

Abstract
Large-scale evolutionary studies often require the automated construction of alignments of a large number of homologous gene families. The majority of eukaryotic genes can produce different transcripts due to alternative splicing or transcription initiation, and many such transcripts encode different protein isoforms. As analyses tend to be gene centered, one single-protein isoform per gene is selected for the alignment, with the de facto approach being to use the longest protein isoform per gene (Longest), presumably to avoid including partial sequences and to maximize sequence information. Here, we show that this approach is problematic because it increases the number of indels in the alignments due to the inclusion of nonhomologous regions, such as those derived from species-specific exons, increasing the number of misaligned positions. With the aim of ameliorating this problem, we have developed a novel heuristic, Protein ALignment Optimizer (PALO), which, for each gene family, selects the combination of protein isoforms that are most similar in length. We examine several evolutionary parameters inferred from alignments in which the only difference is the method used to select the protein isoform combination: Longest, PALO, the combination that results in the highest sequence conservation, and a randomly selected combination. We observe that Longest tends to overestimate both nonsynonymous and synonymous substitution rates when compared with PALO, which is most likely due to an excess of misaligned positions. The estimation of the fraction of genes that have experienced positive selection by maximum likelihood is very sensitive to the method of isoform selection employed, both when alignments are constructed with MAFFT and with Prank+F. Longest performs better than a random combination but still estimates up to 3 times more positively selected genes than the combination showing the highest conservation, indicating the presence of many false positives. We show that PALO can eliminate the majority of such false positives and thus that it is a more appropriate approach for large-scale analyses than Longest. A web server has been set up to facilitate the use of PALO given a user-defined set of gene families; it is available at http://evolutionarygenomics.imim.es/palo.
Keywords: protein isoform, alternative splicing, alignment, evolutionary rate, positive selection
Introduction
The availability of complete genome sequences from many different organisms has stimulated large-scale studies of gene evolution. It has been observed, for example, that evolutionary rates vary greatly across genes, and several studies have focused on the identification of factors that may explain this variability (Duret and Mouchiroud 2000; Pál et al. 2001; Zhang and Li 2004; Albà and Castresana 2005; Drummond et al. 2006; McInerney 2006; Chen et al. 2011). Other works have been centered on the “blind” identification of genes that may have undergone episodes of adaptive selection in different species or lineages using divergence data (Clark et al. 2003; Arbiza et al. 2006; Bakewell et al. 2007; Gibbs et al. 2007; Kosiol et al. 2008; Vilella et al. 2009; Carneiro et al. 2012). The search for signs of positive or adaptive selection is based on the detection of an excess of nonsynonymous substitutions (amino acid altering, dN) versus synonymous substitutions (nonamino acid altering, dS) in a given branch of the tree when compared with the other branches. An approach that has become increasingly popular in these analyses is the branch-site test (Zhang et al. 2005), capable of detecting positive selection even if it is only occurring at a few sites in a sequence.
All the studies mentioned earlier require the construction of multiple sequence alignments from sets of homologous genes. Typically, the alignments are performed at the protein level and subsequently converted to coding sequence alignments for the estimation of dN and dS. The correct estimation of substitutions rates depends on the accuracy of the alignments; ideally, all sites in the same column of the alignment should be homologous. However, alignment programs do not always give the correct alignment from an evolutionary perspective (Wong et al. 2008; Markova-Raina and Petrov 2011). If we are analyzing one or a few gene families, many of these errors can be corrected manually, but this is not the case for large-scale automated analyses, which often involve thousands of gene families. The consequences of alignment errors in evolutionary analyses have generated an important degree of concern in the scientific community. For example, several authors have shown that positive selection tests are very sensitive to alignment inaccuracies (Mallick et al. 2009; Schneider et al. 2009; Fletcher and Yang 2010; Jordan and Goldman 2012). Although the results vary depending on the aligner, many false positives remain even when the best aligners are used (Markova-Raina and Petrov 2011). This means that further improvements in methods for the automated analyses of gene families are highly desirable.
One possible avenue of improvement has to do with the extent of homology shared by the sequences that are included in the alignment. Even if the genes are on the whole homologous (i.e., derived form a common ancestor), there may be regions in the protein sequences used in the comparison that do not have a common origin, and this could negatively affect the quality of the alignment. This will occur, for example, when we mix different protein isoforms for some of the genes we want to compare. Many eukaryotic genes can produce several transcripts as a result of alternative splicing or multiple transcription initiation sites (table 1), and many of these transcripts encode proteins that are partially different. As in most analyses the object of study is the gene, when we compare different genes, only one protein isoform per gene is selected. As a result of this process, the isoforms selected for different genes may contain nonhomologous regions, for example, derived from transcript variant specific exons. Multiple alignment programs should treat these regions as long insertions, but this process is not perfect, and some residues are sometimes spuriously aligned with nonhomologous regions from other sequences (Laurie et al. 2012). As a consequence, the number of inferred nonsynonymous substitutions, and hence the number of sites evolving adaptively is overestimated.
Table 1.
Number of Coding Transcripts for Different Species from ENSEMBL Version 64
| Species | No of Protein Coding Genes | No of Transcripts |
||
|---|---|---|---|---|
| Mean | Median | SD | ||
| Homo sapiens | 21,165 | 4.33 | 3 | 4.06 |
| Takifugu rubripes | 18,523 | 2.58 | 2 | 2.09 |
| Mus musculus | 22,705 | 2.44 | 2 | 2.23 |
| Drosophila melanogaster | 13,781 | 1.59 | 1 | 1.51 |
| Caenorhabditis elegans | 20,389 | 1.47 | 1 | 1.14 |
| Gallus gallus | 16,736 | 1.33 | 1 | 0.80 |
| Equus ferus caballus | 20,436 | 1.11 | 1 | 0.43 |
| Bos taurus | 19,994 | 1.11 | 1 | 0.36 |
Note.—SD, standard deviation.
Ensuring that we are not including isoforms containing sequences derived from exons that are not shared by all genes in the family would help prevent these errors, but this is not a realistic option. First, the annotation of possible transcripts and protein isoforms is still an ongoing task, and transcript coverage remains low and is likely to remain so, for many species (table 1), and thus for some genes, we only have partial transcripts, which means that it is often not possible to find “equivalent” isoforms. Second, even if we had complete knowledge of all protein isoforms expressed by each gene in a family, we may still have many families in which no single-protein isoform is common to all the genes, as splicing patterns are also evolving features. This is illustrated by a recent study reporting that for approximately 13% of human RefSeq annotated genes, an equivalent splicing isoform in mouse cannot be identified (Zambelli et al. 2010).
Given these limitations, how can we choose the best protein isoform combination from all those available? Because the major problem is the misalignment of nonhomologous residues, the combination of isoforms with the highest protein sequence conservation calculated from the alignment, albeit conservative, may be a good approach to minimize the number of false positives in adaptive selection tests. The problem is that the number of possible alignments and the associated computational cost can be prohibitive for many families. The number of known protein isoforms per gene is increasing rapidly with RNA deep sequencing approaches (Martin and Wang 2011; Djebali et al. 2012), and it is important to understand the consequences of choosing a particular protein isoform for evolutionary analyses.
To date most researchers, including ourselves, have addressed the issue by taking the longest protein isoform per gene (Arbiza et al. 2006; Bakewell et al. 2007; Gibbs et al. 2007; Kosiol et al. 2008; Vilella et al. 2009; Toll-Riera et al. 2011; Carneiro et al. 2012; Laurie et al. 2012). This maximizes the amount of sequence information that we use in the alignment, but it also favors the inclusion of sequences that contain low frequency and/or species-specific exons, potentially increasing the number of alignment errors. Trimming low-quality regions from such alignments is one possible solution to reduce the number of false positives in adaptive selection tests (Privman et al. 2012), but it has the disadvantage that, as a byproduct of trimming “bad quality” regions, we are also eliminating genuine fast evolving regions that may include true positively selected sites. Another strategy, which we have applied in the past, is discarding alignments that contain possible badly aligned exons by using an exon-specific similarity threshold, but this has the cost of an important decrease in data set size (Toll-Riera et al. 2011).
To address the problem of choosing a reasonable set of protein isoforms, here we propose a novel approach: The use of the combination of protein isoforms that are most similar in length. Homologous proteins that are more similar in length are also more likely to be more similar from a functional and evolutionary perspective. In addition, alignments of these proteins will tend to contain less gaps, and thus less potential errors, than alignments obtained with proteins of very different length. The analyses we perform using several homologous gene family sets show that this method, which we call PALO (Protein ALignment Optimizer), results in the estimation of a significantly lower number of positively selected genes with the branch-site test, strongly suggesting that it is effective in reducing the number of false positives in this kind of analysis.
Materials and Methods
Data Set Description
Orthologous and paralogous gene sequences and their corresponding encoded protein isoforms from different species were obtained from ENSEMBL version 64 (Flicek et al. 2012). We gathered three data sets of 1 to 1 orthologous genes for species separated at different evolutionary distances (Mammalia, Vertebrata, and Metazoa) and human and mouse orthologous genes with multiple orthology relationships (paralogs data set, corresponding to 1 to many, many to 1, and many to many orthology relationships) (Vilella et al. 2009). We discarded approximately 150 genes in the paralogs data set that had more than 50,000 possible combinations, as the small gain in data set size did not compensate the high computational cost of running all possible alignments for the Cons method (see later). The number of gene families in each data set and the species composition is listed in table 2. Database sequence identifiers for all data sets analyzed and the possible number of protein isoform combinations are available at supplementary file S1, Supplementary Material online.
Table 2.
Homologous Gene Family Data Set Description
| Data Set Name | ENSEMBL Homology | Species | No. of Gene Families | ||||
|---|---|---|---|---|---|---|---|
| Mammalia | 1 to 1 orthologs | Homo sapiens, Mus musculus, Equus ferus caballus, and Bos Taurus | 13,153 | ||||
| Vertebrata | H. sapiens, B. taurus, Gallus gallus, and Takifugu rubripes | 5,551 | |||||
| Metazoa | H. sapiens, G. gallus, Drosophila melanogaster, Caenorhabditis elegans | 1,612 | |||||
| Paralogs | 1 to many orthologs | H. sapiens and M. musculus | 850 | ||||
Algorithm Description
For more than 90% of the gene families in each data set, we could choose among different protein isoform combinations due to the existence of multiple transcripts encoding different proteins in one or more species. After calculating all possible combinations, we applied different methods to select a single combination:
Cons: The protein isoform combination that results in the best-conserved alignment, measured as percentage of amino acid identity over the length of the alignment (Conservation score). Note that this approach requires performing the alignment of all possible combinations, and it was only intended for benchmarking.
Longest: The protein isoform combination that corresponded to the longest protein isoform available for each gene.
PALO: The protein isoform combination that corresponded to the minimum value of the sum of squares of pairwise protein length differences (least squares). For each combination we used the following equation:
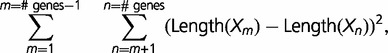 \sum _{m = 1}^{m = {\rm \#\ genes} - 1} \quad\sum _{n = m + 1}^{n = \#\ {\rm genes}} ({\rm Length(}X_m ) - {\rm Length}(X_n ))^2 , \sum _{m = 1}^{m = {\rm \#\ genes} - 1} \quad\sum _{n = m + 1}^{n = \#\ {\rm genes}} ({\rm Length(}X_m ) - {\rm Length}(X_n ))^2 ,
|
where Xi is a given protein isoform of gene i.
Random: A randomly chosen protein isoform combination.
PALO Software Implementation
We developed a Python application that calculates the PALO protein isoform combination given a set of homologous genes and associated proteins. The required gene information can be downloaded from ENSEMBL or other gene databases. The set of homologous genes must be provided in a text file of tab-separated values, each row corresponding to the genes that we want to include in the alignment. The second file should contain the protein information for each gene, namely the gene identifier, protein identifier, and the protein sequence length. To avoid server overload, gene families with more than 1 × 107 combinations are currently not accepted in the web version. The program and a web server application can be accessed at http://evolutionarygenomics.imim.es/palo. The code is also available at http://github.com/egenomics/palo.
Construction of Multiple Sequence Alignments
We obtained multiple sequence alignments for all possible protein isoform combinations in each of the families of the four data sets (table 2) using MAFFT with default parameters (Katoh et al. 2002; Katoh and Toh 2008). Subsequently, we calculated the percentage identity (Conservation score) as the number of columns with identical residues in all sequences divided by the total number of columns in the alignment.
For comparison, we also constructed amino acid sequence alignments with Prank+F (Löytynoja and Goldman 2005, 2008). This program is different from most other alignment programs in that it uses an evolutionary model to place insertions and deletions, with the result of minimizing the overalignment of nonhomologous regions. Prank+F has a much higher computational cost than MAFFT, and for this reason, we did not employ to calculate the alignments of all possible protein isoform combinations. Instead, we used Prank+F to recalculate the alignment corresponding to the Cons combination (as determined using MAFFT alignments) and the alignments corresponding to the combinations selected by Longest, PALO, and Random approaches. Prank+F can use a gene tree with distances for a better assessment of the evolutionary relationships between the different genes. We used the well-established species trees for the 1:1 orthologous data sets. The distances for the trees used in the Mammalia and Vertebrata data sets were extracted from previously described genomic sequence alignment data (Miller et al. 2007). The species tree for Metazoa did not include branch distances, as current estimates are highly variable. Obtaining reliable trees for the gene families in the paralogs data set was in many cases not possible due to the presence of several very close homologs. We ruled out using Prank+F for this data set, as the results would not be fully comparable to those obtained for the other data sets.
Data Sets for the Comparison of Longest and PALO
To understand the differences in the alignments generated when the Longest or PALO combination was used, we extracted all gene families, from the four initial data sets, in which the two methods resulted in a different protein isoform combination. We did not consider alignments in which the Conservation score with Cons was less than 20% as these alignments are of very poor quality. We also discarded families for which we had only one possible protein isoform combination as these are uninformative. These data sets were used in tables 3 and 4.
Table 3.
Characteristics of Alignments Depending on the Method Used to Select a Protein Isoform Combination
| Data Set | Method | % Hit Cons | Conservation Score |
% Indels | ||
|---|---|---|---|---|---|---|
| Mean | Median | SD | ||||
| Mammalia (3,827) | Cons | — | 70.26 | 73 | 18.66 | 12.32 |
| PALO | 71.73 | 68.24 | 72 | 20.78 | 14.33 | |
| Longest | 16.33 | 63.29 | 66 | 19.06 | 19.64 | |
| Random | 16.57 | 43.10 | 43 | 28.25 | 44.29 | |
| Vertebrata (1,836) | Cons | — | 51.40 | 50 | 17.04 | 16.16 |
| PALO | 59.04 | 49.48 | 49 | 18.21 | 18.54 | |
| Longest | 17.70 | 46.75 | 45.5 | 16.60 | 22.56 | |
| Random | 16.39 | 33.93 | 33 | 21.79 | 43.45 | |
| Metazoa (221) | Cons | — | 27.69 | 24 | 13.93 | 17.29 |
| PALO | 63.28 | 26.42 | 23 | 14.44 | 19.31 | |
| Longest | 24.29 | 24.48 | 22 | 12.73 | 24.4 | |
| Random | 22.60 | 18.37 | 16 | 13.72 | 42.71 | |
| Paralogs (154) | Cons | — | 51.89 | 47.5 | 24.81 | 18.91 |
| PALO | 59.09 | 47.04 | 41.5 | 27.83 | 23.73 | |
| Longest | 20.78 | 41.23 | 36 | 22.43 | 31.86 | |
| Random | 14.94 | 31.55 | 23 | 27.29 | 49.77 | |
Note.—Alignments were generated by MAFFT. Data are for gene families in which PALO selected a different combination from Longest. % Hit Cons, percentage of cases in which the protein isoform combination is the same as in Cons. SD, standard deviation.
Table 4.
Estimation of Nucleotide Substitutions in the Human Branch
| dN/dS |
dN |
dS |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | N | Mean | Median | SD | P | Mean | Median | SD | P | Mean | Median | SD | P |
| MAFFT | |||||||||||||
| Cons | 3,711 | 0.158 | 0.098 | 0.214 | — | 0.022 | 0.015 | 0.025 | — | 0.166 | 0.138 | 0.108 | — |
| PALO | 3,640 | 0.162 | 0.100 | 0.211 | 0.283 | 0.025 | 0.015 | 0.040 | 0.074 | 0.174 | 0.140 | 0.127 | 0.132 |
| Longest | 3,652 | 0.172 | 0.110 | 0.208 | 3.43e − 05 | 0.029 | 0.017 | 0.039 | 3.53e − 09 | 0.182 | 0.144 | 0.135 | 2.21e − 05 |
| Random | 2,920 | 0.191 | 0.116 | 0.286 | 3.11e − 06 | 0.036 | 0.018 | 0.059 | 7.42e − 15 | 0.204 | 0.153 | 0.178 | 3.05e − 14 |
| Prank+F | |||||||||||||
| Cons | 3,938 | 0.156 | 0.099 | 0.200 | — | 0.022 | 0.015 | 0.024 | — | 0.165 | 0.138 | 0.103 | — |
| PALO | 3,856 | 0.159 | 0.101 | 0.201 | 0.440 | 0.024 | 0.015 | 0.030 | 0.126 | 0.172 | 0.140 | 0.119 | 0.113 |
| Longest | 3,879 | 0.168 | 0.108 | 0.195 | 1.4e − 04 | 0.029 | 0.017 | 0.038 | 1.64e − 08 | 0.183 | 0.144 | 0.141 | 2.44e − 05 |
| Random | 3,127 | 0.185 | 0.112 | 0.256 | 1.2e − 02 | 0.035 | 0.018 | 0.058 | 4.54e − 13 | 0.207 | 0.152 | 0.191 | 1.37e − 14 |
Note.—MAFFT alignments, Mammalia data set. Data are for gene families in which PALO selected a different combination from Longest. dN, nonsynonymous substitution rate; dS, synonymous substitution rate; SD, standard deviation. P value is for Mann–Whitney test against Cons.
Identification of Indels
We scanned the alignments to calculate the number of columns in each alignment that contained indels in one or more sequences and divided the number of columns with indels by the total length of the alignments (table 3, % indels). We also extracted from each alignment the number of indels and the indel size (supplementary file S2: table S3, Supplementary Material online).
Estimation of Nonsynonymous and Synonymous Substitution Rates
We obtained coding sequence alignments corresponding to the already generated protein sequence alignments using the software pal2nal (Suyama et al. 2006). Then we estimated the number of nonsynonymous substitutions per nonsynonymous site (dN) and the number of synonymous substitution per synonymous site (dS) using the free-ratio model in CodeML (Yang 2007). This program does not consider columns containing gaps in one or more sequences. For the analysis of the data, we used similar filters to those employed in our previous works (Toll-Riera et al. 2011; Laurie et al. 2012). In particular, we discarded genes with branches showing dS < 0.01, as such low dS values may result in inaccurate dN/dS estimates, and branches showing dS or dN > 2 indicating saturation of substitutions. Finally, we also discarded a small number of outlier genes showing abnormally high dN/dS values (dN/dS > 10).
Tests of Positive Selection
For each gene family and branch in the tree, we performed a branch-site test of positive selection (Zhang et al. 2005), as implemented in the phylogenetic analysis by maximum likelihood (PAML) software package (Yang 2007). This test compares the null model where codon-based dN/dS for all branches can only be ≤1, with the alternative model where the labeled foreground branch may include codons evolving at dN/dS > 1. The likelihood ratio test was used to compare the two models. It was calculated as 2 × (L1 − L0), where L1 is the maximum likelihood value of the alternative hypothesis and L0 the maximum likelihood value of the null hypothesis. A χ2 distribution with 1 degree of freedom was used to calculate the P value.
Statistical Data Analyses
We used Python to code the analysis pipeline. Analysis of data, including generation of plots and statistical tests, was done with R (R Development Core Team 2010).
Results and Discussion
Methods to Select a Protein Isoform Combination from a Gene Family
To understand the impact of the method employed to select a protein isoform combination on downstream analyses, we gathered several gene family data sets using orthology and paralogy information from EnsemblCompara (Vilella et al. 2009). Three of the data sets contained 1 to 1 orthologous genes from four different species separated by increasingly larger phylogenetic distances (Mammalia, Vertebrata, and Metazoa) (table 2). The average number of possible combinations was 28 in Mammalia, 25.4 in Vertebrata, and 21.6 in Metazoa. In all data sets, the distribution had a long tail, with median values ranging between 8 and 9, and with some families showing >1,000 possible protein isoform combinations (fig. 1). An additional set of families was constructed that contained paralogous gene copies from human, mouse, or both (data set paralogs) (table 2). These families were much larger, and consequently, the number of possible isoform combinations per family was also in general much higher (average 1,513; median 20). Most of the families in these different data sets (>90%) were associated with more than one possible protein isoform combination.
Fig. 1.—

Number of protein isoform combinations in different data sets. See table 1 for a description of the data sets.
Once we had the gene family data sets, we applied different methods to select one protein isoform combination per family: 1) Cons: The combination resulting in the best-conserved alignment, as measured by the number of identical positions divided by alignment length (Conservation score). This combination was determined after running all possible protein isoform combination alignments with MAFFT (Katoh and Toh 2008). Under this strategy, the number of alignment errors should be kept to a minimum at the cost of losing some combinations for which the alignment includes genuinely rapidly evolving regions. 2) Longest: The combination that corresponded to the longest protein isoform per gene. 3) PALO: The combination that corresponded to the protein isoforms that were more similar in length using least squares (see Materials and Methods). This is a novel method we implemented here for the first time, the code and a web server application are available at http://evolutionarygenomics.imim.es/palo. 4). Random: A randomly chosen protein isoform combination.
Overall, PALO selected a different protein isoform combination than Longest in 33.6% of the gene families for which there was more than one possible combination (6,038 gene families in all data sets taken together). This percentage increased to 53% for gene families having 30 or more possible isoform combinations. To illustrate the difference between Longest and PALO, figure 2 shows a cartoon of a hypothetical family in which the two methods select a different protein isoform combination.
Fig. 2.—

Schematic representation of protein isoform combination selection by Longest and PALO. Hypothetical gene family with four possible protein isoform combinations.
The use of the different methods has different computational costs. Longest only requires one operation for gene (selecting the longest isoform) and is thus really fast. In PALO, we first need to calculate all possible protein isoform combinations and their sequence length distance to select the combination associated with the smallest difference in length. With the application developed here, written in Python, and using a desktop computer processor with 48-Gb RAM, the process is approximately linear for up to 60 million combinations, taking approximately 0.2 s for a Mammalia gene family associated with 10,000 protein isoform combinations. The cost associated with Cons is much higher as we need to compute all the alignments for all protein isoform combinations to select the one with the highest sequence conservation. With MAFFT, which is one of the fastest aligners, 10,000 alignments of four orthologous mammalian protein sequences take approximately 32 min (∼2,000 s) using a single processor. In other words, Cons is approximately 4 orders of magnitude slower than PALO when aligning four sequences. As alignment cost increases in a nonlinear manner with the number of sequences, the Cons approach rapidly becomes prohibitive for larger gene families with many protein isoforms.
Characteristics of the Alignments Depending on the Method
In the 6,038 gene families in which PALO and Longest chose a different protein isoform combination, PALO matched the Cons combination in 60–70% of cases, in marked contrast with Longest and Random, which only matched the Cons combination in approximately 16–21% of cases (table 3). Where the selected combination varied, we performed alignments with MAFFT and examined overall sequence conservation, alignment length, and number of indels.
By definition, Cons alignments were those with the highest Conservation score, which was calculated as the percentage identity over the alignment length. In second place were alignments obtained with the PALO combination, followed by Longest and Random (table 3). These trends were consistently observed across families binned by the number of possible protein isoform combinations (supplementary file S2: table S1, Supplementary Material online), as well as in the initial complete gene family data sets (supplementary file S2: table S2, Supplementary Material online). Longest selects the longest protein isoform per gene, so the alignments should be longer using this method than using any other method. We confirmed this expectation in all four data sets (supplementary file S2: fig. S1, Supplementary Material online), differences with PALO being highly significant for Mammalia, Vertebrata, and paralogs (Mann–Whitney test P < 10−4).
We next evaluated whether there were any differences in the proportion and number of indels in the alignment, measured as gaps in one or more sequences. A priori we expected a larger proportion of indels in the Longest alignments than in the PALO or Cons alignments. This is because PALO selects isoforms that are as similar as possible in length and this should result in less indels, and Cons selects alignments with the maximum number of amino acid identities divided by alignment length (including indels). It is important to note that an increase in the number of indels, both in real alignments and in sequence evolution simulations, has been linked with an increase in the number of misaligned positions and a consequent overestimation of the number of positively selected sites (Fletcher and Yang 2010; Markova-Raina and Petrov 2011; Jordan and Goldman 2012). We confirmed that a significantly larger percentage of the alignment was covered by indels in Longest than in PALO or Cons (table 3). This result was highly significant for all data sets (Mann–Whitney test P < 10−4), and it resulted from both a larger number of indels and a larger mean indel length (supplementary file S2: table S3, Supplementary Material online). Random was the method that resulted in the largest proportion of indels. With Random, the indels tended to be longer than with the other three methods, probably reflecting the fact that some alignments included partial sequences or very short isoforms.
Impact of the Method on the Estimation of Nonsynonymous and Synonymous Substitution Rates
We next investigated the impact of the different methods of selecting a protein isoform combination on the estimation of the number of nonsynonymous substitutions per nonsynonymous site (dN), the number of synonymous substitutions per synonymous site (dS), and the dN/dS ratio. We used the previously obtained MAFFT amino acid sequence alignments converted to coding sequence alignments. We obtained branch-specific dN, dS, and dN/dS estimates using the maximum likelihood-based method codeml in the PAML package (Yang 2007). We focused on the Mammalia data set, as it fulfils the requirements for optimal dN and dS estimation. First, the species are not only sufficiently closely related to avoid saturation of substitutions but also sufficiently distant to obtain reliable dN and dS estimates for all branches in most gene families. Second, because the data set only contains 1:1 orthologs, we can use the known species tree as input for codeml and avoid uncertainties about branch order that can greatly alter the rate estimations.
The lowest dN, dS, and dN/dS values corresponded to Cons (table 4 and supplementary file S2: tables S4 and S5, Supplementary Material online). As Cons selects the alignment with the highest amino acid sequence conservation, we expect lower dN and dN/dS values with this method than with the other methods. There were no significant differences between PALO and Cons in any of the comparisons, indicating that PALO does not overestimate dN, dS, or dN/dS. In contrast, there were significant differences between Cons and Longest in these three parameters in the human branch (P < 10−4 in table 3, P < 0.05 supplementary file S2: table S4, Supplementary Material online, for complete data set) and in dN in the mouse branch (P < 0.05 supplementary file S2: table S5, Supplementary Material online). In the alignments obtained with Random, the substitution rates were clearly overestimated with respect to the other methods, and in this case, we found significant differences in dN, dS, or dN/dS values with respect to Cons not only in the human and mouse branches but also in the horse and cow branches (supplementary file S2: tables S4 and S5, Supplementary Material online). Overall, estimations for the human branch were the most sensitive to the method employed, probably due to the larger number of annotated transcript variants in humans (table 1), causing different methods to more often select a different isoform.
The results described earlier are difficult to explain other than by a higher proportion of misaligned positions in Longest than in PALO. First, taking the four methods together, there is a clear relationship between the proportion of indels and the estimated dN and dS values, which is consistent with previously reported alignment inaccuracies with increasing number of indels (see previous section). Second, the higher dN and dS values in Longest are unlikely to be due to the inclusion of additional rapidly evolving regions, as there are no significant differences in alignment length between Cons, Longest, and PALO when we exclude regions with gaps (supplementary file S2: fig. S2, Supplementary Material online). Third, even if there were differences in the selective regime of the sequences selected by the different methods, this should not affect dS, as this is basically a neutral substitution rate and should not vary in the absence of alignment errors.
Prank+F is a phylogeny aware program that, contrary to most other programs, does not underestimate insertions, which helps reduce the number of misaligned positions (Löytynoja and Goldman 2008; Fletcher and Yang 2010). We generated alignments with this program using the same protein isoform combinations as before. PRANK+F alignments resulted in lower average dN/dS ratio estimates than MAFFT alignments in the human branch (table 4) although the results were less clear for other branches (supplementary file S2: tables S4 and S5, Supplementary Material online). Overall, the relative differences between the methods were essentially maintained when using Prank+F.
Impact on the Estimation of the Fraction of Positively Selected Genes
The identification of genes evolving under positive selection has been reported to be very sensitive to misalignment errors (Mallick et al. 2009; Schneider et al. 2009; Fletcher and Yang 2010; Markova-Raina and Petrov 2011; Jordan and Goldman 2012). What is the effect of the method used to select a protein isoform combination on the estimation of genes under positive selection? We ran the branch-site test, available in the PAML software package (Yang 2007), to test for positive selection in the human or mouse branches using all Mammalia alignments (table 5). The fraction of positively selected genes at a P value < 0.05 increased gradually following the order Cons, PALO, Longest, and Random, both for the human and the mouse branches (table 5). Using Bonferroni correction for multiple testing or the false discovery rate correction of Benjamini and Hochberg, which are two commonly employed adjustments (Anisimova and Yang 2007), the differences between the methods became even more evident. For example, Longest detected 666 genes under positive selection in the human branch and 600 in the mouse branch, compared with 220 and 392, respectively, in the case of Cons. Although Cons may be conservative in some cases—it will always select the best-conserved alignment discarding alternatives that may be genuinely associated with higher divergence—the differences between Longest and PALO were also remarkable (in the same comparison 666 vs. 406 in human, and 600 vs. 495 in mouse), indicating that Longest overestimates the impact of positive selection. Figure 3 shows part of the alignment of brain Kelch-like protein 13 in 1:1 orthologs from human, mouse, horse, and cow. The combination selected by Longest contains an N-terminal extension. The number of aligned positions is the same in both cases but dN/dS is much higher in Longest, indicating that some positions are misaligned. In this case, this leads to incorrect inference of positive selection with Longest.
Table 5.
Number of Estimated Positively Selected Genes in Human and Mouse Branches Using Different Methods
| Software | Method | N | Raw P < 0.05 | Bonferroni P < 0.05 | BH P < 0.05 |
|---|---|---|---|---|---|
| Human | |||||
| MAFFT | Cons | 12.794 | 988 (7.72) | 69 (0.54) | 220 (1.72) |
| PALO | 12.702 | 1.133 (8.91) | 185 (1.45) | 406 (3.19) | |
| Longest | 12.758 | 1.401 (10.98) | 344 (2.69) | 666 (5.21) | |
| Random | 10.365 | 1.977 (19.07) | 742 (7.15) | 1.478 (14.25) | |
| PRANK+F | Cons | 12.800 | 962 (7.52) | 71 (0.55) | 208 (1.62) |
| PALO | 12.708 | 1.086 (8.54) | 176 (1.38) | 374 (2.94) | |
| Longest | 12.758 | 1.341 (10.5) | 331 (2.59) | 617 (4.83) | |
| Random | 10.363 | 1.909 (18.41) | 653 (6.30) | 1.393 (13.43) | |
| Mouse | |||||
| MAFFT | Cons | 12.866 | 1.360 (10.5) | 99 (0.77) | 392 (3.04) |
| PALO | 12.777 | 1.435 (11.23) | 127 (0.99) | 495 (3.87) | |
| Longest | 12.829 | 1.545 (12.04) | 166 (1.29) | 600 (4.67) | |
| Random | 10.573 | 1.428 (13.5) | 206 (1.94) | 701 (6.63) | |
| PRANK+F | Cons | 12.872 | 1.341 (10.82) | 89 (0.69) | 341 (2.65) |
| PALO | 12.770 | 1.396 (11.35) | 121 (0.94) | 454 (3.55) | |
| Longest | 12.827 | 1.495 (12.1) | 161 (1.25) | 561 (4.37) | |
| Random | 10.555 | 1.313 (13.64) | 194 (1.83) | 653 (6.18) | |
Note.—BH: Benjamini and Hochberg false discovery correction. The proportion of genes under positive selection is significantly higher in all methods with respect to Cons by a Fisher test with P value < 10−3.
Fig. 3.—

Example analysis of a gene family using Longest and PALO. The example shown is ENSEMBL gene ENSG00000003096 (brain Kelch-like protein 13), associated with 66 possible protein isoform combinations in 1:1 orthologs from human, mouse, horse, and cow. The first 70 positions of the alignments using the Longest (above) and PALO (below) methods are shown. Longest selects a human protein isoform that shows an extension at the N-terminus. Misalignment of this region results in inflated dN/dS values at the level of the whole protein and an artifactual signal of positive selection (PS) in the human branch.
The method used to select a protein isoform combination seems to have a much larger effect on the estimation of the fraction of positively selected genes than on the estimation of dN/dS. For example, for mouse the inflation of dN/dS values is only significant for Random (supplementary file S2: tables S4 and S5, Supplementary Material online), whereas in this same species, the increase in the fraction of positively selected genes is very evident both for Longest and Random. Another matter of concern is that the scale of the problem is not the same for different branches, which may lead to incorrect conclusions when comparing species. For example, if we use Longest, the human branch appears to have a higher fraction of positively selected genes than the mouse branch, but the opposite is observed if we use PALO or Cons (table 5). All else being equal, we can expect more positively selected genes in the mouse branch simply because it has accumulated 2–3 times more substitutions in the same amount of time (Waterston et al. 2002; Toll-Riera et al. 2011) and so the test should be more sensitive. In fact, the result obtained with Longest is likely to be an artifact caused by the selection of longer isoforms for human compared with the other species, simply because there is a higher number of annotated isoforms in this species.
In a comparison of different multiple alignment programs, which included MAFFT and Prank+F, Fletcher and Yang (2010) reported that for all programs, the false-positive rate decreased as sequences became more divergent. Using the Conservation score to bin the alignments in different groups here, we observed the same trend (supplementary file S2: table S6, Supplementary Material online). In addition, for the most conserved Cons alignments (>86% identity over alignment length) the fraction of human-specific positively selected genes was very similar for Cons and PALO (∼0.7%) but still more than double for Longest (∼1.6%).
The estimated fraction of positively selected genes when using Prank+F alignments was consistently lower than with MAFFT alignments, in agreement with previous results (Fletcher and Yang 2010; Jordan and Goldman 2012). The reduction, which was of approximately 10–12% in most cases, affected all four methods in a similar manner (table 5). This means that many false positives still remained using Prank+F and Longest. In this line, in a recent study focusing on orthologous genes from the 12 Drosophila genomes found that although Prank+F improved the problem of false positives in the estimates of positive selection, still approximately 50% of the positions reported to be under positive selection appeared to be misaligned residues (Markova-Raina and Petrov 2011). It is important to note that in this study, the authors used the longest annotated transcript per gene and observed that in the majority of cases the sites wrongly inferred as positively selected were close to the start or end of an indel. Our results indicate that, by eliminating unnecessary indels—those inserted to accommodate the presence of nonhomologus regions—the problem of overestimation of positive selection can be reduced in a very significant manner.
Conclusions
The number of known transcripts per gene is going to increase very rapidly in the forthcoming years due to the generalized use of RNA ultrasequencing techniques (RNASeq) in a large variety of species. Results from the ENCODE project using such deep RNA sequencing techniques indicate that a typical human gene can encode 10–12 transcript variants, although the majority of them are expressed at low levels (Djebali et al. 2012). It is thus important to establish the most appropriate methods for selecting protein isoforms for comparative and evolutionary analyses.
This problem has traditionally been tackled by taking the longest protein isoform per gene. Here, we have shown that this leads to an important overestimation of the fraction of positively selected sites, due to a higher fraction of misaligned positions in more indel-rich alignments. We propose using instead the protein isoforms that are most similar in length, as this significantly improves the quality of the alignments generated and reduces the likelihood of wrongly identifying positively selected sites. Possible future developments include filtering out possible spurious or very low abundance protein isoforms to reduce the number of combinations to be tested.
Supplementary Material
Supplementary files S1 and S2 (figures S1 and S2 and tables S1–S6) are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org).
Acknowledgments
The authors thank Magda Gayà, Georgios Athanasiadis, and the members of the Evolutionary Genomics Group (UPF-IMIM) for fruitful discussions. They are also grateful to François Serra for help with parallelizing the computations. This work was funded by Ministerio de Economía y Competitividad (FPI BES-2010-038494 to J.L.V.-C., Plan Nacional BIO2009-08160 and BFU2012-36820) and Fundació ICREA to M.M.A.
Literature Cited
- Albà MM, Castresana J. Inverse relationship between evolutionary rate and age of mammalian genes. Mol Biol Evol. 2005;22:598–606. doi: 10.1093/molbev/msi045. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Yang Z. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol Biol Evol. 2007;24:1219–1228. doi: 10.1093/molbev/msm042. [DOI] [PubMed] [Google Scholar]
- Arbiza L, Dopazo J, Dopazo H. Positive selection, relaxation, and acceleration in the evolution of the human and chimp genome. PLoS Comput Biol. 2006;2:e38. doi: 10.1371/journal.pcbi.0020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakewell MA, Shi P, Zhang J. More genes underwent positive selection in chimpanzee evolution than in human evolution. Proc Natl Acad Sci U S A. 2007;104:7489–7494. doi: 10.1073/pnas.0701705104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro M, et al. Evidence for widespread positive and purifying selection across the European rabbit (Oryctolagus cuniculus) genome. Mol Biol Evol. 2012;29:1837–1849. doi: 10.1093/molbev/mss025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SC-C, Chuang T-J, Li W-H. The relationships among microRNA regulation, intrinsically disordered regions, and other indicators of protein evolutionary rate. Mol Biol Evol. 2011;28:2513–2520. doi: 10.1093/molbev/msr068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, et al. Inferring nonneutral evolution from human-chimp-mouse orthologous gene trios. Science. 2003;302:1960–1963. doi: 10.1126/science.1088821. [DOI] [PubMed] [Google Scholar]
- Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Raval A, Wilke CO. A single determinant dominates the rate of yeast protein evolution. Mol Biol Evol. 2006;23:327–337. doi: 10.1093/molbev/msj038. [DOI] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D. Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Mol Biol Evol. 2000;17:68–74. doi: 10.1093/oxfordjournals.molbev.a026239. [DOI] [PubMed] [Google Scholar]
- Fletcher W, Yang Z. The effect of insertions, deletions, and alignment errors on the branch-site test of positive selection. Mol Biol Evol. 2010;27:2257–2267. doi: 10.1093/molbev/msq115. [DOI] [PubMed] [Google Scholar]
- Flicek P, et al. Ensembl 2012. Nucleic Acids Res. 2012;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- Jordan G, Goldman N. The effects of alignment error and alignment filtering on the sitewise detection of positive selection. Mol Biol Evol. 2012;29:1125–1139. doi: 10.1093/molbev/msr272. [DOI] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9:286–298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- Kosiol C, et al. Patterns of positive selection in six Mammalian genomes. PLoS Genet. 2008;4:e1000144. doi: 10.1371/journal.pgen.1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie S, Toll-Riera M, Radó-Trilla N, Albà MM. Sequence shortening in the rodent ancestor. Genome Res. 2012;22:478–485. doi: 10.1101/gr.121897.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 2005;102:10557–10562. doi: 10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science. 2008;320:1632–1635. doi: 10.1126/science.1158395. [DOI] [PubMed] [Google Scholar]
- Mallick S, Gnerre S, Muller P, Reich D. The difficulty of avoiding false positives in genome scans for natural selection. Genome Res. 2009;19:922–933. doi: 10.1101/gr.086512.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markova-Raina P, Petrov D. High sensitivity to aligner and high rate of false positives in the estimates of positive selection in the 12 Drosophila genomes. Genome Res. 2011;21:863–874. doi: 10.1101/gr.115949.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. 2011;12:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- McInerney JO. The causes of protein evolutionary rate variation. Trends Ecol Evol. 2006;21:230–232. doi: 10.1016/j.tree.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Miller W, et al. 28-way vertebrate alignment and conservation track in the UCSC Genome Browser. Genome Res. 2007;17:1797–1808. doi: 10.1101/gr.6761107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pál C, Papp B, Hurst L. Highly expressed genes in yeast evolve slowly. Genetics. 2001;158:927–931. doi: 10.1093/genetics/158.2.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privman E, Penn O, Pupko T. Improving the performance of positive selection inference by filtering unreliable alignment regions. Mol Biol Evol. 2012;29:1–5. doi: 10.1093/molbev/msr177. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R: a language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing; 2010. [Google Scholar]
- Schneider A, et al. Estimates of positive Darwinian selection are inflated by errors in sequencing, annotation, and alignment. Genome Biol Evol. 2009;1:114–118. doi: 10.1093/gbe/evp012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll-Riera M, Laurie S, Albà MM. Lineage-specific variation in intensity of natural selection in mammals. Mol Biol Evol. 2011;28:383–398. doi: 10.1093/molbev/msq206. [DOI] [PubMed] [Google Scholar]
- Vilella AJ, et al. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Wong KM, Suchard MA, Huelsenbeck JP. Alignment uncertainty and genomic analysis. Science. 2008;319:473–476. doi: 10.1126/science.1151532. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Zambelli F, Pavesi G, Gissi C, Horner DS, Pesole G. Assessment of orthologous splicing isoforms in human and mouse orthologous genes. BMC Genomics. 2010;11:534. doi: 10.1186/1471-2164-11-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- Zhang L, Li W-H. Mammalian housekeeping genes evolve more slowly than tissue-specific genes. Mol Biol Evol. 2004;21:236–239. doi: 10.1093/molbev/msh010. [DOI] [PubMed] [Google Scholar]