nerad TA. Reclinomonas americana n. g., n. sp., a new freshwater heterotrophic flagellate. J Eukaryot Microbiol. 1993;40:172–179. doi: 10.1111/j.1550-7408.1993.tb04900.x. [DOI] [PubMed] [Google Scholar]
- Foster PG, Hickey DA. Compositional bias may affect both DNA-based and protein-based phylogenetic reconstructions. J Mol Evol. 1999;48:284–290. doi: 10.1007/pl00006471. [DOI] [PubMed] [Google Scholar]
- Fricova D, et al. The mitochondrial genome of the pathogenic yeast Candida subhashii: GC-rich linear DNA with a protein covalently attached to the 5′-termini. Microbiology. 2010;156:2153–2163. doi: 10.1099/mic.0.038646-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheret D, Lambert A. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J Mol Biol. 2001;313:1003–1011. doi: 10.1006/jmbi.2001.5102. [DOI] [PubMed] [Google Scholar]
- Gray MW, Burger G, Lang BF. The origin and early evolution of mitochondria. Genome Biol. 2001;2:1018.1011–1018.1015. doi: 10.1186/gb-2001-2-6-reviews1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW, Lang BF. Transcription in chloroplasts and mitochondria: a tale of two polymerases. Trends Microbiol. 1998;6:1–3. doi: 10.1016/S0966-842X(97)01182-7. [DOI] [PubMed] [Google Scholar]
- Gray MW, Lang BF, Burger G. Mitochondria of protists. Annu Rev Genet. 2004;38:477–524. doi: 10.1146/annurev.genet.37.110801.142526. [DOI] [PubMed] [Google Scholar]
- Grigoriev A. Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 1998;26:2286–2290. doi: 10.1093/nar/26.10.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev A. Strand-specific compositional asymmetries in double-stranded DNA viruses. Virus Res. 1999;60:1–19. doi: 10.1016/s0168-1702(98)00139-7. [DOI] [PubMed] [Google Scholar]
- Hampl V, et al. Phylogenomic analyses support the monophyly of Excavata and resolve relationships among eukaryotic “supergroups”. Proc Natl Acad Sci U S A. 2009;106:3859–3864. doi: 10.1073/pnas.0807880106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Gott JM, Gray MW. Doing it in reverse: 3′-to-5′ polymerization by the Thg1 superfamily. RNA. 2012;18:886–899. doi: 10.1261/rna.032300.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob Y, Seif E, Paquet P-O, Lang BF. Loss of the mRNA-like region in mitochondrial tmRNAs of jakobids. RNA. 2004;10:605–614. doi: 10.1261/rna.5227904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Ramineni S, Parekh N. IGIPT—integrated genomic island prediction tool. Bioinformation. 2011;7:307–310. doi: 10.6026/007/97320630007307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karzai AW, Roche ED, Sauer RT. The SsrA-SmpB system for protein tagging, directed degradation and ribosome rescue. Nat Struct Biol. 2000;7:449–455. doi: 10.1038/75843. [DOI] [PubMed] [Google Scholar]
- Keiler KC, Shapiro L, Williams KP. tmRNAs that encode proteolysis-inducing tags are found in all known bacterial genomes: a two-piece tmRNA functions in Caulobacter. Proc Natl Acad Sci U S A. 2000;97:7778–7783. doi: 10.1073/pnas.97.14.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komine Y, Kitabatake M, Yokogawa T, Nishikawa K, Inokuchi H. A tRNA-like structure is present in 10Sa RNA, a small stable RNA from Escherichia coli. Proc Natl Acad Sci U S A. 1994;91:9223–9227. doi: 10.1073/pnas.91.20.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laforest M-J, Roewer I, Lang BF. Mitochondrial tRNAs in the lower fungus Spizellomyces punctatus: tRNA editing and UAG “stop” codons recognized as leucine. Nucleic Acids Res. 1997;25:626–632. doi: 10.1093/nar/25.3.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang BF, Burger G. Purification of mitochondrial and plastid DNA. Nat Protoc. 2007;2:652–660. doi: 10.1038/nprot.2007.58. [DOI] [PubMed] [Google Scholar]
- Lang BF, et al. An ancestral mitochondrial DNA resembling a eubacterial genome in miniature. Nature. 1997;387:493–497. doi: 10.1038/387493a0. [DOI] [PubMed] [Google Scholar]
- Lang BF, Goff LJ, Gray MW. A 5 S rRNA gene is present in the mitochondrial genome of the protist Reclinomonas americana but is absent from red algal mitochondrial DNA. J Mol Biol. 1996;261:607–613. doi: 10.1006/jmbi.1996.0486. [DOI] [PubMed] [Google Scholar]
- Lang BF, Lavrov D, Beck N, Steinberg V. Mitochondrial tRNA structure, identity and evolution of the genetic code. In: Bullerwell CE, editor. Organelle genetics. Evolution of organelle genomes and gene expression. Berlin Heidelberg (Germany): Springer; 2011. pp. 431–474. [Google Scholar]
- Lang BF, Seif E, Gray MW, O'Kelly CJ, Burger G. A comparative genomics approach to the evolution of eukaryotes and their mitochondria. J Eukaryot Microbiol. 1999;46:320–326. doi: 10.1111/j.1550-7408.1999.tb04611.x. [DOI] [PubMed] [Google Scholar]
- Lara E, Chatzinotas A, Simpson AG. Andalucia (n. gen.)—the deepest branch within jakobids (Jakobida; Excavata), based on morphological and molecular study of a new flagellate from soil. J Eukaryot Microbiol. 2006;53:112–120. doi: 10.1111/j.1550-7408.2005.00081.x. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21:1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- Leigh J, Lang BF. Mitochondrial 3′ tRNA editing in the jakobid Seculamonas ecuadoriensis: a novel mechanism and implications for tRNA processing. RNA. 2004;10:615–621. doi: 10.1261/rna.5195504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobry JR. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 1996;13:660–665. doi: 10.1093/oxfordjournals.molbev.a025626. [DOI] [PubMed] [Google Scholar]
- Lonergan KM, Gray MW. Editing of transfer RNAs in Acanthamoeba castellanii mitochondria. Science. 1993a;259:812–816. doi: 10.1126/science.8430334. [DOI] [PubMed] [Google Scholar]
- Lonergan KM, Gray MW. Predicted editing of additional transfer RNAs in Acanthamoeba castellanii mitochondria. Nucleic Acids Res. 1993b;21:4402. doi: 10.1093/nar/21.18.4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley JL, Proudfoot NJ. RNA 3′ ends: formation and function—meeting review. Genes Dev. 1994;8:259–264. doi: 10.1101/gad.8.3.259. [DOI] [PubMed] [Google Scholar]
- Martin NC, Lang BF. Mitochondrial RNase P: the RNA family grows. Nucleic Acids Symp Ser. 1997;36:42–44. [PubMed] [Google Scholar]
- Nadimi M, Beaudet D, Forget L, Hijri M, Lang BF. Group I intron-mediated trans-splicing in mitochondria of Gigaspora rosea and a robust phylogenetic affiliation of arbuscular mycorrhizal fungi with Mortierellales. Mol Biol Evol. 2012;29:2199–2210. doi: 10.1093/molbev/mss088. [DOI] [PubMed] [Google Scholar]
- O'Kelly CJ. The jakobid flagellates: structural features of Jakoba, Reclinomonas and Histiona and implications for the early diversification of eukaryotes. J Eukaryot Microbiol. 1993;40:627–636. [Google Scholar]
- O'Kelly CJ, Nerad TA. Malawimonas jakobiformis n. gen., n. sp (Malawimonadidae n. fam.): a Jakoba-like heterotrophic nanoflagellate with discoidal mitochondrial cristae. J Eukaryot Microbiol. 1999;46:522–531. [Google Scholar]
- Palmer JD. The mitochondrion that time forgot. Nature. 1997;387:454–455. doi: 10.1038/387454a0. [DOI] [PubMed] [Google Scholar]
- Patterson DJ. Jakoba libera (Ruinen, 1938), a hetertrophic flagellate from deep oceanic sediments. J Mar Biol Assn U K. 1990;70:381–393. [Google Scholar]
- Pearson WR. Flexible sequence similarity searching with the FASTA3 program package. Methods Mol Biol. 2000;132:185–219. doi: 10.1385/1-59259-192-2:185. [DOI] [PubMed] [Google Scholar]
- Peck-Miller KA, Altman S. Kinetics of the processing of the precursor to 4.5 S RNA, a naturally occurring substrate for RNase P from Escherichia coli. J Mol Biol. 1991;221:1–5. doi: 10.1016/0022-2836(91)80194-y. [DOI] [PubMed] [Google Scholar]
- Rehmsmeier M, Steffen P, Höchsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10:1507–1517. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JP. Rho-dependent termination and ATPases in transcript termination. Biochim Biophys Acta. 2002;1577:251–260. doi: 10.1016/s0167-4781(02)00456-6. [DOI] [PubMed] [Google Scholar]
- Robison K, McGuire AM, Church GM. A comprehensive library of DNA-binding site matrices for 55 proteins applied to the complete Escherichia coli K-12 genome. J Mol Biol. 1998;284:241–254. doi: 10.1006/jmbi.1998.2160. [DOI] [PubMed] [Google Scholar]
- Rochaix J-D. Post-transcriptional regulation of chloroplast gene expression in Chlamydomonas reinhardtii. Plant Mol Biol. 1996;32:327–341. doi: 10.1007/BF00039389. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Ezpeleta N, et al. Toward resolving the eukaryotic tree: the phylogenetic positions of jakobids and cercozoans. Curr Biol. 2007;17:1420–1425. doi: 10.1016/j.cub.2007.07.036. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K. Invertrons, a class of structurally and functionally related genetic elements that includes linear DNA plasmids, transposable elements, and genomes of adeno-type viruses. Microbiol Rev. 1990;54:66–74. doi: 10.1128/mr.54.1.66-74.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, Salzberg AJ, Kerlavage AR, Tomb J-F. Skewed oligomers and origins of replication. Gene. 1998;217:57–67. doi: 10.1016/s0378-1119(98)00374-6. [DOI] [PubMed] [Google Scholar]
- Schardl CL, Lonsdale DM, Pring DR, Rose KR. Linearization of maize mitochondrial chromosomes by recombination with linear episomes. Nature. 1984;310:292–296. [Google Scholar]
- Schuster W, Hiesel R, Isaac PG, Leaver CJ, Brennicke A. Transcript termini of messenger RNAs in higher plant mitochondria. Nucleic Acids Res. 1986;14:5943–5954. doi: 10.1093/nar/14.15.5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scolnick E, Tompkins R, Caskey T, Nirenberg M. Release factors differing in specificity for terminator codons. Proc Natl Acad Sci U S A. 1968;61:768–774. doi: 10.1073/pnas.61.2.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seif E, Cadieux A, Lang BF. Hybrid E. coli—Mitochondrial ribonuclease P RNAs are catalytically active. RNA. 2006;12:1661–1670. doi: 10.1261/rna.52106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seif E, et al. Comparative mitochondrial genomics in zygomycetes: bacteria-like RNase P RNAs, mobile elements and a close source of the group I intron invasion in angiosperms. Nucleic Acids Res. 2005;33:734–744. doi: 10.1093/nar/gki199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seif ER, Forget L, Martin NC, Lang BF. Mitochondrial RNase P RNAs in ascomycete fungi: lineage-specific variations in RNA secondary structure. RNA. 2003;9:1073–1083. doi: 10.1261/rna.5880403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater GSC, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stark BC, Kole R, Bowman EJ, Altman S. Ribonuclease P: an enzyme with an essential RNA component. Proc Natl Acad Sci U S A. 1978;75:3717–3721. doi: 10.1073/pnas.75.8.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DB, Gruissem W. Control of plastid gene expression: 3′-inverted repeats act as mRNA processing and stabilizing elements, but do not terminate transcription. Cell. 1987;51:1145–1157. doi: 10.1016/0092-8674(87)90600-3. [DOI] [PubMed] [Google Scholar]
- Takano H, Kuroiwa T, Kawano S. Mitochondrial fusion promoting plasmid. Cell Struct Funct. 1997;22:299–308. doi: 10.1247/csf.22.299. [DOI] [PubMed] [Google Scholar]
- Tillier ER, Collins RA. The contributions of replication orientation, gene direction, and signal sequences to base-composition asymmetries in bacterial genomes. J Mol Evol. 2000;50:249–257. doi: 10.1007/s002399910029. [DOI] [PubMed] [Google Scholar]
- Williams KP, Martindale KA, Bartel DP. Resuming translation on tmRNA: a unique mode of determining a reading frame. EMBO J. 1999;18:5423–5433. doi: 10.1093/emboj/18.19.5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhabinskaya D, Benham CJ. Theoretical analysis of the stress induced B-Z transition in superhelical DNA. PLoS Comput Biol. 2011;7:e1001051. doi: 10.1371/journal.pcbi.1001051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvereva MI, et al. Complex of transfer-messenger RNA and elongation factor Tu. Unexpected modes of interaction. J Biol Chem. 2001;276:47702–47708. doi: 10.1074/jbc.M106786200. [DOI] [PubMed] [Google Scholar]