

Abstract
Orphan genes are defined as genes that lack detectable similarity to genes in other species and therefore no clear signals of common descent (i.e., homology) can be inferred. Orphans are an enigmatic portion of the genome because their origin and function are mostly unknown and they typically make up 10% to 30% of all genes in a genome. Several case studies demonstrated that orphans can contribute to lineage-specific adaptation. Here, we study orphan genes by comparing 30 arthropod genomes, focusing in particular on seven recently sequenced ant genomes. This setup allows analyzing a major metazoan taxon and a comparison between social Hymenoptera (ants and bees) and nonsocial Diptera (flies and mosquitoes). First, we find that recently split lineages undergo accelerated genomic reorganization, including the rapid gain of many orphan genes. Second, between the two insect orders Hymenoptera and Diptera, orphan genes are more abundant and emerge more rapidly in Hymenoptera, in particular, in leaf-cutter ants. With respect to intragenomic localization, we find that ant orphan genes show little clustering, which suggests that orphan genes in ants are scattered uniformly over the genome and between nonorphan genes. Finally, our results indicate that the genetic mechanisms creating orphan genes—such as gene duplication, frame-shift fixation, creation of overlapping genes, horizontal gene transfer, and exaptation of transposable elements—act at different rates in insects, primates, and plants. In Formicidae, the majority of orphan genes has their origin in intergenic regions, pointing to a high rate of de novo gene formation or generalized gene loss, and support a recently proposed dynamic model of frequent gene birth and death.
Keywords: orphan genes, genome evolution, insects, ants (Formicidae)
Introduction
Genomic comparisons enable us to study the emergence and benefits of new genetic material upon which selection can act. Genomic innovations underlying speciation and adaptation can be caused by a variety of processes that include, but are not limited to, chromosomal rearrangements, changes in gene regulation and emergence of new genes, or addition and loss of gene fragments (Ohno 1970; Lynch and Conery 2000; Noor et al. 2001; Prud’homme et al. 2007; Carroll 2008; Khalturin et al. 2009; Kaessmann 2010; Stevison et al. 2011). The application of comparative genomics has uncovered great variation in the gene content in genomes within the same (Tettelin et al. 2005; Stankiewicz and Lupski 2010) and among different species (Tatusov et al. 2003; Gil et al. 2004). In fact, only a small set of genes seems to be universal across kingdoms, whereas the phylogenetic distribution of all other genes is restricted at different levels (Harris et al. 2003). Depending on its age, evolutionary rate, and the mechanism by which a new gene has emerged, it shares different degrees of similarity to genes in other species. Most extreme are orphan genes, which lack detectable homology to a gene in other lineages but represent up to one-third of all genes in eukaryotes (Khalturin et al. 2009; Tautz and Domazet-Lošo 2011). Here, we will distinguish between taxon-specific orphan genes (TSOGs), which are characterized by the lack of homology to genes outside of a focal taxonomic group, and species-specific orphan genes (SSOGs), which are the subset of TSOGs that appear to be strictly species-specific according to the taxon sampling; true SSOGs lack homology to any gene from any species. Note that any SSOG may become relabeled as a TSOG as soon as taxon sampling becomes denser.
In general, the variability of gene content across species suggests that gene loss (Krylov et al. 2003) and the emergence of new genes (Long et al. 2003) play a significant role in both genome evolution and phenotypic evolution (Long et al. 2003; Khalturin et al. 2009; Chen et al. 2010). The contribution of new genes to important species-specific adaptations can involve both the morphological level (Khalturin et al. 2008; Milde et al. 2009) as well as the genetic basis for responses to changing environmental stimuli (Colbourne et al. 2011; Donoghue et al. 2011; Voolstra et al. 2011). For example, orphan genes can be involved in the creation of morphological innovation as shown in Hydra sp. In Hydra sp., Khalturin et al. (2008) uncovered a new gene that encodes a protein regulating tentacle formation. Other Hydra sp. orphan genes are involved in the evolution of the cnidarian nematocyst (Milde et al. 2009). In human, it has been suggested that orphan genes play an important role in early brain development (Zhang et al. 2011). Environmentally responsive genes in corals, which show traces of positive selection are often TSOGs (Voolstra et al. 2011). In Daphnia, orphan genes make up more than a third of the genome and are the most environmentally responsive genes in the genome (Colbourne et al. 2011). In plants, orphan genes are also enriched for responses to a variety of abiotic stresses (Donoghue et al. 2011).
But where do new genes in general, and orphan genes in particular, come from? Multiple studies have addressed the molecular genetic mechanisms responsible for the formation of new genes (Long et al. 2003; Zhou et al. 2008; Kaessmann 2010; Wu et al. 2011). These studies identified a variety of genetic mechanisms involved in creating new genes, including gene duplication, gene fusion and fission, exon shuffling, recruitment of new exons from mobile element sequences, retroposition, lateral gene transfer, and de novo origination (i.e., from previously noncoding sequence). Systematic studies on the evolutionary origin of orphan genes in primates (Toll-Riera et al. 2009) and the plant Arabidopsis thaliana (Donoghue et al. 2011) indicate that gene duplication and exaptation from transposable elements (TEs) are the major forces driving the emergence of orphan genes. Another study investigating the emergence of new Drosophila genes (not restricted to orphan genes) corroborated the dominant role of gene duplication but also suggested that surprisingly many genes (∼12%) seem to have originated de novo, that is, from previously noncoding sequences or RNA coding sequences (CDS) (Zhou et al. 2008). Similarly, a recent study has estimated that as many as 10 protein-coding genes emerge de novo in humans per 1 Myr (Wu et al. 2011), contradicting the previous assumption that de novo origination of new genes is very rare (Long et al. 2003). The relative contribution of each of the genetic mechanisms to the creation of new genes remains controversial and seems to vary considerable between taxa (discussed later).
We here investigate rates and genetic mechanisms of orphan gene emergence across insects with a focus on the Formicidae (ants) clade that includes seven ant species with recently sequenced genomes. The ecological prevalence and diversity of insects makes them an ideal object for comparative genomics research. Almost 1 million insect species have been described so far, and it is believed that the total number ranges between 2.5 and 10 million (Grimaldi and Engel 2005). Moreover, insects display a huge variety of structural, physiological, and behavioral adaptations (Grimaldi and Engel 2005), resulting in high rates of speciation, diversification, and adaptation. Overall, comparative genomics across insect genomes benefits from a large taxon sampling covering more than 350 Myr (n > 20; supplementary fig. S1, Supplementary Material online) and provides a promising resource for the study of genomic innovations. With the sequencing of seven ant genomes and the honey bee genome, we can now also study genome evolution in the context of social versus solitary insects (discussed later).
Among the “big four” insect orders that account for approximately 80% of all described insect species, Diptera has been the most “genomically” sampled order with 15 available genomes (supplementary fig. S1, Supplementary Material online). Although genomes are still scarce for Coleoptera and Lepidoptera, taxon sampling has strongly increased for Hymenoptera, which are now represented by nine genomes. These nine fully sequenced hymenopteran species comprise the solitary jewel wasp (Nasonia vitripennis), the eusocial honey bee (Apis mellifera), and seven eusocial ants. The large number of available genomes and variation in important life history traits currently renders the Hymenoptera the only insect order that has a number and diversity of taxa comparable with Dipterans/Drosophilidae so that these two insect orders are well suited for cross-taxa comparative genomics analyses. The nine hymenopteran species span a broad evolutionary time scale of approximately 150–200 Myr (Grimaldi and Engel 2005; Werren et al. 2010), with the speciation times of the sequenced ants spanning approximately 100 Myr (Brady et al. 2006; Moreau et al. 2006; Gadau et al. 2012), whereas the Drosophila clade is characterized by a higher density of very closely related species (12 species with a last common ancestor 40–60 Ma; Tamura et al. 2004; Markow and O’Grady 2007).
Here, we concentrate on genomic innovation in Hymenoptera with a focus on orphan genes in ants. The sequenced seven ant species represent important evolutionary transitions including fundamental changes in nutrition (e.g., carnivores, herbivores specialized on seeds, fungivores, or omnivores), expansion into new habitats (from terrestrial to arboreal), or social organization (supplementary text S1, Supplementary Material online, for details and Gadau et al. 2012, for a review). We compare our results to the other available insect genomes (28; supplementary fig. S1, Supplementary Material online). In particular, we compare rates of orphan gene emergence between Hymenoptera/Formicidae and Diptera/Drosophilidae. We also give a comprehensive overview of the structure and genetic origin of SSOGs in the seven recently sequenced ant genomes. Finally, the comparison among insect and arthropod outgroup genomes allows deriving expected frequencies of orphan genes and thus provides a foundation for future insect genome projects. Our analyses also address the role of new genes for the evolution of sociality and social evolution within ants and Hymenoptera. As such, our results will build a baseline for further studies on genomic innovations in insects in general and Hymenoptera in particular.
Materials and Methods
Thirty Arthropod Genome Data Set
Genome data of 30 arthropod species were obtained from different sources (table 1), which included protein, CDS, and gene feature files.
Table 1.
Data Set Overview: Genomes and Selected Features of 30 Arthropod Species
| Species | Genome Version | Source | Abbreviation | Genes | Missing Genes | SSOGs Standard | SSOGs Refined | Distance to MRCA |
|---|---|---|---|---|---|---|---|---|
| Aedes aegypti | L1.2 | [V] | Aaeg | 17,399 | 1,161 | 1,105 | 52 | |
| Atta cephalotes | 1.2 | [H] | Acep | 18,093 | 989 | 2,074 | 1,970 | 10 |
| Acyrthosiphon pisum | 1 | [A] | Acyp | 34,821 | 12,522 | 12,151 | 280 | |
| Acromyrmex echinatior | 2.0/3.8 | [H] | Aech | 17,278 | 1,253 | 2,730 | 2,644 | 10 |
| Anopheles gambiae | P3.6 | [V] | Agam | 14,324 | 840 | 769 | 150 | |
| Apis mellifera | 2 | [H] | Amel | 11,062 | 223 | 431 | 289 | 140 |
| Bombyx mori | 2 | [S] | Bmor | 14,623 | 2,866 | 2,701 | 285 | |
| Camponotus floridanus | 3.3 | [H] | Cflo | 17,064 | 676 | 1,896 | 1,761 | 115 |
| Culex quinquefasciatus | J1.2 | [V] | Cqui | 18,882 | 902 | 821 | 52 | |
| Drosophila ananassae | 1.3 | [F] | Dana | 15,070 | 944 | 810 | 12 | |
| D. erecta | 1.3 | [F] | Dere | 15,048 | 666 | 558 | 4 | |
| D. grimshawi | 1.3 | [F] | Dgri | 14,986 | 811 | 702 | 32 | |
| D. melanogaster | 5.37 | [F] | Dmel | 13,914 | 318 | 230 | 4 | |
| D. mojavensis | 1.3 | [F] | Dmoj | 14,595 | 1,118 | 948 | 24 | |
| D. persimilis | 1.3 | [F] | Dper | 16,878 | 878 | 755 | 1 | |
| D. pseudoobscura | 1.3 | [F] | Dpse | 16,029 | 567 | 464 | 1 | |
| D. pulex | 1.1 | [J] | Dpul | 30,907 | 13,709 | 13,181 | 470 | |
| D. sechellia | 1.3 | [F] | Dsec | 16,471 | 645 | 527 | 1 | |
| D. simulans | 1.3 | [F] | Dsim | 15,415 | 637 | 522 | 1 | |
| D. virulans | 1.2 | [F] | Dvir | 14,491 | 792 | 680 | 24 | |
| D. willistoni | 1.3 | [F] | Dwil | 15,513 | 1,230 | 1,105 | 36 | |
| D. yakuba | 1.3 | [F] | Dyak | 16,082 | 959 | 829 | 4 | |
| Harpegnathos saltator | 3.3 | [H] | Hsal | 18,564 | 622 | 1,919 | 1,391 | 125 |
| Ixodes scapularis | W1.1 | [V] | Isca | 20,486 | 7,007 | 6,677 | 550 | |
| Linepithema humile | 1.2 | [H] | Lhum | 16,116 | 678 | 1,448 | 1,349 | 120 |
| Nasonia vitripennis | 1.2 | [H] | Nvit | 18,822 | 150 | 2,305 | 2,191 | 150 |
| Pogonomyrmex barbatus | 1.2 | [H] | Pbar | 17,189 | 729 | 2,173 | 2,054 | 105 |
| Pediculus humanus | U1.2 | [V] | Phum | 10,774 | 1,176 | 1,096 | 280 | |
| Solenopsis invicta | 2.2.3 | [H] | Sinv | 16,522 | 1,049 | 926 | 885 | 60 |
| Tribolium castaneum | 3 | [B] | Tcas | 16,645 | 3,757 | 3,623 | 300 |
Note.—For each genome, the species name, genome version, and download source are given. A: AphidBase (Legeai et al. 2010); B: BeetleBase (Kim et al. 2010); F: FlyBase (McQuilton et al. 2012); H: Hymenoptera Genome Database (Munoz-Torres et al. 2011); J: DOE Joint Genome Institute (http://www.jgi.doe.gov/); S: SilkDB (Duan et al. 2010); V: VectorBase (Lawson et al. 2009). Abbreviation: Four-letter species abbreviation used throughout this manuscript; Genes: Number of protein-coding genes in the OGS (i.e., excluding possibly missing genes); SSOGs: derived with standard methods (Standard) or comprehensive filtering (Refined); Distance to the MRCA: Evolutionary distance (Myr) to the MRCA node in the phylogenetic tree of these 30 arthropods.
Divergence times (table 1) were obtained from the timetree.org database (Hedges et al. 2006) and manually reconciled if necessary. These estimates served as approximate values, which allowed normalizing for different phylogenetic branch lengths when determining the rate of SSOG emergence.
Identification of Orphan Genes
SSOGs were identified by filtering annotated proteins against two data sets, including 30 arthropod proteomes and all SwissProt taxonomic divisions excluding invertebrates. For all non-ant genes, three filtering stages were used, whereas for the ant genes, a forth filtering step was added, as follows:
Eliminate all proteins that have a BLAST hit with E ≤ 10−3 in any of the other arthropod proteomes, using default settings otherwise.
Eliminate all proteins that have a BLAST hit with E ≤ 10−3 in any of SwissProt taxonomic divisions other than invertebrates, using default settings otherwise.
Eliminate all proteins that have a BLAST hit with E ≤ 10−3 in any of the other arthropod proteomes when low complexity filtering is deactivated.
(For ants only) Accept as SSOG only if no evidence could be found that homologous genes were missed in other genomes (see Materials and Methods: Identification of missing gene models).
TSOGs were identified as genes annotated in any species part of the focal taxonomic group that lack sequence similarity to genes outside of the focal group. The identification was based on an all versus all BLASTP search; sequence similarity was assumed if a BLAST hit with E < 10−3 was found. BLAST hits of genes in any species from the focal group were ignored.
Identification of Missing Gene Models
Tentative ant SSOGs were compared against the DNA sequence of all hymenopteran species and Drosophila melanogaster. We used TBLASTN (low complexity filtering activated, E < 10−5) to identify the most likely position of the missing gene model in the genome. If such a BLAST hit was found, GeneWise v2.4.1 (Birney et al. 2004) was employed to align the protein sequence of the tentative SSOG to the scaffold strand and position specifically. For performance reasons, the candidate genome region determined by TBLASTN was extracted before running GeneWise using the TBLASTN match coordinates plus 50 kb up- and downstream. Only GeneWise models with a score >35, coverage of the query sequence >75%, and zero indels were accepted. By accepting only zero-indel models, putative pseudogenes were not identified as missing gene models and falsely assumed to be homologous functional genes.
Modeling Rate of Orphan Gene Emergence
A linear model was used to relate the distance to the most recent common ancestor (MRCA) node in the insect phylogenetic tree and the number of SSOGs using R and the lm() function. As a base model, all non-formicid insect species from the 30 arthropod genome data set with less than 300 Myr distance to their MRCA node except Amel were used. We excluded Amel due to its current bias in genome annotation, which is depleted for orphan genes. R’s predict() function was used to determine the confidence and prediction intervals.
Origin of SSOGs in Ants
Gene duplication was inferred for gene pairs with significant sequence similarity. If the two encoding proteins shared a local alignment with E < 10−3 as determined by BLASTP, they are considered to be gene duplicates.
Nondeleterious frame shift mutations across ant SSOGs were inferred as follows: Each SSOG was compared against the full-genome CDS of all other hymenopteran species plus Dmel and Tcas using TBLASTN with E < 10−5. Frame shifts were inferred only if both of the following criteria were met: 1) one or more significant BLAST hits in CDS across these reference species on the sense strand and out of the regular frame, that is, +2 or +3; 2) no significant BLAST hit in CDS in frame +1 or on the antisense strand.
Alternative reading frames were screened among ant SSOGs as follows: Each SSOG protein sequence was compared against all CDS within the same species using TBLASTN with E < 10−5. Self-hits, in-frame matches, and matches on the anti-sense strand were ignored. For the remaining candidate protein/CDS pairs, the genomic locations were obtained from the gff, and only candidates, which are encoded by the same genomic locus were accepted as proteins from alternative reading frames.
Overlap with TEs was identified based on CDS overlaps of ant SSOGs with TE coordinates using BEDtools (Quinlan and Hall 2010). TEs were identified with RepeatMasker (v3.3.0, Smit et al. 2010) and default settings using homology to known metazoan TEs part of Repbase (20110920, Jurka et al. 2005).
Horizontal gene transfer (HGT) candidates using the domain-based approach were determined in a three-step process. First, all proteins in the NCBI nonredundant database (from March 30, 2011) were annotated against Pfam-A v25.0 (Punta et al. 2011) (see Identification of protein domains). For each domain, the relative frequency was determined of occurring in proteins from selected taxonomic units within the NCBI Taxonomy. All domains with a relative frequency of ≥95% in any one of these nodes, Bacteria, Archaea, Viridae, Fungi, and Viridiplantae, were saved as a list of candidate alien domains. Second, the proteomes of the 30 arthropods were annotated against Pfam-A. Overlapping domains were resolved by recursively removing the less significant domain(s) in overlap regions. As a second candidate list of alien domains, those domains were determined which fulfilled the following three criteria: 1) the domain was found in at least one ant species and showed a score above gathering threshold; 2) the domain was not found with a score above the gathering threshold in any non-formicid arthropod species; and 3) the domain showed a difference in E-values of at least 10-fold between the ant match and the best non-Formicidae arthropod match. Third, the final list of alien domains in ants was generated by cross-referencing the two lists of candidate alien domains.
Overlapping gene models were inferred as pairs of genes whose CDS overlaps by at least 30 nt on opposite strands (“CDS–CDS different-strand overlaps”). CDS coordinates were obtained from gene feature files and BEDtools (Quinlan and Hall 2010) was used to intersect these CDS features and identify overlaps ≤30 nt.
Intergenic matches in other hymenopteran genomes were determined using the same method as used by Donoghue et al. (2011): Ant SSOGs were screened against all hymenopteran scaffolds with TBLASTN (E < 10−3). BLAST hits were concatenated if they were nonoverlapping and less than 4,000 nt apart. The coverage was determined as the percentage of aligned codons to the full protein length, and only matches with a bit score >30 and a coverage >10% an intergenic match was inferred.
Expression Analysis
Expressed sequence tags (ESTs) were mapped to the scaffolds using GMAP (Wu and Watanabe 2005) with default settings. Mapped ESTs were then intersected with annotated gene models using BEDtools (Quinlan and Hall 2010), and expression support was inferred in case an EST overlaps with a gene model (i.e., no distinction is made between fully and partially supported gene models).
RNA-seq data for Harpegnathos saltator and Camponotus floridanus were downloaded from NCBI GEO using accession number GSE22680 (Bonasio et al. 2010). Raw sequence reads were mapped using Bowtie + Tophat (Langmead et al. 2009) allowing 1 mismatch and up to 50 alignments per read (“-v 1 -k 50 ––best”) and default parameter values otherwise. Expression levels for SSOGs were quantified with these maps using Cufflinks (Trapnell et al. 2010), correcting for fragment bias (“––frag-bias-correct”) and uncertain alignment location (“––multi-read-correct”) and using default parameter values otherwise.
Real-time quantitative polymerase chain reaction (RT-qPCR) was used to test functional gene status of a different-strand CDS-overlapping gene pair involving an SSOG by employing QuantiTect SYBR Green (Qiagen) one-step chemistry. As template, 20 ng of DNase I treated total RNA derived from a pool of Pogonomyrmex barbatus specimens representing queens and workers of several developmental stages was used. Primers were designed to span partially nonoverlapping fragments of antisense-encoded genes to prevent amplification of transcripts derived from the sense strand. To assess target gene expression levels, cycle threshold (Ct) values were compared with β-actin (PB20873, forward primer 5′-TCAAGGTGTCATGGTCGGTA-3′, reverse primer 5′-CCATGCTCGATCGGATATTT-3′) serving as reference gene, and with negative controls excluding reverse transcriptase to correct for DNA contamination.
Identification of Protein Domains
Protein domains were annotated in predicted peptides using the Pfam-A v25.0 database (Punta et al. 2011). Unless otherwise denoted, only domain annotations above the domain-specific gathering threshold provided by Pfam were considered.
Results
Abundance of SSOGs Is Time Dependent
The primary basis of our results is the robust and accurate determination of SSOGs across 28 insect and two arthropod outgroup species (supplementary fig. S1, Supplementary Material online). We first applied a standard method that identifies an SSOG as a gene encoding a protein that lacks homology to any predicted peptide from other arthropod genomes (table 1, SSOGs Standard). We applied three additional filters to improve these preliminary estimates and exclude potentially false-positive SSOGs. First, we discarded proteins that matched potentially missing gene models in Hymenoptera. Second, we discarded proteins that matched a homolog when including low complexity protein regions. Third, we discarded proteins that matched noninvertebrate proteins, assuming that proteins missing in arthropods but present in other taxonomic divisions likely reflect contamination or selective retention and extinction between lineages (but see later for discussion of HGT as an evolutionary origin of SSOGs). These three filtering steps consistently lowered the estimates of SSOGs in all tested genomes (table 1, SSOGs Refined vs. SSOGs Standard) and had the largest impact on SSOG estimates in the ant genomes. The massive reduction of orphan genes in ants after the three filtering steps is in large part due to the fact that no homology information from other ants was originally used in the annotation of the seven ant genomes, with the exception of C. floridanus and H. saltator (Bonasio et al. 2010). Our estimates of SSOG counts in ant genomes are significantly lower than those originally reported by the individual genome papers (Bonasio et al. 2010; Nygaard et al. 2011; Smith CD, et al. 2011; Smith CR, et al. 2011; Suen et al. 2011; Wurm et al. 2011). This decrease in orphan genes stems not only from the addition of missing gene models based on homology and from a more conservative definition of orphan genes in this study (BLAST E > 10−3 in a database of 520,428 proteins) but also from an increased taxon sampling and revisions in the official genome annotation. SSOG numbers changed from 9,361 to 1,970 in Atta cephalotes (Suen et al. 2011), from 5,183 to 2,644 in Acromyrmex echinatior (Nygaard et al. 2011), from 2,982 to 885 in Solenopsis invicta (Wurm et al. 2011), and from 7,184 to 1,349 in Linepithema humile (Smith CD, et al. 2011). Notably, these four studies used only a handful of reference species and included only two other hymenopteran species, Api. mellifera and N. vitripennis, to determine the number of putative orphan genes. Thus, accurate gene annotation benefits significantly from comprehensive comparison of multiple closely related genomes, but at the same time there still remains a significant percentage of SSOGs in each ant species. Note that all subsequent analyses are based on the SSOG Refined data set.
Averaged over all included insect and arthropod outgroup species, approximately 13% of all genes lack a homologous protein in any other species; this estimate falls within the expected range of 10–30% for SSOGs in other studies (Wilson and Hölldobler 2005; Wilson et al. 2005, 2007; Khalturin et al. 2009; Tautz and Domazet-Lošo 2011). SSOGs are not distributed uniformly across the investigated species. Species that are separated by greater phylogenetic distances from their nearest neighbor appear to have more SSOGs compared with species from well-sampled clades (table 1). Orphan genes are defined based on the absence of detectable homology in other species. Therefore, phylogenetic context, that is, the number of species and distance to other species, is a critical parameter in determining orphan genes (Khalturin et al. 2009; Tautz and Domazet-Lošo 2011). We examined the relationship between SSOG abundance and distance to the MRCA and found that, in general, this relationship can be well approximated with a linear model. We first built a linear model using SSOG counts observed in all insects relative to their estimated divergence time to MRCA excluding ants and Api. mellifera. This model explained a significant proportion of the observed variance ([formula: y = 8.464x + 541.709] R2 = 0.85; P = 6.9 × 10−7; fig. 1). We excluded ants in this analysis because we wanted to test whether SSOG counts observed in these recently sequenced genomes fall within the expected range based on previously sequenced insect genomes. We also excluded Api. mellifera because its gene annotations are severely biased toward genes with homology (Weinstock et al. 2006) and may thus be artificially depleted for orphan genes.
Fig. 1.—
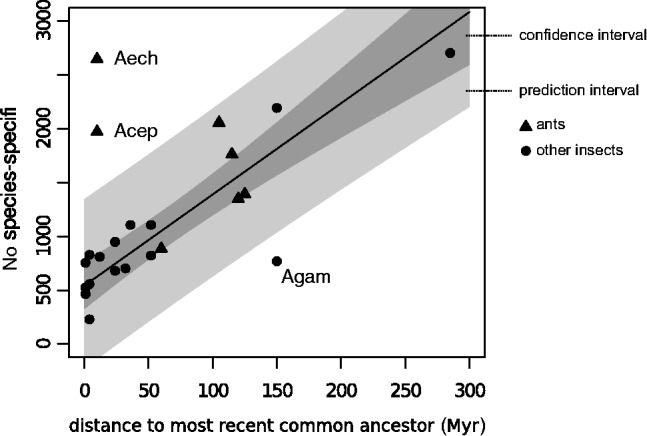
Abundance of SSOGs and their dependence on the distance to the MRCA (table 1). SSOGs per species were plotted against the distance to the MRCA node in the phylogenetic tree. A linear regression (solid black line) was constructed to fit the observed SSOG counts from 12 Drosophilidae, 3 Culicidae, Bombyx, Tribolium, and Nasonia (circles, R2 = 0.85). The confidence interval and the prediction interval of the linear model are shown in dark and light gray, respectively. The ant SSOG data points were added after fitting the linear model and are shown as triangles.
Based on the linear model, we expected to find a minimum of 540 SSOGs for any insect species in our data set, plus an additional 85 SSOGs per every 10 Myr elapsed since the MRCA. Thus, even genomes of recently split species should have accumulated several hundred SSOGs; this prediction is consistent with the observed SSOG counts of closely related Drosophila species (table 1). We then tested whether SSOG counts observed in ants fall within the expected range of this distribution. Most ant genomes contain the expected number of orphan genes given their phylogenetic age. However, the two leaf-cutter ants Att. cephalotes and Acr. echinatior (tribe Attini) clearly fall outside of this distribution and have 1,970 and 2,644 SSOGs, respectively, despite very recent common ancestry (∼10 Ma). In fact, the two leaf-cutter ants exhibit the greatest number of SSOGs among all hymenopteran and dipteran genomes sequenced thus far.
Furthermore, we could show that the trends mentioned earlier do not only apply to SSOGs but are also valid for TSOGs. TSOGs not only include SSOGs but also comprise genes that have homologs only within the considered taxonomic group. Thus, TSOGs are more loosely defined as they additionally comprise genes that have emerged at internal phylogenetic branches. Comparison of the abundance and emergence rate of TSOGs for various taxonomic groups of insects (fig. 2) confirm the trends reported above for SSOGs, supporting 1) a rapid emergence of new genes in Attini (compared to D. melanogaster group with a similar age), and 2) an increased rate of orphan gene gain in Hymenoptera compared with Diptera. Thus, compared with other insects, Hymenoptera in general and leaf-cutter ants in particular appear to exhibit elevated rates of orphan gene gains.
Fig. 2.—

Contrasting the abundance and rate of emergence of orphan genes between partially overlapping taxonomic groups of Hymenoptera and Diptera. Each tested group is highlighted by a rectangle and the associated group data, including number of species, distance to the MRCA, total orphan gene count, and rate of orphan gene emergence, are shown on the right side. Branch lengths in the phylogenetic tree are approximate values and were obtained from the timetree.org database (Hedges et al. 2006).
Origin of Orphan Genes
We utilized two approaches to understand the evolutionary processes underlying the origin of orphan genes. First, we identified the physical location of SSOGs in the ant genomes and asked whether they occur in clusters indicative of genomic hotspots for the emergence of SSOGs. Local differences in mutation rates have been documented before, for example, as an increase of nucleotide substitutions surrounding insertions and deletions in eukaryotic genomes (Tian et al. 2008). Orphan gene clusters could therefore exist as a consequence of hotspots of DNA gain and loss which have been reported for primates (Perry 2006; Mefford and Eichler 2009).
Across the seven ant genomes, we determined SSOG clusters based on gene adjacency. We determined that 24–32% of all SSOGs are located in clusters, depending on whether interruption of direct neighbors was allowed (table 1, see Materials and Methods). The median cluster size is two genes, the biggest perfect cluster contains eight SSOGs, and only eleven clusters (<1%) across all seven species contain more than five SSOGs without interruption. In addition, hardly any SSOGs that are located in clusters (∼0.5%) appear to be tandem duplicates of each other, that is, the clusters of SSOGs typically consist of unrelated genes. These data suggest that most ant-specific SSOGs are uniformly distributed throughout the genomes. Consequently, the frequency of SSOG clusters is influenced by the genome-wide abundance of SSOGs: S. invicta has the lowest number of SSOGs (885) and SSOG clusters (18–24, comprising 4–6% of all SSOGs); at the other extreme, Acr. echinatior has the highest SSOG count (2,644) and SSOG clusters (395–430, comprising 38–48% of all SSOGs; table 1). Future investigations are necessary to clarify whether SSOG clustering contributes to the high frequency of SSOGs observed in the two leaf-cutter species and whether any of 11 SSOG clusters with >5 SSOGs in fact represent genomic locations in which SSOGs emerge more frequently, that is, represent a gene gain/loss hotspot.
Second, to address the phylogenetic history of SSOGs in ants, we determined the relative frequency of the different evolutionary origins of ant SSOGs which are detectable by sequence similarity. We neglect some of the mechanisms previously shown to facilitate gene innovation, such as exonization, exon shuffling, and gene fusion and fission (Long et al. 2003; Zhou et al. 2008; Kaessmann 2010; Schmitz and Brosius 2011; Ranz and Parsch 2012) because such mechanisms would retain short homologous regions among genes of other species and are thus by definition not part of our SSOG data set. Instead, we focused on six scenarios (discussed later) that leave genomic signatures inside or in the immediate proximity to SSOGs, which can be detected using a comparative approach and bioinformatics tools.
Gene Duplication
All metazoans sequenced so far carry a substantial number of genes that exist in multiple copies, and many more have likely emerged by gene duplication but subsequently diverged beyond recognition (Zhang 2003). Gene duplication is assumed to occur frequently (Lynch and Conery 2000), although most of the time, one copy eventually becomes pseudogenized. Alternatively, it is possible that the second copy is retained in the genome, either because higher gene dosage or redundancy is beneficial, or because sub- or neofunctionalization has occurred (Zhang 2003). Therefore, one possible scenario to explain the emergence of SSOGs is gene duplication and subsequent divergence of one copy beyond the threshold of detectable similarity (Domazet-Loso and Tautz 2003).
We identified 1,195 SSOGs (9.9%) in the seven ant genomes with detectable sequence similarity to a paralog, that is, another gene in the same species and where this paralog has in turn detectable homologs in other species (table 2). Therefore, gene duplication and divergence appear to happen less frequently in ants than has been reported for primates and plants (>20%, Toll-Riera et al. 2009; Donoghue et al. 2011).
Table 2.
Inferred Origins of Orphan Genes in Different Data Sets and Investigated in Different Studies
| This Study | This Study | Toll-Riera et al. (2009) | Donoghue et al. (2011) | |
|---|---|---|---|---|
| Study | ||||
| Data set | Formicidae SSOGs | Attini SSOGs | Primate shared TSOGs | Arabidopsis thaliana SSOGs |
| Genome size (Mb) | 250–450 | 300–335 | 1,600–2,870 | 125 |
| Genomic TE content (%) | 8–30 | 25–28 | ∼50 | ∼10 |
| Origins (%) | ||||
| Gene duplication | 9.9 | 6.4 | 24 | 22 |
| Overlap with TE | 12.4 | 10.6 | 53 | 10 |
| Frame shift | 2.2 | 2.2 | NA | 7 |
| Overlapping genes | 11.1 | 13.3 | NA | 1 |
| Intergenic match | 43.5 | 61.2 | 6% de novo | 25 |
| HGT | 0.1 | 0.0 | NA | NA |
| Unexplained | 20.8 | 6.3 | 17 | 35 |
Note.—The Formicidae and Attini data sets consist of 12,054 and 4,614 SSOGss, respectively. Genome stats were obtained from the Arabidopsis Genome Initiative (2000), Lander et al. (2001), Bonasio et al. (2010), Nygaard et al. (2011), Smith CD, et al. (2011); Smith CR, et al. (2011), and Suen et al. (2011).
It is possible that among the duplicated gene pairs, the associated SSOG is a spurious open-reading frame (ORF). SSOG function may have been lost, and the sequence divergence may reflect the degeneration of its CDS. To test for this scenario, we performed RT-qPCR on one pair of genes, which includes an SSOG (PB21732) and the corresponding paralog, which has orthologs in other species (PB13276). For both of these genes, low expression could be detected, indicating that at least in this gene pair, both gene copies are expressed and possibly functional. Of course, high-throughput expression data are necessary to validate the activity of duplicate gene pairs involving SSOGs more comprehensively.
Nondeleterious Frame-Shift Mutations
A nondeleterious frame shift mutation in the N-terminal region of the CDS has the power to produce an almost entirely different protein sequence from a slightly altered CDS (Hahn and Lee 2005; Raes and Van De Peer 2005; Okamura et al. 2006) without affecting the regulatory context of the gene. Therefore, genes which have undergone a nondeleterious frame shift mutation are likely expressed and can immediately contribute to an organism’s phenotype.
Across all seven ant genomes, we identified 268 SSOGs (2.2%) that are candidates for frame shift mutations. These candidate SSOGs match CDS translations in other insect species in a different frame on the same strand of DNA. The contribution of frame shifts to orphan gene emergence has not been investigated in primates (Toll-Riera et al. 2009) but has been predicted to contribute even more frequently to the origin of A. thaliana SSOGs (7%, Donoghue et al. 2011). Thus, the creation of new proteins by frame shifts in the CDS may represent an important, but often neglected, mechanism for the emergence of new proteins. Note that we also tested for another mechanism, which has the potential of producing different-frame proteins: Alternative translation start sites in a transcript allow for the co-existence of multiple alternative reading frames in the same transcript (Kochetov 2008). We could, however, find only 12 cases (0.1%) in which an SSOG protein sequence originates from translation of an alternative reading frame in a non-SSOG, suggesting that alternative reading frames virtually never contribute to SSOG emergence in ants.
Overlap with TEs
TEs (or mobile elements) represent “jumping DNA fragments” that are typically 1–10 kb long and can change the genome in various ways by moving or inserting copies of themselves into new genomic locations (González and Petrov 2009). Although it is generally assumed that random insertion of mobile elements is detrimental to the organism, accumulating evidence suggests that TEs can also contribute to adaptation (Kazazian 2004; Gogvadze and Buzdin 2009; González and Petrov 2009). In the majority of documented cases, such adaptations are achieved by TEs modulating gene expression (Fablet et al. 2007; González and Petrov 2009), but TEs have also been shown to disrupt CDSs and facilitate the generation of new genes (Lockton and Gaut 2009; Toll-Riera et al. 2009).
Across the seven ant genomes, we determined the number of SSOGs whose CDSs overlap with TEs from either of the following four categories: short interspersed elements (SINEs), long interspersed elements (LINEs), long terminal repeats, and DNA TEs. Based on these overlaps, 1,496 ant SSOGs (12.4%) may have formed by domesticating TEs into their CDSs. It should be noted that overlap between a TE and the CDS of an orphan gene does not necessarily qualify them as exaptations as previous studies have sometimes done (Toll-Riera et al. 2009). Employing this previously used definition, the frequency of predicted TE exaptation during orphan gene emergence in ants is comparable to A. thaliana, where TEs have been predicted to affect approximately 10% of the SSOGs (Donoghue et al. 2011). In contrast, Toll-Riera et al. (2009) reported that TEs overlap with more than half of the orphan genes in primates, suggesting that TEs might be more frequently involved in the creation of new genes in primates. An increased rate of TE exaptation may be related to a much higher abundance of TEs in primates than in the investigated plant and insect species (table 2). In particular, Alu elements are a major type of SINEs in primates, and these elements often contain motifs that can readily turn into functional splice sites allowing exonization (Gal-Mark et al. 2008). These conditions may lead to the observed higher frequency of new genes whose generation is driven by mobile elements in primates, compared with other taxa.
Horizontal Gene Transfer
In recent years, evidence has accumulated that HGT is not limited to exchange between bacteria but also occurs in eukaryotic genome evolution (Gladyshev et al. 2008; Dunning Hotopp 2011; Sommer and Streit 2011; Christin et al. 2012). In numerous insect and arthropod genomes, putative HGTs have been found (Dunning Hotopp et al. 2007; Nikoh et al. 2008; Klasson et al. 2009; Moran and Jarvik 2010; Werren et al. 2010; Grbić et al. 2011; Zhu et al. 2011; Acuna et al. 2012). HGT has not been quantified by the other orphan gene studies in primates (Toll-Riera et al. 2009) and Arabidopsis (Donoghue et al. 2011). However, HGT has been suggested as a principal source for orphan genes (Tautz and Domazet-Lošo 2011) and may well have occurred in ant lineages, in particular because some of the sequenced ant species have been living in very close symbioses with other organisms including bacteria (e.g., gut endosymbionts—Blochmannia spp. in C. floridanus) and fungi (both leaf-cutter ants) for millions of years (Gil et al. 2003). In addition, the general insect endosymbiontic bacteria of the genus Wolbachia have been found in many ant species (Russell 2011) including Attine ants (Van Borm et al. 2001) and S. invicta (Bouwma et al. 2006). However, this close association of some ant species with bacteria prevented sampling of pure ant genomic DNA and made subsequent in silico filtering of bacterial sequences necessary (Bonasio et al. 2010; Nygaard et al. 2011), which interfere with the detection of HGT.
We screened all SSOGs against the SwissProt database using BLAST not only to identify possible candidates for HGT but also to exclude potentially spurious SSOGs, which may have arisen by contamination during genome sequencing (table 1; SSOG Standard vs. SSOG Refined). Eight ant genes were identified in filtering steps between the Standard and Refined sets of SSOGs (see Materials and Methods) as potential contaminants or products of HGT (supplementary table S2, Supplementary Material online). In addition, we used a phylogenomic domain-based approach to determine protein domains in the refined set of SSOGs that have not been identified in the other arthropod genomes but could have been transferred from other organisms (see Materials and Methods). This domain-based approach using HMMer complements BLAST-based approaches; it can achieve a higher sensitivity and identify short functional meaningful protein components, even if the rest of the protein has diverged or has been lost. Our domain-based approach identified six additional ant genes containing nonarthropod domains, suggesting that the seven ants have acquired at most 14 (0.1%) ant SSOGs through HGT (supplementary table S2, Supplementary Material online). It is, however, possible that the number of predicted HGTs across the seven ant species is an underestimate considering thorough elimination steps of bacterial sequences before and after genome assembly.
Overlapping Genes
Eukaryotic genomes harbor an abundance of overlapping genes (Sanna et al. 2008; Soldà et al. 2008). In most cases, overlapping genes involve a noncoding gene feature (intron or untranslated region) from one gene and part of the CDS from another gene, whereas only a small fraction of overlapping gene pairs involve two CDSs (“CDS-CDS overlap”). Pairs of genes with CDS overlaps can be located on the same DNA strand (“same-strand”) or on opposite strands (“different-strand”), with different-strand CDS-overlaps being much more abundant (Sanna et al. 2008). In general, it is thought that CDS–CDS overlap is selected against due to additional constraint on sequence evolution because multiple proteins have to be optimized simultaneously (“structural constraint,” Keese et al. [1992]); gene overlap also introduces potential for transcriptional (Prescott and Proudfoot 2002; Osato et al. 2007) or translational (Yu et al. 2007) interference (“regulatory constraint”) between overlapping genes.
We observed a high rate of overlapping gene models (different-strand CDS overlaps) in some of the ant genomes irrespective of their orphan gene status (supplementary table S3, Supplementary Material online). Only 21% of these overlapping genes involve SSOGs, suggesting that different-strand CDS overlaps are a general phenomenon of gene organization in ant genomes. Nonetheless, 1,341 (11.1%) ant-specific SSOGs are involved in different-strand CDS overlaps. Similarly to frame shifts, these data implicate the reuse of existing ORFs in other frames, and our results support the frequent emergence of ant SSOGs in the anti-sense strand of existing CDS. Gene overlaps were not considered as a source of orphan genes in primates (Toll-Riera et al. 2009), but affect approximately 1% of SSOGs in Arabidopsis (table 2). Although gene overlaps are thus rarer in Arabidopsis than in ants, SSOGs were much more frequently involved in gene overlaps in Arabidopsis than non-SSOGs (Donoghue et al. 2011). Thus, existing ORFs may become functionalized in another frame, whereas the original protein function is still conserved, in particular in some ant species.
The high frequency of gene overlaps in ants compared with Arabidopsis and other insects and the possibility of technical artifacts calls for further in depth studies. In fact, the frequency of CDS–CDS overlaps may be influenced by difficulties in the accurate prediction of gene and exon–intron structure in some of the ant genomes. Indeed, we found a high incidence of CDS–CDS overlaps in Att. cephalotes, L. humile, and P. barbatus, whose genomes were all annotated using MAKER (Cantarel et al. 2008). It is possible that overlaps in these genomes are inflated due to technical artifacts where noncoding regions were mistaken for a protein-coding region, suggesting that some gene models in these draft genomes require refinement.
By contrast, the genomes of Acr. echinatior, C. floridanus, and H. saltator were all annotated using the pipeline of BGI (http://www.genomics.cn/), and both C. floridanus and H. saltator genomes were sequenced and computationally processed in the same lab and with exactly the same methods (Bonasio et al. 2010). Hence, it is very unlikely that technical artifacts can explain the discrepancy in the number of overlapping genes between C. floridanus (which compares well with all previously sequenced insects) and the two ant species Acr. echinatior and H. saltator (which have increased frequencies of gene overlaps; supplementary table S3, Supplementary Material online). Furthermore, the H. saltator genome is larger than the C. floridanus genome, suggesting that a putative increase of overlapping genes in H. saltator cannot be explained by overall increase in gene density. With RT-qPCR, we specifically validated the expression of PB27018, a gene with significant different-strand CDS overlap to gene PB23252 in P. barbatus. Additionally, all SSOGs involved in gene overlaps in C. floridanus and H. saltator have RNA-seq support (Bonasio et al. 2010). However, partial EST or RNA-seq support for these genes may be ambiguous because these sequence data are typically not strand specific. Hence, such expression data can only confirm overlapping gene models, if both overlapping genes have also unique exons.
Signals from Conserved Intergenic Regions in Formicidae and Hymenoptera
With the five previously presented scenarios, the origin of 4,306 (36%) of all 12,054 ant SSOGs could be explained. For the remaining 7,748 SSOGs having thus far unknown origin, we tried to find any traces of their ORF in other hymenopteran genomes. Such ORF traces were detected by screening the proteins encoded by the remaining SSOGs against the six-frame translated scaffolds or chromosomes of all available hymenopteran species which, by excluding the five previous scenarios of origin, would have to occur in intergenic regions.
We found significant scaffold matches for 5,239 (43.5%) SSOGs in at least one other hymenopteran species. Among the now six scenarios of origin we considered, this approach identified the largest proportion of SSOGs (table 2). These SSOGs with intergenic matches could represent genes that were actually generated by one of the scenarios of origin tested earlier, but whose signature cannot be unambiguously detected any more. Alternative scenarios comprise generalized gene loss or pseudogenization in other lineages, and de novo gene formation from previously noncoding regions.
We found an abundance of SSOGs for which gene models with a slightly disrupted and possibly nonfunctional ORF could be constructed in other species (supplementary fig. S2A, Supplementary Material online), supporting an abundance of pseudogenes that are homologous to SSOGs. At the same time, recent studies have highlighted the unexpectedly high rate of de novo formation of new genes in non-CDSs in Drosophila (Zhou et al. 2008) and human (Wu et al. 2011). In general, the expected genomic signatures of pseudogenization of orthologs outside a focal lineage and de novo generation of genes are similar (supplementary text S6, Supplementary Material online): Searching tentative SSOGs against genome sequences of related species should result in significant hits in noncoding regions, and the number of hits should decrease as the phylogenetic distance increases because both pseudogenized genes and noncoding regions are not constrained by the necessity to retain a fully functional ORF (Zhang and Gerstein 2004; Donoghue et al. 2011). Hence, an unequivocal inference of either one scenario requires a well-sampled phylogeny and confirmation with transcription and translation evidence (Guerzoni and McLysaght 2011). In some cases, the two processes of pseudogenization and de novo gene formation might even be linked, when a pseudogene acquires a new function (Balakirev and Ayala 2003; Carvunis et al. 2012).
Overall, we expect that most of the SSOGs with intergenic matches derive from de novo formation or generalized gene loss, but we cannot estimate the extent of SSOGs, which were possibly missed in the identification of the previously tested mechanisms due to sequence divergence. As additional ant genomes become available, along with full-genome alignments among all ant genomes, the origin of SSOGs with intergenic matches and the issue of gene loss versus de novo gene formation can be revisited again.
Unexplained Origin
With the aforementioned six mechanisms, the evolutionary origin of 9,545 ant-specific SSOGs (79%) could be explained. Similar to other studies in primates (17%, Toll-Riera et al. 2009) and Arabidopsis (36.67%, Donoghue et al. 2011), we could not predict the evolutionary origin for 21% ant SSOGs (2,509 genes). This suggests a general trend whereby the evolutionary origin of some new genes cannot be reconstructed after a few million years of divergence. Among the ant-specific SSOGs with unexplained origin, the majority are single copy genes (92.1%). Future in-depth studies may want to first focus on the 197 ant SSOGs that exist in multiple copies, because SSOGs with multiple retained copies may have a high chance in having lineage-specific function.
Discussion
Abundance of Orphan Genes among Insect Taxa
The comparative analyses of 28 insect and 2 outgroup arthropod genomes allowed us to identify SSOGs and TSOGs. It should be noted that our analysis relies on existing gene annotations, which tend to be conservative and may leave some genes, in particular orphan genes that lack homologs in reference species, undiscovered. With several filtering steps, however, we tried to maximize the fidelity of orphan gene identification among the annotated arthropod genes. Moreover, our Refined orphan gene data set (table 1) consists only of proteins with no sequence similarity to genes in other species, including similarity in protein low complexity regions. As such, the Refined orphan gene data set includes only genes with truly novel protein sequences. Overall, the presented estimates of orphan genes are conservative and may thus underestimate their true abundance.
In general, much of the observed variation in SSOG counts among the examined insect genomes can be explained by the evolutionary distance of the sampled species (fig. 1). However, three insect genomes significantly deviate from the expected SSOG counts: Genomes of the two leaf-cutter species, Att. cephalotes and Acr. echinatior, appear to be enriched for SSOGs, and the genome of the mosquito Anopheles gambiae appears to be depleted of SSOGs. Even if normalized for differences in evolutionary distances, we found that hymenopteran genomes contain more SSOGs and TSOGs in comparison with dipterans.
As another general trend, our comparison uncovered an accelerated accumulation of SSOGs between younger species pairs or clades in both Hymenoptera and Diptera (fig. 1). Genomes of sister species that split a few million years ago typically contain more than 500 SSOGs, whereas the long-term gain of SSOGs is only approximately 85 genes per 10 Myr (fig. 1). Assuming that at the time of speciation, there are no or very few orphan genes present between the separating populations, the presence of several hundred orphan genes in closely related sister species can only be explained by a strongly accelerated rate of orphan gene emergence immediately after speciation. These results support findings from broad phylogenetic analyses of orphan gene emergence in which a high rate of new gene formation is consistently inferred in the most recent evolutionary history, in particular for mouse, Drosophila, and Arabidopsis (Tautz and Domazet-Lošo 2011). Overall, these findings suggest that new genes may be formed frequently over a very short evolutionary time, but despite rapid sequence evolution and the potential that some of these genes are adaptive, a large number of newly generated genes are likely to be purged over a longer evolutionary time frame. Thus, the imbalance between a rapid generation of orphan genes early in the history of a taxon and lower rate of orphan gene retention over longer phylogenetic distances probably causes the observed lower average rate of net gains of orphan genes in older clades or in longer phylogenetic branches.
In Formicidae, we found a 3-fold difference between the lowest and highest numbers of SSOGs among the seven ant genomes (table 1), indicating that the Formicidae gene content shaped by birth and death of orphan genes is highly variable. Across the ant genomes, no major genomic hot spots, characterized by a high density of neighboring SSOGs, could be identified. Overall, Formicidae SSOGs appear to be uniformly distributed throughout the genome, and if at all, they occur in small clusters of two to three neighboring SSOGs. We note that a uniform distribution of orphan genes across chromosomes has already been reported in Arabidopsis (Donoghue et al. 2011). Similar to other organisms (e.g., Domazet-Loso and Tautz 2003; Donoghue et al. 2011), orphan genes identified among ants are significantly shorter, have longer introns, and a biased GC content compared with nonorphan genes (supplementary text S3, Supplementary Material online).
Previous studies and this one (table 3) have found incomplete expression support for SSOGs using ESTs or RNA-seq short reads mapped to the predicted gene models. This may be attributed to generally lower and possibly more tissue-specific expression of orphan genes in comparison with phylogenetically conserved genes (Levine et al. 2006; Donoghue et al. 2011; Wu et al. 2011); it is also possible that some orphan genes may instead represent spurious ORFs that are not expressed. We argue that the observed trend of a high prevalence of ant SSOG is robust for two reasons:
Table 3.
Expression Support across Formicidae Genes
| Species | Gene Count | Genes Supported | SSOG Count | SSOGs Supported |
|---|---|---|---|---|
| Successful mapping of ESTs to annotated gene models | ||||
| Atta cephalotes | 18,093 | 2,837 (16%) | 1,970 | 217 (11%) |
| Solenopsis invicta | 16,522 | 4,075 (25%) | 885 | 244 (28%) |
| Pogonomyrmex barbatus | 17,189 | 5,920 (34%) | 2,054 | 401 (20%) |
| Camponotus floridanus | 17,740 | 1,480 (8%) | 1,761 | 30 (2%) |
| Linepithema humile | 16,116 | 3,089 (19%) | 1,349 | 136 (10%) |
| Harpegnathos saltator | 18,564 | 1,727 (9%) | 1,391 | 26 (2%) |
| Successful mapping of RNA-seq reads to annotated gene models | ||||
| C. floridanus | 17,064 | 14,407 (84%) | 1,761 | 1,187 (67%) |
| H. saltator | 18,564 | 15,913 (86%) | 1,391 | 859 (62%) |
First, the high SSOG counts in ants are consistent with a high number of SSOGs in the independent lineage of N. vitripennis (2,191 SSOGs; table 1). The current data therefore suggest that a large fraction of the gene content in Hymenoptera is taxon specific. The only exception seems to be Api. mellifera which, based on the current annotation, likely has undergone a significant genome contraction in terms of annotated genes.
Second, we expect that, by and large, the annotated gene contents of the majority of the genomes in our analyses are comparable. Typically, projects annotating recently sequenced genomes use gene annotation of previously published genomes of related species to help identify genes through homology (Yandell and Ence 2012). The most extreme example may be the Drosophila clade: The genome of D. melanogaster was sequenced first (Adams et al. 2000) and has since been further characterized experimentally. When the genomes of 11 closely related Drosophila species were sequenced, much effort was invested to identify genes that are homologous to known D. melanogaster genes (Clark et al. 2007), rendering the D. melanogaster genome the one with the smallest number of expected false-positive orphan gene identifications. For the recently sequenced ant genomes, we replicated a search for homologous missing gene models that, if remained undetected, would inflate the number of orphan genes. Overall, we have taken special care to prevent any systematic biases and artifacts in gene annotation that could affect the identification of ant SSOGs despite the variety of employed sequencing and annotation methods used among the originally published genomes of the seven ant species. Therefore, it is very likely that Hymenoptera have indeed significantly more orphans than Diptera (fig. 2).
What Drives the Creation and Retention of Orphan Genes?
Orphan genes may be created and retained as a result of either adaptive or (nearly) neutral processes. These two processes, adaptive and nearly neutral, are not mutually exclusive, and both might be acting on different subsets of SSOGs and TSOGs, or on the same genes at different time scales.
If driven by positive selection, more orphan genes may be associated with a higher number or complexity of lineage- or taxon-specific transitions such as change in life history components or morphological innovation. A recent study in honey bees, which evolved eusociality independently from ants, indicates that TSOGs contributed to the evolution of eusociality in bees (Johnson and Tsutsui 2011). We could not identify homologs of the implicated honey bee genes in ants, and it is unclear whether these genes have significantly diverged in their sequence or do not exist at all in ants. Using the first caste- and subcaste-specific RNA-seq libraries from H. saltator and C. floridanus (Bonasio et al. 2010), we were able to identify approximately 70 SSOGs that show significant differential gene expression between castes (gamergate–worker, or minor–major worker; supplementary text S2, supplementary tables S4 and S5, Supplementary Material online). These expression patterns indicate that a small number of SSOGs are likely involved in traits associated with the social organization of ant societies, that is, division of labor and caste determination, respectively. It should, however, be noted that orphan genes are depleted among the differentially expressed genes (supplementary text S2, Supplementary Material online), suggesting that quantitatively, this class of genes has not primarily driven the evolution of eusociality in ants.
Although method to predict the subcellular localization of proteins may be error prone, our tests show strong statistical support that proteins encoded by orphan genes, compared with nonorphan genes, are located preferentially extracellular, mitochondrial, or nuclear (supplementary text S3, Supplementary Material online). We speculate that the abundance of SSOGs and TSOGs is partly associated with physiological adaptations and fundamental transitions in life styles across the seven sequenced ant species (supplementary text S3, Supplementary Material online).
On the other hand, a fraction of the orphan genes is probably nonadaptive and will eventually become eliminated or pseudogenized. As such, orphan genes could represent protogenes that are part of a proposed evolutionary continuum ranging from new nongenic ORFs to functional genes (Carvunis et al. 2012). Once nongenic ORFs get transcribed and translated, they are considered protogenes, which are exposed to selection. These protogenes can be retained as functional de novo genes if they are adaptive (Carvunis et al. 2012). Accordingly, the overall trend that hymenopteran genomes contain more orphan genes than dipteran genomes could be a consequence of natural selection being more permissive in retaining superfluous protogenes in Hymenoptera than in Diptera. Clearly, natural selection will influence retention as has been shown for gene duplicate retention in yeast populations (Ames et al. 2010). In eusocial insects, it is assumed that due to their haplo-diploid sex determination mechanism and a small number of reproducing individuals per colony, the effective population size is reduced in comparison to solitary insects with a comparable population size (Crozier 1979; Schmitz and Moritz 1998; Bromham and Leys 2005). Consequently, selection should be less efficient in removing nonadaptive or slightly deleterious genes. At the same time, genetic drift has a higher impact on genome evolution in eusocial Hymenopterans compared with solitary Dipterans (Wright and Andolfatto 2008). However, even (nearly) neutral processes could eventually give rise to functionally relevant orphan genes for lineage-specific adaptation, assuming that longer retention times increase the chance of acquiring rare beneficial mutations. Accordingly, the presence of an increased number of orphan genes in a genome would suggest a higher adaptive potential of this species.
Specifics of Orphan Gene Emergence in Ants
We have investigated in detail through which mechanisms ant orphan genes were formed. Our results indicate that contrary to the long-standing assumption that new genes originate primarily from gene duplications (Ohno 1970; Long et al. 2003; Zhou et al. 2008), the majority of orphan genes in ants resulted from either de novo formation or generalized gene loss (discussed later). Thus, observed patterns of orphan gene emergence in ants rather support a dynamic model of frequent gene birth and death, which has very recently been proposed (Carvunis et al. 2012). Our study represents only the third study we are aware of that systematically investigates the evolutionary origin of orphan genes in eukaryotes. Toll-Riera et al. (2009) studied primate orphan genes, which are conserved between three species over 25 Myr (human, chimp, macaque), but which lack homology in other mammalian species that split ≤41 Ma (Kumar and Hedges 1998). Donoghue et al. (2011) investigated A. thaliana-specific orphan genes and used A. lyrata and non-Brassicaceae species as references with evolutionary distances of 10 and ≤72 Myr (Wikström et al. 2001; Hu et al. 2011). The frequency spectrum of various mechanisms for orphan gene emergence in ant, Arabidopsis, and primate orphan genes are summarized in table 2. Clearly, cross-study comparisons are hampered by various factors, including different definitions of orphan genes, different characteristic of the data sets including phylogenetic distances between focal and outgroup species, and differences in the considered mechanisms and scenarios. However, our analysis is relatively similar to Donoghue et al. (2011) as we address a comparable set of scenarios with similar methods.
Scenarios of Orphan Gene Emergence Inferred from Similarity to Other Genes, Proteins, or TEs
Consistent throughout all three studies, gene duplication followed by sequence divergence is a dominant mechanism bringing about orphan genes. Among ants, primates, and Arabidopsis, the lowest rate of orphan gene formation by gene duplication was found in ants (table 2). Mobile elements seem to contribute equally to orphan gene formation in ants and in Arabidopsis, but are significantly more frequently found in primates. Overall, all three studies confirm previous theoretical expectations and recent empirical data suggesting that the two mechanisms, gene duplication and exaptation from TEs, play an important role in the evolution of genetic innovations (Ohno 1970; Lynch and Conery 2000; Kazazian 2004; Zhou et al. 2008).
New protein sequences can also emerge by exploiting different frames of existing ORFs. In ants and Arabidopsis, a substantial number of SSOG proteins are predicted to be formed from either nondeleterious frame shifts in an ORF or by utilizing parts of the antisense strand of an ORF, thereby generating overlapping genes (table 2). Although both these mechanisms can contribute to the formation of new genes across different taxa, further studies are necessary to validate the high frequency of orphan gene emergence via different-frame functionalization of existing CDS in ants. Mechanisms for generating new proteins have not been addressed in primates (Toll-Riera et al. 2009), possibly due to terminology: Strictly speaking, using different frames of existing genes can create orphan proteins, but the new gene itself may still share significant sequence similarity to the non–frame-shifted or overlapping homolog in other species. Therefore, based on sequence similarity, the protein, but not necessarily the gene, is an orphan.
Scenarios of Orphan Gene Emergence with No Similarity to Other Genes, Proteins, or TEs
A majority of ant SSOGs has at least a partial ORF match in the DNA sequence of other Hymenoptera (“intergenic match”), but their emergence could not be explained by the previously mentioned mechanisms (43.5% in total, or 55% of SSOGs with explained origin; table 2). Such intergenic matches of orphan genes may be indicative for either one of two scenarios: First, intergenic matches may represent remnants of genes, which have been independently lost across all but one lineage in which it has been selectively retained as a functional gene (“generalized gene loss,” Forêt et al. [2010]). Second, intergenic matches may represent precursor sequences of de novo gene generation events (Wu et al. 2011). Both scenarios are feasible, considering that a recent study in human found that approximately 100 disrupted protein-coding genes (loss-of-function variants) exist in genomes within each individual human genome, that is, variation exists already at the population level (MacArthur et al. 2012). It should be noted that this high rate of gene disruption has been reported to be strongly favored toward seemingly dispensable genes. Such dispensable genes include less evolutionarily conserved genes, genes that tend to have highly similar paralogs, and genes that tend to have lower connectivity in gene and protein–protein interactions. Accordingly, dispensable genes will in the long run likely be removed by purifying selection (MacArthur et al. 2012). However, over long evolutionary time, it is possible that pseudogenization and gene loss occurs rapidly and frequently among genomes of species that split several million years ago. This suggests that a substantial amount of pseudogenized genes exists in the ant genomes and lends at least partial support to the generalized gene loss scenario.
At the same time, genes being formed de novo across several lineages with adaptive potential has found much support in recent studies (Cai et al. 2008; Khalturin et al. 2009; Kaessmann 2010; Tautz and Domazet-Lošo 2011; Wilson and Masel 2011; Wu et al. 2011), and de novo formation of new genes may occur even more frequently than gene duplication (Carvunis et al. 2012). 24% of orphan genes (797 genes) with intergenic match are single exon genes. This observed simpler gene structure in humans and ants supports the scenario of de novo gene birth (Guerzoni and McLysaght 2011; Wu et al. 2011). Ant SSOGs with intergenic matches are the best candidates for a confirmation of their de novo formation.
For a number of ant orphan genes, it still remains unclear whether the orphan gene stems from gene losses and lineage-specific retention or from the gain of a previously nonexistent gene, for example, by de novo formation. Although the myrmicine ant species are currently represented by four hierarchically related species, the resolution does not always suffice to unambiguously resolve the issue of gene gain versus gene loss (supplementary text S6, Supplementary Material online), and additional close-species taxon sampling may be necessary to uncover the true genetic origin of such orphan genes.
Finally, for a significant fraction of ant orphan genes (20.8%), which we classified as orphan genes with unexplained origin, the origin of their genetic material could not be determined using sequence similarity. The “unexplained” category may include genes that are not necessarily new but rapidly evolving so that their sequence diverged beyond recognition. We found that 1:1 orthologs that are restricted to Formicidae evolve 2–3 times faster than nonrestricted 1:1 orthologs (supplementary text S4, Supplementary Material online), and that in some cases, such sequence divergence may be driven by positive selection. If the sequence divergence is even higher, genes may eventually appear as orphan genes because their ancestry is no longer identifiable at the sequence level. This divergence scenario is further supported by the strong correlation between the number of SSOGs, the phylogenetic distance to the next closely related species in the data set (fig. 1) and the finding that the frequency of detectable protein domains is negatively correlated with the phylogenetic conservation of genes (supplementary text S5, Supplementary Material online).
Toward the Identification of Taxon-Specific Differences in Orphan Gene Emergence
Systematic studies of orphan gene emergence in ants, primates (Toll-Riera et al. 2009), and plants (Donoghue et al. 2011) pave the way toward the identification of similarities and differences in the mechanistic basis of orphan gene emergence between taxa. Do genetic mechanisms that create orphan genes occur at unequal rates, that is, are rates specific for each clade? We have identified differences in the frequency spectrum of orphan gene emergence between the three clades which could support taxon-specific differences in orphan gene emergence (table 2). For instance, higher relative rates of TE-mediated orphan gene formation in primates (Toll-Riera et al. 2009) could very well relate to taxon-specific genomic characteristics such as the lineage-specific TE content, including many lineage-specific TE families with varying activity levels.
However, we have also outlined earlier that characteristics of the data sets and applied methods vary, which warrants caution in such cross-study comparisons. Inferring gene duplication and frame-shifts in one species requires well-conserved genes in other species, which are used for comparison. In our study, gene duplicates were identified as phylogenetically conserved genes, that is, a paralog of a given orphan gene exists that in turn has orthologs in other species. The resulting prediction rate will decrease with increasing evolutionary distance, and this inherent bias might contribute to lowering the predicted rate of gene duplication and frame shifts accounting for orphan gene creation in our data set, compared with other studies that have used much more closely related genomes. On the other hand, our study did not require ancestors dating back approximately 25 Myr (Toll-Riera et al. 2009), such that very young orphan genes, which seem to make up a significant fraction of the overall orphan gene count, are included in our study (fig. 1).
Outlook
Insects comprise the most diverse group of metazoan species and thus represent great study objects to investigate the role and origin of new genes in genome and phenotypic evolution. We have uncovered a relatively high number of SSOG and TSOG in ants (Formicidae), suggesting that ant genomes and their gene content are highly dynamic. Understanding the roles that new gene classes, such as orphan genes, play in genome evolution might provide insights into the striking reciprocal paradoxon of evolutionary developmental biology: “The conservation of similar developmental genetic toolkits despite a diversity of life forms, and the inverse paradox—the development of similar morphologies despite the phylogenetically variable presence of the genetic tools that are thought to be responsible for those forms” (Cañestro et al. 2007). Our comprehensive annotation of orphan genes with a special focus on the recently sequenced Formicidae undoubtedly represents a valuable resource for further research on genomes of social insects and their evolution. The pending availability of further genomes (Robinson et al. 2011) will not only further boost the powers of comparative genomics but also call for more in-depth functional and molecular investigations on selected genes to resolve issues of mechanisms and implications of gene loss and gain.
Supplementary Material
Supplementary texts S1–S6, tables S1–S5, and figures S1–S5 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the Volkswagen Foundation to L.W. and E.B.-B., the NSF award IOS-0920732 to J.S., a post-doctoral fellowship from the University of Pennsylvania Department of Cell and Developmental Biology to D.F.S., and a Howard Hughes Medical Institute Collaborative Innovation Award #2009005 to D. Reinberg, S. Berger, and J. Liebig.
Literature Cited
- Acuna R, et al. Adaptive horizontal transfer of a bacterial gene to an invasive insect pest of coffee. Proc Natl Acad Sci U S A. 2012;109(11):4197–4202. doi: 10.1073/pnas.1121190109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MD, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287(5461):2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Ames RM, et al. Gene duplication and environmental adaptation within yeast populations. Genome Biol Evol. 2010;2:591–601. doi: 10.1093/gbe/evq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408(6814):796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Balakirev ES, Ayala FJ. Pseudogenes: are they “junk” or functional DNA? Annu Rev Genet. 2003;37:123–151. doi: 10.1146/annurev.genet.37.040103.103949. [DOI] [PubMed] [Google Scholar]
- Birney E, Clamp M, Durbin R. GeneWise and genomewise. Genome Res. 2004;14(5):988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, et al. Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science. 2010;329(5995):1068–1071. doi: 10.1126/science.1192428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwma AM, Ahrens ME, DeHeer CJ, DeWayne Shoemaker D. Distribution and prevalence of Wolbachia in introduced populations of the fire ant Solenopsis invicta. Insect Mol Biol. 2006;15(1):89–93. doi: 10.1111/j.1365-2583.2006.00614.x. [DOI] [PubMed] [Google Scholar]
- Brady SG, Schultz TR, Fisher BL, Ward PS. Evaluating alternative hypotheses for the early evolution and diversification of ants. Proc Natl Acad Sci U S A. 2006;103(48):18172–18177. doi: 10.1073/pnas.0605858103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromham L, Leys R. Sociality and the rate of molecular evolution. Mol Biol Evol. 2005;22(6):1393–1402. doi: 10.1093/molbev/msi133. [DOI] [PubMed] [Google Scholar]
- Cañestro C, Yokoi H, Postlethwait JH. Evolutionary developmental biology and genomics. Nat Rev Genet. 2007;8(12):932–942. doi: 10.1038/nrg2226. [DOI] [PubMed] [Google Scholar]
- Cai J, Zhao R, Jiang H, Wang W. De novo origination of a new protein-coding gene in Saccharomyces cerevisiae. Genetics. 2008;179(1):487–496. doi: 10.1534/genetics.107.084491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–196. doi: 10.1101/gr.6743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell. 2008;134(1):25–36. doi: 10.1016/j.cell.2008.06.030. [DOI] [PubMed] [Google Scholar]
- Carvunis AR, et al. Proto-genes and de novo gene birth. Nature. 2012 doi: 10.1038/nature11184. Advance Access published July 19, 2012, doi: 10.1038/nature11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhang YE, Long M. New genes in Drosophila quickly become essential. Science. 2010;330(6011):1682–1685. doi: 10.1126/science.1196380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christin PA, et al. Adaptive evolution of C(4) photosynthesis through recurrent lateral gene transfer. Curr Biol. 2012;22(5):445–449. doi: 10.1016/j.cub.2012.01.054. [DOI] [PubMed] [Google Scholar]
- Clark AG, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450(7167):203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- Colbourne JK, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331(6017):555–561. doi: 10.1126/science.1197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier R. Genetics of sociality. In: Hermann HR, editor. Social insects. New York: Academic Press; 1979. pp. 223–286. [Google Scholar]
- Domazet-Loso T, Tautz D. An evolutionary analysis of orphan genes in Drosophila. Genome Res. 2003;13(10):2213–2219. doi: 10.1101/gr.1311003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoghue MT, Keshavaiah C, Swamidatta SH, Spillane C. Evolutionary origins of Brassicaceae specific genes in Arabidopsis thaliana. BMC Evol Biol. 2011;11(1):47. doi: 10.1186/1471-2148-11-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J, et al. SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res. 2010;38(Database issue):D453–D456. doi: 10.1093/nar/gkp801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning Hotopp JC. Horizontal gene transfer between bacteria and animals. Trends Genet. 2011;27(4):157–163. doi: 10.1016/j.tig.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning Hotopp JC, et al. Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science. 2007;317(5845):1753–1756. doi: 10.1126/science.1142490. [DOI] [PubMed] [Google Scholar]
- Fablet M, Rebollo R, Biémont C, Vieira C. The evolution of retrotransposon regulatory regions and its consequences on the Drosophila melanogaster and Homo sapiens host genomes. Gene. 2007;390(1–2):84–91. doi: 10.1016/j.gene.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Forêt S, et al. New tricks with old genes: the genetic bases of novel cnidarian traits. Trends Genet. 2010;26(4):154–158. doi: 10.1016/j.tig.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Gadau J, et al. The genomic impact of 100 million years of social evolution in seven ant species. Trends Genet. 2012;28(1):14–21. doi: 10.1016/j.tig.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal-Mark N, Schwartz S, Ast G. Alternative splicing of Alu exons—two arms are better than one. Nucleic Acids Res. 2008;36(6):2012–2023. doi: 10.1093/nar/gkn024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil R, et al. The genome sequence of Blochmannia floridanus: comparative analysis of reduced genomes. Proc Natl Acad Sci U S A. 2003;100(16):9388–9393. doi: 10.1073/pnas.1533499100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil R, Silva FJ, Peretó J, Moya A. Determination of the core of a minimal bacterial gene set. Microbiol Mol Biol Rev. 2004;68(3):518–537. doi: 10.1128/MMBR.68.3.518-537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladyshev EA, Meselson M, Arkhipova IR. Massive horizontal gene transfer in bdelloid rotifers. Science. 2008;320(5880):1210–1213. doi: 10.1126/science.1156407. [DOI] [PubMed] [Google Scholar]
- Gogvadze E, Buzdin A. Retroelements and their impact on genome evolution and functioning. Cell Mol Life Sci. 2009;66(23):3727–3742. doi: 10.1007/s00018-009-0107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González J, Petrov DA. The adaptive role of transposable elements in the Drosophila genome. Gene. 2009;448(2):124–133. doi: 10.1016/j.gene.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grbić M, et al. The genome of Tetranychus urticae reveals herbivorous pest adaptations. Nature. 2011;479(7374):487–492. doi: 10.1038/nature10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi D, Engel MS. Evolution of the insects. Cambridge Evolution Series. Cambridge: Cambridge University Press; 2005. [Google Scholar]
- Guerzoni D, McLysaght A. De novo origins of human genes. PLoS Genet. 2011;7(11):e1002381. doi: 10.1371/journal.pgen.1002381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn Y, Lee B. Identification of nine human-specific frameshift mutations by comparative analysis of the human and the chimpanzee genome sequences. Bioinformatics. 2005;21(Suppl 1):i186–i194. doi: 10.1093/bioinformatics/bti1000. [DOI] [PubMed] [Google Scholar]
- Harris JK, Kelley ST, Spiegelman GB, Pace NR. The genetic core of the universal ancestor. Genome Res. 2003;13(3):407–412. doi: 10.1101/gr.652803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22(23):2971–2972. doi: 10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- Hu TT, et al. The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet. 2011;43(5):476–481. doi: 10.1038/ng.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BR, Tsutsui ND. Taxonomically restricted genes are associated with the evolution of sociality in the honey bee. BMC Genomics. 2011;12(1):164. doi: 10.1186/1471-2164-12-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1–4):462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kaessmann H. Origins, evolution, and phenotypic impact of new genes. Genome Res. 2010;20(10):1313–1326. doi: 10.1101/gr.101386.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian HH. Mobile elements: drivers of genome evolution. Science. 2004;303(5664):1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Keese PK, Gibbs A, Gibbst A. Origins of genes: “big bang” or continuous creation? Proc Natl Acad Sci U S A. 1992;89(20):9489–9493. doi: 10.1073/pnas.89.20.9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalturin K, et al. A novel gene family controls species-specific morphological traits in Hydra. PLoS Biol. 2008;6(11):e278. doi: 10.1371/journal.pbio.0060278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalturin K, Hemmrich G, Fraune S, Augustin R, Bosch TCG. More than just orphans: are taxonomically-restricted genes important in evolution? Trends Genet. 2009; 25(9):404–413. doi: 10.1016/j.tig.2009.07.006. [DOI] [PubMed] [Google Scholar]
- Kim HS, et al. BeetleBase in 2010: revisions to provide comprehensive genomic information for Tribolium castaneum. Nucleic Acids Res. 2010;38(Database issue):D437–D442. doi: 10.1093/nar/gkp807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasson L, Kambris Z, Cook PE, Walker T, Sinkins SP. Horizontal gene transfer between Wolbachia and the mosquito Aedes aegypti. BMC Genomics. 2009;10(1):33. doi: 10.1186/1471-2164-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochetov AV. Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays. 2008;30(7):683–691. doi: 10.1002/bies.20771. [DOI] [PubMed] [Google Scholar]
- Krylov DM, Wolf YI, Rogozin IB, Koonin EV. Gene loss, protein sequence divergence, gene dispensability, expression level, and interactivity are correlated in eukaryotic evolution. Genome Res. 2003;13(10):2229–2235. doi: 10.1101/gr.1589103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Hedges SB. A molecular timescale for vertebrate evolution. Nature. 1998;392(6679):917–920. doi: 10.1038/31927. [DOI] [PubMed] [Google Scholar]
- Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson D, et al. VectorBase: a data resource for invertebrate vector genomics. Nucleic Acids Res. 2009;37(Database issue):D583–D587. doi: 10.1093/nar/gkn857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legeai F, et al. AphidBase: a centralized bioinformatic resource for annotation of the pea aphid genome. Insect Mol Biol. 2010;19(Suppl 2):5–12. doi: 10.1111/j.1365-2583.2009.00930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MT, Jones CD, Kern AD, Lindfors HA, Begun DJ. Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. Proc Natl Acad Sci U S A. 2006;103(26):9935–9939. doi: 10.1073/pnas.0509809103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockton S, Gaut BS. The contribution of transposable elements to expressed coding sequence in Arabidopsis thaliana. J Mol Evol. 2009;68(1):80–89. doi: 10.1007/s00239-008-9190-5. [DOI] [PubMed] [Google Scholar]
- Long M, Betrán E, Thornton K, Wang W. The origin of new genes: glimpses from the young and old. Nat Rev Genet. 2003;4(11):865–875. doi: 10.1038/nrg1204. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- MacArthur DG, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335(6070):823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markow TA, O’Grady PM. Drosophila biology in the genomic age. Genetics. 2007;177(3):1269–1276. doi: 10.1534/genetics.107.074112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuilton P, St Pierre SE, Thurmond J. FlyBase 101—the basics of navigating FlyBase. Nucleic Acids Res. 2012;40(Database issue):D706–D714. doi: 10.1093/nar/gkr1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Eichler EE. Duplication hotspots, rare genomic disorders, and common disease. Curr Opin Genet Dev. 2009;19(3):196–204. doi: 10.1016/j.gde.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milde S, et al. Characterization of taxonomically restricted genes in a phylum-restricted cell type. Genome Biol. 2009;10(1):R8. doi: 10.1186/gb-2009-10-1-r8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, Jarvik T. Lateral transfer of genes from fungi underlies carotenoid production in aphids. Science. 2010;328(5978):624–627. doi: 10.1126/science.1187113. [DOI] [PubMed] [Google Scholar]
- Moreau CS, Bell CD, Vila R, Archibald SB, Pierce NE. Phylogeny of the ants: diversification in the age of angiosperms. Science. 2006;312(5770):101–104. doi: 10.1126/science.1124891. [DOI] [PubMed] [Google Scholar]
- Munoz-Torres MC, et al. Hymenoptera genome database: integrated community resources for insect species of the order Hymenoptera. Nucleic Acids Res. 2011;39(Database issue):D658–D662. doi: 10.1093/nar/gkq1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoh N, et al. Wolbachia genome integrated in an insect chromosome: evolution and fate of laterally transferred endosymbiont genes. Genome Res. 2008;18(2):272–280. doi: 10.1101/gr.7144908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor MA, Grams KL, Bertucci LA, Reiland J. Chromosomal inversions and the reproductive isolation of species. Proc Natl Acad Sci U S A. 2001;98(21):12084–12088. doi: 10.1073/pnas.221274498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard S, et al. The genome of the leaf-cutting ant Acromyrmex echinatior suggests key adaptations to advanced social life and fungus farming. Genome Res. 2011;21(8):1339–1348. doi: 10.1101/gr.121392.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Evolution by gene duplication. London: Allen and Unwin; 1970. [Google Scholar]
- Okamura K, Feuk L, Marquès-Bonet T, Navarro A, Scherer SW. Frequent appearance of novel protein-coding sequences by frameshift translation. Genomics. 2006;88(6):690–697. doi: 10.1016/j.ygeno.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Osato N, Suzuki Y, Ikeo K, Gojobori T. Transcriptional interferences in cis natural antisense transcripts of humans and mice. Genetics. 2007;176(2):1299–1306. doi: 10.1534/genetics.106.069484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH. Hotspots for copy number variation in chimpanzees and humans. Proc Natl Acad Sci U S A. 2006;103(21):8006–8011. doi: 10.1073/pnas.0602318103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott EM, Proudfoot NJ. Transcriptional collision between convergent genes in budding yeast. Proc Natl Acad Sci U S A. 2002;99(13):8796–8801. doi: 10.1073/pnas.132270899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prud’homme B, Gompel N, Carroll SB. Emerging principles of regulatory evolution. Proc Natl Acad Sci U S A. 2007;104(Suppl 1):8605–8612. doi: 10.1073/pnas.0700488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, et al. The Pfam protein families database. Nucleic Acids Res. 2011;40(D1):D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raes J, Van De Peer Y. Functional divergence of proteins through frameshift mutations. Trends Genet. 2005;21(8):428–431. doi: 10.1016/j.tig.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Ranz JM, Parsch J. Newly evolved genes: moving from comparative genomics to functional studies in model systems: how important is genetic novelty for species adaptation and diversification? BioEssays. 2012;34(6):477–483. doi: 10.1002/bies.201100177. [DOI] [PubMed] [Google Scholar]
- Robinson GE, et al. Creating a buzz about insect genomes. Science. 2011;331(6023):1386. doi: 10.1126/science.331.6023.1386. [DOI] [PubMed] [Google Scholar]
- Russell J. The ants are unique and enigmatic hosts of prevalent Wolbachia symbionts. Myrmecol News. 2011;16:7–23. [Google Scholar]
- Sanna CR, Li WH, Zhang L. Overlapping genes in the human and mouse genomes. BMC Genomics. 2008;9(1):169. doi: 10.1186/1471-2164-9-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz J, Brosius J. Exonization of transposed elements: a challenge and opportunity for evolution. Biochimie. 2011;93(11):1928–1934. doi: 10.1016/j.biochi.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Schmitz J, Moritz RF. Sociality and the rate of rDNA sequence evolution in wasps (Vespidae) and honeybees (Apis) J Mol Evol. 1998;47(5):606–612. doi: 10.1007/pl00006417. [DOI] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. RepeatMasker. 2010 Open-3.0 1996–2010. Available from: http://www.repeatmasker.org. [Google Scholar]
- Smith CD, et al. Draft genome of the globally widespread and invasive Argentine ant (Linepithema humile) Proc Natl Acad Sci U S A. 2011;108(14):5673–5678. doi: 10.1073/pnas.1008617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CR, et al. Draft genome of the red harvester ant Pogonomyrmex barbatus. Proc Natl Acad Sci U S A. 2011;108(14):5667–5672. doi: 10.1073/pnas.1007901108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldà G, et al. Non-random retention of protein-coding overlapping genes in Metazoa. BMC Genomics. 2008;9(1):174. doi: 10.1186/1471-2164-9-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer RJ, Streit A. Comparative genetics and genomics of nematodes: genome structure, development, and lifestyle. Annu Rev Genet. 2011;45:1–20. doi: 10.1146/annurev-genet-110410-132417. [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Lupski JR. Structural variation in the human genome and its role in disease. Annu Rev Med. 2010;61:437–455. doi: 10.1146/annurev-med-100708-204735. [DOI] [PubMed] [Google Scholar]
- Stevison LS, Hoehn KB, Noor MAF. Effects of inversions on within- and between-species recombination and divergence. Genome Biol Evol. 2011;3:830–841. doi: 10.1093/gbe/evr081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen G, et al. The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genet. 2011;7(2):e1002007. doi: 10.1371/journal.pgen.1002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Subramanian S, Kumar S. Temporal patterns of fruit fly (Drosophila) evolution revealed by mutation clocks. Mol Biol Evol. 2004;21(1):36–44. doi: 10.1093/molbev/msg236. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz D, Domazet-Lošo T. The evolutionary origin of orphan genes. Nat Rev Genet. 2011;12(10):692–702. doi: 10.1038/nrg3053. [DOI] [PubMed] [Google Scholar]
- Tettelin H, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A. 2005;102(39):13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, et al. Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature. 2008;455(7209):105–108. doi: 10.1038/nature07175. [DOI] [PubMed] [Google Scholar]
- Toll-Riera M, et al. Origin of primate orphan genes: a comparative genomics approach. Mol Biol Evol. 2009;26(3):603–612. doi: 10.1093/molbev/msn281. [DOI] [PubMed] [Google Scholar]
- Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Borm S, Wenseleers T, Billen J, Boomsma J. Wolbachia in leafcutter ants: a widespread symbiont that may induce male killing or incompatible matings. J Evol Biol. 2001;14(5):805–814. [Google Scholar]
- Voolstra CR, et al. Rapid evolution of coral proteins responsible for interaction with the environment. PLoS One. 2011;6(5):e20392. doi: 10.1371/journal.pone.0020392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock GM, et al. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006;443(7114):931–949. doi: 10.1038/nature05260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH, et al. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science. 2010;327(5963):343–348. doi: 10.1126/science.1178028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikström N, Savolainen V, Chase MW. Evolution of the angiosperms: calibrating the family tree. Proc Biol Sci. 2001;268(1482):2211–2220. doi: 10.1098/rspb.2001.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BA, Masel J. Putatively noncoding transcripts show extensive association with ribosomes. Genome Biol Evol. 2011;3:1245–1252. doi: 10.1093/gbe/evr099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EO, Hölldobler B. The rise of the ants: a phylogenetic and ecological explanation. Proc Natl Acad Sci U S A. 2005;102(21):7411–7414. doi: 10.1073/pnas.0502264102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GA, et al. Orphans as taxonomically restricted and ecologically important genes. Microbiology. 2005;151(Pt 8):2499–2501. doi: 10.1099/mic.0.28146-0. [DOI] [PubMed] [Google Scholar]
- Wilson GA, Feil EJ, Lilley AK, Field D. Large-scale comparative genomic ranking of taxonomically restricted genes (TRGs) in bacterial and archaeal genomes. PLoS One. 2007;2(3):e324. doi: 10.1371/journal.pone.0000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Andolfatto P. The impact of natural selection on the genome: emerging patterns in Drosophila and Arabidopsis. Annu Rev Ecol Evol Syst. 2008;39(1):193–213. [Google Scholar]
- Wu DD, Irwin DM, Zhang YP. De Novo origin of human protein-coding genes. PLoS Genet. 2011;7(11):e1002379. doi: 10.1371/journal.pgen.1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21(9):1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- Wurm Y, et al. The genome of the fire ant Solenopsis invicta. Proc Natl Acad Sci U S A. 2011;108(14):5679–5684. doi: 10.1073/pnas.1009690108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell M, Ence D. A beginner’s guide to eukaryotic genome annotation. Nat Rev Genet. 2012;13(5):329–342. doi: 10.1038/nrg3174. [DOI] [PubMed] [Google Scholar]
- Yu JS, Kokoska RJ, Khemici V, Steege DA. In-frame overlapping genes: the challenges for regulating gene expression. Mol Microbiol. 2007;63(4):1158–1172. doi: 10.1111/j.1365-2958.2006.05572.x. [DOI] [PubMed] [Google Scholar]
- Zhang J. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18(6):292–298. [Google Scholar]
- Zhang YE, Landback P, Vibranovski MD, Long M. Accelerated recruitment of new brain development genes into the human genome. PLoS Biol. 2011;9(10):e1001179. doi: 10.1371/journal.pbio.1001179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gerstein M. Large-scale analysis of pseudogenes in the human genome. Curr Opin Genet Dev. 2004;14(4):328–335. doi: 10.1016/j.gde.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Zhou Q, et al. On the origin of new genes in Drosophila. Genome Res. 2008;18(9):1446–1455. doi: 10.1101/gr.076588.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, et al. Horizontal gene transfer in silkworm, Bombyx mori. BMC Genomics. 2011;12(1):248. doi: 10.1186/1471-2164-12-248. [DOI] [PMC free article] [PubMed] [Google Scholar]