

Abstract
Recent studies suggested a role for the human endogenous retrovirus (HERV) group HERV-K(HML-2) in melanoma because of upregulated transcription and expression of HERV-K(HML-2)-encoded proteins. Very little is known about which HML-2 loci are transcribed in melanoma. We assigned >1,400 HML-2 cDNA sequences generated from various melanoma and related samples to genomic HML-2 loci, identifying a total of 23 loci as transcribed. Transcription profiles of loci differed significantly between samples. One locus was found transcribed only in melanoma-derived samples but not in melanocytes and might represent a marker for melanoma. Several of the transcribed loci harbor ORFs for retroviral Gag and/or Env proteins. Env-encoding loci were transcribed only in melanoma. Specific investigation of rec and np9 transcripts indicated transcription of protein encoding loci in melanoma and melanocytes hinting at the relevance of Rec and Np9 in melanoma. UVB irradiation changed transcription profiles of loci and overall transcript levels decreased in melanoma and melanocytes. We further identified transcribed HML-2 loci formed by reverse transcription of spliced HML-2 transcripts by L1 machinery or in a retroviral fashion, with loci potentially encoding HML-2-like proteins. We reveal complex, sample-specific transcription of HML-2 loci in melanoma and related samples. Identified HML-2 loci and proteins encoded by those loci are particularly relevant for further studying the role of HML-2 in melanoma. Transcription of HERVs appears as a complex mechanism requiring specific studies to elucidate which HERV loci are transcribed and how transcribed HERVs may be involved in disease.
Keywords: repetitive DNA, HERV, provirus, transcription, retrotransposition, neoplasms
Introduction
Approximately 8% of the human genome mass are of direct or indirect retroviral origin. So-called human endogenous retroviruses (HERVs) are remnants of ancient retroviruses that infected the genome of germ line cells of human ancestral species millions of years ago. Quite a number of phylogenetically distinct retroviruses invaded the germ line and thus left their traces in the human genome. Approximately 40 different HERV groups were previously cataloged in the human genome sequence. (The term "group" is favorable over the formerly used term "family" because "family" is reserved for Retroviridae [Blomberg et al. 2009; Mayer et al. 2011; Stoye 2012].) Some of those HERV groups significantly amplified in copy numbers, sometimes up to several thousand copies, following initial germ line invasion by reinfection or intracellular formation of new copies. HERV loci consist of so-called proviruses harboring retroviral Gag, Protease, Polymerase, and Envelope-coding sequences and flanked by long terminal repeats (LTRs). HERV loci are often significantly mutated due to long time presence in the genome, displaying deletions of variable sizes and numerous other nonsense mutations. Some of the mutated HERV loci could also serve as template for new copies. Proviral loci also became often reduced to so-called solitary LTRs by homologous recombination between the two proviral LTRs leaving a single LTR behind (for reviews, see Mager and Medstrand 2003; Bannert and Kurth 2006; Ruprecht, Mayer, et al. 2008; Stoye 2012).
HERVs influence the human genome and human biology. Although coding capacity for original retroviral proteins has been impaired by nonsense mutations for most HERVs, many HERV loci nevertheless remain transcriptionally active and a number of studies identified HERV transcripts from different HERV groups in various tissue and cell types. Transcriptional activity of HERV loci differs between cell and tissue types and is therefore regulated in some way (for instance, see Yin et al. 1997; Büscher et al. 2005; Frank et al. 2005, 2008; Seifarth et al. 2005; Hu et al. 2006; Muradrasoli et al. 2006; Oja et al. 2007; Haupt et al. 2011; Perot et al. 2012). Several instances of transcriptionally active HERV loci influencing neighboring genes by providing (alternative) promoters have been documented. HERV loci can furthermore affect transcripts of cellular genes by providing alternative splice and polyadenylation signals (reviewed in Cohen et al. 2009).
Some HERV groups have biological relevance in humans. The syncytin-1/ERVWE1 gene is, in essence, a proviral locus of the HERV-W group. The Env protein of that HERV-W locus, named Syncytin-1, has been selected during evolution for contributing to fusion of cell membranes of trophoblasts to form syncytiotrophoblasts, which is an essential process during human placenta formation (Mallet et al. 2004; Dupressoir et al. 2012). The HERV-K(HML-2) group, in short, HML-2, is exceptional regarding evolution, coding capacity and potential clinical involvement. Besides evolutionarily older loci, the HML-2 group comprises younger proviruses, several of them being human specific and thus having formed after the evolutionary split of human from chimpanzee, resulting in presence/absence of alleles of HML-2 loci (Reus et al. 2001; Hughes and Coffin 2004; Macfarlane and Simmonds 2004; Belshaw et al. 2005). Evolutionarily young HML-2 proviruses often encode one or several of the former retroviral proteins (Kitamura et al. 1996; Schommer et al. 1996; Barbulescu et al. 1999; Mayer et al. 1999; Tönjes et al. 1999). HML-2-encoded Rec and Np9 proteins, the latter being encoded by so-called HML-2 type I proviruses lacking a 292-bp sequence portion within the pol-env boundary, interact with cellular proteins such as promyelocytic leukemia zinc finger protein, ligand of numb protein X, testicular zinc-finger protein, androgen receptor, and family with sequence similarity 21 (FAM21) and might thus be involved in tumorgenesis (Boese et al. 2000; Armbruester et al. 2004; Kramer-Hämmerle et al. 2005; Kaufmann et al. 2010). Nude mice transgenic for the nuclear export factor Rec display disturbed germ cell development and histological lesions reminiscent of carcinoma in situ (Galli et al. 2005). We recently identified a novel HML-2-encoded protein, Env-SP, a stable signal peptide resulting from processing of HML-2 Env, which is very similar in sequence to Rec protein, but displays several biological features different from Rec (Ruggieri et al. 2009).
HML-2 expression has been found upregulated in several tumor types, among them germ cell tumors (GCTs). GCT precursor lesions (carcinoma in situ) already display high amounts of HML-2 RNA (Herbst et al. 1996), and patients with GCT display high antibody titers against HML-2-encoded Gag and Env proteins already at the time of tumor detection (Sauter et al. 1995, 1996; Boller et al. 1997). Previous studies also suggested a role of HML-2 in the etiology of melanoma, the most malignant type of skin cancer with increasing incidence worldwide that arises from uncontrolled proliferation of pigment-producing melanocytes in skin, mucosa, or uvea (Dennis 1999). Some melanoma cell lines were shown to produce retrovirus-like particles (RVLPs) with reverse transcriptase activity that contain HML-2-like sequences and Gag and Env protein (Muster et al. 2003). Those RVLPs were subsequently found to be defective and noninfectious (Büscher et al. 2005). Expression of HML-2 was reported as elevated in melanoma, which may be due to increased promotor activity and demethylation of the HML-2 proviral 5′-LTR (Stengel et al. 2010). HML-2 RNA, as well as Gag, Env, Rec, and Np9 proteins, was identified in melanoma tissue and melanoma cell lines (Büscher et al. 2005, 2006; Reiche et al. 2010). HERV-K-specific antibodies were found in melanoma patients, and serological response against HML-2 Gag and Env protein was furthermore reported and correlated with survival probability (Büscher et al. 2005; Hahn et al. 2008). Adherent melanoma cells were shown to undergo a transition to a more malignant, nonadherent phenotype when exposed to stress, accompanied by HERV-K(HML-2) expression (Serafino et al. 2009). Specific activation of HML-2 expression was furthermore reported in response to UV radiation, the most established environmental risk factor in melanoma development (Schanab et al. 2011).
However, it still remains uncertain whether HML-2 retroviral sequences are activated as a corollary of malignant transformation or if they might actively participate in tumor development and the ability of melanoma cells to escape immunosurveillance.
On the basis of the description of overall transcript levels of HERV groups in cells and tissues, we and others recently established strategies for identifying those HERV loci that actually contribute to HERV-group-specific RNAs. If HERV RNA or HERV-encoded proteins are of biological relevance, solely the transcribed HERV loci should obviously be of relevance and their identification is therefore of primary interest. A recent microarray-based approach aims at identifying transcribed HERV loci by a collection of locus-specific oligoncleotides (Perot et al. 2012). We recently established a strategy that involves generation of HERV-specific cDNA sequences by RT-PCR followed by cloning and sequencing of RT-PCR products. HERV cDNA sequences can then be assigned to genomic HERV loci by sequence comparisons (Flockerzi et al. 2008). We thus obtained transcription profiles of HML-2 loci and loci of other HERV groups in various tissue and cell types, with transcription levels of specific HERV loci differing significantly between examined tissues and cell types (Flockerzi et al. 2008; Frank et al. 2008; Ruprecht et al. 2008; Laufer et al. 2009).
To contribute to a better understanding of the biological role of HML-2 in melanoma, we set out to identify HML-2 loci transcribed in melanoma. We generated HML-2 cDNA sequences from melanoma tissue, melanoma cell lines, and melanocyte cell lines. We derived transcription profiles for HML-2 loci and correlated transcription of HML-2 loci with provirus structures and coding capacity for Gag, Env, Rec, and Np9 proteins. We also quantified HML-2 transcript levels in the various samples and examined differences in transcription profiles following UV irradiation. Finally, we analyzed in more detail transcribed HML-2 loci that appeared to be spliced and subsequently retrotransposed HML-2 messenger RNAs (mRNAs).
Materials and Methods
Melanoma Cell Lines and Human Tissue Samples
Noncommercial WM3734 human melanoma cell lines were obtained in accordance with consent procedures approved by the Internal Review Boards of the University of Pennsylvania School of Medicine and The Wistar Institute. WM3734a (BRAFV600E, NRAS wt) were isolated and routinely maintained in Tu2% medium, consisting of 80% [v/v] MCDB 153 Basal Medium (Biochrom, Berlin, Germany) and 20% [v/v] L-15 Leibovitz Medium (Biochrom, Berlin, Germany) supplemented with 2% [v/v] FBS, 1.68 mM CaCl2, and 2.5 ng/ml insulin, as described previously (Satyamoorthy et al. 1997). SK-Mel-25, SK-Mel-28 (BRAFV600E, CDK4R24C) (Muster et al. 2003; Büscher et al. 2005, 2006; Stengel et al. 2010), and MEWO (Büscher et al. 2005; Reiche et al. 2010) cells were cultured in RPMI 1640 medium (Gibco, Life Technologies, Carlsbad, CA) supplemented with 10% [v/v] FCS. Melanocyte cell lines Benno and Oskar were cultured in Melanocyte Growth Medium (PromoCell, Heidelberg, Germany). All cells were cultured at 37 °C in a humidified 5% [v/v] CO2 atmosphere. The consistency of cellular genotypes was confirmed by DNA fingerprinting at the Department of Human Genetics of The Saarland University Hospital. Human melanoma tissues were obtained in accordance with consent procedures approved by the ethical review committee of the Saarland state chamber of physicians (No. 178/11). Melanoma lymph node metastases were stored at −80 °C immediately following surgical removal at the Department of Dermatology, Medical Faculty, University Clinic of Saarland, Homburg, Germany. Total RNAs from malignant melanoma metastases were obtained from Cambridge Bioscience (Cambridge, UK). Samples contained at least 90% tumor tissue were derived from jejunum (cat# CR562868), lung (cat# CR562901), and lymph node (cat# CR561386). Minimum stage grouping for samples CR562868 and CR562901 was IV and IIIB for sample CR561386.
RNA Isolation, Generation of cDNA, and PCR
RNA was isolated from cells and metastases using the RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. Cultured cells were pelleted by centrifugation for 5 min at 300 × g, resuspended and applied to a QIAshredder column (Qiagen, Hilden, Germany), according to the manufacturer's instructions, before application to an RNeasy Mini spin column. Tissue samples from metastases were homogenized in cold Trizol (Invitrogen, Life Technologies, Carlsbad, CA), applied to a QIAshredder column (Qiagen, Hilden, Germany), centrifuged at 15,000 × g for 2 min, and incubated at 30 °C for 5 min. After addition of 0.4 ml chloroform, samples were mixed by vortexing for 40 s, incubated at 30 °C for 3 min, and centrifuged at 8,600 × g and 4 °C for 20 min. The upper phase was mixed with 1 vol. 70% [v/v] ethanol and applied to an RNeasy Mini spin column. To remove traces of genomic DNA, isolated RNA was treated with Turbo DNA-freeTM Kit (Applied Biosystems, Foster City, CA). RNA (10 µg) was treated in a 50 µl reaction, according to the manufacturer's instructions. Removal of DNA was verified by Alu-element-specific PCR (Klein et al. 1993) and further controls in subsequent experiments (see below).
cDNA was generated using the Omniscript RT Kit (Qiagen, Hilden, Germany) and 10 µM of random hexanucleotide primers in a 30 µl reaction, following the manufacturer's instructions. 6 µl of the DNase reaction containing 1.2 µg RNA and random primers were combined, denatured at 70 °C for 5 min, and cooled to room temperature to allow primer annealing. Two reactions were prepared for each RNA sample, one of them serving as RT(–) negative control. A mastermix was prepared with buffer, dNTPs, and RNase inhibitor, which was distributed to all RNA samples. RT enzyme was subsequently added to the RT reactions and an equal amount of water to corresponding RT(–) controls.
For subsequent gag-specific PCR of cDNA, primers were optimized toward amplification of as many loci as possible in one PCR. An alignment of all genomic HML-2 loci was generated using BLAT results of the HML-2 consensus sequence obtained from RepBase. Coordinates and sequences of complete loci including LTRs, if present, were assembled using the UCSC table browser (Karolchik et al. 2004). Combinations of four forward and three reverse primers, reflecting nucleotide differences in the primer binding regions of the various HERV-K(HML-2) target sequences, were used to amplify an approximately 620-bp portion of the HML-2 gag gene (nt 1,778–2,396 in the HERV-K(HML-2.HOM) reference sequence, GenBank accession no. AF074086.2). Foward primers HML-2_1778_for1: 5′-CCCCCAGAAAGTCAGTATGGA-3′; HML-2_1778_for2: 5′-TCTCCAGAGGTTCAGTATGGA-3′; HML-2_1778_for3: 5′-CCCCCAGAAAATCAGTATGGA-3′; and HML-2_1778_for4: 5′-TCTCCAGAGGTGCAGTATAGA-3′ were combined in a 10:3:2:1 ratio. Reverse primers HML-2_2396_rev1: 5′-TTTCCCAGGCTCTAAGGCAG-3′; HML-2_2396_rev2: 5′-TTCCCAGGCCCTGAGGCAA-3′; and HML-2_2396_rev3: 5′-TTTCCTAGGCTCTAAGGCAG-3′ were combined in a 12:6:1 ratio, corresponding roughly to the number of potential targets of each primer variant. The PCR mix contained forward and reverse primer mixes at 0.5 µM each, 1 U recombinant Taq DNA polymerase (Invitrogen, Life Technologies, Carlsbad, CA), 1.5 mM MgCl2, 0.2 mM of each dNTP, 1× PCR buffer, and 1 µl of cDNA in a 20 µl reaction. A water control was included, as well as a negative control for each sample, containing 1 µl of the respective RT(–) reaction as template. Cycling conditions were as follows: initial denaturation 3 min at 95 °C, 40 cycles 50 s at 95 °C, 50 s at 53 °C, 1 min at 72 °C, and final elongation 10 min at 72 °C. The low-stringency annealing temperature should contribute to amplification of HML-2 loci for which primers do not match perfectly. For amplification of the rec/np9 region, primers spanning nt 6,451–8,728 in the HERV-K(HML-2.HOM) reference sequence were used, generating products of approximately 370 bp for np9 and approximately 580 bp for rec. The forward primers np9-FOR-1: 5′-ATGAACCCATCAGAGATGCAA-3′; np9-FOR-2: 5′-ATGAATCCATCAGAGATGCAA-3′; np9-FOR-3: 5′-GCGAACCCTTCAGAGATGCAA-3′; and np9-FOR-4: 5′-ATGAACCCATCGGAGATGAAA-3′ were combined in a 17:1:1:1 ratio. Reverse primers np9-REV-1: 5′-AGCATCTGTTTAACAAAGCA-3′ and np9-REV-2: 5′-AGCATGTTTAACAAAGCA-3′ were combined in a 19:1 ratio. PCR mix, controls, and cycling conditions were the same as for the gag-amplicon except an annealing temperature of 54 °C. gag and rec/np9 RT-PCR products were separated by agarose gelelectrophoresis and purified using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany) following the manufacturer's instructions. RT-PCR products were subsequently cloned into pGEM-T® Easy vector (Promega, Fitchburg, WI), and ligations were transformed into chemocompetent E. coli DH5α cells. Insert-containing clones were identified by colony PCR using vector-specific M13 primers. Plasmid DNA of positive clones was isolated in a 96-well format using the Agencourt CosMCPrep Kit (Beckman Coulter Genomics, Danvers, MA). Sequences of cloned RT-PCR products were generated by Seq-IT GmbH (Kaiserslautern, Germany) using vector-specific T7 primer and an Applied Biosystems 3730 DNA-Analyzer.
Assignment of cDNA Sequences to Genomic HML-2 Loci
Quality of generated cDNA sequences was checked by eye using FinchTV (Geospiza Inc., PerkinElmer, Seattle, WA), and poor quality reads were excluded from further analysis. Vector and PCR primer sequence portions were removed from cDNA sequences using Geneious software (Biomatters Ltd., Auckland, New Zealand). Trimmed cDNA sequences were assigned to genomic HML-2 loci by BLAT searching the human NCBI36/hg18 reference genome sequence at the UCSC Genome Browser (Kent et al. 2002). An assignment was defined as unambiguous if there was only one best match with less than six mismatches to the corresponding genomic sequence and the second best match displaying at least one more mismatch. Sequences with more than six mismatches were excluded from the analysis, allowing up to 1% difference in sequence due to RT-PCR and sequencing errors and interindividual differences. Unlike K115, the polymorphic K113 provirus in 19p12 is not annotated in the Genome Browser assembly NCBI36/hg18 and could thus not be identified by our BLAT search. The investigated amplicon region of K113 matches that of the HML-2 locus in 1q22 with one nucleotide difference. The relevant nucleotide position of all sequences mapping to 1q22 was thus checked manually to exclude that they are actually derived from K113. ORFs for retroviral proteins Gag, Env, Rec, and Np9 were predicted using Geneious software (Biomatters Ltd., Auckland, New Zealand). In case of Rec and Np9, untrimmed sequences including the primer binding region were used, as the ATG start codon is part of the forward primer used. Sequence identities and similarities were calculated using SIAS (http://imed.med.ucm.es/Tools/sias.html, last accessed January 31, 2013).
UV Irradiation of Cultured Melanoma Cells
Cells were grown to approximately 70–80% confluency. Following the experimantal strategy as described by Schanab et al. (2011), cell culture medium was discarded, and cells were washed with 1× DPBS (Gibco, Life Technologies, Carlsbad, CA). DPBS was removed leaving a small amount of fluid to keep cells moist, while cells were irradiated with 200 mJ/cm2 UVB using a UV409T ultraviolet lamp (Waldmann, Villingen-Schwenningen, Germany). The fluence rate of the lamp at the site of irradiation was measured using a UV-Meter Vario Control (Waldmann, Villingen-Schwenningen, Germany). Cells were subsequently cultured in fresh media at 37 °C/5% CO2 [v/v]. Cells were harvested 24 h later, and total RNA was isolated as described earlier.
qRT-PCR
HML-2 transcription was quantified by Real-Time PCR using the KAPATM SYBR® FAST qPCR MasterMix Universal Kit (Peqlab, Erlangen, Germany) and the StepOnePlusTM Real-Time PCR system (Applied Biosystems, Foster City, CA). The PCR mix contained 1× KAPATM SYBR® FAST qPCR MasterMix Universal, 10 µM of forward and reverse primer each, 1× ROX Reference Dye High, and 1 µl of a 1:10 dilution of cDNA or RT(–) control in a 10 µl reaction. Cycling conditions were as follows. Cycling stage: 1 min at 95 °C, 40 cycles of 3 s at 95 °C, 30 s at 55 °C, and 30 s at 72 °C. Melt curve stage: 15 s 95 °C, 1 min 60 °C, and 15 s 95 °C with reporter signal measured every 0.3 °C during the second 95 °C ramp. For HML-2 gag-specific amplifications, above-described PCR primer mixes were used. HML-2 transcript levels were normalized to transcript levels of housekeeping genes G6PDH (forward primer: 5′-ATCGACCACTACCTGGGCAA-3′, reverse primer: 5′-TTCTGCATCACGTCCCGGA-3′) and RPII (forward primer: 5′-GCACCACGTCCAATGACAT-3′, reverse primer: 5′-GTGCGGCTGCTTCCATAA-3′) (Radonic et al. 2004). For each amplicon, cDNA samples were measured in triplicate and the RT(–) reaction of each sample was measured once. A water control was included for each amplicon. StepOneTM Software version 2.2.2 (Applied Biosystems, Foster City, CA) was employed for normalization and further analysis of measured cDNA levels.
Testing Employed Cell Lines for Polymorphic HML-2 Loci
Presence of HML-2 loci previously described to be polymorphic in the population was examined by long-PCR using primers flanking the HML-2 locus. For the 1p31.1 locus that was formed within L1-elements, a reliable long-PCR strategy could not be obtained. Instead, a second primer located in the gag portion of the proviral sequence was used. Primer sequences are listed in supplementary table S1, Supplementary Material online. The PCR mix contained 0.5 µM of forward and reverse primer each, 200 µM of each dNTP, 1× buffer HF, 0.4 U Phusion® High-Fidelity DNA polymerase (Finnzymes, Thermo Fisher Scientific, Waltham, MA), and 100 ng genomic DNA in a 20 µl reaction. The cycling conditions were as follows: initial denaturation 30 s at 98 °C, 35 cycles of 10 s at 98 °C, 30 s at 62 °C and 5 min at 72 °C, and final elongation 10 min at 72 °C. Presence was verified in a second PCR using the forward primer and a second primer located in the HML-2 LTR. Primer mix and cycling conditions were as described above except an elongation time of 45 s.
Results
Upregulated HERV-K(HML-2) transcription was reported for melanoma, and an involvement of HML-2-encoded proteins in melanoma development was considered before (Büscher et al. 2005, 2006; Reiche et al. 2010; Schanab et al. 2011). Transcribed HML-2 loci may be regarded as of greater biological relevance than nontranscribed loci. We identified in this study HML-2 loci being transcribed in melanoma and thus actually generating the HML-2 RNA detected in previous studies.
Strategy for Identification of Transcribed HML-2 Loci
To identify transcribed HML-2 loci in melanoma, we performed HML-2-specific RT-PCR on total RNA isolated from various melanoma-derived cell lines, melanoma tumor samples, and melanocyte cell lines followed by assignment of cDNA sequences to particular HML-2 loci. HML-2-specific PCR primers were located in the central portion of the HML-2 gag gene (fig. 1a). Primers were optimized toward amplification of as many loci as possible in one PCR. A complete alignment of genomic HML-2 loci was generated as described in the Materials and Methods section. The alignment comprised a total of 80 HML-2 loci, including the polymorphic proviruses HERV-K113 and HERV-K115. Thirty-six HML-2 loci were close to full length with internal sequences >7,000 bp (supplementary fig. S1, Supplementary Material online). For cDNA amplification, a region of approximately 620 bp in the central portion of the gag gene was chosen (nt 1,778–2,396 in the HERV-K(HML-2.HOM) reference sequence [GenBank accession no. AF074086.2]) that is present in 35 (97.22%) of the full-length loci, including HERV-K113 and HERV-K115, and 13 shorter loci with internal sequences between 3,889 and 6,579 bp. We chose this gag region because it shows high sequence similarity in the primer regions but still allows discrimination of loci due to a relatively high number of nucleotide differences within the amplicon. All but two loci, both located in 1p36.21, can be distinguished by at least one nucleotide sequence difference, as well as various indel positions (fig. 1b).
Fig. 1.—

HERV-K(HML-2) provirus structure and sequence comparison of the investigated gag amplicon. (a) Schematic representation of the HML-2 provirus structure and splice products. Splice donor (SD) and acceptor (SA) sites for env and the additionally spliced rec mRNA in type II proviruses are indicated. In type I proviruses, which lack a 292 bp sequence portion comprising the rec splice donor site (SD2) at the pol/env boundary, an alternative splice donor site upstream the rec SD2 is used to produce the shorter np9 mRNA. The position of the gag amplicon used here to investigate HML-2 transcription is indicated. (b) Neighbor joining tree depicting the absolute number of differences (excluding indel positions) between HML-2 loci for the proviral gag portion amplified in this study. Primer-binding regions were deleted before sequence comparisons. Taxon names give the chromosomal band, and, if applicable, HGNC approved names of loci. The scale bar depicts 1 nt sequence difference. All but two loci, both located in 1p36.21, can be distinguished by at least one nucleotide difference.
With respect to a previous study by Flockerzi et al. (2008), which employed a partially overlapping HML-2 region located approximately 150 bp upstream of our amplicon, we further improved the amplification strategy. We increased the number of HML-2 loci recognized by using mixes of several forward and reverse primers considering nucleotide differences between primer and template sequence in the various HML-2 loci. In total, we designed four different forward and three different reverse primers. Twenty-eight HML-2 loci were identical in sequence for both primer regions for one of the primer variants, compared with 13 loci in the study by Flockerzi et al. (2008) that did not yet employ such primer variants. Another 11 HML-2 loci display one or two mismatches in one or both primers with mismatches being located away from the primers' 3′-ends. Another six HML-2 loci show more than two mismatches, and three loci have a nucleotide variation in the 3′-end of the primer binding region, which likely inhibits amplification. Taken together, use of primer variants based on sequence variations of HML-2 loci significantly increases the number of amplifiable loci.
In addition, PCR was performed under low stringency annealing conditions, further contributing to amplification of HML-2 loci for which primers do not match perfectly. This was confirmed subsequently as eight of the loci amplified from genomic and/or cDNA showed one or two mismatches to the primer variants (see later).
PCR primers were verified on genomic DNA. Fifty-five generated sequences could be unambiguously assigned to genomic HML-2 loci. In total, 18 different loci located on 11 different chromosomes were amplified, with four loci being located on chromosome 3 and three loci each on chromosomes 5 and 7 (supplementary material, Supplementary Material online). Five of those loci show one or two mismatches to the employed forward and/or reverse primer sequences, showing that low stringency PCR conditions also allow for amplification of such loci.
RT-PCR products were cloned, sequenced, and assigned to genomic HML-2 loci based on characteristic nucleotide differences between loci. A cDNA/HML-2 locus assignment was regarded as unambiguous if there was only one BLAT's best match of a cDNA sequence to an HML-2 locus, and second, third, and so on ranking matches displayed more differences (see also the Materials and Methods section). Sequences with more than six mismatches were excluded from the analysis, allowing up to approximately 1% difference in sequence due to RT-PCR and sequencing errors and interindividual sequence differences (single-nucleotide polymorphisms [SNPs]). According to previous work, cDNA sequences with unusually high numbers of mismatches most likely arise due to ex vivo recombination between transcripts/cDNA from various transcribed loci (Flockerzi et al. 2007). The ERVK-6 locus in 7p22.1 (HERV-K(HML-2.HOM)), which is known to be allelic in that it is present as a single provirus or as two tandem proviruses sharing a central LTR, was treated differently. As both proviral portions differ only by a few nucleotides along approximately 7 kb, the generated cDNA sequences mapping to 7p22.1 could not be assigned unambiguously to either one of the two proviruses. The tandem provirus was thus regarded as one locus.
Identification of Transcribed HML-2 Loci in Melanoma and Related Samples
To identify transcribed HML-2 loci in melanoma, we analyzed melanoma cell lines SK-Mel-25, SK-Mel-28, MEWO, WM3734a, three melanoma RNA samples, two melanoma lymph node metastases, and the two melanocyte cell lines Benno and Oskar for transcribed HML-2 loci.
In total, 991 gag gene-derived cDNA sequences of sufficient quality were generated and assigned to HML-2 loci by sequence comparisons. An average of four sequences (5%) per sample displayed more than one best match in BLAT searches and were excluded from analysis because they could not be assigned unambiguously to one particular HML-2 locus. As mentioned earlier, the tandem proviral HERV-K(HML-2.HOM) locus was regarded as one locus. The remaining unambiguously assignable 945 sequences showed, on average, 1.15 mismatches to their best BLAT match. Altogether, 918 (97.14%) of those sequences could be assigned unambiguously to a genomic locus with less than six mismatches, with 489 (51.75%) and 259 (27.41%) sequences showing no or a single, respectively, mismatch to the best BLAT match (fig. 2a). Ninety-five (10.05%), 39 (4.13%), 13 (1.38%), 15 (1.59%), and 8 (0.85%) sequences showed 2, 3, 4, 5, and 6 mismatches, respectively. The remaining 2.86% of sequences with more than six mismatches were excluded from further analysis.
Fig. 2.—

Summary of mismatches of generated gag cDNA sequences to their best BLAT match. (a) Gag sequences amplified from cDNA from various melanoma cell lines and samples, as well as melanocyte cell lines, were assigned to genomic HML-2 loci using BLAT. Given are absolute numbers of mismatches for 945 unambiguously assignable sequences to their best BLAT match. Greater than 10 mismatches were summarized. cDNA sequences with more than six mismatches were excluded from analysis. (b) Mismatches of HML-2 gag cDNA sequences generated from melanoma and melanocyte cell lines to their best BLAT match, before (black) and 24 h after (gray) irradiation with 200 mJ/cm2 UVB. Given are the relative number of mismatches of 527 sequences generated from cell line cDNAs before and of 479 sequences after irradiation.
For each sample, an average of 83.45 cDNA sequences could be assigned unambiguously to an HML-2 locus, thus identifying that locus as transcribed in the particular sample. We identified a total of 22 different HML-2 loci to be transcribed in the investigated samples (table 1). Four of these loci, located on human chromosomes 8p23.1 (ERVK-26), 11q12.3 (ERVK-27), 19q12 (ERVK-28), and 19q13.12 (ERVK-29), have not been described as transcribed before. Note that the 8p23.1 locus is different from HERV-K115 that is located in the same chromosomal band. In accord with a recent initiative for assigning names to transcribed HERV loci (Mayer et al. 2011), cDNA sequences corresponding to the four HML-2 loci so far not described as transcribed were submitted as EST sequences to Genbank (JZ164910, JZ164909, JZ164911, and JZ164912), and accession numbers were linked to HUGO Gene Nomenclature Committee approved designations for those HML-2 loci (ERVK-26 to ERVK-29 in table 1). On average, eight different HML-2 loci were transcribed in each sample. The transcribed loci were located on 12 different chromosomes, with five loci being located on chromosome 3 and three loci each on chromosomes 1 and 7. Six of those loci show one or two mismatches to the forward or reverse primer variants employed for amplification.
Table 1.
Relative Cloning Frequencies of HML-2 Loci Identified as Transcribed in This Studya
| Name | HML-2 Locus |
Melanoma Cell Lines |
Melanoma RNA |
Lymph Node Metastases |
Melanocytes |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr | Start | End | Band | ORFs | SK-Mel-25 | SK-Mel-28 | MEWO | WM3734a | RNA1 | RNA2 | RNA3 | LNM1 | LNM2 | Benno | Oskar | |
| ERVK-1 | 1 | 75,615,359 | 75,621,731 | 1p31.1 | n | 29.17 | ||||||||||
| ERVK-7 | 1 | 153,863,081 | 153,872,260 | 1q22 | n | 48.08 | 6.94 | 2.25 | 6.98 | 5.56 | 9.88 | 53.57 | 2.56 | 1.49 | ||
| ERVK-18 | 1 | 158,927,199 | 158,936,430 | 1q23.3 | 1.12 | |||||||||||
| ERVK-5b | 3 | 102,893,427 | 102,902,549 | 3q12.3 | g/n | 9.62 | 6.00 | 2.25 | 6.74 | 12.79 | 31.94 | 1.23 | 1.19 | 14.10 | 5.97 | |
| ERVK-3 | 3 | 114,225,814 | 114,234,972 | 3q13.2 | n | 1.16 | ||||||||||
| ERVK-4 | 3 | 127,091,992 | 127,101,129 | 3q21.2 | r | 10.58 | 4.00 | 3.37 | 1.16 | 9.72 | 4.94 | 7.69 | ||||
| ERVK-13 | 3 | 149,764,167 | 149,768,086 | 3q24 | 0.96 | 3.57 | ||||||||||
| ERVK-11 | 3 | 186,763,030 | 186,772,209 | 3q27.2 | n | 1.39 | 1.00 | 4.49 | 6.98 | 2.78 | 4.94 | 1.49 | ||||
| ERVK-10 | 5 | 156,017,295 | 156,026,474 | 5q33.3 | g/n | 5.56 | 1.00 | 2.47 | 14.93 | |||||||
| ERVK-9 | 6 | 78,483,381 | 78,492,802 | 6q14.1 | g/r | 1.19 | ||||||||||
| ERVK-6b | 7 | 4,588,583 | 4,606,557 | 7p22.1 | g/e/r | 21.15 | 74.99 | 6.00 | 5.82 | 1.39 | 20.99 | 23.81 | ||||
| ERVK-14b | 7 | 104,175,604 | 104,180,502 | 7q22.1 | 4.17 | 79.00 | 92.13 | 85.39 | 60.47 | 4.17 | 45.68 | 1.19 | 69.23 | 61.19 | ||
| ERVK-15b | 7 | 141,097,395 | 141,102,407 | 7q34 | 0.96 | 1.00 | 2.25 | 1.23 | 5.13 | |||||||
| ERVK-26c | 8 | 12,360,863 | 12,370,378 | 8p23.1 | 5.56 | |||||||||||
| ERVK-16 | 10 | 6,906,147 | 6,915,609 | 10p14 | r | 1.92 | ||||||||||
| ERVK-17 | 10 | 101,570,559 | 101,577,735 | 10q24.2 | r | 2.78 | 1.23 | 11.94 | ||||||||
| ERVK-27b,c | 11 | 61,892,538 | 61,907,139 | 11q12.3 | 1.00 | 2.47 | ||||||||||
| ERVK-21 | 12 | 57,007,509 | 57,016,965 | 12q14.1 | e/r | 14.29 | ||||||||||
| ERVK-28b,c | 19 | 32,820,338 | 32,829,201 | 19q12 | g/e/r | 1.39 | ||||||||||
| ERVK-29c | 19 | 42,289,389 | 42,298,906 | 19q13.12 | r | 1.92 | 4.17 | 2.33 | 8.33 | 3.70 | 1.28 | 1.49 | ||||
| ERVK-23 | 21 | 18,855,530 | 18,863,833 | 21q21.1 | n | 2.33 | ||||||||||
| ERVK-24 | 22 | 17,306,187 | 17,315,361 | 22q11.21 | g/n | 4.81 | 1.00 | 1.23 | 1.19 | 1.49 | ||||||
| Total number of sequences | 104 | 72 | 100 | 89 | 89 | 86 | 72 | 81 | 84 | 78 | 67 | |||||
| Number of different loci | 9 | 7 | 9 | 4 | 5 | 9 | 10 | 12 | 8 | 6 | 8 | |||||
aHML-2 gag-specific RT-PCR was performed on total RNA isolated from various melanoma cell lines and samples, as well as from melanomctes. Given in the first columns are the chromosome positions of the different HML-2 loci found as transcribed in the investigated samples. The name in the first column refers to the official locus name according to Mayer et al. (2011), if the locus has been described as transcribed before (see supplementary table S2, Supplementary Material online, for alternative names and GenBank accession numbers of loci). Given are the relative cloning frequencies (%) for the different loci in each sample (number of sequences unambiguously assigned to a specific locus divided by total number of sequences generated from the particular sample) and the number of different loci found in each sample. Presence of ORFs for Gag (g), Env (e), Rec (r), or Np9 (n) proteins is indicated.
bLoci displaying one or two mismatches to the forward or reverse primer variants employed for amplification.
cLoci identified as transcribed only in this study.
As a higher transcription rate of a particular HML-2 locus (and thus higher amounts of the particular cDNA) increases its probability of being cloned and sequenced, relative cloning frequency of cDNA from a particular HML-2 locus roughly correlates with relative transcription rate of that locus. In each sample, few loci showed high relative cloning frequencies of up to 92%, suggesting a much higher level of transcription of those HML-2 loci compared with other transcribed HML-2 loci (table 1). Especially the ERVK-14 locus in 7q22.1 shows high cloning frequencies ranging from 46% to 92% in five melanoma samples and both melanocyte cell lines. The ERVK-6 locus in 7p22.1 shows high cloning frequencies ranging from 21% to 75% in four melanoma samples. Loci ERVK-1, ERVK-7, and ERVK-5 show frequencies ranging from 29% to 53% in up to two melanoma samples. Most other HML-2 loci appear to be transcribed at relatively low levels.
Transcription profiles of HML-2 loci were different for each investigated sample (fig. 3). None of the transcribed loci was found active in all samples. Nine of the HML-2 loci were detected exclusively in one of the melanoma samples, and all these loci but the ones in 1p31.1 (ERVK-1) and 12q14.1 (ERVK-21) seem to be transcribed at relatively low levels. Loci displaying high relative cloning frequencies, for example, ERVK-14 in 7q22.1, ERVK-7 in 1q22, or ERVK-5 in 3q12.3, were detected in melanoma samples and in melanocytes. Exceptions were the aforementioned ERVK-1 locus in 1p31.1 that was found in only one of the melanoma samples with a relative cloning frequency of 29%, and the ERVK-6 locus in 7p22.1 with relative cloning frequencies of up to 75% in 7 of the melanoma samples, but ERVK-6 was not found transcribed in both melanocyte cell lines.
Fig. 3.—

Transcription profiles of HERV-K(HML-2) loci in melanoma. HML-2 gag cDNA sequences were amplified from melanoma cell lines SK-Mel-25, SK-Mel-28, MEWO, and WM3734a, three melanoma RNA samples, two lymph node metastases, and melanocyte cell lines Benno and Oskar. HML-2 loci are indicated on the x axis by chromosomal band and, if available, HGNC approved names. Samples are indicated on the y axis. l. n., lymph node; mel., melanoma. Given on the z axis are relative cloning frequencies as percentages of cDNA sequences that could be assigned unambiguously to particular HML-2 loci, approximately corresponding to transcription levels of those loci in the particular sample. See table 1 for further details.
Taken together, up to 12 different HML-2 loci were found to be transcribed in melanoma and melanocytes, and overall transcription patterns differed significantly between the various samples. Some loci, among them ERVK-6 in 7p22.1, were found transcribed exclusively in melanoma.
Provirus Structure and Coding Capacity of HML-2 Loci Transcribed in Melanoma and Related Samples
Regarding the proviral structure of transcribed HML-2 loci, most of them consist of two LTRs and internal proviral sequences (table 2). Fifteen loci have intact 5′- and 3′-LTRs, eight loci harbor internal deletions, and/or at least one LTR is missing. Seventeen loci exhibit internal sequences of >7,000 bp. ERVK-15 harbors the shortest internal sequence of approximately 3,900 bp.
Table 2.
Characteristics of HERV-K(HML-2) Loci Identified as Transcribed in This Studya
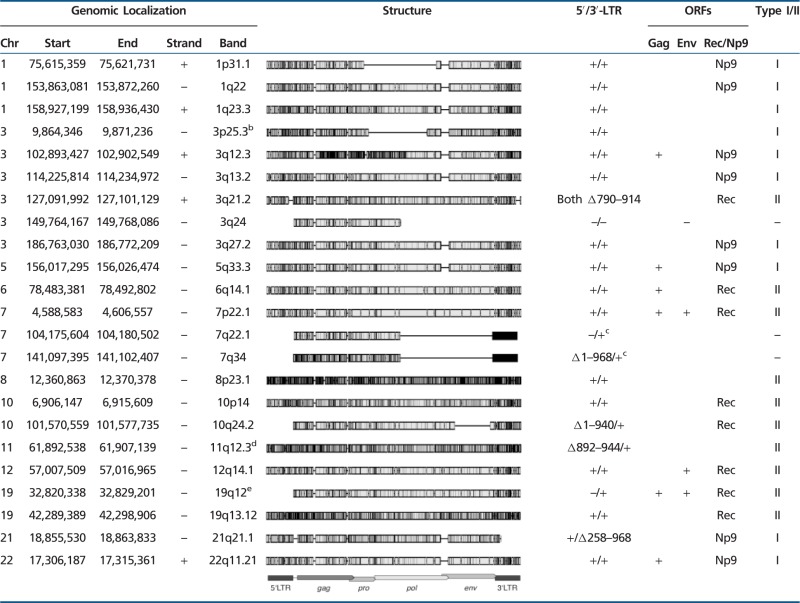 |
aThe genomic localization of the complete HML-2 locus with LTRs, if present, is given in the first five columns (assembly March 2006 NCBI36/hg18). The structure column shows an alignment of the complete loci compared with the HERV-K(HML-2.HOM) reference sequence (GenBank accession no. AF074086.2). Non-HML-2 repeat insertions in the loci in 11q12.3 and 19q12 were deleted before alignment (see later). Gray boxes represent the HERV sequence with black marks representing differences to the reference. Indel positions are indicated by horizontal lines. Presence (+) or absence (–) of 5′- and 3′-LTRs is given, as well as deletions (Δ in the LTR sequences, compared with the human LTR5 consensus sequence as provided by RepBase [http://www.girinst.org/repbase/update/browse.php, last accessed January 31, 2013]). ORFs for gag and env were predicted using geneious software (Biomatters Ltd.). A complete ORF coding for proteins of 666 aa (±1) for Gag or 699 aa (±1) for Env is indicated by "+." Complete deletion of the coding region is indicated by "–." The last column gives the classification as type I or II, based on a 292 bp deletion within the pol-env boundary in type I proviruses (see text). Larger truncations in that region allowing no discrimination are indicated by "–." ORFs for accessory proteins Np9 or Rec, encoded by type I or II proviruses, respectively, are given (see also Mayer et al. 2004).
bThe locus in 3p25.3 was only found after irradiation with UVB (see Results for details).
cThe 3′-LTR of the 7q21.1 and 7q34 locus is annotated on the opposite strand.
dThe 5′-LTR is interrupted by AluY/AuYb8/AluY between nucleotides 891 and 945, the internal sequence shows an insertion of AluSg between 5,717 and 5,718, and (TA)n/AluY/(CAG)n, as well as an additional 63-bp fragment of LTR5 after bp 5,998, all annotated on the opposite strand.
eThe internal HERV sequence is interrupted by an AluYa5 element between nucleotides 4,583 and 4,584.
As for coding capacity of transcribed HML-2 loci for clinically relevant proteins, potential ORFs in gag and env genes were predicted (table 2). Six loci harbor an ORF coding for a Gag protein of the expected size of 667 (±1) aa. Gag encoding loci were found transcribed in melanoma samples and in melanocytes with cloning frequencies ranging from <5% for the ERVK-24 locus in 22q11.21 up to 75% for the ERVK-6 locus in 7p22.1. Three HML-2 loci encoding an env ORF of the expected size of 700 (±1) aa (ERVK-6 in 7p22.1, ERVK-21 in 12q14.1, ERVK-28 in 19q12) were only found transcribed in melanoma. HML-2 loci ERVK-21 and ERVK-28 were transcribed at relatively low frequencies of 14% and 1% in one melanoma RNA sample and one metastasis, respectively, whereas the ERVK-6 locus showed higher cloning frequencies of up to 75%. The two loci encoding both Gag and Env proteins of expected sizes, namely ERVK-28 in 19q12 and the ERVK-6 locus in 7p22.1, were both found transcribed exclusively in melanoma. Some more loci show ORFs for shorter or longer potential Gag and Env proteins, but it is difficult to predict whether those loci are translated into proteins at all.
HML-2 proviruses encode two other proteins with interesting biological features, namely Rec and Np9, that can be encoded by so-called HML-2 type II or type I, respectively, proviruses that differ by a 292-bp sequence at the pol-env boundary, with type I loci lacking that sequence. Out of the 10 transcribed type I loci, eight show an intact ORF for the Np9 protein (unpublished data). Out of the 10 transcribed type II loci, eight show ORFs for Rec protein (Mayer et al. 2004). Three other HML-2 loci lack the type I/II discriminating region.
In summary, most HML-2 loci transcribed in melanoma feature both LTRs and close to complete internal proviral sequences. Six and three, respectively, of those HML-2 loci show ORFs coding for Gag and/or Env proteins. Two loci encoding both Gag and Env were exclusively found transcribed in melanoma. Eight loci each encoding Rec or Np9 proteins were found transcribed in both melanoma and melanocytes.
Identification of Rec and Np9 mRNA Producing HML-2 Loci
We specifically examined in an additional analysis transcription of HML-2 loci producing rec or np9 mRNA. To do so, we performed RT-PCR on total RNA isolated from melanoma cell lines SK-Mel-25, SK-Mel-28, MEWO, and WM3734a and from melanocyte cell lines Benno and Oskar. The RT-PCR amplicon was based on previously reported rec and np9 exonic regions (Löwer et al. 1995; Armbruester et al. 2004) and was improved regarding unambiguous assignment of generated cDNA sequences to HML-2 loci compared with a previous study (Mayer et al. 2004). The 5′-end of the forward primer was located right on the rec and np9 start codon. The primer sequence was as reported in Armbruester et al. (2004), but additional primer variants considering sequence variations between HML-2 loci were included in PCR amplifications (see Materials and Methods). The reverse primer was located approximately 180 nt downstream from the reported np9 and rec stop codons. PCR products were approximately 580 bp and approximately 370 bp in size for rec and np9 mRNA-derived cDNA.
We generated a total of 91 cDNA sequences from the six cell lines. We could unambiguously assign 84 cDNA sequences to genomic HML-2 loci. Four type II and three type I HML-2 loci were identified as producing rec or np9 transcript in the investigated samples (table 3). The Rec encoding ERVK-6 locus in 7p22.1 showed highest relative cloning frequencies of up to 88% in all cell lines but Benno. Although 7p22.1 gag-derived cDNAs were exclusively found in melanoma, rec transcript was also found in both Benno and Oskar melanocyte cell lines. Np9 transcript from loci 1q22 (ERVK-7), 3q12.3 (ERVK-5), and 22q11.21 (ERVK-24) was found in both the melanoma and melanocyte cell lines.
Table 3.
Relative Cloning Frequencies of Type I/II HML-2 Loci Identified as Transcribed in This Studya
 |
aWe specifically investigated transcription of type I and II HML-2 loci coding for accessory proteins Np9 and Rec, respectively, performing Np9/Rec-specific RT-PCR on total RNA isolated from melanoma and melanocyte cell lines. Given in the first column are the official locus names according to Mayer et al. (2011) and the chromosome positions of the different HML-2 loci found as transcribed in the investigated samples. Given are the absolute number of sequences (#) and the relative cloning frequencies (%) for the different loci in each sample (number of sequences unambiguously assigned to a specific locus divided by the total number of sequences generated from the particular sample).
More than 90% of the generated rec and np9 cDNA sequences showed ORFs encoding proteins of expected sizes of 104 aa or 105 aa for Rec and 75 aa for Np9. The encoded proteins showed no or only a few amino acid changes compared with reported Rec and Np9 protein sequences (UniProt accession no. Q69383, P61571–61576, P61578–61583). One rec cDNA sequence mapping to 7p22.1 (ERVK-6, HERV-K(HML-2.HOM)), generated from SK-Mel-28, showed a 1-bp deletion in the 5′-region of the rec ORF resulting in a frameshift. Two more sequences mapping to 7p22.1, obtained from cell line WM3734a, showed a nonsense mutation resulting in a premature stop codon. However, we interpret those three sequences as mutant sequences generated in vitro during cDNA generation or PCR.
Taken together, rec and np9 mRNA from, in total, seven HML-2 loci were found transcribed in both melanoma and melanocytes. Rec and Np9 protein encoding loci were found transcribed in both tumor and normal samples.
Identification of Reverse-Transcribed HML-2 Transcripts
Of further note, a significantly mutated HML-2 type II locus in 2q32.1, with less than 500 bp of internal sequence, was found to be transcribed in the Benno melanocyte cell line. The locus consists of three short internal portions, representing approximately 100 bp upstream of the env splice donor site immediately downstream from the 5′-LTR, approximately 280 bp from between the env splice acceptor site and the rec splice donor site, and approximately 95 bp downstream from the rec/np9 splice acceptor site, with all splice sites being incomplete (fig. 4a). The locus harbors approximately 90 bp from the 5′-LTR's 3′-end and a 3′-LTR lacking the 3′ most approximately 150 bp. Nevertheless, both primer regions are present in that locus. More detailed sequence comparisons indicate that this particular locus is a retrotransposed HML-2 mRNA that was spliced similar to rec mRNA except for a short nucleotide stretch. It displays hallmarks typical for reverse transcription of mRNA by L1 machinery. The locus is flanked at the 3′-end by an 8-nt-long poly-A stretch located approximately 90 bp downstream from the HML-2 3′-LTR's poly-A signal, and it is encompassed by a target site duplication (5′: AATCTGAATTCTT; 3′: AATCTGATTTCTT) that displays similarities to the reported L1 target site consensus sequence (Jurka 1997; Ostertag and Kazazian 2001). Notably, this locus was not annotated as a processed pseudogene in pseudogene-related annotation tracks at UCSC Genome Brower. The locus was very likely formed before the split of Hominoidea. Primate genome comparisons at the UCSC Genome Browser (Kent et al. 2002) indicate that nearly identical genomic regions including the HML-2 locus are present in the genomes of chimpanzee, gorilla, orangutan, and gibbon, but the HML-2 locus is missing in rhesus and marmoset (fig. 4b). The latter species belongs to new world monkeys that entirely lack HERV-K(HML-2) sequences. A potential ORF of 408 bp (136 aa) was predicted for that retrotransposed HML-2 locus. The N-terminal 60 aa of the resulting hypothetical protein show 90% sequence identity (93.33% similarity) to previously described Rec and Env protein sequences (UniProt accession no. Q69383, Q69384), which are identical with each other in the N-terminal 85 aa. The central and C-terminal portions lack significant similarity to Rec or Env proteins.
Fig. 4.—

A HERV-K(HML-2) locus in 2q32.1 transcribed in melanocyte cell line Benno shows typical features of retrotransposition by L1 machinery. (a) Depicted in the center-right part of the figure is a dot matrix comparison (window size: 30; minimum score: 50%; jump: 1) of the 2q32.1 locus with the HERV-K(HML-2.HOM) proviral locus (see text) as a reference. Locations of retroviral LTRs, gag, pro, pol, and env gene,s and splice donor (SD) and splice acceptor (SA) sites are indicated for the reference sequence next to the y axis. Subregions in the 2q32.1 locus (chr2 locus), as indicated in the dot matrix comparison, were compared in more detail with a cDNA assigned to this locus (cDNA), a previously reported rec mRNA sequence (Rec) (see text) and the sequence of HERV-K(HML-2.HOM) (HOM). Numbers above the alignment indicate nucleotide positions with respect to the HOM sequence. More detailed sequence comparison shows that the locus represents a retrotransposed HML-2 mRNA that was spliced similar to rec mRNA, with splice donor and acceptor sites resembling the ones in rec mRNA (ii–v). The 2q32.1 locus is flanked by target site duplications (TSDs) and a poly(A)-signal in the 3′-end (i, vi, and vii). The position of the forward primer used for RT-PCR is indicated in (iii). (b) Graphical depiction of the HERV-K(HML-2) locus in human chromosome 2q32.1 as provided by UCSC Genome Browser (Kent et al. 2002). The retrotransposed HML-2 sequence was detected and annotated by Repeatmasker v.3.2.7, indicated by a black horizontal bar. The same genome region encompassing the HML-2 locus is present in other primate genomes as indicated by greenish or black-colored horizontal boxes. Rhesus and marmoset genomes harbor the same genome region but lack exactly the HML-2 portion as indicated by thin horizontal lines. (b) Compiled from annotation tracks provided in hg18 and hg19 (http://genome.ucsc.edu, last accessed January 31, 2013).
Two cDNA sequences generated from melanocytes Benno mapped to an HML-2 type II locus in 10q24.2 that carries a 1,372 bp deletion within the env gene (fig. 5a). The locus was formed within an intron of the ABCC2 gene that is transcribed in antisense orientation. Notably, the 10q24.2 locus lacks the intron portion of the rec/np9 splice acceptor site. The 5′-end of the missing env portion resembles a splice donor site with only the intron portion missing. It appears that this locus resembles on the DNA level another or a partial splice variant of HML-2 transcript. This locus was likely formed recently in human evolution as it is only present in the human genome but missing in the corresponding genome region of non-human primates (fig. 5b). Therefore, we speculate that this locus represents a (partially) spliced HML-2 transcript that was reverse transcribed into a DNA copy in a retroviral fashion after the evolutionary split of human from chimpanzee. Furthermore, a transcript is generated from that locus, and an ORF for a hypothetically larger protein of 214 aa is encoded that is identical to HML-2 Env protein for the N-terminal 200 aa (and thus to Rec for aa 1–85) and to Rec protein for the C-terminal 14 aa. In other words, the encoded protein is a chimera of Env and Rec protein portions.
Fig. 5.—

An HML-2 type II locus in 10q24.2, transcribed in melanocyte cell line Benno, shows a 1,372 bp deletion in env. (a) Depicted in the center-right part of the figure is a dot matrix comparison of the ERVK-17 locus in 10q24.2 with HERV-K(HML-2.HOM) and detailed alignments of indicated splice sites of the 10q24.2 genomic sequence (chr10 locus) with two cDNAs assigned to this locus (cDNA1, cDNA2), a rec mRNA sequence (Rec) and HERV-K(HML-2.HOM) sequence (HOM). Numbers above the alignment indicate nucleotide positions in HOM (see also the legend of fig. 4). The position of the forward primer used for RT-PCR is indicated in (i). The 10q24.2 locus lacks the intron portion of the rec splice acceptor site (iv). The 5′-end of the missing env portion resembles an alternative splice donor site with the corresponding intron portion missing (iii) approximately 330 bp downstream the regular rec splice donor site (ii). The locus appears to resemble, on the DNA level, another splice variant of HML-2 transcript, and it might represent a spliced HML-2 transcript that was reverse transcribed into a DNA copy in a retroviral fashion. (b) The reverse-transcribed locus is present in the human genome but missing in the genomes of non-human primates. See legend of figure 4 for further details.
One type II cDNA sequence generated from RNA from melanocytes Benno matched best to an HML-2 locus in 3q12.3 (ERVK-5) but nevertheless displayed a relatively high number of six mismatches to that locus. Sequences showing the same characteristic nucleotide differences when compared with ERVK-5 were also found in melanoma-unrelated samples investigated by us in a separate study. We were able to isolate the identical sequence from corresponding genomic DNA. Therefore, some isolated cDNA sequences point to a hitherto unidentified allelic variant of an existing HERV-K(HML-2) locus, alternatively an, as of yet, unidentified HML-2 locus (details to be published elsewhere).
Taken together, we identified two transcribed HML-2 loci that very likely were generated by reverse transcription of spliced HML-2 transcript either by L1 machinery or in a retroviral fashion. The HML-2 locus on chromosome 2q32.1 may encode a protein half Rec-like, and the chromosome 10q24.2 locus ERVK-17 may encode a chimeric Env/Rec protein. We also obtained evidence for a novel HML-2 type II-like allele that displays several nucleotide differences within the Rec coding sequence.
Polymorphic Loci Do Not Account for Differences in Transcription Profiles
Several HERV-K(HML-2) loci have been described as polymorphic in the human population (Barbulescu et al. 1999; Hughes and Coffin 2004; Macfarlane and Simmonds 2004; Mayer et al. 2005). Of those polymorphic loci, the ERVK-6 locus in 7p22.1 (HERV-K(HML-2.HOM)) and four loci located in 1p31.1 (ERVK-1), 3p25.3 (ERVK-2), 3q21.2 (ERVK-3), and 6q14.1 (ERVK-9) were identified as transcribed in one or several of the investigated samples. As a lack of cDNA, thus transcript, from one or several of those loci might be explained by absence of the particular locus in a sample's genomic DNA, we tested the genomic DNA of the available cell lines for presence of those HML-2 loci. We furthermore included in this analysis the HERV-K113 locus in 19p12 and the HERV-K115 locus in 8p23.1, which were not found transcribed in our samples. Polymorphic HML-2 loci can consist of a complete provirus, a solitary LTR resulting from homologous recombination between the 5′- and 3′-LTR, or an empty site if the particular HML-2 insertion is not fixed in the population. Using specific PCR primers flanking an HML-2 locus and a flanking primer plus a primer within the LTR, sizes of the generated PCR products were indicative of the different alleles.
For most examined polymorphic HML-2 loci, an approximately 10-kb-long PCR product could be generated in most of the samples, indicating presence of at least one copy of the particular HML-2 loci in those samples (supplementary fig. S3, Supplementary Material online). The HERV-K113 locus could not be amplified from any of the samples, and the HERV-K115 locus (at least one copy) was identified only in melanocyte cell line Benno. This finding is not unexpected in the light of previously reported low allele frequencies for those two loci (Burmeister et al. 2004; Macfarlane and Simmonds 2004; Moyes et al. 2005).
Therefore, presence of at least one copy of most polymorphic proviruses in nearly all the samples argues against differences in HML-2 transcription profiles being due to lack of the respective HML-2 loci in the investigated samples.
Quantitation of HML-2 Transcription and HML-2 Transcription Profiles Following UVB Irradiation
We quantified overall HML-2 gag transcription levels in the investigated melanoma and melanocyte samples and in GCT cell lines NCCIT and Tera-1, which both display upregulated HML-2 transcription (Florl et al. 1999; Ruprecht et al. 2008), by quantitative real-time RT-PCR. HML-2 transcript levels were normalized to transcript levels of G6PDH and RPII housekeeping genes, and HML-2 transcript level in melanocyte cell line Benno before UV treatment was taken as reference. Melanocyte cell line Oskar and melanoma cell lines SK-Mel-25, SK-Mel-28 and MEWO showed HML-2 transcription levels similar to that of melanocyte cell line Benno (fig. 6a). WM3734a cells, the three melanoma RNA samples and the lymph node metastasis showed HML-2 transcript levels 5–23 times higher than Benno. Still, HML-2 transcript levels in GCT cell lines NCCIT and Tera-1 were 733-fold and 77-fold higher compared with Benno.
Fig. 6.—

Quantitation of HERV-K(HML-2) gag transcription by qRT-PCR. (a) Relative HML-2 gag transcript levels in melanoma cell lines SK-Mel-25, SK-Mel-28, MEWO, and WM3734a, three melanoma RNA samples, a melanoma lymph node metastasis (LM), and melanocyte cell lines Benno and Oskar, as well as GCT cell lines NCCIT and Tera-1. HML-2 transcript levels were normalized to transcript levels of G6PDH and RPII housekeeping genes. Gag transcript level in melanocyte cell line Benno before UV treatment was taken as reference. Black bars indicate minimum and maximum relative levels of gene expressionas calculated by StepOne software (see Materials and Methods). Note the different scale for NCCIT and Tera-1 cells. (b) Relative HML-2 gag transcription in melanoma and melanocyte cell lines before (dark gray bars) and 24 h after irradiation (light gray bars) with 200 mJ/cm2 UVB.
Previous studies reported deregulation of HML-2 transcription in melanoma cell lines following UVB irradiation (Schanab et al. 2011). Such deregulation may be caused by deregulation of transcription of particular HML-2 loci or by general deregulation of all transcribed HML-2 loci. We therefore investigated changes in transcription profiles of HML-2 loci following UV irradiation. Following the experimental strategy described in Schanab et al. (2011), melanoma and melanocyte cell lines were irradiated with 200 mJ/cm2 of UVB, and total RNA was isolated from irradiated cells 24 h later. HML-2 transcription profiles were investigated as described earlier. In total, 528 sequences were generated of which 49 (9.28%) showed more than one best match in BLAT searches and were thus excluded from further analysis. As mentioned earlier, the HERV-K(HML-2.HOM) tandem provirus was regarded as one locus. The remaining 479 (90.72%) sequences could be unambiguously assigned to a genomic HML-2 locus with an average mismatch count of 2.27. Four hundred thirty-six (91.02%) of those sequences could be assigned unambiguously to a genomic locus with less than six mismatches (fig. 2b) and were analyzed further.
Overall, HML-2 gag transcript levels were also quantified in UV-irradiated cell lines 24 h after exposure to 200 mJ/cm2 UVB. Each of the cell lines showed a considerable decrease in relative HML-2 expression compared with nonirradiated cells (fig. 6b). The strongest effect was observed in melanocyte cell line Benno and melanoma cell line WM3734a, which showed highest relative HML-2 transcript levels before treatment. In both cell lines, relative HML-2 transcript levels were reduced to approximately 14% of the transcription in nonirradiated cells.
UV irradiation modified transcription profiles of HML-2 loci (fig. 7). Except for SK-Mel-25, the total number of transcribed loci slightly increased after irradiation. On average, 10 loci were transcribed after irradiation, in contrast to seven loci in untreated cells, with no specific activation of particular loci. The relative cloning frequency of cDNA (thus relative transcription level) of the often dominantly transcribed ERVK-14 locus in 7q22.1, which showed frequencies up to 92% before treatment, was reduced to a frequency of, at most, 39% following irradiation. The ERVK-2 locus in 3p25.3 was found transcribed in UV-irradiated SK-Mel-28 and MEWO cells but had not been found transcribed in untreated cells (table 2). Of further note, melanoma and melanocyte cell lines did not display characteristic differences in changing transcription profiles.
Fig. 7.—

Influence of UV irradiation on HERV-K(HML-2) transcription. Melanoma cell lines SK-Mel-25, SK-Mel-28, MEWO, and WM3734a and melanocyte cell lines Benno and Oskar were irradiated with 200 mJ/cm2 UVB (see text). Given are relative cloning frequencies as percentages of gag-derived cDNA sequences that could be unambiguously assigned to particular HML-2 loci before (dark gray bars) and 24 h after (light gray bars) UVB irradiation of each cell line.
Taken together, all melanoma samples and a melanoma-derived cell line show higher HML-2 gag transcript levels compared with melanocytes. UVB irradiation decreased HML-2 transcript levels in all irradiated melanoma and melanocyte cell lines. Transcription levels of several HML-2 loci appear to be altered by UV irradiation in melanoma and melanocyte cell lines, with nonuniform responses.
Discussion
Several previous studies suggested an involvement of HERV-K(HML-2) in the etiology of melanoma. If HML-2 sequences turn out to be of relevance in the development of melanoma, it will certainly be of interest to identify the transcribed HML-2 loci, which actually account for HML-2 RNA and proteins detected in previous studies (Büscher et al. 2005, 2006; Reiche et al. 2010; Stengel et al. 2010). We therefore identified in this study for the first time transcribed HML-2 loci in melanoma and biologically related samples. We were thus able to address whether transcription of certain HML-2 loci distinguishes melanoma from healthy samples and whether previously reported increased HML-2 expression in melanoma is due to specific activation of certain HML-2 loci or a more or less even upregulation of several loci.
On the basis of a previous study (Flockerzi et al. 2008), we improved the HML-2 cDNA amplification strategy by increasing the number of amplifiable loci using mixes of primer variants considering nucleotide differences in the primer binding regions of HML-2 loci. In contrast to Flockerzi et al. (2008), who identified transcribed HML-2 loci based on amplicons in the gag and env gene regions, we restricted our analysis to an amplicon in the gag gene. All loci detected as gag- or env-derived cDNAs in the previous study can also be detected by the gag primer variants used in this study. Furthermore, all HML-2 loci with a perfect match to the env primers used in the previous study are also recognized by primer variants in this study, with the exception of one locus harboring a deletion in gag (but not described as transcribed so far). A second amplicon located in env would thus not have increased the number of amplifiable loci but instead would have only resulted in twice the amount of work and expense.
Generated HML-2 cDNA sequences were assigned to genomic loci based on characteristic (private) nucleotide differences distinguishing the various HML-2 loci in the human genome from each other. As many of the HML-2 loci represent evolutionarily young HERV loci, the number of random mutations accumulated over time is lower than for other, (much) older HERV groups. We therefore designed the PCR product amplified from cDNA to be >600 bp to ensure as much as possible that cDNA sequences generated by Sanger sequencing harbor sufficient numbers of private nucleotide differences to allow reliable assignment of cDNA sequences to specific loci. Although a next-generation sequencing-based approach would have allowed generation of much higher numbers of cDNA sequence reads, maximum read lengths would have been rather short, which in the case of HML-2 likely complicates reliable cDNA assignment or makes it impossible for many HML-2 loci. For a few HML-2 loci, there was only one nucleotide along the investigated 620 bp distinguishing one from the other locus (fig. 1b). Even for this study's much longer Sanger sequencing reads, cDNA assignment was sometimes complicated by sequence differences introduced by RT-PCR/sequencing errors, SNPs, or artifactual cDNA sequences due to recombination of transcripts from different HML-2 loci during cDNA generation (Flockerzi et al. 2007) resulting in mismatches of cDNA sequences to the genomic reference. However, the best BLAT match to one particular HML-2 locus is expected to be correct, if matches to other HML-2 loci display more differences. We excluded cDNA sequences if unambiguous assignment to a particular HML-2 locus was not possible. All in all, less than 3% and 9% of the sequences generated from samples before and after UV irradiation, respectively, had to be omitted due to >6 mismatches or unambiguous assignments.
Our strategy for cDNA generation by random priming explicitly does not differentiate between sense and antisense transcripts of HML-2 loci. This may be of importance for HML-2 loci lacking the 5′-LTR but having a 3′-LTR. Because the HML-2 LTR can exert bidirectional promoter activity (Domansky et al. 2000), such mutated HML-2 loci may be transcribed by the 3′-LTR in antisense. For HML-2 loci lacking both LTRs transcripts may stem from upstream or downstream promoters and the HML-2 loci may therefore be transcribed in sense or antisense, respectively (see later). However, we aim at identifying all the HML-2 loci transcribed in some way because antisense transcribed loci may exert some biological function. For instance, HML-2 antisense RNA might regulate expression of genes by RNAi. Overall, for most HML-2 loci having a 5′-LTR and HML-2 5′-LTRs usually displaying promoter activity, HML-2 loci only transcribed in antisense very likely comprise a minority (Vinogradova et al. 2001; Buzdin et al. 2006).
We verified our cDNA amplification strategy by PCR on genomic DNA. Eighteen different HML-2 loci were identified from unambiguous assignment of 55 sequences to genomic HML-2 loci. As a total of 39 HML-2 loci show sufficient sequence similarity to the primer variants and could thus be amplified in theory, the 18 different cloned HML-2 might initially suggest a somewhat preferential amplification of certain loci. However, that observed distribution did not correlate with HML-2 loci subsequently cloned from cDNA. For instance, HML-2 loci in 3q27.2 (ERVK-11), 19q12 (ERVK-28), and 22q11.21 (ERVK-24) showing somewhat higher cloning frequencies when amplified from genomic DNA (supplementary fig. S2, Supplementary Material online) were identified at relatively low frequencies when amplified from cDNA (table 1). In subsequent amplifications from cDNA, besides 13 of the loci amplified from genomic DNA, we identified an additional nine loci not amplified from genomic DNA showing that, in fact, more than the aforementioned 18 loci can be amplified. We wish to mention that 50 more sequences generated from genomic DNA had to be omitted from analysis as they showed greater than 6 and up to 26 mismatches to their best BLAT match. Such high numbers of unassignable sequences were not observed for sequences generated from cDNA, where >90% of the sequences could be unambiguously assigned. We suspect that the high number of unassignable sequences from genomic DNA is because of recombinants that were created during PCR because of an excess of potential recombination partners present in the genomic DNA, which is in contrast to the situation in cDNA where much less such recombination partners are present. This observation may be of importance for other studies involving PCR amplification of high-copy number repetitive elements. A relatively high number of recombinations may be produced during PCR in such experiments, and it may be advisable to verify the desired sequence by sequencing. Also, a very low amount of template DNA might decrease the number of recombinations during PCR.
As for the identification of transcribed HML-2 loci, 23 transcribed HML-2 loci is very similar to the number previously described for GCTs and brain tissues (Flockerzi et al. 2008). Nineteen loci were detected in both studies. Four loci (ERVK-25 to ERVK-28, see table 1) have not been described as transcribed before and were therefore added to databases of the HGNC that also assigns locus designations to transcribed HERV loci (Mayer et al. 2011). When comparing our results with results from a recent microarray-based study (Perot et al. 2012), 16 loci identified in our study were also reported as transcribed in Perot et al. (2012), whereas seven loci were not identified in that study, though, melanoma was not specifically investigated in Perot et al. Overall transcription profiles of HML-2 loci seem different in melanoma and melanocytes when compared with those found for GCTs (Flockerzi et al. 2008). In GCTs, especially transcription of ERVK-24 in 22q11.21, ERVK-23 in 21q21.1, and ERVK-20 in 11q23.3 appears specifically upregulated, yet those loci appear only low level transcribed in melanoma based on cloning frequencies, or were not detected as transcribed at all, specifically ERVK-20. Furthermore, overall HML-2 transcription profiles appear clearly different for the various samples, that is, each sample displayed a particular set of transcribed HML-2 loci that overlaps only partially with those of other samples. As for HML-2 loci distinguishing melanoma from melanocytes, locus ERVK-6 in 7p22.1 appears as an interesting candidate because it was exclusively detected in seven of the melanoma-derived samples but not in the melanocyte cell lines. More specific and larger scale analysis might reveal the ERVK-6 locus as a biomarker for melanoma.
Observed differences in transcription profiles or lack of transcripts from particular HML-2 loci is not explained simply by lack of those loci in investigated samples. When testing for presence of 7 HML-2 loci described to be polymorphic in the human population (Barbulescu et al. 1999; Hughes and Coffin 2004; Macfarlane and Simmonds 2004; Mayer et al. 2005), we found at least one copy of each locus in nearly all the investigated cell lines.
Most of the transcribed HML-2 loci harbor more or less full-length internal sequences and 5′- and 3′-LTRs, with the former harboring the classical proviral promotor. For 5′-LTR lacking HML-2 loci, transcription might have been initiated by bidirectional promotor activity of the 3′-LTR (see earlier). ERVK-14 in 7q22.1 and ERVK-17 in 10q24.2, both lacking the 5′-LTR, are located intronically within genes, thus different splicing might account for the transcript of the corresponding HML-2 locus. ERVK-14 is located intronically and in antisense orientation within the LHFPL3 gene. It has been reported that the ERVK-14 5′-portion contributes an internal exon for 14 annotated antisense ESTs (Kim and Hahn 2010). ERVK-17 is located intronically in antisense orientation in the MRP2 gene, a member of the MRP subfamily, mediating transport of anticancer agents and contributing to drug resistance (Kruh and Belinsky 2003). For other HML-2 loci yet unidentified promotors located nearby might have initiated transcription, or readthrough transcription from neighboring genes might have produced the HML-2 transcript, for example, ERVK-15 in 7q34 is located approximately 1.6 kb upstream of the housekeeping gene SSBP1 that has been reported as being involved in mitochondrial biogenesis and as being a subunit of a single-stranded DNA-binding complex involved in the maintenance of genome stability (Tiranti et al. 1995; Huang et al. 2009).
Several HML-2 loci identified as transcribed harbor full-length ORFs for retroviral Gag and Env proteins. The three loci theoretically capable of producing full-length Env protein were exclusively found transcribed in melanoma. Although ERVK-21 in 12q14.1 and ERVK-28 in 19q12 were each found transcribed in only one of the samples, the ERVK-6 locus in 7p22.1 seems to be of particular interest, as it was found transcribed in seven melanoma samples but was not found transcribed in melanocytes. A previous study detected spliced env mRNA and Env protein in cell lines SK-Mel-28 and MEWO (Büscher et al. 2005). In both cell lines, our study identified the ERVK-6 locus as transcribed, thus previously detected mRNA and protein might have originated from this locus. Moreover, ERVK-6-locus-encoded Env protein might account for Env-directed antibodies detected in sera from melanoma patients (Hahn et al. 2008). For GCTs, there was no obvious correlation between expression of Env-encoding HML-2 loci and presence of antibodies (Flockerzi et al. 2008). As the ERVK-6 locus also harbors an ORF for a full-length Gag protein, ERVK-6 might also account for Gag protein detected in melanoma metastases and Gag-directed antibodies detected in sera from melanoma patients (Büscher et al. 2005; Hahn et al. 2008). Anyway, additional studies investigating more samples will be required to elucidate the biological significance of the ERVK-6 locus in melanoma. Two more loci harboring gag ORFs, ERVK-9 in 6q14.1 and ERVK-28 in 19q12, are expressed in only one of the melanoma samples and are transcribed at very low levels based on cDNA cloning frequencies. The remaining three loci with gag ORFs are expressed in both melanoma and melanocytes and show no striking difference concerning relative cloning frequencies thus transcript levels. The same was found for GCTs; Gag-encoding loci were found transcribed in tumor samples from antibody positive and negative patients (Flockerzi et al. 2008). Generation of antibodies against HML-2 Gag and Env protein in melanoma and GCT patients appears as a more complex mechanism that is determined not just by activation of Gag and/or Env encoding HML-2 loci.
Previous studies suggested an involvement of HML-2-encoded accessory proteins Rec and Np9 in tumorgenesis (Boese et al. 2000; Armbruester et al. 2004; Galli et al. 2005; Denne et al. 2007). We found Rec and np9 transcripts in both melanoma and melanocyte cell lines. A previous study by Büscher et al. (2006) detected rec and np9 mRNA in melanoma as well as rec mRNA in melanocytes, yet Rec and Np9 proteins were detected in only a small number of tumors. Thus, presence of rec and np9 mRNA does not necessarily imply presence of corresponding proteins at detectable levels. We did not investigate protein expression in our study or specifically quantify rec and np9 transcription. However, overall Rec and/or Np9 expression in melanocytes may be lower than in melanoma or mRNA or proteins may be degraded more quickly.
A type II locus in 2q32.1 was found transcribed in melanocytes Benno. That locus is of particular interest because it presents features of a processed pseudogene generated by L1 retrotransposition machinery. Retrotransposed elements of the HERV-H and HERV-W group have been described before (Goodchild et al. 1995; Pavlícek et al. 2002). To the best of our knowledge, this is the first reported locus of the HERV-K(HML-2) group that was formed by retrotransposition and that is transcribed despite its truncated structure. Additional evidence for this locus being transcribed is provided by EST sequence entries AW968573, AA167225, BF921025, GD144208, and AA491129 that locate just upstream of or partially overlap with the locus. Analysis of primate genome sequences furthermore indicated that the locus was formed just before the evolutionary split of Hominoidea. The HML-2 locus in chromosome 10 likely formed by reverse transcription and integration in a retroviral fashion of an HML-2 proviral transcript from which only an intron within env had been removed. That locus was very likely formed relatively recently in human evolution as it is not found in non-human primates. Moreover, both loci harbor open reading frames for HML-2 Rec and Env-like proteins. It is currently not known whether any of those proteins is expressed, so that they must be deemed hypothetical at this point. However, we note that according to dbSNP 137, there is only one synonymous SNP known for the chromosome 2q32.1 locus (rs2099848), and two nonsynonymous SNPs (rs61870457 and rs9633711) and one synonymous SNP (rs9633710) for the chromosome 10q24.2 locus. However, there are no nonsense sequence variations known. Of further note, the retrotransposed HML-2 rec locus in 2q32.1 was not identified in our initial BLAT search for compiling HML-2 sequences from the human reference genome nor was it included in a recent study that described full-length and partial HML-2 loci in the human genome (Subramanian et al. 2011). Even a spliced rec mRNA sequence as BLAT query did not identify the 2q32.1 locus. More specific analysis may thus identify other retrotransposed HML-2 transcripts in the human (or other Old World primate) genomes.
Another type II locus in 10q24.2 (ERVK-17) found transcribed in melanocytes Benno might represent another HML-2 mRNA splice variant that was reverse transcribed and reintegrated into the genome yet rather in a retroviral fashion. This locus harbors a 1,372 bp deletion within the env gene that is similar to the intronic portion spliced from rec mRNA, except that a different splice donor site somewhat downstream from the actual rec splice donor site had been used. In contrast to a protein hypothetically encoded by the 2q32.1 locus, which is similar to previously described Rec and Env protein sequences only for its N-terminus, the 10q24.2 locus hypothetically encodes a chimeric protein consisting of Env and Rec protein portions. In the light of the HML-2 Np9 protein being an HML-2 protein variant resulting from deletion of a 292 bp sequence within the HML-2 pol-env region (Armbruester et al. 2002), it is conceivable that those proteins are translated and of biological significance.
Quantitation by qRT-PCR revealed upregulated HML-2 gag transcript levels in about half of the melanoma samples. Melanoma cell line WM3734a, three melanoma RNA samples, and one melanoma lymph node metastasis showed upregulated HML-2 transcript levels compared with melanocytes Benno and Oskar (no qRT-PCR results of sufficient quality could be obtained for the second lymph node metastasis for unknown reasons, which was thus omitted from analysis). Although relative transcript levels in these melanoma samples were significantly lower than in GCT cell lines NCCIT and Tera-1 that display strongly upregulated HML-2 transcription (Florl et al. 1999; Ruprecht, Mayer, et al. 2008), still markedly elevated HML-2 transcription levels in melanoma and other cancers might suffice for detectable expression of HML-2 proteins (Büscher et al. 2005; Reiche et al. 2010).
A previous study reported upregulated levels of HML-2 RNA and proteins in melanoma cell lines following irradiation with UVB (Schanab et al. 2011). Following irradiation of cells with the same dose of UVB, we observed changing HML-2 transcription profiles for each of the investigated melanoma and melanocyte cell lines. Apart from the ERVK-14 locus in 7q22.1 dominantly transcribed before UVB treatment but displaying reduced transcript levels afterward in five of the melanoma cell lines and both melanocyte cell lines, no specific (de)activation of HML-2 loci became obvious, including melanoma cell lines versus melanocytes.
We note that our quantitation of HML-2 transcript levels produced results in conflict with the study by Schanab et al. (2011) that reported activation of HML-2 transcription as a specific response to UVB in melanoma. That study, employing a qRT-PCR amplicon located within pol, reported upregulated HML-2 transcription in melanoma and lower transcription in nonmelanoma cells. In contrast, we found significantly lowered HML-2 transcription in melanoma and melanocyte cell lines. Our results are based on a different qRT-PCR amplicon located in gag. However, when comparing the loci recognized by our primers located in gag and those recognized by the pol primers employed in Schanab et al., both primer pairs should be able to amplify about the same HML-2 loci. That is, differences in measured transcript levels are not due to selective amplification of deregulated HML-2 loci by one or the other primer pair. In the same line, we exclude that the Schanab et al. primers amplified partially deleted HML-2 loci that were not amplifiable in our study but that were (strongly) upregulated by UVB. At present, we do not have a good explanation for the observed differences. Additional studies, such as analysis of HML-2 transcripts amplified by the Schanab et al. primers, will be required to potentially explain this discrepancy.
We revealed in this study HML-2 loci transcribed in melanoma. The identified HML-2 loci are of immediate relevance if HML-2 transcript or encoded proteins are considered to play a role in melanoma. The presented data reveal rather complex transcription profiles of HML-2 loci in melanoma in that various HML-2 loci display variable transcriptional activities in different melanoma-derived samples. CpG methylation, histone modifications, and transcription factors very likely regulate in concert transcription of HML-2 LTRs. The epigenetic status of HML-2 loci may be different in every melanoma sample investigated in our study, resulting in different transcriptional activities of HML-2 LTRs. However, although changes in methylation and chromatin status are often observed in cancer cells, only specific and detailed epigenetic studies will be able to explain (in)activity of HML-2 loci in the various melanoma samples. As for a biological role of Rec and Np9 proteins in melanoma, our data indicate transcription of Rec and Np9 encoding loci both in melanoma and normal melanocytes. Provided that the proteins are also found at corresponding levels, our findings imply that Rec and/or Np9 may rather have indirect effects during melanoma development. Detailed molecular and cell biology studies will be required to elucidate the role of those proteins in melanoma development. However, our study provides important information as to which Rec and Np9 protein sequences from which HML-2 loci should be considered in such studies. Our study furthermore points toward additional studies regarding the ERVK-6 locus as it may represent a biomarker for melanoma. Last but not least, our study revealed, for the first time, HML-2 loci that were generated by reverse transcription of spliced HML-2 RNA. Moreover, both loci are transcribed and potentially encode Rec- and Env-like proteins, the function and relevance of which is currently unknown. The data presented here and previous investigations demonstrate that transcription of HERV loci is complex. Many more specific studies are required to further elucidate when and how HERV loci are transcribed and how transcribed HERV loci could be involved in disease.
Supplementary Material
Supplementary figures S1–S3 and tables S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors acknowledge Alexandra Stark and Heike Palm for maintaining and providing melanocyte cell cultures. This work was supported by grants from Deutsche Forschungsgemeinschaft to J.M. and E.M.
Literature Cited
- Armbruester V, et al. A novel gene from the human endogenous retrovirus K expressed in transformed cells. Clin Cancer Res. 2002;8:1800–1807. [PubMed] [Google Scholar]
- Armbruester V, et al. Np9 protein of human endogenous retrovirus K interacts with ligand of numb protein X. J Virol. 2004;78:10310–10319. doi: 10.1128/JVI.78.19.10310-10319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannert N, Kurth R. The evolutionary dynamics of human endogenous retroviral families. Annu Rev Genomics Hum Genet. 2006;7:149–173. doi: 10.1146/annurev.genom.7.080505.115700. [DOI] [PubMed] [Google Scholar]
- Barbulescu M, et al. Many human endogenous retrovirus K (HERV-K) proviruses are unique to humans. Curr Biol. 1999;9:861–868. doi: 10.1016/s0960-9822(99)80390-x. [DOI] [PubMed] [Google Scholar]
- Belshaw R, et al. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): implications for present-day activity. J Virol. 2005;79:12507–12514. doi: 10.1128/JVI.79.19.12507-12514.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg J, Benachenhou F, Blikstad V, Sperber G, Mayer J. Classification and nomenclature of endogenous retroviral sequences (ERVs): problems and recommendations. Gene. 2009;448:115–123. doi: 10.1016/j.gene.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Boese A, et al. Human endogenous retrovirus protein cORF supports cell transformation and associates with the promyelocytic leukemia zinc finger protein. Oncogene. 2000;19:4328–4336. doi: 10.1038/sj.onc.1203794. [DOI] [PubMed] [Google Scholar]
- Boller K, Janssen O, Schuldes H, Tönjes RR, Kurth R. Characterization of the antibody response specific for the human endogenous retrovirus HTDV/HERV-K. J Virol. 1997;71:4581–4588. doi: 10.1128/jvi.71.6.4581-4588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister T, et al. Insertional polymorphisms of endogenous HERV-K113 and HERV-K115 retroviruses in breast cancer patients and age-matched controls. AIDS Res Hum Retroviruses. 2004;20:1223–1229. doi: 10.1089/aid.2004.20.1223. [DOI] [PubMed] [Google Scholar]
- Büscher K, et al. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 2005;65:4172–4180. doi: 10.1158/0008-5472.CAN-04-2983. [DOI] [PubMed] [Google Scholar]
- Büscher K, et al. Expression of the human endogenous retrovirus-K transmembrane envelope, Rec and Np9 proteins in melanomas and melanoma cell lines. Melanoma Res. 2006;16:223–234. doi: 10.1097/01.cmr.0000215031.07941.ca. [DOI] [PubMed] [Google Scholar]
- Buzdin A, Kovalskaya-Alexandrova E, Gogvadze E, Sverdlov E. At least 50% of human-specific HERV-K (HML-2) long terminal repeats serve in vivo as active promoters for host nonrepetitive DNA transcription. J Virol. 2006;80:10752–10762. doi: 10.1128/JVI.00871-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CJ, Lock WM, Mager DL. Endogenous retroviral LTRs as promoters for human genes: a critical assessment. Gene. 2009;448:105–114. doi: 10.1016/j.gene.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Denne M, et al. Physical and functional interactions of human endogenous retrovirus proteins Np9 and rec with the promyelocytic leukemia zinc finger protein. J Virol. 2007;81:5607–5616. doi: 10.1128/JVI.02771-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis LK. Analysis of the melanoma epidemic, both apparent and real: data from the 1973 through 1994 surveillance, epidemiology, and end results program registry. Arch Dermatol. 1999;135:275–280. doi: 10.1001/archderm.135.3.275. [DOI] [PubMed] [Google Scholar]
- Domansky AN, et al. Solitary HERV-K LTRs possess bi-directional promoter activity and contain a negative regulatory element in the U5 region. FEBS Lett. 2000;472:191–195. doi: 10.1016/s0014-5793(00)01460-5. [DOI] [PubMed] [Google Scholar]
- Dupressoir A, Lavialle C, Heidmann T. From ancestral infectious retroviruses to bona fide cellular genes: role of the captured syncytins in placentation. Placenta. 2012;33:663–671. doi: 10.1016/j.placenta.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Flockerzi A, et al. Expression pattern analysis of transcribed HERV sequences is complicated by ex vivo recombination. Retrovirology. 2007;4:39. doi: 10.1186/1742-4690-4-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flockerzi A, et al. Expression patterns of transcribed human endogenous retrovirus HERV-K(HML-2) loci in human tissues and the need for a HERV Transcriptome Project. BMC Genomics. 2008;9:354. doi: 10.1186/1471-2164-9-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florl AR, Löwer R, Schmitz-Dräger BJ, Schulz WA. DNA methylation and expression of LINE-1 and HERV-K provirus sequences in urothelial and renal cell carcinomas. Br J Cancer. 1999;80:1312–1321. doi: 10.1038/sj.bjc.6690524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank O, et al. Human endogenous retrovirus expression profiles in samples from brains of patients with schizophrenia and bipolar disorders. J Virol. 2005;79:10890–10901. doi: 10.1128/JVI.79.17.10890-10901.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank O, et al. Variable transcriptional activity of endogenous retroviruses in human breast cancer. J Virol. 2008;82:1808–1818. doi: 10.1128/JVI.02115-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli UM, et al. Human endogenous retrovirus rec interferes with germ cell development in mice and may cause carcinoma in situ, the predecessor lesion of germ cell tumors. Oncogene. 2005;24:3223–3228. doi: 10.1038/sj.onc.1208543. [DOI] [PubMed] [Google Scholar]
- Goodchild NL, Freeman JD, Mager DL. Spliced HERV-H endogenous retroviral sequences in human genomic DNA: evidence for amplification via retrotransposition. Virology. 1995;206:164–173. doi: 10.1016/s0042-6822(95)80031-x. [DOI] [PubMed] [Google Scholar]
- Hahn S, et al. Serological response to human endogenous retrovirus K in melanoma patients correlates with survival probability. AIDS Res Hum Retroviruses. 2008;24:717–723. doi: 10.1089/aid.2007.0286. [DOI] [PubMed] [Google Scholar]
- Haupt S, et al. Human endogenous retrovirus transcription profiles of the kidney and kidney-derived cell lines. J Gen Virol. 2011;92:2356–2366. doi: 10.1099/vir.0.031518-0. [DOI] [PubMed] [Google Scholar]
- Herbst H, Sauter M, Mueller-Lantzsch N. Expression of human endogenous retrovirus K elements in germ cell and trophoblastic tumors. Am J Pathol. 1996;149:1727–1735. [PMC free article] [PubMed] [Google Scholar]
- Hu L, et al. Expression of human endogenous gammaretroviral sequences in endometriosis and ovarian cancer. AIDS Res Hum Retroviruses. 2006;22:551–557. doi: 10.1089/aid.2006.22.551. [DOI] [PubMed] [Google Scholar]
- Huang J, Gong Z, Ghosal G, Chen J. SOSS complexes participate in the maintenance of genomic stability. Mol Cell. 2009;35:384–393. doi: 10.1016/j.molcel.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Coffin JM. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution. Proc Natl Acad Sci U S A. 2004;101:1668–1672. doi: 10.1073/pnas.0307885100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J. Sequence patterns indicate an enzymatic involvement in integration of mammalian retroposons. Proc Natl Acad Sci U S A. 1997;94:1872–1877. doi: 10.1073/pnas.94.5.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, et al. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004;32:D493–D496. doi: 10.1093/nar/gkh103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann S, et al. Human endogenous retrovirus protein Rec interacts with the testicular zinc-finger protein and androgen receptor. J Gen Virol. 2010;91:1494–1502. doi: 10.1099/vir.0.014241-0. [DOI] [PubMed] [Google Scholar]
- Kent WJ, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DS, Hahn Y. Human-specific antisense transcripts induced by the insertion of transposable element. Int J Mol Med. 2010;26:151–157. [PubMed] [Google Scholar]
- Kitamura Y, Ayukawa T, Ishikawa T, Kanda T, Yoshiike K. Human endogenous retrovirus K10 encodes a functional integrase. J Virol. 1996;70:3302–3306. doi: 10.1128/jvi.70.5.3302-3306.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein V, et al. Identification of chromosome-specific sequence-tagged sites by Alu-PCR. Genet Anal Tech Appl. 1993;10:6–9. doi: 10.1016/1050-3862(93)90018-e. [DOI] [PubMed] [Google Scholar]
- Kramer-Hämmerle S, et al. Identification of a novel Rev-interacting cellular protein. BMC Cell Biol. 2005;6:20. doi: 10.1186/1471-2121-6-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruh GD, Belinsky MG. The MRP family of drug efflux pumps. Oncogene. 2003;22:7537–7552. doi: 10.1038/sj.onc.1206953. [DOI] [PubMed] [Google Scholar]
- Laufer G, Mayer J, Mueller BF, Mueller-Lantzsch N, Ruprecht K. Analysis of transcribed human endogenous retrovirus W env loci clarifies the origin of multiple sclerosis-associated retrovirus env sequences. Retrovirology. 2009;6:37. doi: 10.1186/1742-4690-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löwer R, Tönjes RR, Korbmacher C, Kurth R, Löwer J. Identification of a Rev-related protein by analysis of spliced transcripts of the human endogenous retroviruses HTDV/HERV-K. J Virol. 1995;69:141–149. doi: 10.1128/jvi.69.1.141-149.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane C, Simmonds P. Allelic variation of HERV-K(HML-2) endogenous retroviral elements in human populations. J Mol Evol. 2004;59:642–656. doi: 10.1007/s00239-004-2656-1. [DOI] [PubMed] [Google Scholar]
- Mager DL, Medstrand P. Retroviral repeat sequences. In: Cooper D, editor. Nature encyclopedia of the human genome. Hampshire (United Kingdom): Macmillan Publishers Ltd; 2003. pp. 57–63. [Google Scholar]
- Mallet F, et al. The endogenous retroviral locus ERVWE1 is a bona fide gene involved in hominoid placental physiology. Proc Natl Acad Sci U S A. 2004;101:1731–1736. doi: 10.1073/pnas.0305763101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer J, Blomberg J, Seal RL. A revised nomenclature for transcribed human endogenous retroviral loci. Mob DNA. 2011;2:7. doi: 10.1186/1759-8753-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer J, et al. An almost-intact human endogenous retrovirus K on human chromosome 7. Nat Genet. 1999;21:257–258. doi: 10.1038/6766. [DOI] [PubMed] [Google Scholar]
- Mayer J, et al. Human endogenous retrovirus HERV-K(HML-2) proviruses with Rec protein coding capacity and transcriptional activity. Virology. 2004;322:190–198. doi: 10.1016/j.virol.2004.01.023. [DOI] [PubMed] [Google Scholar]
- Mayer J, et al. Haplotype analysis of the human endogenous retrovirus locus HERV-K(HML-2.HOM) and its evolutionary implications. J Mol Evol. 2005;61:706–715. doi: 10.1007/s00239-005-0066-7. [DOI] [PubMed] [Google Scholar]
- Moyes DL, et al. The distribution of the endogenous retroviruses HERV-K113 and HERV-K115 in health and disease. Genomics. 2005;86:337–341. doi: 10.1016/j.ygeno.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Muradrasoli S, Forsman A, Hu L, Blikstad V, Blomberg J. Development of real-time PCRs for detection and quantitation of human MMTV-like (HML) sequences HML expression in human tissues. J Virol Methods. 2006;136:83–92. doi: 10.1016/j.jviromet.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Muster T, et al. An endogenous retrovirus derived from human melanoma cells. Cancer Res. 2003;63:8735–8741. [PubMed] [Google Scholar]
- Oja M, Peltonen J, Blomberg J, Kaski S. Methods for estimating human endogenous retrovirus activities from EST databases. BMC Bioinformatics. 2007;8(2 Suppl):S11. doi: 10.1186/1471-2105-8-S2-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Kazazian HH., Jr Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001;35:501–538. doi: 10.1146/annurev.genet.35.102401.091032. [DOI] [PubMed] [Google Scholar]
- Pavlícek A, Paces J, Elleder D, Hejnar J. Processed pseudogenes of human endogenous retroviruses generated by LINEs: their integration, stability, and distribution. Genome Res. 2002;12:391–399. doi: 10.1101/gr.216902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perot P, et al. Microarray-based sketches of the HERV transcriptome landscape. PLoS One. 2012;7:e40194. doi: 10.1371/journal.pone.0040194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radonic A, et al. Guideline to reference gene selection for quantitative real-time PCR. Biochem Biophys Res Commun. 2004;313:856–862. doi: 10.1016/j.bbrc.2003.11.177. [DOI] [PubMed] [Google Scholar]
- Reiche J, Pauli G, Ellerbrok H. Differential expression of human endogenous retrovirus K transcripts in primary human melanocytes and melanoma cell lines after UV irradiation. Melanoma Res. 2010;20:435–440. doi: 10.1097/CMR.0b013e32833c1b5d. [DOI] [PubMed] [Google Scholar]
- Reus K, et al. Genomic organization of the human endogenous retrovirus HERV-K(HML-2.HOM) (ERVK6) on chromosome 7. Genomics. 2001;72:314–320. doi: 10.1006/geno.2000.6488. [DOI] [PubMed] [Google Scholar]
- Ruggieri A, et al. Human endogenous retrovirus HERV-K(HML-2) encodes a stable signal peptide with biological properties distinct from Rec. Retrovirology. 2009;6:17. doi: 10.1186/1742-4690-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht K, et al. Human endogenous retrovirus family HERV-K(HML-2) RNA transcripts are selectively packaged into retroviral particles produced by the human germ cell tumor line Tera-1 and originate mainly from a provirus on chromosome 22q11.21. J Virol. 2008;82:10008–10016. doi: 10.1128/JVI.01016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht K, Mayer J, Sauter M, Roemer K, Mueller-Lantzsch N. Endogenous retroviruses: endogenous retroviruses and cancer. Cell Mol Life Sci. 2008;65:3366–3382. doi: 10.1007/s00018-008-8496-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyamoorthy K, et al. Melanoma cell lines from different stages of progression and their biological and molecular analyses. Melanoma Res. 1997;7(2 suppl):S35–S42. [PubMed] [Google Scholar]
- Sauter M, et al. Human endogenous retrovirus K10: expression of Gag protein and detection of antibodies in patients with seminomas. J Virol. 1995;69:414–421. doi: 10.1128/jvi.69.1.414-421.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter M, et al. Specificity of antibodies directed against Env protein of human endogenous retroviruses in patients with germ cell tumors. Cancer Res. 1996;56:4362–4365. [PubMed] [Google Scholar]
- Schanab O, et al. Expression of human endogenous retrovirus K is stimulated by ultraviolet radiation in melanoma. Pigment Cell Melanoma Res. 2011;24:656–665. doi: 10.1111/j.1755-148X.2011.00860.x. [DOI] [PubMed] [Google Scholar]
- Schommer S, Sauter M, Kräusslich HG, Best B, Mueller-Lantzsch N. Characterization of the human endogenous retrovirus K proteinase. J Gen Virol. 1996;77 (Pt 2):375–379. doi: 10.1099/0022-1317-77-2-375. [DOI] [PubMed] [Google Scholar]
- Seifarth W, et al. Comprehensive analysis of human endogenous retrovirus transcriptional activity in human tissues with a retrovirus-specific microarray. J Virol. 2005;79:341–352. doi: 10.1128/JVI.79.1.341-352.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafino A, et al. The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Exp Cell Res. 2009;315:849–862. doi: 10.1016/j.yexcr.2008.12.023. [DOI] [PubMed] [Google Scholar]
- Stengel S, Fiebig U, Kurth R, Denner J. Regulation of human endogenous retrovirus-K expression in melanomas by CpG methylation. Genes Chromosomes Cancer. 2010;49:401–411. doi: 10.1002/gcc.20751. [DOI] [PubMed] [Google Scholar]
- Stoye JP. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol. 2012;10:395–406. doi: 10.1038/nrmicro2783. [DOI] [PubMed] [Google Scholar]
- Subramanian RP, Wildschutte JH, Russo C, Coffin JM. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology. 2011;8:90. doi: 10.1186/1742-4690-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, et al. Chromosomal localization of mitochondrial transcription factor A (TCF6), single-stranded DNA-binding protein (SSBP), and endonuclease G (ENDOG), three human housekeeping genes involved in mitochondrial biogenesis. Genomics. 1995;25:559–564. doi: 10.1016/0888-7543(95)80058-t. [DOI] [PubMed] [Google Scholar]
- Tönjes RR, Czauderna F, Kurth R. Genome-wide screening, cloning, chromosomal assignment, and expression of full-length human endogenous retrovirus type K. J Virol. 1999;73:9187–9195. doi: 10.1128/jvi.73.11.9187-9195.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova TV, et al. Solitary human endogenous retroviruses-K LTRs retain transcriptional activity in vivo, the mode of which is different in different cell types. Virology. 2001;290:83–90. doi: 10.1006/viro.2001.1134. [DOI] [PubMed] [Google Scholar]
- Yin H, et al. Transcription of human endogenous retroviral sequences related to mouse mammary tumor virus in human breast and placenta: similar pattern in most malignant and nonmalignant breast tissues. AIDS Res Hum Retroviruses. 1997;13:507–516. doi: 10.1089/aid.1997.13.507. [DOI] [PubMed] [Google Scholar]