

Abstract
Secondary dengue viral infection can produce capillary leakage associated with increased mortality known as dengue hemorrhagic fever (DHF). Because the mortality of DHF can be reduced by early detection and intensive support, improved methods for its detection are needed. We applied multidimensional protein profiling to predict outcomes in a prospective dengue surveillance study in South America. Plasma samples taken from initial clinical presentation of acute dengue infection were subjected to proteomics analyses using ELISA and a recently developed biofluid analysis platform. Demographics, clinical laboratory measurements, nine cytokines, and 419 plasma proteins collected at the time of initial presentation were compared between the DF and DHF outcomes. Here, the subject's gender, clinical parameters, two cytokines, and 42 proteins discriminated between the outcomes. These factors were reduced by multivariate adaptive regression splines (MARS) that a highly accurate classification model based on eight discriminant features with an area under the receiver operator curve (AUC) of 0.999. Model analysis indicated that the feature–outcome relationship were nonlinear. Although this DHF risk model will need validation in a larger cohort, we conclude that approaches to develop predictive biomarker models for disease outcome will need to incorporate nonparametric modeling approaches. Clin Trans Sci 2012; Volume #: 1–13
Keywords: infectious disease, hemorrhagic disorders and therapies, plasma, proteins, viral infection, host response
Introduction
Dengue infection remains an international public health problem affecting urban populations in tropical and subtropical regions, where it is currently estimated that about 2.5 billion people are at risk of infection. Dengue viruses (family: Flaviviridae; genus: Flavivirus) are transmitted among humans primarily by Aedes aegypti mosquitoes. In humans, dengue infection can produce a spectrum of diseases ranging from asymptomatic to a flu‐like state (dengue fever [DF]) to a hemorrhagic form (dengue hemorrhagic fever [DHF]). 1 The latter, DHF, is a life threatening complication characterized by high fever, coagulopathy, vascular leakage, and hypovolemic shock.
Due to a number of factors, including increasing urbanization, globalization of travel, and reduction in the use of the pesticide dichlorodiphenyltrichloroethane, dengue disease is reemerging in the Americas. Here, an estimated 890,000 cases of DF were reported in 2007 representing a significant increase from historical trends. 2 The mortality of DHF is age dependent, primarily affecting both children and the elderly. 3 In Southeast Asia, a disproportionate amount of DHF hospitalizations are of children whereas in the Americas, there is a more even distribution across ages.
The risk factors and etiology of DHF are not fully understood. Dengue viruses occur in four distinct serotypes, with hyperendemic regions having more than one circulating serotype at a time. Many epidemiological studies have found an increased risk of DHF after a second infection with a different serotype. 3 , 4 , 5 This observation has led to the “antibody‐dependent enhancement (ADE) theory,” which proposes that neutralizing antibodies generated during the adaptive immune response cross‐react, but do not neutralize, a second infecting dengue virus serotype. These antibody‐viral complexes are taken up by monocytes by binding their cell‐surface Fc receptors, resulting in increased intracellular viral load. As a result, highly activated monocytes release enhanced cytokines and factors involved in vascular leakage. Other evidence points to DHF being the result of an interplay between host and viral factors. 1
Currently, there is no drug therapy for DHF. However, while DHF fatality rates can exceed 20%, early and intensive supportive therapy has reduced it to less than 1%. 6 Consequently, clinical features, biochemical assays, and gene expression profiling have been used to identify DHF risk.
Recent advances in global‐scale proteomics technologies enable the detection of candidate protein biomarkers—these include proteins, peptides, or metabolites that can be measured alone (or in a combination) and would reliably indicate disease outcome. 7 With the advancement of multidimensional profiling techniques, the systematic identification of predictive proteins associated with DHF is now feasible. To identify differentially expressed proteins associated with DHF, we have developed a reproducible, novel preseparation fractionation method, termed the biofluid analysis platform (BAP), that takes advantage of high recovery and quantitative size exclusion fractionation, followed by quantitative saturation fluorescence labeling and two‐dimensional (2D) gel electrophoresis (2‐DE), and tandem liquid chromatographytandem mass spectrometry (LC‐MS/MS) protein identification to identify differentially expressed proteins associated with DHF. Statistical analysis of discriminant proteins indicates that the proteins are not normally distributed, precluding conventional parametric modeling approaches. Application of these protein profiles to associate with disease outcome was accomplished by multivariate adaptive regression splines (MARS) modeling, where a highly accurate classifier of the sample set was obtained using cross‐validation. These findings suggest optimal approaches for modeling predictive biomarker panels using discovery proteomics approaches in human host response to infectious disease.
Methods
Study population
An active surveillance for dengue diseases study was conducted in Maracay, Venezuela. Febrile subjects with signs and symptoms consistent with dengue virus infection who presented at participating clinics and hospitals, or who were identified by community‐based active surveillance, were included in the study. 8 On the day of presentation, a blood sample was collected for dengue virus real time‐polymerase chain reaction (RT‐PCR) confirmation and plasma preparation. The subjects were monitored for clinical outcome, and DF and DHF cases were scored following WHO case definitions. 9 An additional blood sample was collected on study day 30 for plasma preparation. Plasma specimens were stored at −70°C until proteomic processing. Numbers of patients and disease characteristics are shown in Table 1 . A Fisher's exact test performed on this study population to examine diagnosis by sex indicated that the number of males and females with each disease are not significantly different from each other. A similar analysis was performed to examine diagnosis by age, and the results indicate that there was no difference between age and disease.
Table 1.
Clinical characteristics of study population.
| Phenotype | Characteristic | No. of men = 23 (42%) | No. of women = 32 (58%) | All subjects = 55 |
|---|---|---|---|---|
| DHF (n = 13) | n = 3 (23%) | n = 10 (77%) | n = 13 | |
| Age (years) | 24 ± 22 | 18 ± 11 | 19 ± 13.4 | |
| Weight (kg) | 46 ± 6.6 | 42 ± 9.3 | 45 ± 14 | |
| Temp max (ºC) | 39.1 ± 1.04 | 39 ± 0.65 | 39 ± 0.70 | |
| Fever (days) | 6 ± 1.73 | 5 ± 0.66 | 5 ±1b | |
| Hemoglobin (gm%) | 12.83 ± 0.83 | 12 ± 0.97 | 12 ± 0.93a | |
| Hematocrit (%) | 41.16 ± 1.89 | 39 ± 3.68 | 39 ± 3.5 | |
| Platelets (103/μL) | 125.33 ± 13 | 99 ± 35 | 105 ± 33c | |
| RBC (×106/μL) | 2.6 ± 0.6 | 4 ± 1.48 | 3 ± 1.37a | |
| Lymphocytes (103/μL) | 29.5 ± 11 | 39 ± 15.6 | 37 ± 14.8 | |
| Neutrophils (103/μL) | 66.1 ± 7.25 | 59 ± 14.98 | 61 ± 13.65 | |
| Diarrhea | 67% | 40% | 46%a | |
| DF (n = 42) | n = 20 (47%) | n = 22 (52%) | n = 42 | |
| Age (years) | 14.35 ± 7.05 | 16.7 ± 7.9 | 15.59 ± 7.5 | |
| Weight (kg) | 42.5 ± 17.67 | 33.4 ± 12.4 | 36 ± 13 | |
| Temp max (ºC) | 39.07 ± 0.66 | 38.72 ± 0.65 | 38.8 ± 0.67 | |
| Fever (days) | 4.5 ± 1.05 | 4.08 ± 1.11 | 4.2 ± 1 | |
| Hemoglobin (gm%) | 13.96 ± 1.73 | 13.22 ± 1.32 | 13.57 ± 1.56 | |
| Hematocrit (%) | 42.7 ± 4.53 | 40.27 ± 4.24 | 41.42 ± 4.5 | |
| Platelets (103/μL) | 167.25 ± 35.7 | 155.4 ± 45 | 161 ± 40.7 | |
| RBC (X106/μL) | 4.70 ± 1.88 | 4.46 ± 2.1 | 4.56 ± 1.98 | |
| Lymphocytes (103/μL) | 42.45 ± 12.25 | 48.45 ± 14.5 | 45.6 ± 13.68 | |
| Neutrophils (103/μL) | 56.1 ± 12.62 | 50.54 ± 14.44 | 53.19 ± 13.73 | |
| Diarrhea | 10% | 18% | 14% |
DHF = dengue hemorrhagic fever; DF = dengue fever; n = number; RBC = red blood cell count.ap< 0.05; bp< 0.01; cp< 0.001.
RT‐PCR
Viral RNA was prepared from 140 μL sera using QIAamp Viral RNA Mini Kits following the manufacturer's instructions (Qiagen Inc., Valencia, CA, USA). Nested dengue virus RT‐PCR was performed on serum samples for virus detection as described. 10
Multiplex bead‐based cytokine measurements
Plasma samples were analyzed for the concentrations of nine human cytokines (IL‐6, IL‐10, IFN‐γ, IP‐10, MIP‐1α, TNFα, IL‐2, VEGF, and TRAIL; Bioplex, Bio‐Rad, Hercules, CA, USA). Briefly, plasma samples were thawed, centrifuged at 1,900 XG for 3 minutes at 4°C, and incubated with microbeads labeled with antibodies specific to each analyte for 30 minutes. Following a wash step, the beads were incubated with the detection antibody cocktail, each bead specific to a single cytokine. After another wash step, the beads were incubated with streptavidin‐phycoerythrin for 10 minutes and washed, and then the analyte concentrations determined using the array reader. For each analyte, a standard curve was generated using recombinant proteins to estimate protein concentration in the unknown sample.
BAP preseparation fractionation
The BAP preseparation fractionation system is a semiautomated and custom‐designed device consisting of four 1 × 30 cm columns fitted with upward flow adapters and filled with Superdex S‐75 (GE Healthcare, Pittsburgh PA, USA) size‐exclusion beads. Plasma samples were injected into each of the columns through four HPLC injectors, and buffer flow was controlled by a high performance liquid chromatography (HPLC) pump (Model 305, Gilson, Middleton, WI, USA). The effluent from each column was monitored by individual UV/Vis monitors (Model 251, Gilson) that each control individual fraction collectors (Model 203B, Gilson). The columns were equilibrated with running buffer (50 mM (NH4)2CO3, pH 8.0), and up to 300 μL of plasma, containing 3 mg of protein and 8 M urea spiked with 3 μg of purified Alexa‐488 labeled thaumatin (Sigma‐Aldrich, St. Louis, MO, USA), were pumped into the columns at an upward flow rate of 20 mL/h. The eluent was monitored at 493 nm by the UV/Vis monitor that was programmed to detect a predetermined signal of 0.1 mV in the detector output that designated the start and end of the fluorescent thaumatin peak, and signaled the fraction collector to change collection tubes after an appropriate delay. The fractions preceding the end of the thaumatin peak were pooled and designated the “protein pool,” while the fractions subsequent to the peak up to the free dye peak were pooled and designated the “peptide pool.”
After size exclusion chromatography (SEC), the protein pools were incubated at 4°C overnight to permit further renaturation. They were then loaded onto antibody (IgY) depletion columns per the manufacturer's instructions (Phenomenex, Torrance, CA, USA) that deplete 14 of the most highly abundant proteins found in plasma or serum. The flow‐through was collected and rerun through the columns a second time. The proteins obtained from the second flow‐through were concentrated and resuspended in 2‐DE buffer for quantitative saturation fluorescence labeling.
Saturation fluorescence labeling
We developed a saturation fluorescence approach using uncharged BODIPY FL‐maleimide (BD) that reacts with protein thiols at a dye‐to‐protein thiol ratio of greater than 50:1 to give an uncharged product, with no nonspecific labeling. BD‐labeled protein isoelectric points are unchanged and mobilities were identical to those in the unlabeled state. 11 , 12 Using the ProExpress 2D imager (PerkinElmer, Cambridge, United Kingdom), BD protein labeling (ex: 460/80 nm; em: 535/50 nm) has a dynamic range over four log orders of magnitude, and can detect 5 fmol of protein at a signal‐to‐noise ratio of 2:1. This saturation fluorescence labeling method has yielded high accuracy (>91%) in quantifying blinded protein samples. 13 To ensure saturation labeling, protein extracts or pools to be labeled were analyzed for cysteine (cysteic acid) content by amino acid analysis (Model L8800, Hitachi High Technologies America, Pleasanton, CA, USA) and sufficient dye added to achieve the desired excess of dye to thiol.
BD‐labeled proteins were separated by 2‐DE, employing an IPGphor multiple sample isoelectric focusing (IEF) device (Pharmacia, Piscataway, NJ, USA) in the first dimension, and Protean Plus and Criterion Dodeca cells (Bio‐Rad) in the second dimension. 11 Sample aliquots were first loaded onto 11 cm dehydrated precast immobilized pH gradient (IPG) strips (Bio‐Rad), and rehydrated overnight. IEF was performed at 20°C with the following parameters: 50 V, 11 hours; 250 V, 1 hour; 500 V, 1 hour; 1,000 V, 1 hour; 8,000 V, 2 hours; 8,000 V, 6 hours. The IPG strips were then incubated in 4 mL of equilibration buffer (6 M urea, 2% sodium dodecyl sulfate (SDS), 50 mM Tris‐HCl, pH 8.8, 20% glycerol) containing 10 μL/mL tri‐2 (2‐carboxyethyl) phosphine (Geno Technology Inc., St. Louis, MO, USA) for 15 minutes at 22°C with shaking. The samples were incubated in another 4 mL of equilibration buffer with 25 mg/mL iodoacetamide for 15 minutes at 22°C with shaking in order to ensure protein S‐alkylation. Electrophoresis was performed at 150 V for 2.25 hours, 4°C with precast 8–16% polyacrylamide gels in Tris‐glycine buffer (25 mM Tris‐HCl, 192 mM glycine, 0.1% SDS, pH 8.3).
Protein fluorescence staining
After electrophoresis, the gels were directly imaged at 100 μm resolution using the PerkinElmer ProXPRESS 2D Proteomic Imaging System to quantify BD‐labeled proteins (>90% of human proteins contain at least one cysteine 14 ). A gel containing the most common features was selected by Nonlinear SameSpots software (Nonlinear Dynamics, Ltd. Newcastle Upon Tyne, United Kingdom) as the reference gel for the entire set of gels, and this gel was then fixed in buffer (10% methanol, 7% acetic acid in ddH20), and directly stained with SyproRuby stain (Invitrogen, Carlsbad, CA, USA), and destained in buffer. SyproRuby is an ionic dye that typically labels proteins with multiple fluors, including a Sypro‐stained gel in the analysis ensures that the maximum number of proteins were detected and quantified. The destained gel were scanned at 555/580 nm (ex/em). The exposure time for both dyes was adjusted to achieve a value of approximately 55,000–63,000 pixel intensity (16‐bit saturation) from the most intense protein spots on the gel.
Measurement of relative spot intensities
The 2D gel images were analyzed using Progenesis/SameSpots software. The reference gel was selected according to quality and number of spots. Once “landmarks” were defined the program performed automatic spot detection on all images. The SyproRuby stained reference gel was used to define spot boundaries, however, the gel images taken under the BD‐specific filters were used to obtain the quantitative spot data. This strategy ensures that spot numbers and outlines were identical across all gels in the experiment, eliminating problems with unmatched spots 15 , 16 as well as ensuring that the greatest number of protein spots and their spot volumes were accurately detected and quantified. Spot volumes were normalized using a software‐calculated bias value assuming that the great majority of spot volumes did not change in abundance.
Protein identification
Selected 2‐DE spots were picked robotically, trypsin‐digested, and peptide masses identified by MALDI TOF/TOF (AB Sciex 4800, Applied Biosystems, Foster City, CA, USA). Data were analyzed with the Applied Biosystems software package included 4000 Series Explorer (v. 3.6 RC1), Data Version (3.80.0) to acquire both MS and MS/MS spectral data. The instrument was operated in positive ion reflectron mode, mass range was 850–3000 Da, and the focus mass was set at 1,700 Da. For MS data, 2,000–4,000 laser shots were acquired and averaged from each sample spot. Automatic external calibration was performed using a peptide mixture with reference masses 904.468, 1,296.685, 1,570.677, and 2,465.199.
Following MALDI MS analysis, MALDI MS/MS was performed on several (5–10) abundant ions from each sample spot. A 1 kV positive ion MS/MS method was used to acquire data under postsource decay (PSD) conditions. The instrument precursor selection window was ±3 Da. For MS/MS data, 2,000 laser shots were acquired and averaged from each sample spot. Automatic external calibration was performed using reference fragment masses 175.120, 480.257, 684.347, 1,056.475, and 1,441.635 (from precursor mass 1,570.700).
Applied Biosystems GPS ExplorerTM (v. 3.6) software was used in conjunction with MASCOT to search the respective protein database using both MS and MS/MS spectral data for protein identification. Protein match probabilities were determined using expectation values and/or MASCOT protein scores. MS peak filtering included the following parameters: mass range 800–4,000 Da, minimum S/N filter = 10, mass exclusion list tolerance = 0.5 Da, and mass exclusion list (for some trypsin and keratin‐containing compounds) included masses 842.51, 870.45, 1,045.56, 1,179.60, 1,277.71, 1,475.79, and 2,211.1. For MS/MS peak filtering, the minimum S/N filter = 10.
For protein identification, the Homo sapiens taxonomy was searched in the NCBI database. Other parameters included the following: selecting the enzyme as trypsin; maximum missed cleavages = 1; fixed modifications included carbamidomethyl (C) for 2D gel analyses only; variable modifications included oxidation (M); precursor tolerance was set at 0.2 Da; MS/MS fragment tolerance was set at 0.3 Da; mass = monoisotopic; and peptide charges were only considered as + 1.
Protein identification was performed using a Bayesian algorithm 17 where matches were indicated by expectation score, an estimate of the number of matches that would be expected in that database if the matches were completely random. Confirmation of the protein identification was performed by LC‐MS/MS (Orbitrap Velos, ThermoFinnegan, San Jose, CA, USA).
Statistical analysis
Statistical comparisons were performed using SAS®, version 9.1.3 (SAS Inc., Cary, NC, USA) and PASW Statistics 17.0, Release 17.0.2 (SPSS Inc., Chicago, IL, USA).
Multivariate analysis of variance (MANOVA)
The MANOVA model is a popular statistical model used to determine whether significant mean differences exist among disease and gender groups. One advantage of MANOVA is that the correlation structure is taken into consideration between each cytokine. The Wilks’ lambda statistics as a MANOVA‐based score were used to analyze data where there is more than one dependent variable (SAS 9.2 PROC GLM).
Mars
MARS is a nonparametric regression method that uses piecewise linear spline functions (basis functions) as predictors. The basis functions are combinations of independent variables and so this method allows detection of feature interactions and performs well with complex data structures. 18 MARS uses a two‐stage process for constructing the optimal classification model. The first half of the process involves creating an overly large model by adding basis functions that represent either single variable transformations or multivariate interaction terms. The model becomes more flexible and complex as additional basis functions are added. The process is complete when a user‐specified number of basis functions have been added. In the second stage, MARS deletes basis functions in order, starting with the basis function that contributes the least to the model until an optimum model is reached. By allowing the model to take on many forms as well as interactions, MARS can reliably track the very complex data structures that are often present in high‐dimensional data. Cross‐validation techniques were used within MARS to avoid overfitting the classification model. Log‐transformed cytokine and normalized spot intensities from 2‐DE were modeled using 10‐fold cross validation and a maximum of 126 functions (Salford Systems Inc., San Diego, CA, USA).
Generalized additive models (GAMs)
GAMs were estimated by a backfitting algorithm within a Newton–Raphson technique. We used SAS 9.2 PROC GAM and STATISTICA 8.0 (StatSoft, Tulsa, OK, USA) to fit the GAM fittings with binary logit link function that provided multiple types of smoothers with automatic selection of smoothing parameters.
Results
Clinical demographics
The initial clinical parameters were compared for the 55 volunteers (42 DF, 13 DHF) at the time of initial presentation ( Table 1 ). Here, the number of days of fever (4.2 ± 1 days vs. 5 ± 1 days, p < 0.01), initial platelet counts (161 ± 40.7 × 103/μL vs. 105 ± 33 × 103/μL, p < 0.001), red blood count (4.56 ± 13.68 × 106/μL vs. 3 ± 1.37 × 106/μL, p < 0.05). and frequency of diarrhea (46% vs. 14%, p < 0.05) were statistically different between DF and DHF, respectively.
Our study design was intended to include patients with all four dengue serotypes. The distribution of dengue viral serotypes in the study population are shown in Table 2 . Although dengue 1 was the most predominant serotype in DF patients in this study, accounting for 50% of the DF infections, dengue 2 was the most predominant for the subset with DHF, with 62% being infected with that serotype. This difference in serotypes are significantly different (p value = 0.0085, Fisher's exact test).
Table 2.
Distribution of dengue serotypes by disease.
| Disease | Dengue 1 | Dengue 2 | Dengue 3 | Dengue 4 | Total |
|---|---|---|---|---|---|
| DF (%) | 21 (50) | 6 (14) | 10 (24) | 5 (12) | 42 |
| DHF (%) | 2 (15) | 8 (62) | 2 (15) | 1 (8) | 13 |
| Total | 23 | 14 | 12 | 5 | 55 |
DF = dengue fever, DHF = dengue hemorrhagic fever. Percentages for each serotype are given by disease (row).
Cytokine analyses
Plasma proteins were isolated from subjects obtained during the initial clinic visit. Focused proteomics analyses were performed using bead‐based immunoplex to measure cytokines that have been associated with DHF in previous studies. 19 , 20 Analysis of the plasma concentrations of the cytokines indicated that their distributions were highly skewed; despite logarithmic transformation of the data, they remained nonnormally distributed. As a result, the cytokines were compared between the two outcomes using the Wilcoxon rank‐sum test. Also, we adopted a permutation test to derive p values since the violation of normal assumption does not affect this method. Only two cytokines retained significance between DF and DHF, IL‐6 (p= 0.002) and IL‐10 (p < 0.001) ( Figures 1A and B ). For both cytokines, the median value of the logarithm base two‐transformed concentration was greater in DHF than that of DF subjects.
Figure 1.
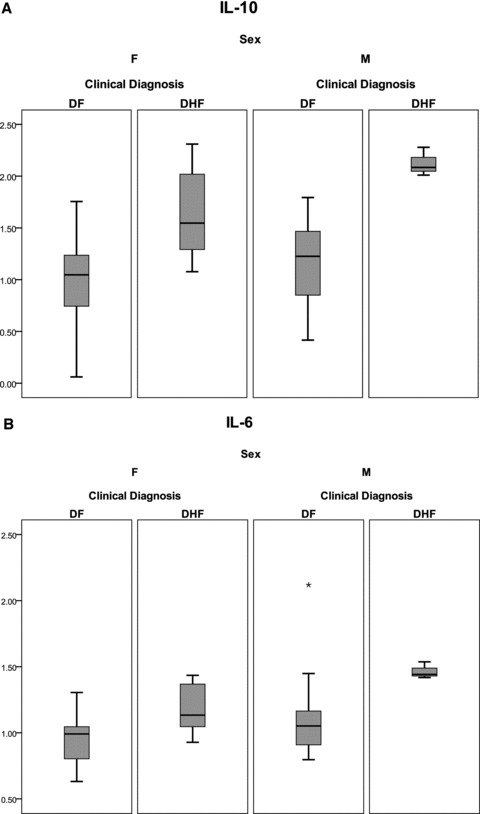
Shown is a box‐plot comparison of log2‐transformed cytokine values for IL‐6 (A), and IL‐10 (B) by diagnosis and gender. DF = dengue fever; DHF = dengue hemorrhagic fever. Horizontal bar = median value; shaded box = 25–75 % interquartile range (IQR); error bars = median ± 1.5 (IQR); *= outlier.
Differences between cytokines were analyzed as a function of gender using two‐factor ANOVA. For IL‐6 and IL‐10, MIP‐1α, and TRAIL, we identified a significant gender and diagnosis (DF vs. DHF) effect ( Table 3 ). To correct for correlated cytokines, we also applied a MANOVA test to the overall data set. In this analysis, both gender (p= 0.0165) and diagnosis (p < 0.0001) had significant Wilks–Lamba p values. Together, these analyses indicate that gender is an important confounding variable in the cytokine response to dengue infection (also plotted in Figures 1A and B ).
Table 3.
Two‐way ANOVA for detection of interactions between gender and disease.
| Cytokine | Source | Type III sum of squares | Df | Mean square | F | Sig. |
|---|---|---|---|---|---|---|
| IL‐6 | Disease | 0.637 | 1 | 0.637 | 11.034 | 0.002 |
| Gender | 0.335 | 1 | 0.335 | 5.795 | 0.020 | |
| Disease × gender | 0.032 | 1 | 0.032 | 0.559 | 0.459 | |
| Error | 2.715 | 47 | 0.058 | |||
| Total | 3.557 | 50 | ||||
| IL‐10 | Disease | 4.643 | 1 | 4.643 | 28.675 | 0.000 |
| Gender | 0.667 | 1 | 0.667 | 4.182 | 0.046 | |
| Disease × gender | 0.231 | 1 | 0.231 | 1.428 | 0.238 | |
| Error | 7.610 | 47 | 0.162 | |||
| Total | 12.531 | 50 |
Df = degrees of freedom; Sig. = significance.
BAP
The BAP, a discovery‐based sample prefractionation method with 2‐DE using saturation fluorescence labeling, was applied more comprehensively to identify proteins associated with the development of DHF. The BAP combines a high recovery Superdex S‐75 size‐exclusion chromatography (SEC) of plasma with electronically triggered fraction collection to create protein and peptide pools for subsequent separation and analysis. An important feature of the BAP is the utilization of deionized urea to initially dissociate protein/peptide complexes in the plasma prior to SEC. Generating reproducible run‐to‐run fractionation was accomplished by spiking Alexa 488‐labeled thaumatin (approximately 23 kDa) into the urea‐treated samples before SEC. Multiple side‐by‐side columns can thus collect virtually identical protein fractions of proteins with 95–100% recoveries (measured in over 200 test samples, not shown) ensuring accurate differential analyses. The protein fraction was then depleted off 14 of the most high‐abundance plasma proteins, and this fraction was subsequently labeled with saturating ratios of a cysteine‐reactive dye to protein thiols followed by 2‐DE. 11 , 12 , 21
One hundred six serum samples, representing acute and convalescent samples from 53 subjects were analyzed by BAP (two samples from the 55 enrolled subjects were not analyzed by BAP due to sample limitation). Four hundred and nineteen spots were mapped and the normalized spot intensities were compared. For the purposes of biomarker panel development, normalized spot intensities were compared between DF and DHF in the acute samples. From this analysis, 34 spots met statistical cutoff criteria (p < 0.05, t‐test).
MARS‐based modeling for predictors of DHF
Because the proteomic quantifications are not normally distributed and included outliers, we evaluated nonparametric modeling methods. MARS is a robust, nonparametric, piecewise linear approach that establishes relationships within small intervals of independent variables, detects feature interactions, and is generally resistant to the effects of outlier influence. 22 To identify features important in DHF, gender, logarithm‐transformed cytokine expression values (IL‐6 and IL‐10), and 34 2‐DE protein spots were modeled using 10‐fold cross‐validation and a maximum of 126 basis functions, schematically diagrammed in Figure 2 . Because of the small sample size represented in the study population for dengue serotypes 1, 3, and 4 ( Table 2 ), dengue serotype was excluded from the modeling. The optimal model was selected on the basis of the lowest cross‐validation error.
Figure 2.
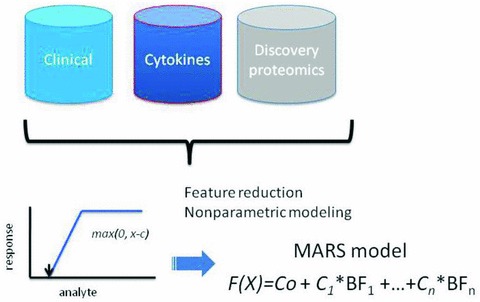
Schematic diagram of modeling strategy to identify predictors of DHF using different data types. Data sources include: clinical demographics, normalized spot intensities by 2‐DE analysis, and log2‐transformed cytokine measurements. MARS produces a linear combination of basis functions (BFs), each represented by the value of the maximum of (0, x‐c), where x is the analyte concentration.
The optimal discriminant model selected one cytokine (IL‐10) and seven protein spots. The proteins that corresponded to each predictive spot were identified by LC‐MS/MS analysis ( Table 4 ). Here, the confidence for identification of each protein was high, given as the expectation score. The proteins identified included tropomyosin, complement 4A, immunoglobulin G, fibrinogen, and three isoforms of albumin. The location of the seven proteins spots on 2‐DE and the effect of disease on their abundance is shown in Figure 3 . Here, the 2‐DE analysis provided additional information not accessible by shotgun‐based mass spectrometry. For example, the albumin isoforms were distinct isoforms of albumin as indicated by their unique isoelectric points ( Table 4 , Figure 3 ). Moreover, two of the albumin isoforms, represented as spots 505 and 507, were much larger than native albumin, suggesting that they were cross‐linked proteins.
Table 4.
MARS basis functions. Shown are the basis functions (BF) for the MARS model for dengue hemorrhagic fever.
| Bm | Definition | am | Variable descriptor |
|---|---|---|---|
| BF1 | (IL‐10–1.15) + | 5.83E‐03 | IL‐10 |
| BF3 | (20873 ‐ fibrinogen) + | 5.42E‐05 | Fibrinogen |
| BF5 | (437613 ‐ albumin) + | 1.39E‐06 | Albumin*1 |
| BF6 | (C4A–385932) + | −4.90E‐06 | Complement 4A |
| BF8 | (C4A–256959) + | 3.25E‐06 | Complement 4A |
| BF11 | (469259 ‐ albumin) + | 2.48E‐06 | Albumin*2 |
| BF17 | (122218 ‐ TPM4) + | 5.27E‐06 | TPM4 |
| BF19 | (Immunoglobulin gamma–57130) + | −1.35E‐06 | Immunoglobulin gamma‐chain, V region |
| BF23 | (657432 ‐ albumin) + | −9.97E‐07 | Albumin*3 |
Bm = each individual basis function, am = coefficient of the basis function; (y) += max(0,y); *= variable isoforms likely due to posttranslational modification and/or proteolysis.
Figure 3.
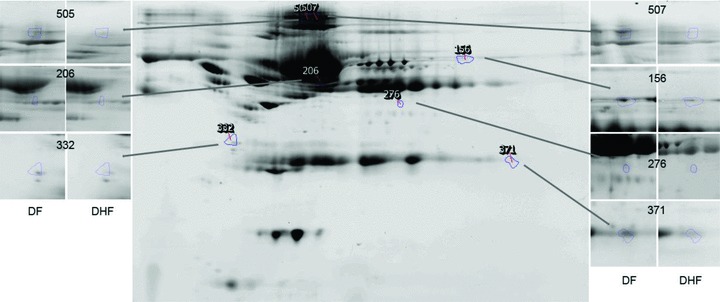
Shown is a reference gel of 2‐DE of BAP fractionated and IgY depleted plasma from the study subjects. The location of protein spots that contribute to the prediction of DHF are indicated. Insets, spot appearances for reference gels for DHF and DF. Spot 156 (C4A), 206 (albumin * 1), 276 (fibrinogen), 332 (tropomyosin), 371 (immunoglobulin gamma‐variable region), 506 (albumin * 2), and 507(albumin * 3).
A comparison of the normalized spot intensities for the seven discriminant proteins were plotted by the outcome of dengue disease ( Figure 4 ). Similar to the cytokine analysis, although the proteins differ by median value, the values were highly overlapping for the two populations, indicating that any singular protein would have poor ability to discriminate between disease types.
Figure 4.
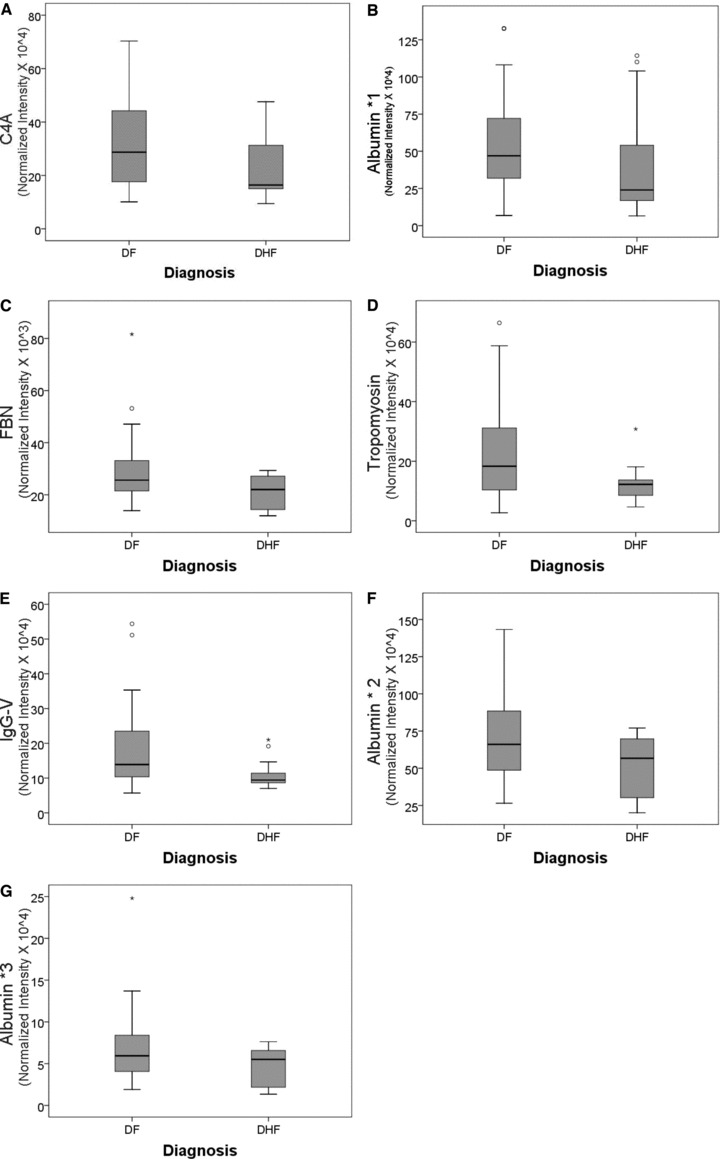
Shown is a box‐plot comparison of 2‐DE spot expression values for C4A (A), Albumin * 1 (B), fibrinogen (FBN, C), tropomyosin (D), IgG‐V (E) and albumin * 2 (F) by diagnosis. DF = dengue fever; DHF = dengue hemorrhagic fever; horizontal bar = median value; shaded box = 25–75 % interquartile range (IQR); error bars = median ± 1.5(IQR); *= outlier.
The optimal MARS model is represented by nine basis functions, whose values are shown in Table 5 . The model is represented by a linear combination of basis functions, where each basis function is a range over which the individual protein's concentration contributes to the classification. Also of note, the basis functions are composed of single features, indicating that interactions between the features do not contribute significantly to the discrimination.
Table 5.
Confusion matrix for MARS classifi er of DHF. For each disease (class), the prediction success of the MARS classifi er is shown.
| Class | Total | Prediction | |
|---|---|---|---|
| DF (n = 38) | DHF (n = 13) | ||
| DF | 38 | 38 | 0 |
| DHF | 13 | 0 | 13 |
| Total | 51 | Correct = 100% | Correct = 100% |
To determine which of these features contribute the most information to the model, variable importance was assessed. Variable importance is a relative indicator (from 0% to 100%) for the contribution of each variable to the overall performance of the model ( Figure 5 ). The variable importance computed for the top three proteins was IL‐10 (100%), with albumin*1 (50%), followed by fibrinogen (40%).
Figure 5.
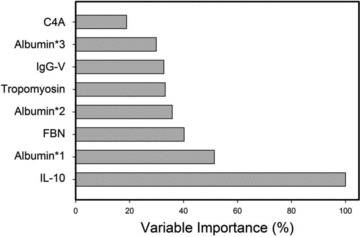
Variable importance was computed for each feature in the MARS model. Y‐axis = percent contribution for each analyte.
Model diagnostics
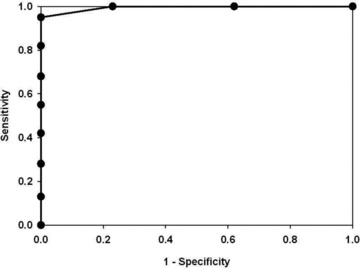
The performance of the MARS predictor of DHF was assessed using several approaches. First, the overall accuracy of the model on the data set was analyzed by minimizing classification error using cross‐validation. The model accuracy produced 100% accuracy for both DHF and DF classification ( Table 6 ). Another evaluation of the model performance is seen by analysis of the area under the receiver operating characteristic (ROC) curve (AUC), where sensitivity versus one‐specificity was plotted. In the ROC analysis, a diagonal line starting at zero indicating that the output was a random guess, whereas an ideal classifier with a high true positive rate and low false positive rate will curve positively and strongly toward the upper left quadrant of the plot. 23 The AUC is equivalent to the probability that two cases, one chosen at random from each group, are correctly ordered by the classifier. 24 In the DHF MARS model, an AUC of >0.999 is seen ( Figure 6 ), indicating it performs as a highly accurate classifier on these samples.
Table 6.
Protein identifi cation of MARS features. Shown are the protein identifi cations for the 2‐DE proteins identifi ed that contribute to the MARS predictive classifi er for DHF.
| No. | Protein name | GI Accession no. | UniProt accession no. | Gel spot no. | pI | MW (Da) | MS ID expectation value |
|---|---|---|---|---|---|---|---|
| 1 | C4A | 239740686 | XP_002343974 | 156 | 8.18 | 71 | 5.00E‐10 |
| 2 | Albumin* | 168988718 | P02768 | 206 | 6.28 | 52 | 2.51E‐57 |
| 3 | Fibrinogen | 237823914 | P02671 | 276 | 7.35 | 40 | 9.98E‐38 |
| 4 | Tropomyosin | 10441386 | AAG17014 | 332 | 5.08 | 29 | 1.58E‐41 |
| 5 | Immunoglobulin gamma V | 567146 | AAA52924 | 371 | 8.81 | 24 | 7.92E‐04 |
| 6 | Albumin* | 168988718 | P02768 | 506 | 6.19 | 263 | 5.00E‐47 |
| 7 | Albumin* | 168988718 | P02768 | 507 | 6.23 | 263 | 6.29E‐32 |
Figure 6.
C4‐2 tumor cell proliferation and effects of digoxin. C4‐2 cell xenograft tumors were excised after 7 days of treatment with digoxin or control. Two hours prior to sacrifice, mice were injected with BrdU labeling reagent and stained for BrdU. Photomicrographs are representative of both control‐ and digoxin‐treated tissue with five mice each.
Post hoc GAM analysis
To confirm that a nonlinear method was the most appropriate modeling approach for these discriminant proteins, the predictive variables were subjected to a GAM analysis. GAMs are data‐driven modeling approaches used to identify nonlinear relationships between predictive features and clinical outcome when there are a large number of independent variables. 25 , 26 Inspection of the residual plots for tropomyosin, complement 4, and albumin isoforms *1–*3 shows nonlinear relationships, indicating that these variables do not satisfy classical assumptions for the use of linear modeling ( Figure 7 ). By contrast, IL‐10 and immunoglobulin gamma approximate a global linear relationship. We interpreted this analysis to indicate that modeling approaches that assume global linear relationships, such as logistic regression, are not generally suited to relate information in proteomics measurements to clinical phenotypes or outcomes.
Figure 7.
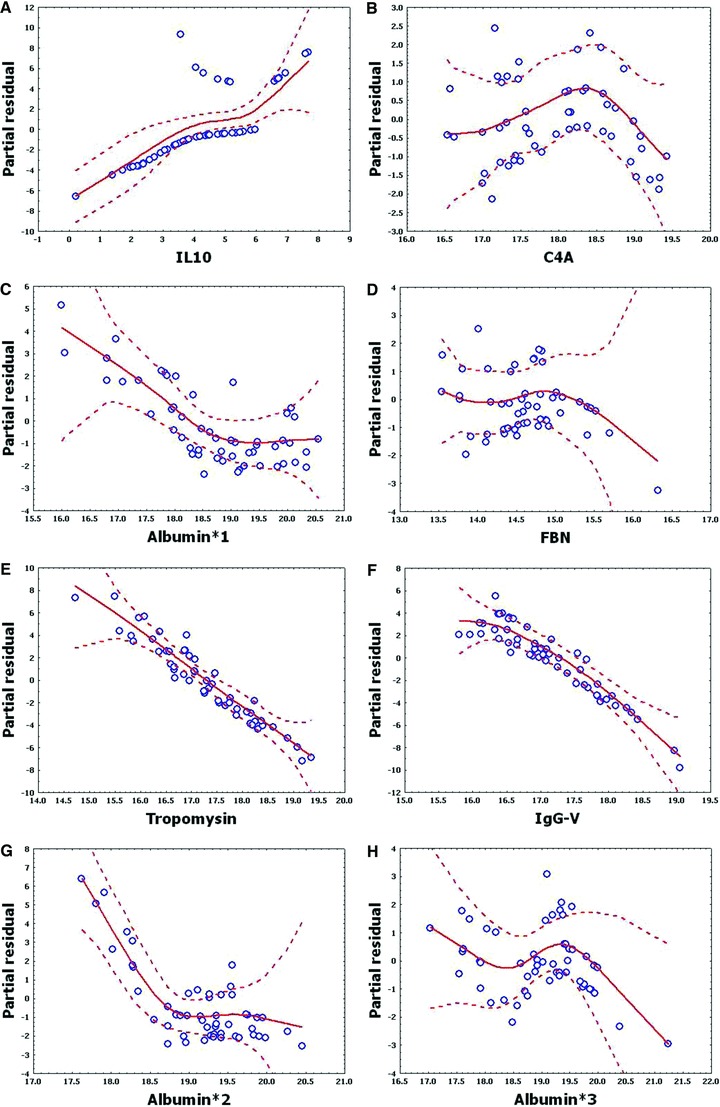
Shown are the partial residual plots for log‐transformed values of eight proteins important in MARS classifi er. Y‐axis, partial residuals of generalized additive model; X‐axis, log of respective feature. Note that regional deviations from classical linear model assumptions are seen.
Discussion
Because previous work has shown that the mortality of DHF is improved with early detection and intensive treatment, 6 the identification of predictive models that aid in early detection of DHF will have an important translational impact into the clinic. In this study, we applied BAP discovery and nonparametric modeling in a prospective study of hyperendemic dengue infections to identify a panel of differentially expressed plasma proteins that associate with the clinical outcome of DHF. Identification of predictive biomarkers in complex biofluids, such as plasma, have been challenging for proteomics technologies. Plasma is a complex biofluid, with its constituent proteins present in a broad dynamic concentration range spanning 12 log orders of magnitude or more. 27 , 28 Moreover, the tendency of high‐abundance proteins to adsorb lower abundance proteins and peptides, 29 , 30 the presence of proteases that may produce peptide fragments, 31 , 32 and the individual variation in plasma protein abundances serve to compound the difficulties in comprehensive proteomic analyses of plasma.
To partially circumvent these difficulties of plasma protein discovery, we developed a hybrid prefractionation 2‐DE mass spectrometry‐based platform coupled with high recovery sample prefractionation. The initial denaturation of the plasma prior to rapid SEC fractionation avoids the pitfall of peptide loss through its binding to high‐abundance plasma carrier proteins. 29 , 30 Moreover, SEC is a nonadsorptive, high recovery prefractionation approach that achieves 95–100% recovery of the input protein. Downstream of SEC, antibody depletion results in significant increase in proteome coverage, enhancing detection of low‐abundance proteins. 33 Finally, our development of a quantitative saturation fluorescence labeling approach results in accurate, quantitative 2D‐E to identify differentially expressed proteins. 12
Another major challenge in biomarker panel development is the combination of discriminant proteins into a robust predictive model. The challenges for model building include reduction of highly correlated features and selection of appropriate statistical models for the underlying data structures. Our analysis of the distribution of normalized and logarithm‐transformed protein concentrations, derived either from quantitative bead‐based ELISA or normalized spot intensities from the saturation fluorescence‐labeled 2‐DE analysis, indicated that the distribution of protein concentrations were highly overlapping ( Figures 1 and 4 ). Consequently, these individual features used alone would not result in robust separation between DF and DHF. Moreover, the protein concentrations were not normally distributed and therefore demand analysis by nonparametric methods. Therefore, we have applied MARS as a robust nonparametric modeling approach for feature reduction and model building. MARS is a nonparametric, multivariate regression method that can estimate complex nonlinear relationships by a series of spline functions of the predictor variables. Regression splines seek to find thresholds and breaks in relationships between variables and are very well suited for identifying changes in the behavior of individuals or processes over time. Some of the advantages of MARS are that it can model predictor variables of many forms, whether continuous or categorical, it can tolerate large numbers of input predictor variables, and is able to model missing values. As a nonparametric approach, MARS does not make any underlying assumptions about the distribution of the predictor variables of interest. This characteristic is extremely important in our DHF modeling because many of the cytokine and protein expression values are not normally distributed, as would be required for the application of classical modeling techniques such as logistic regression. The basic concept behind spline models is to model using potentially discrete linear or nonlinear functions of any analyte over differing intervals. The resulting piecewise curve, referred to as a spline, is represented by basis functions within our model. Other studies have shown that MARS is a superior method in the prediction of nonparametric data sets to phenotypes. 34 One disadvantage of MARS is data overfitting. For this reason, we have chosen to restrict our models to those that incorporate one or fewer interaction terms.
Using a combined BAP‐nonparametric MARS modeling approach, our most accurate model for the prediction of DHF was based on IL‐10, C4A, fibrinogen, trypomoyosin, immunoglobulin G, and several albumin isoforms. The presence of FBN, IgG, and albumin isoforms in 2‐DE despite IgY depletion suggests that these forms represent denatured or posttranslationally modified proteins that do not interact with the depletion antibodies. The exact nature of these modifications will require further investigation. This model was able to accurately predict DHF in 100% of the cases, and evaluation of the sensitivity–specificity relationship by ROC analysis indicated a very good fit of the model to our data. The model diagnostics using GAM further provide support that nonlinear approaches were appropriate to associate disease state with protein expression patterns.
The etiology of DHF is a complex event determined by host–viral interactions. Dengue virus circulates as one of four distinct serotypes; the cocirculation of multiple serotypes is characteristic of hyperendemic transmission. Although serotype 2 was enriched in the subset of patients with DHF in this study, it is important to note that the sample size of DHF is small, and we interpret this to be the result of random selection bias. Previous epidemiological studies have shown that a sequential heterotypic dengue virus infection is an important risk factor for DHF. In adults, almost all cases of DHF occur in secondary heterologous infections, leading to the ADE theory, 35 although host immunological status, including MHC expression are thought to modify the expression of the disease. 36 ADE is thought to increase the mass and tropism of dengue infection, where dengue virus–antibody complexes are taken up by monocytes in an Fc receptor‐dependent manner. As a result, activated monocytes induce a cytokine storm whose effect may result in endothelial dysfunction and vascular leakage. Previous work has shown that soluble mediators, including IL‐2, IL‐4, IL‐6, IL‐10, IL‐13, and IFN‐γ are found in plasma in increased concentrations in patients with severe dengue infections. 19 In a prospective study of a single serotype outbreak in Cuba, IL‐10 was observed to be higher in individuals with secondary dengue infections. 20 We also note that dengue loading into monocytes in vitro resulted in enhanced IL‐6 and IL‐10 production. 37 The identification of IL‐10 in our study, as increased in DHF, is a partial validation of our modeling.
Previous work has shown that immunological responses to viral vaccines, including arthropod born yellow fever are significantly affected by gender. 38 Interestingly, our two‐factor ANOVA is the first observation to our knowledge that shows gender‐specific cytokine response in acute DF infections. This gender effect confounds the statistical analysis of mixed gender population studies. Recognition of this finding will be important to guide the design of subsequent biomarker verification studies.
The analysis of clinical parameters measured upon initial entry into the study showed that the platelet concentration is significantly reduced in subjects with DHF versus DF. Thrombocytopenia is a well‐established feature of DHF, responsible in part for increased tendency for cutaneous hemorrhages. The origin of thrombocytopenia in DHF is thought to be the consequence of both bone marrow depression and accelerated antibody‐mediated platelet sequestration by the liver. 39 Despite its statistical association with DHF, platelet counts do not contribute as strongly to an overall classifier of DHF as do circulating IL‐10, immunoglobulin, and albumin isoforms.
In addition to the tropism of dengue virus for monocytes and dendritic cells, severe dengue infections also involve viral‐induced liver damage. 40 In this regard, increases in liver enzymes (LDH, AST) as well as decreases in albumin concentration have been observed. 41 These phenomena probably represent leakage of hepatocyte cytoplasm and impairment in hepatic synthetic capacity, respectively. In this study, 2‐DE fractionation of plasma proteins provided an additional dimension of information not accessible by clinical assays. For example, the alternative migration of albumin isoforms (albumin *1–*3, Figure 3 ), differing in molecular weight and isoelectric points, would not be detectable by mass spectrometry or by clinical assays. We suspect that these proteins were detected by BAP despite our antibody depletion step because the proteins were in a form not recognized by the antibody. In this regard, albumin is a target for nonenzymatic glycosylation and ischemia‐induced oxidation, which could partially explain its presence on the 2‐DE. The biochemical processes underlying these changes in albumin in dengue infections are presently unknown and will need to be investigated in future studies.
We note that fibrinogen is an important predictor in the MARS model, whose circulating concentration is reduced as a result of DHF ( Figure 6 ). Fibrinogen is a major component of the classical coagulation cascade. In this regard, coagulation defects, similar to mild disseminated intravascular coagulation, are seen in DHF. In fact, isotopic studies indicated a rapid turnover of fibrinogen, 42 thereby explaining its reduction in patients with DHF measured by our analysis. Previous work using a 2D differential fluorescence gel approach comparing individuals with DF versus normal controls, identified reduced fibrinogen γ expression. 43 However, from the design of this study, comparing DHF versus controls, the use of fibrinogen to differentiate DF from DHF could not be assessed.
Activation of the complement cascade is thought to mediate the process of capillary leakage in DHF by inducing direct endothelial damage. 44 Antibody‐antigen complexes are initiators of the classical complement cascade by binding and activating the C1q protein. Subsequently, C4 is cleaved by the activated convertase, whose product becomes part of the activated C3 convertase (C4AC2) to produce a membrane attack complex. In this regard, other studies have found that the dengue nonstructural protein (NS)‐1 activates the complement cascade, and NS‐1 levels are associated with DHF. 45 Our findings of decreased complement C4A association with DHF would be consistent with a mechanism of NS1‐mediated complement consumption and endothelial leakage.
Finally, we were surprised by the detection of plasma tropomyosin in our study. Tropomyosin is a cytoskeletal actin binding protein, typically associated with muscle regeneration and cardiac contractility. Previous high‐resolution plasma proteome analysis using LC prefractionation and immunoaffinity depletion and 2‐DE also identified tropomyosin among 372 unique proteins in normal human plasma. 46 The mechanisms how circulating tropomyosin is affected by DHF are unknown to us.
In summary, we focused on modeling discriminant proteins that differentiate between individuals with DF versus those with DHF. Using nonparametric methods for developing predictive classifiers using a high‐resolution focused and discovery‐based approach we have identified a highly accurate classifier of DHF based on IL‐10, fibrinogen, C4A, immunoglobulin G, tropomyosin, and three isoforms of albumin. Most of these proteins can be linked to the biological processes underlying that of DHF, including cytokine storm, capillary leakage, hepatic injury, and antibody consumption, suggesting that these predictors may have biological relevance. More work will be required to verify this model and analyze the biological pathways affected in severe dengue virus infections.
Sources of Funding
Research funding support was provided by the NIAID Clinical Proteomics Center, HHSN272200800048C (ARB), NHLBI Proteomics Center, HHSN268201000037C (A Kurosky, UTMB, PI) and 1U54RR029876 UTMB CTSA (ARB), and the Military Infectious Diseases Research Program work unit 6000RAD1.S.B0302.
Ethical Approval
This study was conducted under a human subjects study protocol number NMRCD.2005.0007 (Active Dengue Surveillance and Predictors of Disease Severity in Maracay, Venezuela) approved by the Centro de Investigaciones Biomedicas de la Universidad de Carabobo (BIOMED‐UC), Maracay, Venezuela, and the Naval Medical Research Center institutional review boards in compliance with all applicable federal regulations governing the protection of human subjects.
Disclosure
The views expressed in this paper are those of the author and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, or the US Government.
Josefina Garcia, Eric S. Halsey, Patrick J. Blair, Claudio Rocha, Isabel Bazan, and Tadeusz J. Kochel are military service members or employees of the US Government. This work was prepared as part of their official duties. Title 17 U.S.C. §105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a US Government work as a work prepared by a military service member or employee of the US Government as part of that person's official duties.
Conflict of Interest
None.
Acknowledgments
The authors thank Leny Curico Manihuari, Juan Flores Michi, Nora Mar ín Romero, Nadia Tereshkova, Montes Criollo, Johnni Mozombite Flores, Lucy Navarro Sánchez, Magaly Ochoa Isuiza, Geraldine Ocmín Galán, Iris Reátegui Carrión, Zoila Martha Reategui Chota, Rubiela Nerza Rubio Briceño, Ysabel Ruiz Berger, Zenith Tamani Guerrero, Clara Chávez López, Junnelhy Mireya Flores López, Xiomara Mafaldo García, Sandra Ivonne Muñoz Perez, Myriam Ojaicuro Pashanaste, Zenith María Pezo Villacorta, Liliana Rios López, Rosana Magaly Sotero Jiménez, Sarita Del Pilar Tuesta Dávila, Joel Cahuachi Tuesta, Moises Tanchiva Tuanama, Stalin Fran Vilcarromero Llaja, Diana Maritza Bazan Ferrando, Alex Jaime Vasquez Valderrama, Gabriela Vasquez La Torre, Leslye Angulo Melendez, Patricia del Carmen Barrera Bardales, Guadalupe Flores Ancajima, Zaira Hellen Villa Galarce, Rebeca Salome Carrion Torres, Regina Rosa Fernandez Montano, C Guevara, CE Vidal Oré, and C Manrique del Lara Estrada (DISA) for technical support.
Other members of the Venezuelan Dengue Fever Working Group are: Gloria Sierra, M.D., Iris Villalobos, M.D., M.P.H., and Carlos Espino, Ph.D.
References
- 1. Martina BEE, Koraka P, Osterhaus ADME. Dengue virus pathogenesis: an integrated view. Clin Microbiol Rev. 2009;22(4): 564–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Martin JL San, Brathwaite O, Zambrano B, Solorzano JO, Bouckenooghe A, Dayan GH, Guzman MG. The epidemiology of dengue in the Americas over the last three decades: a worrisome reality. Am J Trop Med Hyg. 2010;82(1): 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Guzman MG, Kouri G, Bravo J, Valdes L, Vazquez S, Halstead SB. Effect of age on outcome of secondary dengue 2 infections. Int J Infect Dis. 2002;6(2): 118–124. [DOI] [PubMed] [Google Scholar]
- 4. Graham RR, Juffrie M, Tan R, Hayes CG, Laksono I, Ma‘roef C, Erlin S, Porter KR, Halstead SB. A prospective seroepidemiologic study on dengue in children four to nine years of age in Yogyakarta, Indonesia I. Studies in 1995–1996. Am J Trop Med Hyg. 1999;61(3): 412–419. [DOI] [PubMed] [Google Scholar]
- 5. Thomas L, Verlaeten O, Cabie A, Kaidomar S, Moravie V, Martial J, Najioullah F, Plumelle Y, Fonteau C, Dussart P, Cesaire R. Influence of the dengue serotype, previous dengue infection, and plasma viral load on clinical presentation and outcome during a dengue‐2 and dengue‐4 co‐epidemic. Am J Trop Med Hyg. 2008; 78(6): 990–998. [PubMed] [Google Scholar]
- 6. Ranjit S, Kissoon N, Jayakumar I. Aggressive management of dengue shock syndrome may decrease mortality rate: a suggested protocol *. Pediatric Critical Care Medicine 2005; 6(4): 412–419. [DOI] [PubMed] [Google Scholar]
- 7. Zhao Y, Brasier AR. Methods for biomarker verification and assay development. Current Proteomics. 2011; 8: 138–152. [Google Scholar]
- 8. Forshey BM, Guevara C, Laguna‐Torres VA, Cespedes M, Vargas J, Gianella A, Vallejo E, Madrid C, Aguayo N, et al. Arboviral etiologies of acute febrile illnesses in Western South America, 2000–2007. PLoS Negl Trop Dis. 2010; 4(8): e787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dengue haemorrhagic fever: diagnosis, treatment, prevention and control. Geneva : World Health Organization; 1997: 12–23. [Google Scholar]
- 10. Lanciotti RS, Calisher CH, Gubler DJ, Chang GJ, Vorndam AV. Rapid detection and typing of dengue viruses from clinical samples by using reverse transcriptase‐polymerase chain reaction. J Clin Microbiol. 1992; 30(3): 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jamaluddin M, Wiktorowicz JE, Soman KV, Boldogh I, Forbus J, Spratt H, Garofalo RP, Brasier AR. Role of peroxiredoxin‐1 and −4 in protection of RSV‐induced cysteinyl‐oxidation of nuclear cytoskeletal proteins. J Virol. 2010; 84: 9533–9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pretzer EP, Wiktorowicz JE. Saturation fluorescence labeling of proteins for proteomic analyses. Anal Biochem. 2008; 374: 250–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Turck CW, Falick AM, Kowalek JA, Lane WS, Lilley KS, Phinney BS, Weintraub ST, Wikowska HE, Yates NA. ABRF‐PRG06: Relative Protein Quantification. Long Beach , CA : Association of Biomolecular Resource Facilities; 2006. [DOI] [PubMed] [Google Scholar]
- 14. Miseta A, Csutora P. Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol Biol Evol. 2000; 17: 1232–1239. [DOI] [PubMed] [Google Scholar]
- 15. Dowsey AW, Morris JS, Gutstein HB, Yang GZ. Informatics and statistics for analyzing 2‐D gel electrophoresis images. Methods Mol Biol. 2010; 604: 239–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Karp NA, Feret R, Rubtsov DV, Lilley KS. Comparison of DIGE and post‐stained gel electrophoresis with both traditional and SameSpots analysis for quantitative proteomics. Proteomics. 2008; 8(5): 948–960. [DOI] [PubMed] [Google Scholar]
- 17. Zhang W, Chait BT. ProFound: an expert system for protein identification using mass spectrometric peptide mapping information. Anal Chem. 2000; 72: 2482–2489. [DOI] [PubMed] [Google Scholar]
- 18. Friedman JH. Multivariate adaptive regression splines. Annal Statist. 1991; 19(1): 1–67. [DOI] [PubMed] [Google Scholar]
- 19. Bozza F, Cruz O, Zagne S, Azeredo EL, Nogueira RM, Assis EF, Bozza PT, Kubelka CF. Multiplex cytokine profile from dengue patients: MIP‐1beta and IFN‐gamma as predictive factors for severity. BMC Infect Dis. 2008; 8(1): 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perez AB, Garcia G, Sierra B, Alvarez M, Vazquez S, Cabrera MV, Rodriguez R, Rosario D, Martinez E, Denny T, Guzman MG. IL‐10 levels in dengue patients: some findings from the exceptional epidemiological conditions in Cuba. J Med Virol. 2004; 73(2): 230–234. [DOI] [PubMed] [Google Scholar]
- 21. Tyagarajan K, Pretzer EP, Wiktorowicz JE. Thiol‐reactive dyes for fluorescence labeling of proteomic samples. Electrophoresis. 2003; 24: 2348–2358. [DOI] [PubMed] [Google Scholar]
- 22. Cook NR, Zee RYL, Ridker PM. Tree and spline based association of gene‐gene interaction models for ischemic stroke. Statist Med. 2005; 23: 1439–1453. [DOI] [PubMed] [Google Scholar]
- 23. Fawcett T. An introduction to ROC analysis. Pattern Recog Lett. 2006; 27: 861–874. [Google Scholar]
- 24. Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic curve. Radiology. 1982; 143: 29–36. [DOI] [PubMed] [Google Scholar]
- 25. Austin PC. A comparison of regression trees, logistic regression, generalized additive models, and multivariate adaptive regression splines for predicting AMI mortality. Stat Med. 2007; 26(15): 2937–2957. [DOI] [PubMed] [Google Scholar]
- 26. Hastie T, Tibshirani R. Generalized additive models for medical research. Stat Methods Med Res. 1995; 4(3): 187–196. [DOI] [PubMed] [Google Scholar]
- 27. Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002; 1(11): 845–867. [DOI] [PubMed] [Google Scholar]
- 28. Rifai N, Gerszten RE. Biomarker discovery and validation. Clin Chem. 2006; 52(9): 1635–1637. [DOI] [PubMed] [Google Scholar]
- 29. Gundry RL, Fu Q, Jelinek CA, Van Eyk JE, Cotter RJ. Investigation of an albumin‐enriched fraction of human serum and its albuminome. Proteomics Clin Appl. 2007; 1(1): 73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seferovic MD, Krughkov V, Pinto D, Han VK, Gupta MB. Quantitative 2‐D gel electrophoresis‐based expression proteomics of albumin and IgG immunodepleted plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2008; 865(1–2): 147–152. [DOI] [PubMed] [Google Scholar]
- 31. Villanueva J, Philip J, Chaparro CA, Li Y, Toledo‐Crow R, DeNoyer L, Fleisher M, Robbins RJ, Tempst P. Correcting common errors in identifying cancer‐specific serum peptide signatures. J Proteome Res. 2005; 4(4): 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Villanueva J, Nazarian A, Lawlor K, Yi SS, Robbins RJ, Tempst P. A sequence‐specific exopeptidase activity test (SSEAT) for “functional” biomarker discovery. Mol Cell Proteomics. 2008; 7(3): 509–518. [DOI] [PubMed] [Google Scholar]
- 33. Tu C, Rudnick PA, Martinez MY, Cheek K, Stein SE, Slebos RJC, Liebler D. Depletion of abundant plasma proteins and limitations of plasma proteomics. J Proteome Res. 2010; 9(10): 4982–4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brasier AR, Victor S, Ju H, Busse WW, Curran‐Everett D, Bleecker ER, Castro M, Chung KF, Gaston B, Israel E, et al. Predicting intermediate phenotypes in asthma using bronchoalveolar lavage‐derived cytokines. Clin Transl Sci. 2010; 13: 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Endy TP, Nisalak A, Chunsuttitwat S, Vaughn DW, Green S, Ennis FA, Rothman AL, Libraty DH. Relationship of preexisting dengue virus (DV) neutralizing antibody levels to viremia and severity of disease in a prospective cohort study of DV infection in Thailand. J Infect Dis. 2004; 189: 990–1000. [DOI] [PubMed] [Google Scholar]
- 36. Green S, Rothman A. Immunopathological mechanisms in dengue and dengue hemorrhagic fever. Curr Opin Infect Dis. 2006; 19(5): 429–436. [DOI] [PubMed] [Google Scholar]
- 37. Chareonsirisuthigul T, Kalayanarooj S, Ubol S. Dengue virus (DENV) antibody‐dependent enhancement of infection upregulates the production of anti‐inflammatory cytokines, but suppresses anti‐DENV free radical and pro‐inflammatory cytokine production, in THP‐1 cells. J Gen Virol. 2007; 88(2): 365–375. [DOI] [PubMed] [Google Scholar]
- 38. Klein SL, Jedlicka A, Pekosz A. The Xs and Y of immune responses to viral vaccines. Lancet Infect Dis. 2010; 10(5): 338–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mitrakul C, Poshyachinda M, Futrakul P, Sangkawibha N, Ahandrik S. Hemostatic and platelet kinetic studies in dengue hemorrhagic fever. Am J Trop Med Hyg. 1977; 26: 975–984. [DOI] [PubMed] [Google Scholar]
- 40. Seneviratne SL, Malavige GN, de Silva HJ. Pathogenesis of liver involvement during dengue viral infections. Trans R Soc Trop Med Hyg. 2006; 100(7): 608–614. [DOI] [PubMed] [Google Scholar]
- 41. Villar‐Centeno LA, Diaz‐Quijano FA, Martinez‐Vega RA. Biochemical alterations as markers of dengue hemorrhagic fever. Am J Trop Med Hyg. 2008; 78(3): 370–374. [PubMed] [Google Scholar]
- 42. Srichaikul T, Nimmanitaya S, Artchararit N, Siriasawakul T, Sungpeuk P. Fibrinogen metabolism and disseminated intravascular coagulation in dengue hemorrhagic fever. Am J Trop Med Hyg. 1977; 26: 525–532. [DOI] [PubMed] [Google Scholar]
- 43. Albuquerque LM, Trugilho MRO, Chapeaurouge A, Jurgilas P, Bozza PT, Bozza FA, Perales J, Neves‐Ferreira AGC. Two‐dimensional difference gel electrophoresis (DiGE) analysis of plasmas from dengue fever patients. J Proteome Res. 2009; 8(12): 5431–5441. [DOI] [PubMed] [Google Scholar]
- 44. Avirutnan P, Punyadee N, Noisakran S, Komoltri C, Thiemmeca S, Auethavornanan K, Jairungsri A, Kanlaya R, Tangthawornchaikul N, Puttikhunt C, et al. Vascular leakage in severe dengue virus infections: a potential role for the nonstructural viral protein NS1 and complement. J Infect Dis. 2006; 193(8): 1078–1088. [DOI] [PubMed] [Google Scholar]
- 45. Thayan R, Huat TL, See LL, Tan CP, Khairullah NS, Yusof R, Devi S. The use of two‐dimension electrophoresis to identify serum biomarkers from patients with dengue haemorrhagic fever. Trans R Soc Trop Med Hyg. 2009; 103(4): 413–419. [DOI] [PubMed] [Google Scholar]
- 46. Pieper R, Gatlin CL, Makusky AJ, Russo PS, Schatz C, Miller SS, Su Q, McGrath A, Estock MA, Parmar P, et al. The human serum proteome: display of nearly 3700 chromatographically separated protein spots on two‐dimensional electrophoresis gels and identification of 325 distinct proteins. Proteomics. 2003; 3(7): 1345–1364. [DOI] [PubMed] [Google Scholar]