

SUMMARY
The pentaspan membrane glycoprotein CD133 marks lineage-specific cancer progenitor cells and is associated with poor prognosis in a number of tumor types. Despite its utility as a cancer progenitor cell marker, CD133 protein regulation and molecular function remain poorly understood. We find that the deacetylase HDAC6 physically associates with CD133 to negatively regulate CD133 trafficking down the endosomal-lysosomal pathway for degradation. We further demonstrate that CD133, HDAC6, and the central molecule of the canonical Wnt signaling pathway, β-catenin, can physically associate as a ternary complex. This association stabilizes β-catenin via HDAC6 deacetylase activity, which leads to activation of β-catenin signaling targets. Downregulation of either CD133 or HDAC6 results in increased β-catenin acetylation and degradation, which correlates with decreased proliferation in vitro and tumor xenograft growth in vivo. Given that CD133 marks progenitor cells in a wide range of cancers, targeting CD133 may be a means to treat multiple cancer types.
INTRODUCTION
The CD133 (prominin-1) epitope AC133 has been associated with cancer progenitor cells in brain, colon, pancreas, liver, lung, skin, prostate, and ovarian cancers (Ferrandina et al., 2009). AC133 was first discovered as a marker of CD34+ human hematopoietic progenitors (Yin et al., 1997). Characterization of AC133 found it to be specific to CD133, a 120 kDa pentaspan transmembrane protein (Miraglia et al., 1997), which has eight predicted N-glycosylation sites on its two extracellular loops (Yin et al., 1997). CD133 is primarily localized to plasma membrane protrusions (Corbeil et al., 2000; Weigmann et al., 1997), where it can bind to cholesterol and associate with membrane microdomains (Röper et al., 2000). Importantly, cancer cells marked by AC133 are thought to be more resistant to traditional cancer treatments, including some chemotherapies and radiotherapy, and this may explain some instances of cancer relapse (Ferrandina et al., 2009).
Insights into CD133 function come from the discovery of a single-nucleotide frameshift mutation in the CD133 gene, which leads to autosomal-recessive retinal degenerative disease (Maw et al., 2000). Consistent with these observations, the CD133 knockout mouse displays retinal degeneration leading to blindness (Zacchigna et al., 2009). Together, these observations establish a physiological role for CD133 in proper retinal development; however, it does not address its functional significance in cancer.
This study presents evidence for physical interactions between CD133, the deacetylase HDAC6, and β-catenin, the central regulator of canonical Wnt signaling. We demonstrate that HDAC6 governs CD133 trafficking into the endosomal-lysosomal degradation pathway. Moreover, decreasing CD133 levels or HDAC6 activity correlates with expression of mesenchymal-to-epithelial differentiation markers in colon and ovarian cancer cell lines. Our results show that CD133 is important for cell proliferation and anchorage-independent growth in vitro and tumor xenograft growth in vivo. Finally, we present a model whereby CD133, HDAC6, and β-catenin form a ternary complex to contextually stabilize β-catenin through HDAC6 deacetylase activity.
RESULTS
CD133 Physically Associates with HDAC6
To identify a function for CD133, we employed the mammalian affinity purification and lentiviral expression (MAPLE) system coupled to liquid chromatography and tandem mass spectrometry (LC/MS) (Mak et al., 2010) to uncover CD133-associated protein interaction partners. We previously generated a human embryonic kidney (HEK) 293 cell line that stably expresses CD133 with a C-terminal versatile affinity (VA) tag (hereafter referred to as HEK293/CD133-VA cells) (Mak et al., 2011). HEK293/CD133-VA lysates were subjected to a single round of Flag purification, and the purified proteins were subsequently analyzed by LC/MS. After using strict selection criteria to determine protein hits with high confidence (see Experimental Procedures), HDAC6 emerged as the only candidate interaction partner for CD133 (Table S1). Unlike other histone deacetylases, HDAC6 is primarily cytoplasmic, associated with microtubules and dynein motors, and directly regulates the acetylation state of α-tubulin at lysine 40 through its deacetylase activity (Hubbert et al., 2002; Zhang et al., 2003, 2006).
The CD133-HDAC6 physical interaction was confirmed by coimmunoprecipitation (co-IP) followed by western blotting in HEK293 cells (data not shown) and Caco-2 cells (Figure 1A) engineered to express VA-tagged CD133. To further validate the interaction between CD133 and HDAC6, we performed endogenous co-IPs with cell lysates from unmodified Caco-2 cells and found that HDAC6 could specifically be pulled down with immunoprecipitated CD133 protein (Figure S1A). To map the residues in CD133 that are important for its interaction with HDAC6, we created CD133 truncation mutants and generated a series of Caco-2 cell lines stably expressing these constructs. We found that a truncation mutant that contained the first intracellular region of CD133 (CD133(1–445)) maintained binding to HDAC6, whereas a truncation mutant lacking the part of the first intracellular region of CD133 (CD133(1–132)) had significantly reduced binding to HDAC6 (Figure 1B). Therefore, we suggest that the first intracellular region of CD133 is required for its interaction with HDAC6.
Figure 1. CD133 Interacts with HDAC6.

(A) Whole-cell lysates (input) and IPs of VA-tagged CD133 or GFP from engineered Caco-2 cells followed by anti-Flag and anti-HDAC6 western blots.
(B) Whole-cell extracts (input) from Caco-2 cell lines stably expressing the CD133-VA truncations 1–132 AA or 1–445 AA were used in anti-Flag IPs followed by western analysis using anti-Flag and anti-HDAC6 antibodies.
(C) Lysates from Caco-2 cell lines stably expressing either VA-HDAC6-WT, VA-HDAC6-DD, VA-HDAC truncations (1–503, 1–840, ΔBUZ), or VA-GFP were used in anti-Flag IPs followed by western analyses using anti-Flag and anti-AC133 antibodies.
(D) Lysates from Caco-2 cells (untransduced) or Caco-2 cell lines stably expressing wild-type HDAC6 or a specific HDAC6 point mutant (S568E, S568A, Y570E, or Y570F) were used in anti-Flag IPs and analyzed by western blot for interaction with endogenous CD133 using anti-AC133 antibodies.
(E) MFα-CD133-C-T or the control MFα-CD4(TM)-C-T were expressed in the yeast strain THY.AP4 along with NubG-HDAC6 and assayed for growth on selective media. MFα-CD133-C-T was also expressed in THY.AP4 along with positive controls Ost1-NubI and Fur4-NubI and the negative controls Ost1-NubG and Fur4-NubG to demonstrate absence of self-activation and proper membrane integration.
See also Figure S1.
As additional evidence of the CD133-HDAC6 interaction in Caco-2 cells, we confirmed that endogenous HDAC6 protein could specifically immunoprecipitate CD133 from Caco-2 cell lysates (Figure S1A). To map the HDAC6 region that interacted with CD133, we generated a series of HDAC6 mutants and stably introduced them into Caco-2 cells by using the MAPLE system described above. A wild-type, deacetylase-deficient (DD), and a BUZ-domain-deficient HDAC6 interacted with endogenous CD133, as well as a truncated form of HDAC6 containing both catalytic domains (1–840 amino acids [AA]) (Figure 1C, lanes 2–5). In contrast, a truncation mutant of HDAC6 that only contained the first catalytic domain of HDAC6 (1–503 AA) had reduced binding to endogenous CD133 (Figure 1C, lane 6). These findings suggest that the second HDAC6 catalytic domain (504–840) is required to bind CD133 in its first intracellular loop.
Interestingly, HDAC6 protein regulation occurs through phosphorylation of S568 and/or Y570, which leads to inhibition of the deacetylase activity of HDAC6 (Deribe et al., 2009). Given that S568 and Y570 reside within the second HDAC6 catalytic domain, we hypothesized that CD133 binds this region of HDAC6 to competitively inhibit HDAC6 phosphorylation and, hence, deactivation. Indeed, phospho-mimicking HDAC6 mutants S568E or Y570E were unable to immunoprecipitate endogenous CD133 in Caco-2 cells, whereas the wild-type HDAC6 or phospho-null HDAC6 mutants S568A and Y570F could (Figure 1D).
To further validate the CD133-HDAC6 interaction by using an independent approach, we used the membrane yeast two-hybrid (MYTH) assay (Gisler et al., 2008; Snider et al., 2010). In the event of a protein-protein interaction, the MYTH assay relies on the cleavage of a reconstituted split ubiquitin moiety by an endogenous yeast protease, resulting in the release of the LexA transcription factor (TF) for ADE2/HIS3/β-galactosidase reporter activation (Gisler et al., 2008). Expression of CD133 in the MYTH system, where the signal sequence of the α-mating pheromone precursor (MFα) is fused to the C-terminal ubiquitin (Cub) fragment, which is in turn fused to the LexA(VP-16) transcription factor (MFα-CD133-C-T), was confirmed by western blot analyses in the MYTH reporter yeast strain THY.AP4 (Figure S1B). MFα-CD133-C-T was properly integrated into yeast membrane compartments as determined by N-terminal ubiquitin (Nub) I13G/NubI controls (Gisler et al., 2008) (Figures 1E and S1C). Expression of the MFα-CD133-C-T bait and the NubGf-used HDAC6 prey resulted in activation of the MYTH reporter in THY.AP4 cells (Figure 1E). CD133-C-T interacted specifically with HDAC6 as it did not interact with another histone deacetylase, HDAC10 (Figure S1C). Furthermore, two negative control baits, MFα-CD4 transmembrane (TM)-C-T or MFα-SLC22A1-C-T, did not activate the MYTH reporter when expressed with NubG-HDAC6 (Figures 1E and S1D). Overall, our data indicate that CD133 and HDAC6 can physically associate by using multiple protein interaction assays.
HDAC6 Negatively Regulates CD133 Trafficking into the Endosomal-Lysosomal Pathway
Based on the recent reports demonstrating that HDAC6 regulates EGFR stability (Deribe et al., 2009; Gao et al., 2010), we hypothesized that HDAC6 may also regulate the stability of CD133. To examine this idea, we stably knocked down HDAC6 by using lentiviral-based short hairpin RNA (shRNA) (Moffat et al., 2006) in cell lines that endogenously express CD133 (Figure 2A). HDAC6 knockdown resulted in a dramatic reduction of CD133 protein in the colon adenocarcinoma cell lines Caco-2 and HT-29, a retinoblastoma cell line Weri-Rb-1, an ovarian serous adenocarcinoma cell line OVCAR-8 (Figures 2A and S2A), and an acute lymphoblastic leukemia cell line SEM-K2 (Figure S4A).
Figure 2. HDAC6 Negatively Regulates CD133 Trafficking Endosomes via Its Deacetylase Activity.

(A) Caco-2, HT-29, Weri-Rb-1, and OVCAR-8 cancer cell lines were transduced with two independent shRNAs targeting HDAC6 or a control shRNA targeting LacZ, and target knockdown was determined by western blot analyses using anti-HDAC6 antibodies. CD133, acetylated tubulin, and tubulin levels were all monitored by western blot.
(B) Quantitative RT-PCR for HDAC6 and CD133 mRNA expression following knockdown of HDAC6 or CD133 using two independent shRNAs targeting each transcript. Error bars represent SD (n = 3).
(C) Cancer cell lines described in (A) and (B) were treated with either vehicle only (DMSO), the HDAC6-specific inhibitor tubacin, or its inactive counterpart niltubacin at 100 μM for Caco-2 and 20 μM for the remaining cell lines for a duration of 24 hr. Western blot analyses were performed as described in (A).
(D) MFα-CD133-C-T or the control MFα-CD4(TM)-C-T were expressed in the yeast strain THY.AP4 along with NubG-CD63 and assayed for growth on selective media.
(E) AC133 and CD63 immunofluorescence of Caco-2 cells treated with tubacin or niltubacin, as described above, were imaged by confocal microscopy. Nuclei were stained with Hoechst 33342. Scale bar, 50 μm.
(F) Number of cells with a CD133-CD63 colocalization pattern as seen in (E) were counted by eye from a minimum of 200 cells treated with either tubacin or niltubacin. Error bars represent SD (n = 3). *p < 0.05 as determined by a Student’s t test.
See also Figures S2 and S3.
Given that HDAC6 was first identified to deacetylate α-tubulin, we monitored acetylated α-tubulin levels to demonstrate that our HDAC6 knockdown was sufficient to affect its functional impact. As expected, the decrease in HDAC6 transcript levels by knockdown with two independent shRNAs correlated with an increase in acetylated α-tubulin (Figure 2A). By contrast, no change in HDAC6 protein or transcript levels was observed with CD133 knockdown by using two independent shRNAs targeting CD133 (Figures 2B and 6C). Given that HDAC6 can participate in regulating gene transcription, we investigated the impact of HDAC6 knockdown on CD133 transcript levels. We did not observe any significant differences in CD133 transcript in the presence of HDAC6 knockdown (Figure 2B), suggesting that HDAC6 exerts its regulatory activity on CD133 at the protein level.
Figure 6. CD133 Stabilizes β-Catenin Expression.

(A) Endogenous CD133 and β-catenin and control IgG were immunoprecipitated from Caco-2 lysates and analyzed by western blot for associated proteins, including AC133, HDAC6, and β-catenin.
(B) MFα-CD133-C-T or the control MFα-CD4(TM)-C-T were expressed in the yeast strain THY.AP4 along with NubG-β-catenin, and either HDAC6 or empty vector and yeast transformants were assayed for growth on selective media. MFα-CD133-C-T and either HDAC6 or empty vector were also expressed in THY.AP4 along with positive controls Ost1-NubI and Fur4-NubI and the negative controls Ost1-NubG and Fur4-NubG to demonstrate absence of self-activation and proper membrane integration.
(C) Caco-2 and OVCAR-8 cells infected with shRNAs targeting CD133 or HDAC6 and a panel of signaling events were analyzed by western blot, including phospho-β-catenin (S33/S37/T41), acetylated β-catenin (K49), phospho-GSK3β, and acetylated α-tubulin, as well as CD133, HDAC6, β-catenin, GSK3β, and α-tubulin.
See also Figure S6.
To investigate whether the deacetylase activity of HDAC6 was responsible for the downregulation of CD133 and to provide chemical genetic evidence for our observations, we used the small molecule trichostatin A (TSA), which is an inhibitor of class I and class II HDACs, including HDAC6. Consistent with the HDAC6 knockdown data described above, we observed a noticeable decrease in CD133 protein levels by western blot analysis (Figure S2B) but no significant difference in CD133 transcript (Figure S2C; p > 0.05) following TSA treatment. To demonstrate that the effects of TSA were specifically due to its inhibition of HDAC6, we used the HDAC6-specific small-molecule inhibitor tubacin, as well as its inactive counterpart niltubacin as a control (Haggarty et al., 2003). Treatment of Caco-2, HT-29, Weri-Rb-1, OVCAR-8, or SEM-K2 cells with tubacin resulted in a decrease in CD133 (Figures 2C and S4B), but not HDAC6, protein levels, compared to treatment with niltubacin or vehicle alone (Figure 2C). Similar to shRNA-mediated knockdown of HDAC6, tubacin treatment did not have a statistically significant (p > 0.05) effect on CD133 transcript levels relative to cells treated with niltubacin (Figure S2D).
In contrast to inhibition of HDAC6 activity, overexpression of wild-type HDAC6 in Caco-2 cells by the MAPLE system (Figure S2E) or into HT-29 cells by transient transfection (Figure S2F) led to a slight increase in endogenous CD133 levels, whereas overexpression of HDAC6-DD was comparable to expression of green fluorescent protein (GFP). Together, our results are consistent with a model whereby HDAC6 deacetylase activity is required for the stabilization of CD133 protein in cell lines from multiple cancer types.
To further support our RNA interference (RNAi) knockdown and small-molecule inhibition of HDAC6, we observed a destabilization of total CD133 protein, but not transcript, levels in Caco-2 cells when the lysine residue positioned at amino acid 129 in the first intracellular loop of CD133 was mutated to an alanine (Figures S2G and S2H). Given our model and supporting data that HDAC6 binds to the first intracellular loop of CD133 and that its deacetylase activity is required for CD133 stability, we suggest that the K129A mutation may block the interaction between CD133 and HDAC6, leading to CD133 degradation. Notably, we were only able to detect lysine acetylation on CD133 by mass spectrometry on lysines predicted to reside extracellularly (A.B.M., M. Pehar, L. Puglielli, and J.M., unpublished data).
In an effort to elucidate the mechanism of CD133 turnover, we treated Caco-2 cells with the proteasome inhibitor MG132 but observed no significant changes in CD133 protein expression levels (Figure S3A). Moreover, inhibition of proteasome activity by MG132 treatment did not rescue expression of CD133 in Caco-2 cells depleted of HDAC6 activity using shRNAs targeting HDAC6 or the small-molecule inhibitor tubacin (Figures S3B and S3C). These results suggest that CD133 downregulation is not through proteasomal degradation.
In parallel to our MS studies to identify putative CD133-interacting proteins, we also performed a CD133 MYTH screen against a complementary DNA (cDNA) library isolated from adult human kidney to identify CD133 interaction partners (Table S2). Notably, the endosomal- and lysosomal-associated marker CD63 was found to interact with CD133 (Figure 2D). Given that CD63 is an endosomal marker, we hypothesized that CD133 degradation may occur through an endocytic cycle. Removal of HDAC6 activity resulted in loss of CD133 expression at the plasma membrane and a strong colocalization with CD63 in Caco-2 cells (Figures 2E, 2F, and S3D). Moreover, we observed that CD133 protein levels persist in the presence of the endosomal-lysosomal pathway inhibitor ammonium chloride following inhibition of HDAC6 activity by shRNA-mediated knockdown (Figure S3B) or the small-molecule inhibitor tubacin (Figure S3C). To further investigate whether trafficking of CD133 into the endosomal-lysosomal pathway required CD63, we knocked down CD63 with two independent shRNAs and observed no change in CD133 stability (Figures S3F and S3G). Furthermore, CD63 knockdowns did not impair the downregulation of CD133 when Caco-2 cells were treated with tubacin (data not shown). Together, these observations are consistent with the model that HDAC6 regulates CD133 stability through the endosomal-lysosomal pathway.
CD133 Potentiates Cancer Cell Proliferation and Tumorigenesis
In contrast to a previous report showing that siRNA-mediated knockdown of CD133 elicited no effect on the proliferation of Caco-2 cells (Horst et al., 2009), we found that knocking down CD133 by shRNAs in multiple cell lines, including Caco-2 and OVCAR-8 cells, significantly reduced proliferation (Figure 3A; p < 0.05). Notably, knockdown of HDAC6 also significantly reduced proliferation of Caco-2 and OVCAR-8 cells (Figure 3A; p < 0.05). The reduced proliferation observed in Caco-2 and OVCAR-8 cells following knockdown of CD133 or HDAC6 was due to a reduction in clonogenicity (Figure 3B).
Figure 3. CD133 Promotes Cell Proliferation and Tumorigenesis.

(A) Cell number relative to day 0 for Caco-2 and OVCAR-8 cells treated with control, CD133, or HDAC6 shRNAs. Error bars represent SD (n = 3).
(B) Clonogenic assay of Caco-2 and OVCAR-8 cells treated with control, CD133, or HDAC6 shRNAs. Error bars represent SD (n = 3).
(C) Soft agar colony forming assay to measure anchorage-independent growth of Caco-2 and OVCAR-8 cells treated with control, CD133, or HDAC6 shRNAs. Error bars represent SD (n = 3). *p < 0.05 and #p < 0.01 as determined by a Student’s t test.
(D) Tumor volume 40 days after 5 × 105 shRNA-treated OVCAR-8 cells were subcutaneously implanted in NOD/SCID/IL2Rγ−/− mice. Errors bars represent SEM (n = 10). ***p < 0.001 as determined by analysis of variance (ANOVA).
See also Figure S4.
To extend the idea that CD133 and HDAC6 may be collaborating to promote cell proliferation, we assessed the ability of CD133- and HDAC6-depleted cells to grow in an anchorage-independent fashion in soft agar colony forming assays and observed a significant reduction in the number of colonies relative to control cells (Figure 3C; p < 0.05). As anchorage-independent growth is a well-known hallmark of cancer, we investigated the impact of CD133 and HDAC6 depletion on tumor growth by subcutaneously implanting OVCAR-8 cells stably expressing a control shRNA or shRNAs targeting CD133 or HDAC6 into NOD/SCID/IL2Rγ−/− (NSG) mice and observed no significant difference in tumor size between uninfected and control infected OVCAR-8 cells (Figure S4C; p = 0.43). Notably, OVCAR-8 tumors with stable knockdown of either CD133 or HDAC6 were significantly smaller than control tumors (Figure 3D; p < 0.001). Together, these data indicate that CD133 and HDAC6 each promote proliferation in some cancer cell lines.
Consistent with a previous study in the FEMX-I melanoma cell line (Rappa et al., 2008), we found that CD133 is required for the survival of SEM-K2 cells (Mak et al., 2012). Consistent with the idea that HDAC6 is stabilizing CD133 levels, inhibition of HDAC6 activity by shRNA knockdown or tubacin treatment also resulted in significant apoptosis in SEM-K2 as measured by Annexin V staining (Figures S4A and S4B). Despite these findings, there is controversy surrounding how CD133 influences cell proliferation, as others have found that CD133 expression suppresses differentiation in a subset of neuroblastoma cell lines (Takenobu et al., 2011). Given these discrepancies, we next set out to determine the mechanism by which CD133 and HDAC6 promote proliferation in certain colon and ovarian cancer cell lines.
CD133 Suppresses Cancer Cell Differentiation and Mesenchymal-to-Epithelial Transition
Consistent with previous reports (Elsaba et al., 2010), apoptosis was not evident in Caco-2 cells after CD133 knockdown (data not shown). Therefore, we examined a panel of differentiation markers in Caco-2 cells to determine whether CD133 was functioning to suppress cell differentiation and found that upregulation of genes associated with epithelial differentiation (CK20, MUC2, and FABP2) (Figure 4A; p < 0.05), as well as activity of the enterocytic differentiation marker alkaline phosphatase (Figure 4B; p < 0.01), correlated with knockdowns of CD133 or HDAC6 in Caco-2 cells. Further analyses revealed upregulation of genes involved in colon-specific differentiation (Table S3). Loss of CD133 in OVCAR-8 cells leads to changes in differentiation markers as evidenced by loss of the high-mobility group AT-hook 2 (HMGA2) oncogene (Figure 4C; p < 0.01), a marker of poorly differentiated, advanced cancers (Shell et al., 2007).
Figure 4. CD133 Suppresses Cancer Cell Differentiation.

(A) Relative transcript levels of epithelial differentiation markers CK20, MUC2, and FABP2 as measured by quantitative PCR of shRNA-treated Caco-2 cells. Error bars represent SD (n = 3).
(B) Activity of the enterocytic marker alkaline phosphatase (AP) following knockdown of HDAC6 or CD133 in Caco-2 cells. Error bars represent SD (n = 4).
(C) Relative transcript levels of the ovarian differentiation markerHMGA2 as measured by quantitative PCR of shRNA-treated OVCAR-8 cells. Error bars represent SD (n = 3).
(D) Western blot analyses for epithelial (E-cadherin) and mesenchymal (N-cadherin and vimentin) markers in OVCAR-8 cells where CD133 or HDAC6 has been knocked down with two independent shRNAs. CD133 and HDAC6 knockdowns are shown in Figure 4D. GAPDH immunoblots served as a loading control.
(E) Quantitative PCR of EMT-associated and Wnt signaling target genes following knockdown ofCD133orHDAC6inOVCAR-8cells. Error bars represent SD(n= 3).
(F) Invasion of basement membrane extract (BME) of OVCAR-8 cells (24 hr assay) treated with shRNAs to knock down CD133 or HDAC6. Cell invasion values are relative to cell invasion of OVCAR-8 cells treated with the control shRNA targeting a nonhuman transcript, LacZ. Error bars represent SD (n = 3). *p < 0.05 and #p < 0.01 as determined by a Student’s t test.
See also Figure S5.
HMGA2 was demonstrated to be a prognostic marker for ovarian cancer alongside classical markers associated with epithelial-mesenchymal transition (EMT), E-cadherin (CDH1), and vimentin (VIM) (Shell et al., 2007). In OVCAR-8 cells, downregulation of CD133 resulted in an increase of the epithelial marker E-cadherin (CDH1) and a decrease of the mesenchymal markers N-cadherin (CDH2) and vimentin (VIM) (Figures 4D and S5A). Furthermore, significant reduction of the EMT transcription factor program was observed by quantitative PCR, including downregulation of ZEB2, TWIST, SNAIL, and SLUG (Figure 4E; p < 0.05), suggesting that loss of CD133 in OVCAR-8 cells promotes a mesenchymal-to-epithelial transition (MET). This transition was further observed with epithelial-like morphological changes and the reduction of mesenchymal-associated stress fibers (Figure S5B). Therefore, downregulation of CD133 expression through the deacetylase activity of HDAC6 is a trigger for differentiation.
As it has been established that EMT is involved in migration and invasion in cancer metastasis (Thiery et al., 2009), we investigated the impact of CD133 and HDAC6 in migration and in cell invasion in OVCAR-8 cells and observed a significant decrease in their ability to invade basement membrane extract relative to control (Figure 4F; p < 0.05). In further support of decreased cell invasion in OVCAR-8 cells depleted for CD133 or HDAC6, we observed a reduction in expression of mesenchymal- and metastasis-associated genes, including SLUG, LAMC1, and MMP7 (Figure 4E). Interestingly, these genes are reported targets of the Wnt/β-catenin signaling pathway (Brabletz et al., 2009), raising the possibility that CD133 is directly involved in promoting Wnt/β-catenin signaling.
CD133 Expression Promotes β-Catenin Signaling in Colon and Ovarian Cancer Cells
To test whether Wnt/β-catenin signaling was active in Caco-2 and OVCAR-8 cells and whether CD133 is important for maintaining a β-catenin signaling circuit, we carried out two sets of experiments. First, we treated these cells with small molecules— IWP-2 and IWR-1—that target the canonical Wnt pathway (Chen et al., 2009). The effects of IWP-2 and IWR-1 on Caco-2 and OVCAR-8 cells were confirmed as the bona fide TCF/LEF target AXIN2 was significantly decreased (Figure 5A; p < 0.05). IWP-2 or IWR-1 treatment of Caco-2 cells also caused an upregulation of the differentiation markers MUC2 and FABP2, whereas treatment of OVCAR-8 cells responds by downregulating the mesenchymal markers HMGA2 and VIM (Figure 5A; p < 0.05). Second, we used the TOP-destabilized GFP (dGFP) β-catenin signaling reporter and the control FOP-dGFP (Reya et al., 2003) to assess CD133 involvement in the β-catenin signaling. A significant decrease of TOP-dGFP, but not FOP-dGFP, reporter expression in Caco-2 and OVCAR-8 cells was observed with the loss of CD133 (Figure 5B; p < 0.05). In addition, Wnt/ β-catenin signaling negative feedback targets TCF-1, LEF-1, Axin2, and NKD1 were also significantly decreased (Figure 5C; p < 0.05).
Figure 5. CD133 Supports β-Catenin Signaling.

(A) Quantitative PCR to measure transcript levels of differentiation markers for Caco-2 and OVCAR-8 cells treated for 24 hr with 5 μM of IWP-2 or 10 μM of IWR-1. Cells treated with DMSO only acted as a control. Error bars represent SD (n = 3).
(B) Destabilized GFP (dGFP) transcript levels were measured by quantitative PCR for Caco-2 and OVCAR-8 cells stably expressing TOP- or FOP-dGFP treated with shRNAs. Error bars represent SD (n = 3).
(C) Quantitative PCR was performed to measure Wnt signaling target genes in Caco-2 and OVCAR-8 cells treated with control, CD133, or HDAC6. Error bars represent SD (n = 3). *p < 0.05 and #p < 0.01 as determined by a Student’s t test.
CD133 Stabilizes β-Catenin via HDAC6 Deacetylase Activity
Because HDAC6 has previously been shown to associate with β-catenin (Li et al., 2008), we performed endogenous co-IPs with lysates from Caco-2 cells to determine whether CD133, HDAC6, and β-catenin could physically associate in a complex. Indeed, IPs of endogenous CD133, HDAC6, and β-catenin followed by western blotting demonstrated that these proteins are capable of interacting at endogenous levels in Caco-2 cells (Figures 6A and S1A). Interestingly, β-catenin appears to interact exclusively with a lower-molecular-weight form of CD133, which we (Mak et al., 2011) and others have previously identified as a CD133 molecule that is N-glycosylated but lacks complex N-glycosylation.
To further dissect the configuration of this complex, we used the MYTH assay to test digenic interactions between each of these proteins. The CD133-C-T bait and NubG-β-catenin did not appear to interact by MYTH (data not shown and Figure 6B). Therefore, we hypothesized that their association may be mediated by HDAC6, and we introduced a vector encoding HDAC6 under a constitutive yeast promoter or an empty vector into the system. Notably, expression of HDAC6 or the empty vector alone in yeast cells did not influence the Ost1 NubG/NubI controls. Also, NubG-β-catenin did not interact with the CD4(TM)-C-T control bait in the presence or absence of HDAC6 (Figure 6B). Consistent with the model that HDAC6 bridges the interaction between CD133 and β-catenin, expression of HDAC6, but not the empty vector, resulted in activation of the MYTH reporter (Figure 6B), thus providing additional evidence that CD133-C-T, HDAC6, and NubG-β-catenin physically associate in a complex. To visualize the localization of the CD133-HDAC6-β-catenin complex in Caco-2 and OVCAR-8 cells, we performed immunofluorescence costaining experiments and found a striking colocalization of CD133 and β-catenin (Figure S6A; Pearson’s correlation = 0.82). By contrast, only a small fraction of HDAC6 colocalized with CD133 and β-catenin (Figure S6A).
We next monitored levels of β-catenin by western blot analysis and observed a striking downregulation of total β-catenin protein levels when CD133 or HDAC6 was each downregulated by two independent shRNAs in Caco-2 or OVCAR-8 cells (Figure 6C) and also from OVCAR-8 tumors generated in NSG mice (Figure S6B). Because HDAC6 has previously been shown to deacetylate β-catenin at lysine 49 to inhibit phosphorylation and subsequent degradation (Li et al., 2008), we hypothesized that CD133 regulated β-catenin via HDAC6. In support of this and consistent with previous findings, the level of acetylated β-catenin was increased when directly targeting CD133 or when targeting HDAC6 (Figures 6C and S6B). Consistent with a previous study (Li et al., 2008), this correlated with an increase of phosphorylated β-catenin relative to total levels of β-catenin (Figure 6C). However, in stark contrast to β-catenin levels, we found no significant differences in the levels of total or phospho-glycogen synthase kinase (GSK) 3-β (Figure 6C).
As stabilized β-catenin translocates to the nucleus to target TCF/LEF target genes, we tracked β-catenin localization by immunofluorescence and by immunohistochemistry in the presence and absence of CD133 and HDAC6. A striking decrease in cytoplasmic and nuclear localized β-catenin was observed in CD133 and HDAC6 knockdowns in vivo (Figure S6C) and in vitro (Figure S6D). Given the above, we suggest that CD133 regulates β-catenin acetylation via HDAC6 and, thus, its phosphorylation, stability, and nuclear localization to influence β-catenin signaling activity.
DISCUSSION
CD133 has been used as a molecular marker for stem cells and cancer stem cells for more than a decade, yet its molecular function has remained mysterious. Much work has been done to link CD133-expressing cells to underlying molecular cancer signatures and phenotypes with little insight into the posttranslational regulation and molecular function of CD133. Our study uncovers a protein-protein interaction between CD133 and HDAC6, establishes a role for HDAC6 in CD133 turnover, reveals a contextual role for CD133 and HDAC6 in the suppression of cancer cell differentiation and expression of Wnt target genes, and directly links CD133, HDAC6, and β-catenin in a protein complex. Together, these findings have broader implications for regulation and turnover of multispanning cell surface proteins and cell signaling through the canonical Wnt pathway.
HDAC6 becomes phosphorylated at Y570 by EGFR following activation by the EGF ligand (Deribe et al., 2009). This phosphorylation event correlates with decreased HDAC6 deacetylase activity, increased acetylation of K40 on α-tubulin, and increased EGFR endocytosis (Deribe et al., 2009; Gao et al., 2010). We explored the nature of the CD133-HDAC6 interaction by testing whether HDAC6 phospho-mimicking mutants (Y570E and S568E) could interact with CD133 and found that they could not, whereas nonphosphorylatable HDAC6 mutants (Y570F and S568A) still interacted with CD133. These results provide a model to explore the signaling relationship between CD133 and HDAC6; for example, which kinase(s) and phosphatase(s) regulate this interaction? Because phosphorylation of HDAC6 correlates with decreased HDAC6 deacetylase activity, we propose that CD133 is not a substrate of HDAC6 but rather promotes HDAC6 activity by protecting it from dephosphorylation. Our hypothesis is that this function creates a microenvironment for linking extracellular cues with cell surface features that can communicate important signaling components like β-catenin.
HDAC6 has previously been shown to be required for oncogenic Ras- and ErbB2-induced transformation of fibroblast cells and for anchorage-independent growth (Lee et al., 2008). These results are consistent with our own finding that HDAC6 knockdown results in decreased proliferation, colony formation, and invasion in Caco-2 and OVCAR-8 cells as well as decreased tumor growth in an OVCAR-8 xenograft model. Interestingly, knockdown of CD133 had similar effects on proliferation, anchorage independence, invasion, and xenograft growth in Caco-2 and OVCAR-8 cells in our hands, raising the possibility that CD133 and HDAC6 form a signaling module that positively regulates cell state. These results are also consistent with a recent report showing that CD133 knockdown caused decreased proliferation and tumorigenic growth in the neuroblastoma cell line TGW (Takenobu et al., 2011) and decreased anchorage-independent growth and colony formation in HT-29 cells (Elsaba et al., 2010). These findings are inconsistent with previous observations showing that targeting CD133 in SEMK2 (Mak et al., 2012) or FEMX-I cells (Rappa et al., 2008) results in apoptosis. Thus, the cellular role of CD133 appears to be cancer cell type specific.
CD133 is associated with Wnt signaling in this study as well as in an independent study examining the role of CD133 in FEMX-1 cells (Rappa et al., 2008), suggesting that cell-type-specific responses to Wnt signaling activity may explain the differences observed in the cellular role of CD133. Importantly, HDAC6 appears to be dispensable in the normal mouse physiology and development (Zhang et al., 2008), as HDAC6 knockout mice are viable and healthy and do not show any malignant predispositions. Similarly, CD133 is not essential for mouse development or viability (Zacchigna et al., 2009). Together, these results imply that HDAC6 and CD133 are required for cancer cell proliferation—not normal cell proliferation—in certain cellular contexts. The recent report showing that inhibition of HDAC6 induces DNA damage and sensitizes transformed cells to anticancer agents raises the possibility that inhibition of CD133 may also sensitize transformed cells to anticancer agents (Namdar et al., 2010).
Consistent with our findings, HDAC6 is required for transforming growth-factor-β1-induced EMT through SMAD3-activating HDAC6 deacetylation of α-tubulin (Shan et al., 2008). HDAC6 was also shown to increase cell motility via its interaction with the F-actin binding protein cortactin (Zhang et al., 2007). Previous observations demonstrate that CD133 primarily localizes to plasma membrane protrusions in migrating hematopoietic progenitor cells, implying that CD133 also has a role in cell migration (Giebel et al., 2004). Indeed, targeting CD133 in HT-29 cells results in decreased cell migration (Elsaba et al., 2010). In the present study, we extend these findings to OVCAR-8 cells and show that CD133 and HDAC6 promote the mesenchymal state, which is linked to motility and invasion. These findings support a previous study that observed decreased metastasis following depletion of CD133 by RNAi in the melanoma cell line FEMX-I (Rappa et al., 2008). Together, these observations suggest that CD133 and HDAC6 have a role in promoting and maintaining a mesenchymal state, which is associated with stem cell properties (Mani et al., 2008; Thiery et al., 2009), and suggest that the CD133-HDAC6 signaling module may have a role in establishing and maintaining such properties in certain cancer cell contexts.
We relied on our identification of the CD133-HDAC6 interaction and the previous report that demonstrated deacetylation of lysine 49 of β-catenin by HDAC6 (Li et al., 2008) to identify the CD133-HDAC6-β-catenin complex at the plasma membrane (Figure 7A). Knockdown of CD133 caused a dramatic increase in acetylated β-catenin, but not acetylated α-tubulin (Figure 7B). Given that HDAC6 is primarily localized to the cytoplasm, we suggest that the HDAC6 activity responsible in deacetylating α-tubulin and β-catenin is local to the minute fraction associated with CD133 at the plasma membrane. We propose that the molecular mechanism surrounding the role of CD133 in β-catenin signaling is through the recruitment of HDAC6 to deacetylate β-catenin for its stabilization and nuclear localization. Total and phosphorylated GSK3β levels show no obvious change when CD133 and HDAC6 are depleted from cells, suggesting that CD133 regulation of β-catenin is via an independent pathway of the upstream Wnt signaling pathway components involved in GSK3β activity regulation.
Figure 7. Proposed Model Summary.
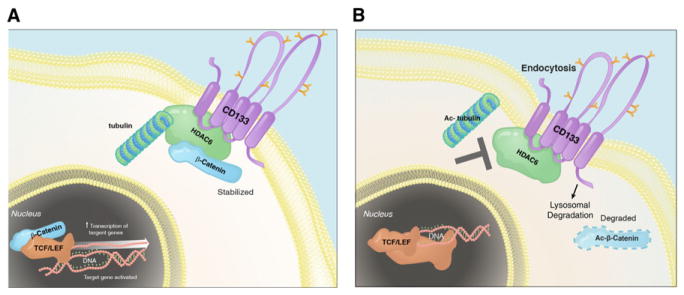
(A) HDAC6 physically interacts with CD133 and negatively regulates its trafficking into endosomes by deacetylating α-tubulin. This allows for the positive regulation of β-catenin signaling by CD133 via recruitment of HDAC6 to deacetylate β-catenin, resulting in its stabilization. Collectively, this results in maintaining a primitive cancer cell state.
(B) Inhibition of HDAC6 results in increased acetylated tubulin, CD133 endocytosis, destabilization of β-catenin, and, in some contexts, cancer cell differentiation.
Our results suggest that one mechanism by which CD133 governs cancer cell identity and state is through the β-catenin signaling pathway (Figure 7). This is consistent with the finding that the Wnt pathway is tightly linked to colon cancer and Wnt signaling, and expression of AC133 is required for maintenance of the colon cancer stem cell compartment (Vermeulen et al., 2010). Furthermore, β-catenin is required for CD133+ leukemia stem cells to drive acute myelogenous leukemia in mice (Wang et al., 2010). Taken together, inhibiting CD133, HDAC6 deacetylase activity, or the β-catenin signaling pathway may be a means to targeting certain cancer progenitor cells.
EXPERIMENTAL PROCEDURES
Affinity Purification and Mass Spectrometry Analysis
Cell extracts from HEK293/CD133-VA were incubated with Flag M2-agarose beads (Sigma-Aldrich, Inc.) at 4°C for 4 hr, followed by washing and elution with 500 mM ammonium hydroxide (pH > 11). The eluate was lyophilized in a SpeedVac and resuspended in 50 mM ammonium bicarbonate (pH 8–8.3) with trypsin for 2 hr at 37°C. The ammonium bicarbonate was removed by SpeedVac, and the samples were suspended in 2% acetonitrile, 0.1% formic acid. Samples were independently and directly loaded onto capillary columns packed with Magic C18AQ (5 μm, 100 A° ). Data were acquired over a 2 hr 2%–40%acetonitrile gradient on a ThermoFinnigan LTQ equipped with a Proxeon NanoSource and an Agilent 1100 capillary pump. RAW files were converted to mgf format and were searched with the Mascot search engine (Matrix Sciences) against the human RefSeq database (release 24). To determine specific interactions with high confidence, we filtered out hits also detected in the control sample (i.e., HEK293/VA-GFP) as well as hits detected with >20% frequency in the internal Samuel Lunenfeld Research Institute database of human anti-Flag interactors. Only protein hits with a minimum of one unique peptide and a MASCOT score greater than 60 and that were detected in all biological replicates (n = 3) are reported.
Membrane Yeast Two-Hybrid Assay and Screen
MYTH analysis and screen were performed as previously described (Deribe et al., 2009). Briefly, the MYTH screen for CD133 was performed by using a cDNA library containing 2.6 × 106 independent clones. Approximately 8 × 106 transformants were screened by growth on selective media, and plasmids were isolated from 384 positive clones and retested in the MYTH system for specificity toward CD133 versus the CD4(TM) control bait in independent quadruplet (n = 4). Clones that were specific to CD133 were analyzed by DNA sequencing and analyzed by bioinformatics to identify corresponding gene hits. 25 unique gene hits from the MYTH screen were identified.
Proliferation and Clonogenic Assays
Total cell numbers were determined by a Z2 Coulter counter (Beckman Coulter, Inc.) at indicated time intervals. Cell numbers were relative to the number of cells plated per well at time 0, which was constant between samples and replicates. For the clonogenic assay, single-cell suspensions of Caco-2 and OVCAR-8 stable lines were plated into 6-well plates at a density of 1,000 cells per well. After 2 weeks of culturing under conditions as described above, cells were fixed in 4% paraformaldehyde in PBS for 15 min, stained with 0.05% crystal violet for 15 min, and washed with water. Colonies visible by eye and possessing >50 cells were counted.
Alkaline Phosphatase Assay
Alkaline phosphatase activity was determined by a fluorometric kit as per manufacture’s protocol (ab83371, Abcam Inc.).
Soft Agar Colony Formation Assay
Approximately 1,000 Caco-2 or OVCAR-8 cells were resuspended in 0.5 ml of 0.35% Bacto agar (214050, BD, Sparks, MD) in the appropriate media and plated on 0.5 ml of 0.5% Bacto agar bases that had settled for 30 min at room temperature in 12-well plates. Appropriate liquid media were added to each well and were changed weekly. The number of colonies was scored after 4 weeks of incubation for Caco-2 samples and after 2 weeks of incubation for OVCAR-8 samples by staining with 0.005% crystal violet and counting by eye under a dissection light microscope. Colonies possessing >50 cells were counted.
Cell Invasion Assay
Cell invasion was measured by using the Cultrex Basement Membrane Extract cell invasion assay (3455-096-K, R&D Systems, Inc.) as per manufacture’s protocol. A one-in-ten dilution of Basement Membrane Extract was used to coat the inner-well chamber overnight, and OVCAR-8 stables were incubated for 24 hr prior to quantitative measurements.
TOP/FOP Assays
Lentiviral constructs encoding the TOP-dGFP and FOP-dGFP reporters were kindly provided by Dr. Laurie Ailles. Lentivirus was produced as described above and used to stably transduce Caco-2 and OVCAR-8 cells at a MOI >1. Stable cells were infected with shRNAs, and dGFP transcript was measured by qRT-PCR.
Animals and Subcutaneous Xenografts
Eight-week-old female NOD/SCID/IL2Rγ−/− (NSG) mice were used and maintained according to Canadian Council of Animal Care (CCAC) guidelines. Subcutaneous injection of 5 × 105 OVCAR-8 cells treated with or without shRNAs into both hind flanks was performed. The inoculation volume was 40 μl in a 1:1 mixture of HBSS:Matrigel. Five mice (ten tumors total) per condition were used.
Plasmids and Antibodies
Primers and antibodies used in this study to generate plasmids, monitor transcript levels by quantitative RT-PCR, and perform western blots are described in Tables S4, S5, and S6.
For a more detailed version of Experimental Procedures please see Extended Experimental Procedures.
Supplementary Material
Acknowledgments
We thank P. Mero, Y. Fedyshyn, B. Fedyshyn, J. Snider, P-A. Penttila, and V. Wong for technical assistance and S-P. Ip for assistance with Figure 7. We thank Drs. B. Gallie and R. Rottapel for providing reagents and J. Woodgett for comments on the manuscript. A.B.M. was supported by a doctoral Canada Graduate Scholarship from the Natural Sciences and Engineering Research Council. This work was supported by funds from the Stem Cell Network to J.M., Canadian Cancer Research Society to I.S., Canadian Institutes of Health Research to I.S. and J.M., Ontario Institute for Cancer Research to J.M., the Canadian Foundation for Innovation to J.M., and the Ontario Ministry of Research and Innovation to J.M. J.M. is a Tier II Canada Research Chair in Functional Genomics of Cancer and is a Research Fellow in the Canadian Institute for Advanced Research.
Footnotes
Supplemental Information includes Extended Experimental Procedures, six figures, and six tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2012.09.016.
LICENSING INFORMATION
This is an open-access article distributed under the terms of the Creative Commons Attribution 3.0 Unported License (CC-BY; http://creativecommons.org/licenses/by/3.0/legalcode).
References
- Brabletz S, Schmalhofer O, Brabletz T. Gastrointestinal stem cells in development and cancer. J Pathol. 2009;217:307–317. doi: 10.1002/path.2475. [DOI] [PubMed] [Google Scholar]
- Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW, Wei S, Hao W, Kilgore J, Williams NS, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5:100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbeil D, Röper K, Hellwig A, Tavian M, Miraglia S, Watt SM, Simmons PJ, Peault B, Buck DW, Huttner WB. The human AC133 hematopoietic stem cell antigen is also expressed in epithelial cells and targeted to plasma membrane protrusions. J Biol Chem. 2000;275:5512–5520. doi: 10.1074/jbc.275.8.5512. [DOI] [PubMed] [Google Scholar]
- Deribe YL, Wild P, Chandrashaker A, Curak J, Schmidt MH, Kalaidzidis Y, Milutinovic N, Kratchmarova I, Buerkle L, Fetchko MJ, et al. Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signal. 2009;2:ra84. doi: 10.1126/scisignal.2000576. [DOI] [PubMed] [Google Scholar]
- Elsaba TM, Martinez-Pomares L, Robins AR, Crook S, Seth R, Jackson D, McCart A, Silver AR, Tomlinson IP, Ilyas M. The stem cell marker CD133 associates with enhanced colony formation and cell motility in colorectal cancer. PLoS ONE. 2010;5:e10714. doi: 10.1371/journal.pone.0010714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandina G, Petrillo M, Bonanno G, Scambia G. Targeting CD133 antigen in cancer. Expert Opin Ther Targets. 2009;13:823–837. doi: 10.1517/14728220903005616. [DOI] [PubMed] [Google Scholar]
- Gao YS, Hubbert CC, Yao TP. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J Biol Chem. 2010;285:11219–11226. doi: 10.1074/jbc.M109.042754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebel B, Corbeil D, Beckmann J, Höhn J, Freund D, Giesen K, Fischer J, Kögler G, Wernet P. Segregation of lipid raft markers including CD133 in polarized human hematopoietic stem and progenitor cells. Blood. 2004;104:2332–2338. doi: 10.1182/blood-2004-02-0511. [DOI] [PubMed] [Google Scholar]
- Gisler SM, Kittanakom S, Fuster D, Wong V, Bertic M, Radanovic T, Hall RA, Murer H, Biber J, Markovich D, et al. Monitoring protein-protein interactions between the mammalian integral membrane transporters and PDZ-interacting partners using a modified split-ubiquitin membrane yeast two-hybrid system. Mol Cell Proteomics. 2008;7:1362–1377. doi: 10.1074/mcp.M800079-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horst D, Scheel SK, Liebmann S, Neumann J, Maatz S, Kirchner T, Jung A. The cancer stem cell marker CD133 has high prognostic impact but unknown functional relevance for the metastasis of human colon cancer. J Pathol. 2009;219:427–434. doi: 10.1002/path.2597. [DOI] [PubMed] [Google Scholar]
- Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- Lee YS, Lim KH, Guo X, Kawaguchi Y, Gao Y, Barrientos T, Ordentlich P, Wang XF, Counter CM, Yao TP. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008;68:7561–7569. doi: 10.1158/0008-5472.CAN-08-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhang X, Polakiewicz RD, Yao TP, Comb MJ. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J Biol Chem. 2008;283:12686–12690. doi: 10.1074/jbc.C700185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak AB, Ni Z, Hewel JA, Chen GI, Zhong G, Karamboulas K, Blakely K, Smiley S, Marcon E, Roudeva D, et al. A lentiviral functional proteomics approach identifies chromatin remodeling complexes important for the induction of pluripotency. Mol Cell Proteomics. 2010;9:811–823. doi: 10.1074/mcp.M000002-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak AB, Blakely KM, Williams RA, Penttilä PA, Shukalyuk AI, Osman KT, Kasimer D, Ketela T, Moffat J. CD133 protein N-glycosylation processing contributes to cell surface recognition of the primitive cell marker AC133 epitope. J Biol Chem. 2011;286:41046–41056. doi: 10.1074/jbc.M111.261545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak AB, Nixon AM, Moffat J. The mixed lineage leukemia (MLL) fusion-associated gene AF4 promotes CD133 transcription. Cancer Res. 2012;72:1929–1934. doi: 10.1158/0008-5472.CAN-11-3589. [DOI] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelialmesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maw MA, Corbeil D, Koch J, Hellwig A, Wilson-Wheeler JC, Bridges RJ, Kumaramanickavel G, John S, Nancarrow D, Röper K, et al. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum Mol Genet. 2000;9:27–34. doi: 10.1093/hmg/9.1.27. [DOI] [PubMed] [Google Scholar]
- Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, Holden JT, Bray RA, Waller EK, Buck DW. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood. 1997;90:5013–5021. [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral highcontent screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- Namdar M, Perez G, Ngo L, Marks PA. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc Natl Acad Sci USA. 2010;107:20003–20008. doi: 10.1073/pnas.1013754107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappa G, Fodstad O, Lorico A. The stem cell-associated antigen CD133 (Prominin-1) is a molecular therapeutic target for metastatic melanoma. Stem Cells. 2008;26:3008–3017. doi: 10.1634/stemcells.2008-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in selfrenewal of haematopoietic stem cells. Nature. 2003;423:409–414. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- Röper K, Corbeil D, Huttner WB. Retention of prominin in microvilli reveals distinct cholesterol-based lipid micro-domains in the apical plasma membrane. Nat Cell Biol. 2000;2:582–592. doi: 10.1038/35023524. [DOI] [PubMed] [Google Scholar]
- Shan B, Yao TP, Nguyen HT, Zhuo Y, Levy DR, Klingsberg RC, Tao H, Palmer ML, Holder KN, Lasky JA. Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J Biol Chem. 2008;283:21065–21073. doi: 10.1074/jbc.M802786200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shell S, Park SM, Radjabi AR, Schickel R, Kistner EO, Jewell DA, Feig C, Lengyel E, Peter ME. Let-7 expression defines two differentiation stages of cancer. Proc Natl Acad Sci USA. 2007;104:11400–11405. doi: 10.1073/pnas.0704372104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider J, Kittanakom S, Damjanovic D, Curak J, Wong V, Stagljar I. Detecting interactions with membrane proteins using amembrane two-hybrid assay in yeast. Nat Protoc. 2010;5:1281–1293. doi: 10.1038/nprot.2010.83. [DOI] [PubMed] [Google Scholar]
- Takenobu H, Shimozato O, Nakamura T, Ochiai H, Yamaguchi Y, Ohira M, Nakagawara A, Kamijo T. CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene. 2011;30:97–105. doi: 10.1038/onc.2010.383. [DOI] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelialmesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Vermeulen L, De Sousa E, Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–1653. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigmann A, Corbeil D, Hellwig A, Huttner WB. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci USA. 1997;94:12425–12430. doi: 10.1073/pnas.94.23.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–5012. [PubMed] [Google Scholar]
- Zacchigna S, Oh H, Wilsch-Bräuninger M, Missol-Kolka E, Jászai J, Jansen S, Tanimoto N, Tonagel F, Seeliger M, Huttner WB, et al. Loss of the cholesterol-binding protein prominin-1/CD133 causes disk dysmorphogenesis and photoreceptor degeneration. J Neurosci. 2009;29:2297–2308. doi: 10.1523/JNEUROSCI.2034-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gilquin B, Khochbin S, Matthias P. Two catalytic domains are required for protein deacetylation. J Biol Chem. 2006;281:2401–2404. doi: 10.1074/jbc.C500241200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, Olashaw N, Parsons JT, Yang XJ, Dent SR, et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27:197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Li N, Cheng HL, Chua K, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.