

Abstract
The effector T-cell subset, Th17, plays a significant role in the pathogenesis of multiple sclerosis as well as other autoimmune diseases. The signature cytokine, IL-17, engages the IL-17R and recruits the E3-ligase Act1 upon stimulation. In this study we examined the role of TRAF4 in IL-17 signaling and Th17-mediated autoimmune encephalomyelitis. Primary cells from TRAF4-deficient mice displayed markedly enhanced IL-17-activated signaling pathways and induction of chemokine mRNA. Adoptive transfer of MOG 35–55 specific wild-type Th17 cells into TRAF4-deficient recipient mice induced an earlier onset of disease. Mechanistically, we found that TRAF4 and TRAF6 utilized the same TRAF-binding sites on Act1, allowing the competition of TRAF4 with TRAF6 for the interaction with Act1. Taken together, this study reveals the necessity of a unique role of TRAF4 in restricting the effects of IL-17 signaling and Th17-mediated disease.
Introduction
The signature cytokine of Th17 cells, IL-17 (IL-17A), has been linked to the pathogenesis of numerous autoimmune diseases. Indeed, IL-17 levels are elevated in patients with multiple sclerosis and rheumatoid arthritis and IL-17-deficient mice are resistant to experimental autoimmune encephalomyelitis (EAE) (1) and collagen-induced arthritis (2). The IL-17 receptor family consists of five members, of which, IL-17A signals through a receptor complex composed of IL-17RA and IL-17RC (3). Both IL-17RA and IL-17RC belong to a SEF/IL-17R (SEFIR) protein family (4).
Act1 (NF-κB-activator-1 or CIKS), also a member of the SEFIR protein family, is an essential E3-ligase in IL-17 signaling and is required for IL-17-dependent immune responses (5, 6). Upon IL-17 stimulation Act1 binds different TRAF proteins. For example, Act1 exerts K63-linked polyubiquitination of TRAF6 (7), leading to the activation of NF-κB. Additionally, IL-17 can promote the stability of numerous mRNA encoding cytokines and chemokines in a TRAF6-independent manner (8, 9). Following IL-17 stimulation, a phosphorylated form of Act1 forms a complex with TRAF2 and TRAF5 to stabilize chemokine mRNA (10, 11). Recently we showed that binding of TRAF3 to the IL-17R interferes with the formation of the IL-17R–Act1–TRAF6 complex, hence dampening IL-17-induced inflammation (12). In the present study, we identified a unique TRAF member, TRAF4, as a novel negative regulator in IL-17-mediated signaling and inflammatory responses via a distinctive mechanism.
Materials and Methods
Mice, cell culture and reagents
TRAF4-deficient C57/BL6 mice were generated as described previously (13). Experiments were performed with gender-matched mice aged 6–8 weeks. The Institutional Animal Care and Use Committee (IACUC) of the Cleveland Clinic Foundation approved all animal procedures. Hela, MEF, HEK293 and primary kidney cells were maintained in DMEM, supplemented with 10% FBS, penicillin G (100 µg/ml) and streptomycin (100 µg/ml). Primary mouse astrocytes were isolated as described previously (6) and maintained in DMEM supplemented with 10% FBS. Isolation of primary kidney cells was described previously (10). TRAF-binding mutants, Flag-TRAF6 and MYC-Act1 were described previously (7). Derek Abbott, CWRU, provided the Omni-tagged TRAF4 expression vector and retroviral-TRAF4 (used for MEF transfection) described in ref. (14). Antibodies to phosphorylated- ERK1/2, JNK, IκBα and total IκBα were from Cell Signaling Technology. Goat anti-TRAF4 and the TRAF6, TRAF3, and Act-1 (all rabbit) antibodies were from Santa Cruz Biotechnology and the rabbit anti-TRAF4 antibody was from Epitomics. Antibodies to TAK1 and mAct1 were described previously (6, 15). The murine-rIL-17A and human-rIL-17A were from R&D Systems. Primer sequences for real time PCR have been described previously (16). Act1-deficient MEF cells reconstituted with wild-type Act1 and coimmunoprecipitation with these cells and primary cells were described in (7). For Co-IP the cells were pelleted in 4°C centrifuge (2000× RPM for 5 minutes) and the pellet was lysed in Co-IP buffer (0.5% Triton X-100, 20 mM HEPES (pH 7.4), 150 mM NaCl, 12.5 mM β-glycerophosphate, 1.5 mM MgCl2, 10 mM NaF, 1 mM sodium orthovanadate, and protease inhibitor cocktail (Roche). siRNA against human-TRAF4 was from Qiagen FlexiTube siRNA (cat# SI0302015)
Induction and assessment of EAE
Passive EAE was induced and assessed as previously described (6, 16). Brains were collected and homogenized as described previously (16). Fluorescence-conjugated CD4, CD8, CD11b, CD45, Ly6G antibodies and isotype controls were purchased from BD Biosciences and the F4/80 antibody was from Serotech.
Statistics
The p values of clinical scores were determined by one-way multiple-range analysis of variance (ANOVA) for multiple comparisons. Other p-values were determined by Student’s t tests.
Results
IL-17-induced signaling and gene expression are increased in TRAF4-deficient cells
Following IL-17 stimulation and Act1 recruitment, TRAF family members TRAF-2, -5 and -6 are required for mRNA stability and NF-κB activation respectively. To test whether TRAF4 plays any role in IL-17-mediated signaling, we isolated primary kidney epithelial cells from TRAF4-deficient and littermate control mice. We observed higher baseline activation of ERK1/2 and JNK in the TRAF4-deficient cells. Although IL-17-induced phosphorylation of ERK1/2 and JNK was only slightly increased, IL-17-induced IκBα degradation was significantly enhanced in the absence of TRAF4 (Fig. 1A and Supplemental Fig. 1A). Furthermore, overexpression of TRAF4 attenuated IL-17-induced and basal-level phosphorylation of ERK1/2, JNK and IκBα and degradation of IκBα (Fig. 1B and Supplemental Fig. 1B). Consistent with this, IL-17-induced expression of CXCL1 (KC), G-CSF and GM-CSF were substantially increased in the TRAF4-deficient primary kidney epithelial cells (Fig. 1C). It is important to note that IL-17R expression levels were comparable between wild-type and TRAF4-deficient cells (data not shown). Furthermore, TRAF4 deficiency did not have any significant impact on IL-1β- or LPS-induced gene expression in kidney epithelial cells or bone-marrow macrophages, respectively (data not shown). Taken together, these observations suggest a specific suppressive role of TRAF4 in IL-17-mediated inflammatory gene expression.
Figure 1. The Role of TRAF4 in IL-17 induced signaling and gene expression.
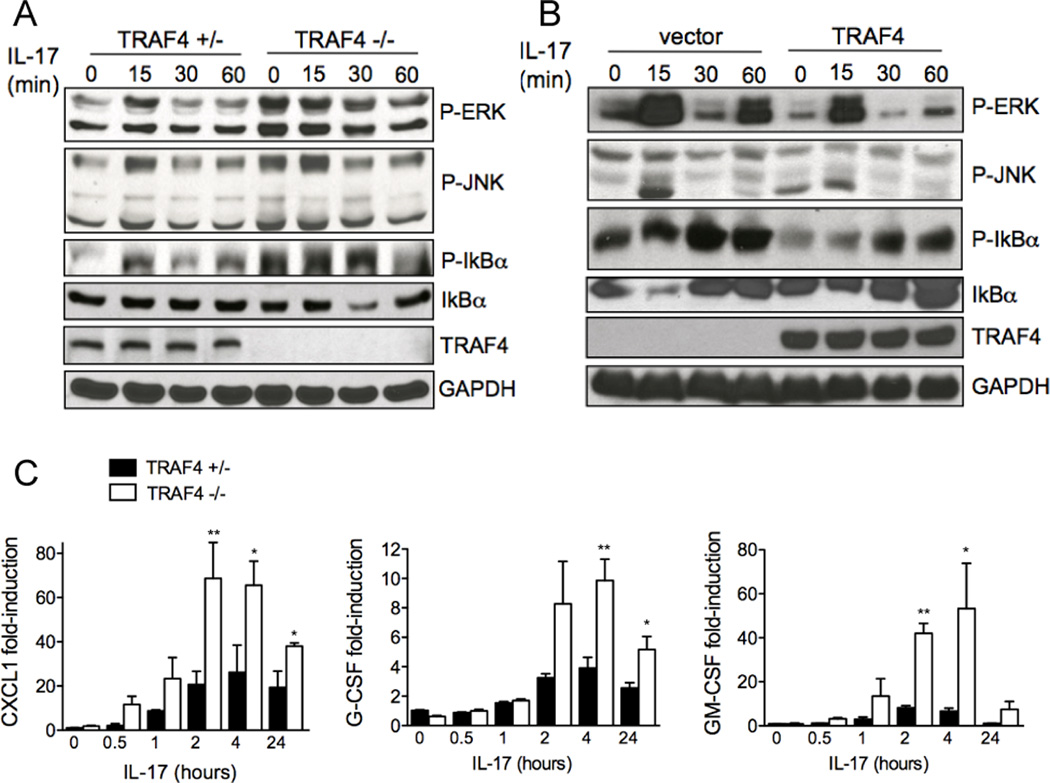
(A) Western analyses of lysates of primary kidney cells from TRAF4-deficient mice and littermate controls. (B) Western analyses of lysates of Hela cells transduced with retrovirus encoding hTRAF4. (C) Quantitative real-time PCR was performed on primary kidney cells from TRAF4-deficient mice and littermate controls. Gene expression levels relative to β-actin, n=5, means ± SEM, *p<0.05, **p<0.01 for control versus stimulated. The data shown represents experiments repeated three times.
TRAF4 deficiency exacerbates EAE severity
To determine the contribution of TRAF4 in IL-17-mediated responses in vivo we administered recombinant IL-17 by intraperitoneal injections into TRAF4-deficient and control mice. We found that the serum concentration of CXCL1 was significantly elevated in the TRAF4-deficient mice (Fig. 2A). We have reported that Act1 deficiency rescues mice from disease pathology in EAE (6, 16). We generated MOG-specific Th17 cells from wild-type C57/BL6 mice and then transferred them into irradiated TRAF4-deficient and littermate control mice. TRAF4-deficient mice exhibited an earlier onset of disease compared to littermate controls (Fig. 2B). Consistent with the clinical severity of disease, the TRAF4-deficient mice had increased numbers of immune cell infiltration in the brain (Fig. 2C) as well as a significantly higher expression of pro-inflammatory genes compared to that in the control mice (Fig. 2D).
Figure 2. TRAF4 deficiency exacerbates Th17 mediated EAE.
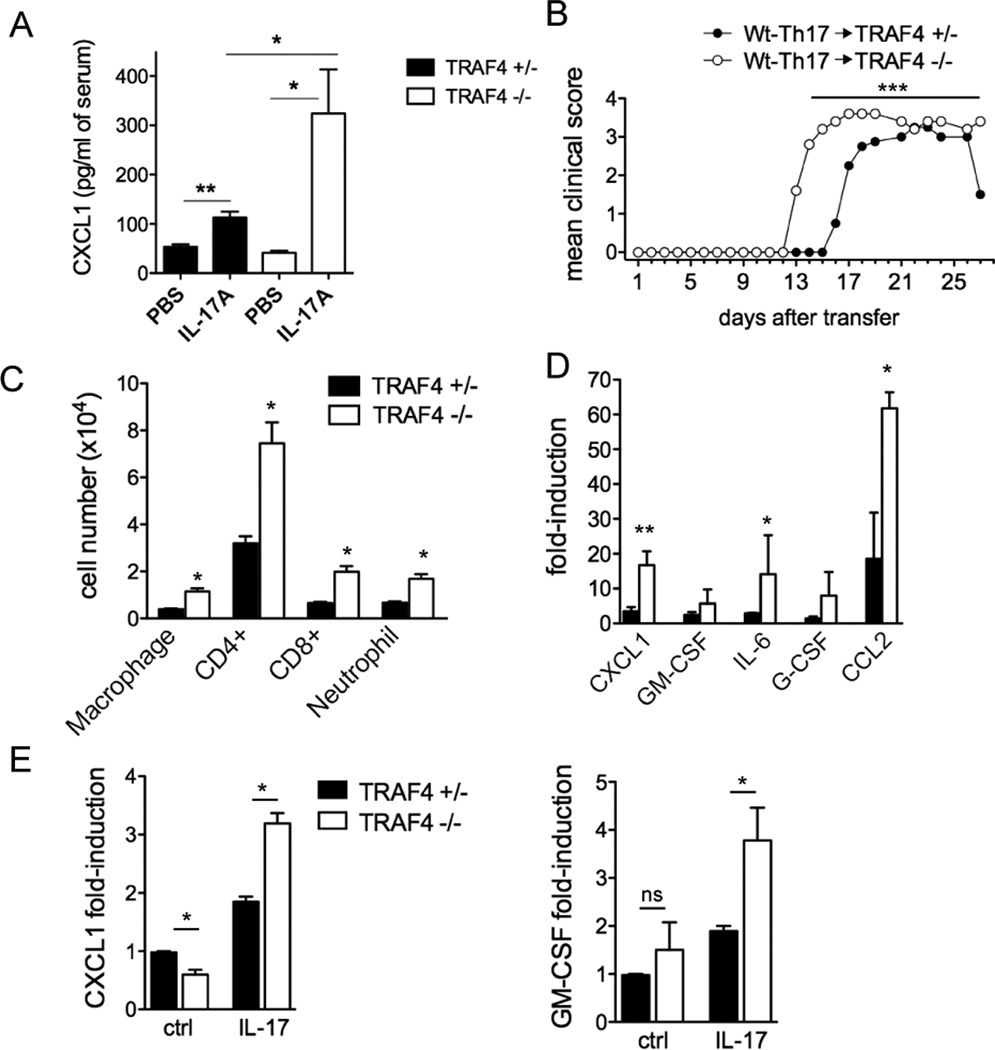
(A) Serum cytokine concentration in TRAF4 deficient mice following intraperitoneal administration of IL-17 (2 µg/mouse for two days) (B) Clinical score of EAE severity. (C) Flow-cytometry of infiltrated immune cells in the brain at the peak of the disease. (D) Spinal cord gene expression at peak of disease relative to naïve TRAF4-deficient and control spinal cord RNA (E) Gene expression from primary astrocytes stimulated with IL-17 (50 ng/ml) for 16 hours. For (A) n=4 mice per group, for (D–E) quantitative real-time PCR was performed, gene expression is relative to β-actin. n=5 per group for (D) and n=2 (E), means ± SEM, *p<0.05, **p<0.01 for littermate control versus TRAF4-deficient. For adoptive transfer experiments n=5 per group, shown here is one of two independently performed experiments, ***p<0.001 determined by one-way ANOVA.
We reported that Act1 expression in the CNS resident cells, including astrocytes, was important for induction of EAE (16, 17). TRAF4-deficient astrocytes indeed exhibited markedly increased expression of CXCL1 and GM-CSF compared to the heterozygous control (Fig. 2E). Taken together, these data suggest that TRAF4 expression can attenuate IL-17-mediated signaling events and lack of this suppressive effect of TRAF4 in vivo results in an enhanced pathological response to IL-17 as well as accelerated EAE.
Act1/TRAF4 complex is distinct from other Act1/TRAF complexes
Considering the importance of TRAF4 in modulating IL-17 signaling, we examined TRAF4 complex formation in response to IL-17 stimulation. As shown in Figure 3A, IL-17 stimulation induced the interaction of Act1 with TRAF4, in addition to its interaction with TRAF6, TRAF3 and TAK1. Interestingly, we found that while neither TRAF3 nor TRAF4 was associated with the IL-17-induced TRAF6 complex (Fig. 3B), Act1 but not TRAF3, TRAF6 or TAK1 was detected in the TRAF4 immunoprecipitate from cell lysates of IL-17-treated MEFs (Fig. 3C). These data indicate that the IL-17-induced Act1-TRAF4 complex is distinct from other Act1/TRAF complexes (Act1-TRAF6 and Act1-TRAF3).
Figure 3. IL-17-induced Distinct Act1-TRAF interactions.
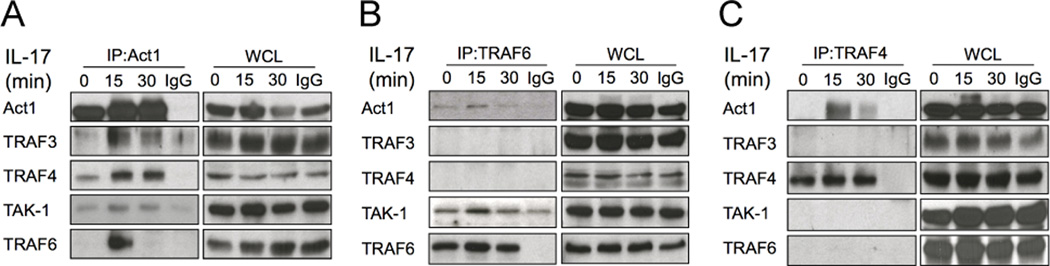
Lysates from Act1-deficient MEFs restored with wild-type Act1 untreated or treated with IL-17 (50 ng/ml) were immunoprecipitated with anti-Act1 (A), anti-TRAF6 (B) and anti-TRAF4 (C), followed by Western analyses for the indicated proteins. Shown are representative of three independently performed assays.
TRAF4 competes with TRAF6 for Act1 TRAF-binding-sites
Since enhanced IL-17-dependent IκBα degradation was observed in TRAF4-deficient cells, we focused on the mechanistic role of TRAF4 in modulating the NF-κB pathway. Since Act1-TRAF4 and Act1-TRAF6 are two distinct complexes, it is possible that TRAF4 competes with TRAF6 for the binding with Act1. We observed markedly enhanced modified TRAF6 bands in the Act1 immunoprecipitate from IL-17-treated TRAF4-deficient cells compared to that in the control cells (Fig. 4A). There was also an increased association of Act1 with TRAF6 in the TRAF4-deficient cells compared to that in control cells (Fig. 4B). These data suggest that TRAF4 deficiency results in more Act1-TRAF6 interaction and TRAF6 modification (previously shown as ubiquitination). Additionally, TRAF6/Act1 interaction was reduced when TRAF4 was overexpressed (Fig. 4C and Supplemental Fig. 2A). Act1 contains two putative TRAF-binding sites, both of which are required for binding to TRAF6 (7). Through immunoprecipitation of TRAF4 we observed that interaction was limited to Act1-wild-type but it was abolished in the Act1-TBm (Fig. 4D). Of note, single mutations of either one of the TRAF-binding sites still retained interaction with TRAF4 and TRAF6 (data not shown) demonstrating that TRAF4 and TRAF6 interact with Act1 via the same TRAF-binding domains.
Figure 4. TRAF4 restricts Act1/TRAF6 interaction.
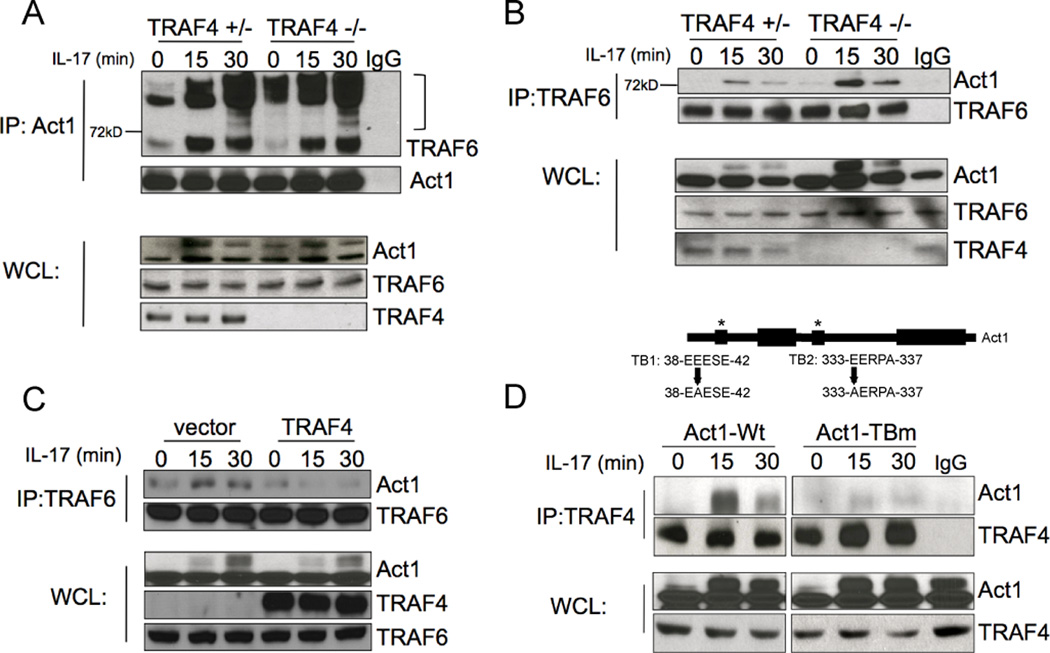
Lysates of primary kidney cells untreated and treated with IL-17 (50 ng/ml) were immunoprecipitated with anti-Act1 (A) and anti-TRAF6 (B), followed by Western analyses for the indicated proteins. (C) Lysates of MEFs transfected with TRAF4 or vector control, untreated or stimulated with IL-17 (50 ng/ml) were immunoprecipitated with anti-TRAF6, followed by Western analyses for the indicated proteins. (D) Lysates of Act1-deficient MEFs expressing WT Act1 or Act1-TBm untreated or stimulated with IL-17 (50 ng/ml) were immunoprecipitated with anti-TRAF4, followed by Western analyses for the indicated proteins. Shown are representative of three independently performed assays.
Discussion
In the present study, we identified a member of the TRAF family, TRAF4, as a novel interaction partner of Act1. TRAF4 competes with TRAF6 for binding Act1, resulting in reduced TRAF6-Act1 complex formation and attenuated inflammatory responses. These results are the first to indicate the critical role of TRAF4 in modulating IL-17-mediated signaling and pathogenesis. We recently described TRAF3 as an IL-17R proximal regulator that modulates IL-17–mediated inflammatory responses and autoimmune diseases like EAE (12). Although both TRAF3 and TRAF4 negatively regulate IL-17-mediated inflammatory responses, the underlying mechanisms are quite different. TRAF3 binds to IL-17R via a putative TB site and hence interferes with the formation of the activation complex IL-17R–Act1–TRAF6 (12). In contrast, TRAF4 competes with TRAF6 for TB sites on Act1. We found that these TB sites are also critical for TRAF4 binding, implying that TRAF4 and TRAF6 occupy the same binding sites on Act1. In the presence of overexpressed TRAF4, the formation of Act1-TRAF6 complex and downstream signaling is greatly reduced. Since dysregulation of IL-17-mediated signaling can be highly detrimental, TRAF3 and TRAF4 represent multiple layers of modulation (at the receptor versus the adaptor Act1) to ensure the appropriate strengths of signaling in a specific immune response. Future studies will be to determine whether IL-17 induces post-translational modifications of TRAF4, which might be how IL-17 modulates TRAF4’s negative regulation during inflammatory responses. While we show that TRAF4 regulates IL-17-induced activities, it is important to note that TRAF4 deficiency can also affect basal activation of signaling pathways, Act1 activation and even cytokine production. Future studies are required to define TRAF4’s ability to affect the basal activity of Act1. Furthermore, TRAF4 has also been shown to inhibit NOD2-RIP2-mediated NF-κB activation via binding to NOD-Like Receptor, NOD2 (14). Although TRAF4 exerts its negative regulation via distinct mechanisms in different pathways, TRAF4 may act as a general inhibitor restricting inflammatory responses.
Supplementary Material
Acknowledgments
We would like to thank Dr. Derek Abbott and Jill Marinis for providing reagents and for insightful discussions. This work was supported by Senior Investigator Award to Xiaoxia Li from the American Asthma Foundation, National Institutes of Health grants to XL, R01NS071996 and P01HL103453.
References
- 1.Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- 2.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 3.Hu Y, Ota N, Peng I, Refino CJ, Danilenko DM, Caplazi P, Ouyang W. IL-17RC is required for IL-17A- and IL-17F-dependent signaling and the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4307–4316. doi: 10.4049/jimmunol.0903614. [DOI] [PubMed] [Google Scholar]
- 4.Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. 2003;28:226–229. doi: 10.1016/S0968-0004(03)00067-7. [DOI] [PubMed] [Google Scholar]
- 5.Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- 6.Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, Kordula T, Zhang QW, Vallance B, Swaidani S, Aronica M, Tuohy VK, Hamilton T, Li X. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 7.Liu C, Qian W, Qian Y, Giltiay NV, Lu Y, Swaidani S, Misra S, Deng L, Chen ZJ, Li X. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci Signal. 2009;2:ra63. doi: 10.1126/scisignal.2000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. 2007;179:4135–4141. doi: 10.4049/jimmunol.179.6.4135. [DOI] [PubMed] [Google Scholar]
- 9.Hartupee J, Liu C, Novotny M, Sun D, Li X, Hamilton TA. IL-17 signaling for mRNA stabilization does not require TNF receptor-associated factor 6. J Immunol. 2009;182:1660–1666. doi: 10.4049/jimmunol.182.3.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bulek K, Liu C, Swaidani S, Wang L, Page RC, Gulen MF, Herjan T, Abbadi A, Qian W, Sun D, Lauer M, Hascall V, Misra S, Chance MR, Aronica M, Hamilton T, Li X. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol. 2011;12:844–852. doi: 10.1038/ni.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF) Nat Immunol. 2011;12:853–860. doi: 10.1038/ni.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu S, Pan W, Shi P, Gao H, Zhao F, Song X, Liu Y, Zhao L, Li X, Shi Y, Qian Y. Modulation of experimental autoimmune encephalomyelitis through TRAF3-mediated suppression of interleukin 17 receptor signaling. J Exp Med. 2010;207:2647–2662. doi: 10.1084/jem.20100703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiels H, Li X, Schumacker PT, Maltepe E, Padrid PA, Sperling A, Thompson CB, Lindsten T. TRAF4 deficiency leads to tracheal malformation with resulting alterations in air flow to the lungs. Am J Pathol. 2000;157:679–688. doi: 10.1016/S0002-9440(10)64578-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marinis JM, Homer CR, McDonald C, Abbott DW. A novel motif in the Crohn's disease susceptibility protein, NOD2, allows TRAF4 to down-regulate innate immune responses. J Biol Chem. 2010;286:1938–1950. doi: 10.1074/jbc.M110.189308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao H, Qian W, Staschke K, Qian Y, Cui G, Deng L, Ehsani M, Wang X, Qian YW, Chen ZJ, Gilmour R, Jiang Z, Li X. Pellino 3b negatively regulates interleukin-1-induced TAK1-dependent NF kappaB activation. J Biol Chem. 2008;283:14654–14664. doi: 10.1074/jbc.M706931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, Liu L, Qian W, Ransohoff RM, Bergmann C, Stohlman S, Tuohy VK, Li X. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zepp J, Wu L, Li X. IL-17 receptor signaling and T helper 17-mediated autoimmune demyelinating disease. Trends Immunol. 2011;32:232–239. doi: 10.1016/j.it.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.