

Abstract
Trying to kill cancer cells by generating DNA damage is by no means a new idea. Radiotherapy and genotoxic drugs are routinely used in cancer therapy. More recent developments also explored the potential of targeting the DNA damage response (DDR) in order to increase the toxicity of radio‐ and chemo‐ therapy. Chk1 inhibitors have pioneered studies in this regard. Interestingly, early studies noted that Chk1 inhibitors were particularly toxic for p53‐deficient cells. The model proposed for this observation was that this effect was due to the simultaneous abrogation of the G2 (Chk1) and G1 (p53) checkpoints. We here challenge this view, and propose a model where the toxicity of Chk1 inhibitors is rather due to the fact that these compounds generate high loads of replicative stress (RS) during S‐phase, which are further boosted by the less restrictive S‐phase entry found in p53‐deficient cells. This new model implies that the particular toxicity of Chk1 inhibitors might not be restricted to p53‐deficient cells, but could be extended to other mutations that promote a promiscuous S‐phase entry. In addition, this rationale also implies that the same effect should also be observed for other molecules that target the RS‐response (RSR), such as inhibitors of the Chk1‐activating kinase ATR.
Keywords: ATR, Chk1, Cancer therapy
Highlights
The ATR‐dependent RSR is distinct from the ATM‐dependent DDR, and should be studied independently.
RSR inhibition might be particularly toxic for a subset of tumors.
ATR inhibitors promote p53‐independent cell death.
1. The RSR: time to fly solo from the DDR
DNA double‐strand breaks (DSB) are amongst the most deleterious lesions that cells can suffer. Their presence can trigger genome rearrangements and the loss of genetic information at the break site. As a consequence, the presence of DSBs is very cytotoxic, a property that has been exploited for cancer treatment most notoriously by radiotherapy. In order to limit the impact of DSB, cells are equipped with a transduction cascade that coordinates the signaling and repair of these genomic lesions, while at the same time limits the expansion of the damaged cells through the activation of cytostatic or apoptotic responses. This cellular response is what is generally quoted under the broad term “DNA damage response” (DDR) (Harper and Elledge, 2007; Jackson and Bartek, 2009). Whereas other post‐translational modifications such as Ubiquitinylation or SUMOylation are now known to be involved in the DDR (Polo and Jackson, 2011), most of our current knowledge is based on phosphorylation‐based signaling events.
Pioneering work from Yossi Shiloh and colleagues led to the identification of a kinase that was responsible for the radiosensitivity observed in patients of a rare hereditary disease known as Ataxia Telangiectasia (AT) (Savitsky et al., 1995). Whereas related to the phosphatydil‐inositol‐3kinase (PI3 K), the Ataxia Telangiectasia‐Mutated (ATM) kinase phosphorylates proteins and no lipids. One of the first ATM targets discovered was the tumor suppressor p53 (Siliciano et al., 1997). Previous work had shown that AT patients had a deficient upregulation of p53 levels in response to DNA damage, which was associated with a weaker G1/S checkpoint (Kastan et al., 1992). Besides ATM‐dependent phosphorylation, the upregulation of p53 in response to DSBs is also stimulated by further phosphorylations made by Chk2, a kinase that is itself phosphorylated and activated by ATM (Chehab et al., 2000; Hirao et al., 2000; Shieh et al., 2000; Tominaga et al., 1999). This linear cascade provides a simple model to understand the toxicity of DSB, which would be due to the activation of a DSB‐ATM‐Chk2‐p53 apoptotic response. In any case, the activation of apoptosis is only one of the many roles of ATM, and p53 is not always necessary for the activation of apoptosis.
Soon after the link of ATM with radiation responses was established, Karlene Cimprich cloned an ATM and Rad‐3 related kinase known as ATR (Cimprich et al., 1996). The role of ATR was also soon linked to DSB, since the overexpression of a kinase dead mutant version of ATR led to radiosensitization and deficient DNA damage induced checkpoints (Cliby et al., 1998; Wright et al., 1998). Moreover, ATR was also shown to phosphorylate p53 (Lakin et al., 1999; Tibbetts et al., 1999). To complete the analogy with the ATM response, ATR signaling is reinforced by the phosphorylation and activation of Chk1, a Chk2 homologous kinase (Liu et al., 2000). Hence, the original and still widely spread view was that the role of ATR was similar to that of ATM and that a DSB‐ATR‐Chk1‐p53 response would be complementary to the DSB‐ATM‐Chk2‐p53 response. Later work revealed that the activation of ATR in response to DSB was ATM‐dependent, once again reinforcing the view of a coordinated ATM‐ and ATR‐dependent DDR (Cuadrado et al., 2006; Jazayeri et al., 2006). However, and beyond the DDR, there were many evidences suggesting that ATR and Chk1 had a life on their own, which was unrelated to ATM and the response to DSB.
Whereas ATM is only activated by DSB, ATR is activated by the presence of single‐stranded DNA (ssDNA), which is present at processed DSB ends but also at stalled replication forks (reviewed in (Cimprich and Cortez, 2008; Lopez‐Contreras and Fernandez‐Capetillo, 2010)). The actual signal for ATR activation is Replication Protein A (RPA)‐coated ssDNA (Zou and Elledge, 2003), which provided an explanation for previous yeast data that had identified ssDNA and RPA as important mediators of the checkpoint response (Garvik et al., 1995; Lee et al., 1998). In cells, ATR exists in a constitutive complex with its binding partner ATRIP, which brings the complex to ssDNA through its association with ATR (Cortez et al., 2001). Finally, and in order to activate ATR, it has to be brought in close proximity to its allosteric activator TopBP1 (Kumagai et al., 2006). This occurs independently from ATR recruitment. The clamp loader Rad17 loads the PCNA‐like heterotrimeric ring 9‐1‐1 (Rad9‐Rad1‐Hus1) to the neighborhood of ssDNA (Zou et al., 2002). The Rad17/9‐1‐1 complex then recruits TopBP1 completing the activation of ATR (Lee et al., 2007). At the same time, Rad17 is also responsible for bringing Claspin to ssDNA (Wang et al., 2006), a mediator molecule that enables the phosphorylation of Chk1 by ATR (Kumagai and Dunphy, 2000). A model of ATR activation is depicted in Figure 1.
Figure 1.
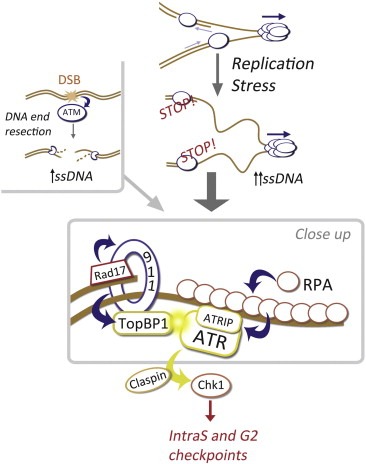
ATR activation: From ssDNA to Chk1. ATM is directly activated by the free and unprocessed DNA ends that arise at DSB. In contrast, ssDNA is the signal for ATR activation. This can also be generated at DSB after a 5′to 3′nucleolytic degradation of one of the chains, which is also necessary to provide the substrate for homologous recombination. However, the most important source of ssDNA occurs at stalled replication forks, in what is known as RS. Upon exposure of ssDNA this is rapidly coated by RPA, which directly binds ATRIP and therefore recruits the ATRIP/ATR complex to ssDNA. At the same time, Rad17 loads the 9‐1‐1 clamp, which then brings the alosteric activator TopBP1 in close proximity to ATR unleashing its kinase activity. In order for ATR to phosphorylate Chk1, a mediator protein named Claspin is still needed that finally enables the interaction of ATR with Chk1, leading to the phosphorylation of Chk1 and a full activation of the RSR.
In contrast to ATR, ATM is not activated by ssDNA. Therefore, whereas DSB activate ATM/Chk2 and ATR/Chk1, the ssDNA‐response relies exclusively on ATR and Chk1. This is best exemplified by the fact that ionizing radiation induces both Chk1 and Chk2 phosphorylation, but only Chk1 is phosphorylated in response to reagents that promote ssDNA accumulation such as hydroxyurea (Cuadrado et al., 2006). A frequent confusion comes from the fact that a persistent stalling of replication forks (or a persistent exposure to HU) ultimately derives into “fork collapse”, which means that DSBs are generated at the forks (Tercero and Diffley, 2001). In mammals, the breakage of stalled forks is mediated by the Mus81 nuclease (Hanada et al., 2007). Noteworthy, Mus81 deficient cells are sensitive to a prolonged exposure to hydroxyurea, suggesting that this breakage of the forks is not pathological but rather a controlled event that allows stalled forks to progress by recombinogenic events. Once DSB are formed at replication forks then a normal ATM/ATR‐dependent DSB‐response ensues. Therefore, the low amounts of Chk2 phosphorylation that are seen after hydroxyurea exposure are due to the secondary DSB that are generated at broken forks, and therefore made by ATM and not ATR (Cuadrado et al., 2006). In fact, ATR is unable to phosphorylate Chk2 even when its activity is artificially unleashed by promoting its interaction with TopBP1 (Toledo et al., 2008). In summary, whereas ATM and ATR cooperate in the response to DSB, the ATR/Chk1 response that safeguards the genome in the context of an excess of ssDNA is independent from ATM and Chk2.
The main endogenous source of exposed ssDNA fragments does not come from resected DSB but rather from what is now loosely defined as “replication stress” (RS). Importantly, a number of evidences indicate that RS is not only a pathological condition, but that every replication concurs with certain degree of RS. This explains why, in contrast to ATM (Barlow et al., 1996; Elson et al., 1996; Xu et al., 1996) or Chk2 (Hirao et al., 2002), ATR (Brown and Baltimore, 2000; de Klein et al., 2000) and Chk1 (Liu et al., 2000; Takai et al., 2000) are essential genes in the mouse. Moreover, ATR elimination in adult mice is essential for replicating cells (Ruzankina et al., 2007), and constitutive ATR hypomorphism leads to increased levels of RS, particularly during embryogenesis (Murga et al., 2009). Hence, every replication demands a proficient ATR/Chk1‐response to prevent the accumulation of cell‐lethal levels or RS.
What RS really means is still to be determined, and most efforts in trying to understand the nature of RS derive from genomic 2D Southern blots in yeast which are often difficult to equate with actual structures (Branzei and Foiani, 2010). Importantly, the Costanzo group has started to visualize the presence of several proteins at stalled replication forks by electron‐microscopy (Hashimoto et al., 2010), a technology which promises to reveal important insights into the DNA structures that are formed during RS. A common view from yeast and vertebrate analysis of RS is that, whatever the structures that are formed at stalled replication forks, they all involve an accumulation of ssDNA. Since ssDNA activates ATR but not ATM or DNA‐PKcs, this already explains why, in contrast to the DSB‐response that is coordinated by the three PIKKs, the response to RS is only dependent on ATR. In summary, and whereas historically ATR and ATM have often only been considered as members of the DDR, we here propose the term RS‐response (RSR) should be more frequently introduced when dealing with the functions of ATR or Chk1. We believe that this simple distinction could be clarifying for a better understanding of the different roles that ATR/Chk1 and ATM/Chk2 play on mammalian health.
2. Targeting ATR and Chk1 in cancer: RS overload
The principle of using DNA damage to kill tumor cells has been applied for decades. In fact, it took less than one year from the discovery of x‐rays in 1895 to the first attempts to treat cancers with these “new kind of rays” (as originally named by Roentgen) were made (Rockwell, 1998). Today, radiotherapy is one of the most consolidated treatments for tumors. We now know that the effect of radiotherapy is due to the large amounts of genomic lesions, perhaps most importantly–but not only–DSB, which are generated by ionizing radiation. In addition to radiation, a large fraction of current cancer chemotherapies are also based on genotoxic chemicals. Rapidly growing cells are more prone to enter apoptosis in response to DNA breaks, and this is the rationale behind these strategies. In this context, the higher load of DNA damage that can be given to cancer cells, the better. An extended version of this strategy is to combine DNA damaging agents with inhibitors of the DDR. This would lead to a further accumulation of DNA damage and therefore an increased toxicity of the therapy. Inhibitors of Chk1 were one of the first DDR inhibitors available and have pioneered studies in this regard (reviewed in (Ma et al., 2011) and references therein). However, the problem behind radio‐ and chemo‐ therapy is still the same that it was one century ago. How do we kill the tumor and not the normal cells?
The solution to this problem came from revisiting the now very popular but old concept of synthetic lethality, which is a household tool for yeast geneticists. The idea is to develop drugs that will be particularly toxic for cells harboring cancer‐associated mutations (Hartwell et al., 1997). For instance, the toxicity of inhibitors of poly‐ADP‐rybosil polymerase (PARP) for cells deficient in homologous recombination is currently being exploited as a therapeutic strategy for BRCA1/2 deficient tumors (Bryant et al., 2005; Farmer et al., 2005). Beyond specific deficiencies in repair pathways, a more general feature that might be associated with cancer is the existence of DNA damage. 6 years ago, two laboratories found evidences of an activated DNA damage response (DDR) in early stages of tumor progression (Bartkova et al., 2005; Gorgoulis et al., 2005). These led them to propose a model in which oncogene activation would generate DNA damage, which, by activating the DDR, would limit cancer development in its early stages. Subsequent works confirmed that, indeed, a wide variety of oncogenes generate DNA damage making the oncogene‐induced DDR model one of the most currently discussed in cancer research (reviewed in (Halazonetis et al., 2008)).
To date, much of the work in this model has been dedicated to understand how oncogenes generate DNA damage, or to what extent the enzymes from the DDR protect us from cancer development. However, it is important to note that whereas DNA breaks might ultimately activate the DDR in the tumor, the available evidences suggest that the initial lesion generated by oncogenes is not DSB but rather RS (Bartkova et al., 2005; Gorgoulis et al., 2005). Strong back up for this idea is given by the fact that cancer‐associated insertion and deletions are preferentially present at fragile sites (Dereli‐Oz et al., 2011), which are endogenous loci that are prone to genomic aberrations in the presence of RS. In the context of this model, for the last years we have been considering a very simple hypothesis. If oncogenes generate RS, which is normally suppressed by the RSR, is it possible that targeting ATR or Chk1 would be particularly toxic for cancer cells presenting considerable amounts of RS? The idea here is very similar to the combination of Chk1 inhibitors with external sources of DNA damage; the difference being that here the source of DNA damage would be intrinsic to the tumor, thereby offering a possibility to preferentially kill the cancer cell (see model in Figure 2).
Figure 2.
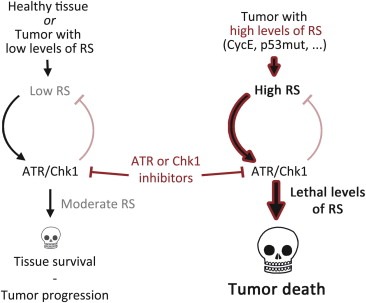
ATR or Chk1 inhibitors in cancer chemotherapy. A certain degree of RS occurs every cell division, where it is detected and suppressed by the ATR‐ and Chk1‐dependent RSR. Inhibitors of ATR or Chk1 exacerbate the levels of RS, which can ultimately promote cell killing by p53‐independent means. In this context, the rationale outlined here is rather simple: Targeting the RSR could be particularly toxic for those cells carrying higher endogenous levels of RS. The key here is that, whereas all tumors might concur with certain degree of RS, these inhibitors should only be toxic for those tumors harboring distinctly high levels of RS. In contrast, healthy tissues and tumors with minimal levels of RS might be largely non‐responsive to ATR or Chk1 inhibitors.
Whereas formal in vivo proof is still missing, several lines of evidence now support this hypothesis. For instance, we have never observed a tumor on ATR‐Seckel mice (Murga M, unpublished observations), which indicates that a severely compromised RSR is largely incompatible with tumor development. Even in vitro, ATR‐Seckel MEF were not able to spontaneously transform (Murga et al., 2009). Moreover, a common cancer event such as the loss of p53 worsened the ageing phenotype of ATR‐Seckel mice (Murga et al., 2009), and also aggravated the severity of ATR elimination in adult mice (Ruzankina et al., 2009). Importantly, the loss of p53 was associated with increased levels of RS and apoptosis on ATR‐Seckel embryos and cells, indicating that low levels of ATR were particularly toxic for p53‐deficient cells. This situation was reminiscent of earlier observations made with Chk1 inhibitors, which were reported to be particularly toxic for p53‐deficient cancer cells (Wang et al., 1996). The important distinction is that the original model proposed that this synthetic lethality was due to the loss of the G2 checkpoint by Chk1 inhibitors, which when combined with the loss of the G1 checkpoint linked to p53 deficiency, could led cells into mitotic catastrophe. We now rather believe that the true explanation to this phenomenon lies on the massive S‐phase damage that is observed in the presence of Chk1 inhibitors (Syljuasen et al., 2005). When combined with the less restrictive S‐phase entry linked to p53 deficiency, this would lead to even higher amounts of RS and cell death. In agreement with this view, we have recently shown that Chk1 and ATR inhibitors generate S‐phase damage, which is further enhanced in p53‐deficient cells (Toledo et al., 2011). A similar toxicity for p53‐deficient cells was also reported with an independent ATR inhibitor (Reaper et al., 2011).
If our model is correct, then other cancer‐associated mutations (besides p53 deficiency) that promote a promiscuous S‐phase entry and RS could also be sensitive to ATR or Chk1 inhibitors. In agreement with this model, we recently observed that ATR and Chk1 inhibitors are also particularly toxic for cells overexpressing cyclin E (Toledo et al., 2011), and RNAi‐mediated depletion of ATR was also found to be very toxic for human cells overexpressing a mutant version of Ras (Gilad et al., 2010). Still, all of the above are based on in vitro findings and the question is: can these ideas be translated into actual cancer therapy? We have a number of unpublished observations in mice that support the validity of this strategy. However, when trying to publish these observations we have invariably confronted a common question. If this model is true, how is it possible that Chk1 inhibitors have failed in curing cancer when tested in clinical trials? To us, the explanation is rather trivial. Chk1 (or ATR) inhibitors might have failed as a general anti‐cancer strategy, but we believe that their efficacy could be much better if the treatment is directed to those tumors that present high loads of RS. Promising drugs such as Imatinib of Olaparib would have also been considered a failure if tested as generic “anti‐cancer” drugs. However, when these therapies are directed to tumors presenting ABL or BRCA1/2 mutations, respectively, they are very efficient. We are currently working to demonstrate that this strategy is useful for the treatment of tumors with high levels of RS. If the in vivo experiments support our hypothesis, we believe that these ideas could be used to develop a more rational use of Chk1 and ATR inhibitors in the clinic.
New and better anti‐cancer drugs are constantly being made, and there is great academic and financial interest behind these efforts. However, we strongly believe that the most important transition to be made in cancer treatment is to learn “who should be given what”. It is very likely that drugs that we already have at hand might be very efficient for the treatment of cancers, but only when directed to those patients that will be mostly sensitized to them. One example of this might be UCN‐01, a Chk1 inhibitor and an old derivative of staurosporine, which was originally discovered as a PKC inhibitor and which at some point was one of the most promising antineoplastic compounds available (Takahashi et al., 1987). However, the poor efficacy in clinical trials and the off‐target effects of the drug dampened the interest in it. Still, it is one of the most potent Chk1 inhibitors available which works in vivo. There is no need to fully abandon these kinds of drugs, once very promising and which when properly administered might end up working very efficiently. We would want to end up by providing one quite striking example of these ideas. Pancreatic adenocarcinomas are one of the most aggressive tumor types, with survival being marginal beyond 6 months from diagnosis (Hidalgo, 2010). Strikingly, a recent report revealed that a patient had survived for more than 3 years upon continues chemotherapy (Villarroel et al., 2011). The drug was not one of the new magic bullets, but rather something as common as Mitomycin C. This sensitivity was found to be due to the fact that the tumor was carrying mutations in PALB2. To us, the message is quite clear. We might already have at hand many compounds that, not necessarily sophisticated or patentable, but that when administered to the proper patient, could be very effective for the treatment of tumors. We believe that the now largely neglected UCN‐01 might fall into this category, as an example of the potential that ATR and Chk1 inhibitors can have for the treatment of tumors presenting high loads of RS. The model awaits experimental confirmation, to which we hope to contribute in the near future.
Acknowledgements
Work in O.F.s laboratory is supported by grants from the Spanish Ministry of Science (CSD2007‐00017 and SAF2008‐01596), Pfizer Foundation Award, EMBO Young Investigator Programme and the European Research Council (ERC‐210520).
Toledo Luis I., Murga Matilde, and Fernandez‐Capetillo Oscar, (2011), Targeting ATR and Chk1 kinases for cancer treatment: A new model for new (and old) drugs, Molecular Oncology, 5, doi: 10.1016/j.molonc.2011.07.002.
References
- Barlow, C. , Hirotsune, S. , Paylor, R. , Liyanage, M. , Eckhaus, M. , Collins, F. , Shiloh, Y. , Crawley, J.N. , Ried, T. , Tagle, D. , 1996. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 86, 159–171. [DOI] [PubMed] [Google Scholar]
- Bartkova, J. , Horejsi, Z. , Koed, K. , Kramer, A. , Tort, F. , Zieger, K. , Guldberg, P. , Sehested, M. , Nesland, J.M. , Lukas, C. , 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 434, 864–870. [DOI] [PubMed] [Google Scholar]
- Branzei, D. , Foiani, M. , 2010. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol.. 11, 208–219. [DOI] [PubMed] [Google Scholar]
- Brown, E.J. , Baltimore, D. , 2000. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes. Dev.. 14, 397–402. [PMC free article] [PubMed] [Google Scholar]
- Bryant, H.E. , Schultz, N. , Thomas, H.D. , Parker, K.M. , Flower, D. , Lopez, E. , Kyle, S. , Meuth, M. , Curtin, N.J. , Helleday, T. , 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Chehab, N.H. , Malikzay, A. , Appel, M. , Halazonetis, T.D. , 2000. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes. Dev.. 14, 278–288. [PMC free article] [PubMed] [Google Scholar]
- Cimprich, K.A. , Cortez, D. , 2008. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol.. 9, 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich, K.A. , Shin, T.B. , Keith, C.T. , Schreiber, S.L. , 1996. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc. Natl. Acad. Sci. U S A. 93, 2850–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliby, W.A. , Roberts, C.J. , Cimprich, K.A. , Stringer, C.M. , Lamb, J.R. , Schreiber, S.L. , Friend, S.H. , 1998. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J.. 17, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez, D. , Guntuku, S. , Qin, J. , Elledge, S.J. , 2001. ATR and ATRIP: partners in checkpoint signaling. Science. 294, 1713–1716. [DOI] [PubMed] [Google Scholar]
- Cuadrado, M. , Martinez-Pastor, B. , Murga, M. , Toledo, L.I. , Gutierrez-Martinez, P. , Lopez, E. , Fernandez-Capetillo, O. , 2006. ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J. Exp. Med.. 203, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Klein, A. , Muijtjens, M. , van Os, R. , Verhoeven, Y. , Smit, B. , Carr, A.M. , Lehmann, A.R. , Hoeijmakers, J.H. , 2000. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol.. 10, 479–482. [DOI] [PubMed] [Google Scholar]
- Dereli-Oz, A. , Versini, G. , Halazonetis, T.D. , 2011. Studies of genomic copy number changes in human cancers reveal signatures of DNA replication stress. Mol. Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elson, A. , Wang, Y. , Daugherty, C.J. , Morton, C.C. , Zhou, F. , Campos-Torres, J. , Leder, P. , 1996. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc. Natl. Acad. Sci. U S A. 93, 13084–13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, H. , McCabe, N. , Lord, C.J. , Tutt, A.N. , Johnson, D.A. , Richardson, T.B. , Santarosa, M. , Dillon, K.J. , Hickson, I. , Knights, C. , 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Garvik, B. , Carson, M. , Hartwell, L. , 1995. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell Biol.. 15, 6128–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad, O. , Nabet, B.Y. , Ragland, R.L. , Schoppy, D.W. , Smith, K.D. , Durham, A.C. , Brown, E.J. , 2010. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer. Res.. 70, 9693–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis, V.G. , Vassiliou, L.V. , Karakaidos, P. , Zacharatos, P. , Kotsinas, A. , Liloglou, T. , Venere, M. , Ditullio, R.A. , Kastrinakis, N.G. , Levy, B. , 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 434, 907–913. [DOI] [PubMed] [Google Scholar]
- Halazonetis, T.D. , Gorgoulis, V.G. , Bartek, J. , 2008. An oncogene-induced DNA damage model for cancer development. Science. 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Hanada, K. , Budzowska, M. , Davies, S.L. , van Drunen, E. , Onizawa, H. , Beverloo, H.B. , Maas, A. , Essers, J. , Hickson, I.D. , Kanaar, R. , 2007. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat. Struct. Mol. Biol.. 14, 1096–1104. [DOI] [PubMed] [Google Scholar]
- Harper, J.W. , Elledge, S.J. , 2007. The DNA damage response: ten years after. Mol. Cell. 28, 739–745. [DOI] [PubMed] [Google Scholar]
- Hartwell, L.H. , Szankasi, P. , Roberts, C.J. , Murray, A.W. , Friend, S.H. , 1997. Integrating genetic approaches into the discovery of anticancer drugs. Science. 278, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Hashimoto, Y. , Chaudhuri, A.R. , Lopes, M. , Costanzo, V. , 2010. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol.. 17, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo, M. , 2010. Pancreatic cancer. N. Engl. J. Med.. 362, 1605–1617. [DOI] [PubMed] [Google Scholar]
- Hirao, A. , Cheung, A. , Duncan, G. , Girard, P.M. , Elia, A.J. , Wakeham, A. , Okada, H. , Sarkissian, T. , Wong, J.A. , Sakai, T. , 2002. Chk2 is a tumor suppressor that regulates apoptosis in both an ataxia telangiectasia mutated (ATM)-dependent and an ATM-independent manner. Mol. Cell Biol.. 22, 6521–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao, A. , Kong, Y.Y. , Matsuoka, S. , Wakeham, A. , Ruland, J. , Yoshida, H. , Liu, D. , Elledge, S.J. , Mak, T.W. , 2000. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 287, 1824–1827. [DOI] [PubMed] [Google Scholar]
- Jackson, S.P. , Bartek, J. , 2009. The DNA-damage response in human biology and disease. Nature. 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri, A. , Falck, J. , Lukas, C. , Bartek, J. , Smith, G.C. , Lukas, J. , Jackson, S.P. , 2006. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol.. 8, 37–45. [DOI] [PubMed] [Google Scholar]
- Kastan, M.B. , Zhan, Q. , el-Deiry, W.S. , Carrier, F. , Jacks, T. , Walsh, W.V. , Plunkett, B.S. , Vogelstein, B. , Fornace, A.J. , 1992. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 71, 587–597. [DOI] [PubMed] [Google Scholar]
- Kumagai, A. , Dunphy, W.G. , 2000. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol. Cell. 6, 839–849. [DOI] [PubMed] [Google Scholar]
- Kumagai, A. , Lee, J. , Yoo, H.Y. , Dunphy, W.G. , 2006. TopBP1 activates the ATR-ATRIP complex. Cell. 124, 943–955. [DOI] [PubMed] [Google Scholar]
- Lakin, N.D. , Hann, B.C. , Jackson, S.P. , 1999. The ataxia-telangiectasia related protein ATR mediates DNA-dependent phosphorylation of p53. Oncogene. 18, 3989–3995. [DOI] [PubMed] [Google Scholar]
- Lee, J. , Kumagai, A. , Dunphy, W.G. , 2007. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem.. 282, 28036–28044. [DOI] [PubMed] [Google Scholar]
- Lee, S.E. , Moore, J.K. , Holmes, A. , Umezu, K. , Kolodner, R.D. , Haber, J.E. , 1998. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 94, 399–409. [DOI] [PubMed] [Google Scholar]
- Liu, Q. , Guntuku, S. , Cui, X.S. , Matsuoka, S. , Cortez, D. , Tamai, K. , Luo, G. , Carattini-Rivera, S. , DeMayo, F. , Bradley, A. , 2000. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes. Dev.. 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Lopez-Contreras, A.J. , Fernandez-Capetillo, O. , 2010. The ATR barrier to replication-born DNA damage. DNA Repair (Amst). 9, 1249–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, C.X. , Janetka, J.W. , Piwnica-Worms, H. , 2011. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends. Mol. Med.. 17, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murga, M. , Bunting, S. , Montana, M.F. , Soria, R. , Mulero, F. , Canamero, M. , Lee, Y. , McKinnon, P.J. , Nussenzweig, A. , Fernandez-Capetillo, O. , 2009. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet.. 41, 891–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo, S.E. , Jackson, S.P. , 2011. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes. Dev.. 25, 409–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaper, P.M. , Griffiths, M.R. , Long, J.M. , Charrier, J.D. , Maccormick, S. , Charlton, P.A. , Golec, J.M. , Pollard, J.R. , 2011. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. [DOI] [PubMed] [Google Scholar]
- Rockwell, S. , 1998. Experimental radiotherapy: a brief history. Radiat. Res.. 150, S157–S169. [PubMed] [Google Scholar]
- Ruzankina, Y. , Pinzon-Guzman, C. , Asare, A. , Ong, T. , Pontano, L. , Cotsarelis, G. , Zediak, V.P. , Velez, M. , Bhandoola, A. , Brown, E.J. , 2007. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 1, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzankina, Y. , Schoppy, D.W. , Asare, A. , Clark, C.E. , Vonderheide, R.H. , Brown, E.J. , 2009. Tissue regenerative delays and synthetic lethality in adult mice after combined deletion of Atr and Trp53. Nat. Genet.. 41, 1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky, K. , Bar-Shira, A. , Gilad, S. , Rotman, G. , Ziv, Y. , Vanagaite, L. , Tagle, D.A. , Smith, S. , Uziel, T. , Sfez, S. , 1995. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 268, 1749–1753. [DOI] [PubMed] [Google Scholar]
- Shieh, S.Y. , Ahn, J. , Tamai, K. , Taya, Y. , Prives, C. , 2000. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes. Dev.. 14, 289–300. [PMC free article] [PubMed] [Google Scholar]
- Siliciano, J.D. , Canman, C.E. , Taya, Y. , Sakaguchi, K. , Appella, E. , Kastan, M.B. , 1997. DNA damage induces phosphorylation of the amino terminus of p53. Genes. Dev.. 11, 3471–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syljuasen, R.G. , Sorensen, C.S. , Hansen, L.T. , Fugger, K. , Lundin, C. , Johansson, F. , Helleday, T. , Sehested, M. , Lukas, J. , Bartek, J. , 2005. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell Biol.. 25, 3553–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, I. , Kobayashi, E. , Asano, K. , Yoshida, M. , Nakano, H. , 1987. UCN-01, a selective inhibitor of protein kinase C from Streptomyces. J. Antibiot.. 40, 1782–1784. (Tokyo) [DOI] [PubMed] [Google Scholar]
- Takai, H. , Tominaga, K. , Motoyama, N. , Minamishima, Y.A. , Nagahama, H. , Tsukiyama, T. , Ikeda, K. , Nakayama, K. , Nakanishi, M. , Nakayama, K. , 2000. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes. Dev.. 14, 1439–1447. [PMC free article] [PubMed] [Google Scholar]
- Tercero, J.A. , Diffley, J.F. , 2001. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 412, 553–557. [DOI] [PubMed] [Google Scholar]
- Tibbetts, R.S. , Brumbaugh, K.M. , Williams, J.M. , Sarkaria, J.N. , Cliby, W.A. , Shieh, S.Y. , Taya, Y. , Prives, C. , Abraham, R.T. , 1999. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes. Dev.. 13, 152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo, L.I. , Murga, M. , Gutierrez-Martinez, P. , Soria, R. , Fernandez-Capetillo, O. , 2008. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes. Dev.. 22, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo, L.I. , Murga, M. , Zur, R. , Soria, R. , Rodriguez, A. , Martinez, S. , Oyarzabal, J. , Pastor, J. , Bischoff, J.R. , Fernandez-Capetillo, O. , 2011. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga, K. , Morisaki, H. , Kaneko, Y. , Fujimoto, A. , Tanaka, T. , Ohtsubo, M. , Hirai, M. , Okayama, H. , Ikeda, K. , Nakanishi, M. , 1999. Role of human Cds1 (Chk2) kinase in DNA damage checkpoint and its regulation by p53. J. Biol. Chem.. 274, 31463–31467. [DOI] [PubMed] [Google Scholar]
- Villarroel, M.C. , Rajeshkumar, N.V. , Garrido-Laguna, I. , De Jesus-Acosta, A. , Jones, S. , Maitra, A. , Hruban, R.H. , Eshleman, J.R. , Klein, A. , Laheru, D. , 2011. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol. Cancer Ther.. 10, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Fan, S. , Eastman, A. , Worland, P.J. , Sausville, E.A. , O'Connor, P.M. , 1996. UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53. J. Natl. Cancer Inst.. 88, 956–965. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Zou, L. , Lu, T. , Bao, S. , Hurov, K.E. , Hittelman, W.N. , Elledge, S.J. , Li, L. , 2006. Rad17 phosphorylation is required for claspin recruitment and Chk1 activation in response to replication stress. Mol. Cell. 23, 331–341. [DOI] [PubMed] [Google Scholar]
- Wright, J.A. , Keegan, K.S. , Herendeen, D.R. , Bentley, N.J. , Carr, A.M. , Hoekstra, M.F. , Concannon, P. , 1998. Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc. Natl. Acad. Sci. U S A. 95, 7445–7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, Y. , Ashley, T. , Brainerd, E.E. , Bronson, R.T. , Meyn, M.S. , Baltimore, D. , 1996. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes. Dev.. 10, 2411–2422. [DOI] [PubMed] [Google Scholar]
- Zou, L. , Cortez, D. , Elledge, S.J. , 2002. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes. Dev.. 16, 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, L. , Elledge, S.J. , 2003. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 300, 1542–1548. [DOI] [PubMed] [Google Scholar]