

Abstract
Human copper transporter 1 (hCTR1) is the major high affinity copper influx transporter in mammalian cells that also mediates uptake of the cancer chemotherapeutic agent cisplatin. A low resolution structure of hCTR1 determined by cryoelectron microscopy was recently published. Several protein structure simulation techniques were used to create an all-atom model of this important transporter using the low resolution structure as a starting point. The all-atom model provides new insights into the roles of specific residues of the N-terminal extracellular domain, the intracellular loop, and C-terminal region in metal ion transport. In particular, the model demonstrates that the central region of the pore contains four sets of methionine triads in the intramembranous region. The structure confirms that two triads of methionine residues delineate the intramembranous region of the transporter, and further identifies two additional methionine triads that are located in the extracellular N-terminal part of the transporter. Together, the four triads create a structure that promotes stepwise transport of metal ions into and then through the intramembranous channel of the transporter via transient thioether bonds to methionine residues. Putative copper-binding sites in the hCTR1 trimer were identified by a program developed by us for prediction of metal-binding sites. These sites correspond well with the known effects of mutations on the ability of the protein to transport copper and cisplatin.
Keywords: CTR1, Molecular model, Copper, Cisplatin, Transporter
Introduction
Copper (Cu) is an essential micronutrient required for the function of numerous intracellular enzymes including superoxide dismutase, cytochrome c oxidase, lysyl oxidase, and dopamine β-hydrolase [1, 2]. Since Cu is not always abundant in the environment, cells have evolved a complex system of Cu transporters and chaperones that accumulate Cu. On the other hand, Cu is toxic due to its ability generate reactive dioxygen species by cycling between Cu(I) and Cu(II) under physiologic conditions. As a result, cellular mechanisms must prevent oxidation of Cu(I) while making free intracellular Cu ions available. This is achieved by a complex set of management strategies that maintain intracellular Cu at vanishingly low concentrations (<10−18 M). The major high-affinity mammalian Cu influx transporter in this system is CTR1, which is responsible for passage of Cu(I) through the plasma membrane and its transfer to one of three distinct intracellular chaperones. The chaperones, in turn, deliver the Cu(I) ions to Cu-dependent enzymes. CTR1 is of interest in cancer chemotherapy because it is also the major influx transporter for Pt ions in the form of cisplatin (cDDP) and carboplatin in tumor cells [3]. These Pt therapeutic agents interact with the N-terminal methionines of CTR1; this interaction plays a significant role in the transport of cDDP and carboplatin into the cell [4, 5]. Knockout of the gene coding for CTR1 markedly reduces the influx of these Pt-containing drugs and renders cells resistant to their cytotoxic effect both in vitro and in vivo [6–8].
Functional human copper transporter 1 (hCTR1) is a homotrimer in the plasma membrane, where each constituent monomer contains 190 amino acids. Folded monomers contain an extracellular N-terminal domain of 67 amino acids, three transmembrane domains, an intracellular loop of 46 amino acids that connects the first and second transmembrane domains and a 15-amino acid C-terminal intracellular tail. The N-terminal domain of hCTR1 contains histidine- and methionine-rich motifs similar to those found in other ion transport proteins, and is glycosylated at ASN15 and THR27. While the exact mode of Cu(I) internalization by hCTR1 is not clear, the trimer appears to form a central pore that functions as a channel [3]. CTR1 is specific for transport of Cu(I). Influx is energy independent [9], but is influenced by temperature, pH, and K+ [10]. The specificity for Cu(I) suggests the involvement of thioether coordination to methionine in the transport process [11]. It has also been proposed that the N-terminal domain acts to concentrate Cu ions, while C189 at the C-terminal end functions as a switch to open and close the pore [12]. High concentrations of Cu, and low concentrations of cDDP trigger removal of CTR1 from the membrane by endocytosis. In some types of cells this is accompanied by degradation [13–15], whereas in others it is not [16, 17]. It has been hypothesized that degradation may serve to prevent accumulation of toxic levels of the metal [17–20]. In mammalian cells, where cDDP induces CTR1 degradation, the protein becomes ubiquitinated [21], while inhibition of proteasome function can block the degradation [14] and enhance the uptake and cytotoxicity of cisplatin [15].
Despite the importance of hCTR1 in both physiological Cu transport and in the cellular pharmacology of the Pt-containing drugs, information about its structure has only recently become available. Cryoelectron microscopic studies [22, 23] have recently provided a 7 Å resolution structure of the hCTR1 transporter [24]. The trimer appears to form a narrow pore at the outer leaflet of the plasma membrane that widens into a vestibule on the intracellular side [24]. Using this structure as a starting point, we have used contemporary protein structure prediction tools to create a model of the entire hCTR1 protein that is consistent with the above-mentioned crystal structure. The all-atom model of hCTR1 provides new insights into the mechanism of Cu and cDDP transport that are consistent with existing experimental data on Cu transport by this transporter. This validation by experimental mutational analysis provides confidence of the validity of the all-atom model in identifying important binding sites and as a starting point for computer-based drug design.
Methods
Modeling of hCTR1 Structure
The structure of the hCTR1 transporter was modeled using the following steps. The regions of the molecule that appeared to contain helical structure based on the results of cryoelectron microscopy were modeled using the low-resolution structure and the model of transmembrane regions that was obtained using both the low-resolution structure and the results of tryptophan scanning analysis [21–23]. The last has been supplied by Unger (personal communication). We used the homology modeling program from the InsightII program package (Accelrys, San Diego). Three dimensional comparison of our model and Unvin’s structures was conducted based on CE algorithm [25], the analysis gave an RMSD = 0.785 Å between the C-alpha atoms in the region of superposition. The three extracellular parts of the structure were then modeled separately. The N-terminal extracellular domain was modeled by Robetta sever in ab initio mode [26, 27]. Positions of the N-terminal domain on the membrane surface and the composite configuration of the N-terminal of the hCTR1 trimer were modeled by our program MAPAS [28] that predicts the protein-membrane contacting surface (Fig. 1a). The combined MAPAS scores for this prediction, including a number of membrane contacting residues and membranephilic area score, was best for the N-terminal domain model shown in Fig. 1c. Location of the residues that connect the N-terminal extracellular domain with the transmembrane helices modeled by cryoelectron structure [24] (residues around 67), along with the symmetric configuration of the N-terminal domains (Fig. 1b), creates a unique configuration and membrane positioning of the trimer N-terminal parts (Fig. 1c). The small regions of adjacent residues were refined using the fragments in the PDB databank substitution module of the InsightII/Homology program (Accelrys, San Diego).
Fig. 1.

Positioning of the N-terminal domains of the hCTR1 based on the membrane-contacting surface prediction and geometrical restrains. a Initial position of the N-terminal domain predicted by the MAPAS server, b unique positions of the N-terminal domains restrained by the contacts with the transmembrane helices (defined by the cryoelectron microscopy blue spheres). The pink spheres correspond to the C-alpha atoms of the residue, c hCTR1 model with the N-terminal domain superimposed with the ribbon diagram of its predicted position versus membrane green, predicted membrane surface (Color figure online)
Figure 2 shows the alignments with predicted secondary structure of the hCTR cytoplasmic loop and C-terminal tail residues with the first threading templates selected by I-Tasser program [29, 30]. Threading [29] identifies template proteins in solved structure databases with a similar structural motif to the query protein sequence. The first template structure proposed by the program threading template usually has the top score in further structure prediction. We used the I-Tasser selections: protein 3BBO as a template for modeling the cytoplasmic loop, and 3CMA protein (a representative of the set of proteins with the identical sequences 3CMA, 3CC2, 1QVF, and 1S72) for modeling the C-terminal tail. The templates were aligned with the density map of the cryoelectron microscopy structure [22] of the CTR1. When superimposed on the cryoelectron microscopy structure, the N-terminal end of this loop model clearly continues the transmembrane helices of each hCTR1 monomer, while the C-terminal ends are located at the beginning of the next transmembrane helices. Moreover, the map of electron density from the cryoelectron microscopy structure has maximums close to the “edge” points of this loop part of the CTR1 model when the two are superimposed (Fig. 3).
Fig. 2.
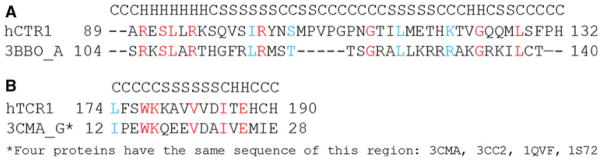
Alignment and secondary structure prediction created by Tasser. a hCTR1 cytoplasmic loop; b C-terminal tail
Fig. 3.

Superimposition of the electron density map of the cryoelectron microscopy structure of the CTR1 with the all-atom model (orange wire line). The “edge” points of the cytoplasmic loop model have the corresponding maximums of the electron density (Color figure online)
Molecular Dynamics (MD) Simulations
We prepared the system including CTR1 and POPC membrane with 256 lipids, with the water box having a height of 15 Å on the axis perpendicular to the membrane. The system included 8,784 protein atoms, 32,964 lipids atoms, and 59,817 water atoms. The NAMD molecular dynamics program (version 2.5 [31] was used with the CHARMM27 force-field parameters [32]. The temperature was maintained at 310 K by means of Langevin dynamics using a collision frequency of 1 ps−1. A fully flexible cell at constant pressure (1 atm) was employed by the Nosé-Hoover Langevin Piston algorithm [33, 34] as used in the NAMD software package. We conducted unrestrained (MD) for 20 ns with a time step of 2 fs using mesh Ewald for electrostatics, grid spacing of 1 Å, with periodic boundary conditions, and a cutoff of 12 Å. The corner 16 lipids at the membrane boundaries were fixed.
Prediction of Metal Location Points on the Surface of CTR1
There are currently 636 protein structures containing Cu in the PDB. To identify possible Cu-binding sites in CTR1 we developed a program called METBIND that identifies possible metal-binding sites in proteins METBIND Tsigelny, personal communication. The program is based on a bioinformatics approach where the predictions are calculated by the statistically significant correspondences to the typical structures of metal–protein complexes contained in PDB. We initially created a database of all possible binding sites for various metals present in the selected part of the non-redundant PDB. We then tested the program on proteins that were not used to develop the adaptive scores for Cu and Pt-binding sites. The program predicted more than 80 % of all Cu-binding sites in this part of the PDB with a Cα RMSD of 1 Å. Below, we briefly describe the program for Cu ions. The database of Cu and PT-binding proteins was divided into sub-groups according to the number of side chains (pairs, triads, and quadruples) that make close contact with Cu ions.
The search algorithm identifies protein structures that are structurally similar to those known to bind Cu ions. Initially, the program checks for possible binding sites on the surface of a protein. The program identifies sites having the specified residue-Cu combination pairs, then the selected sites are filtered with an RSMD filter to find atoms that can be placed within 3.5 Å of a hypothetical Cu ion. Atoms that pass this filter are then considered as possible metal-contacting protein sites. Table 1 lists the information obtained. The data in Table 2 presents a list of the most frequent Cu-residue contacts in the PDB databank and the scoring functions for the binding sites prediction.
Table 1.
Samples of metal-contacting atoms of proteins
| Protein and chain | Atom serial number of CU | Contacting residue | Contacting pair: residue-atom | Distance metal- residue atom Å |
|---|---|---|---|---|
| 3sod_B | 152 | HHHH | H_ND1 H_NE2 H_NE2 H_NE2 | 2.04 2.09 2.24 1.99 |
| 2idq_A | 107 | HCH | H_ND1 C_SG H_ND1 | 1.94 2.14 2.00 |
| 4paz_A | 124 | HCHM | H_ND1 C_SG H_ND1 M_SD | 1.98 2.14 1.95 2.76 |
| 3nsc_A | 602 | HHH | H_ND 1 H_NE2 H_NE2 | 1.93 2.03 2.1 |
| 3nsc_A | 603 | HHH | H_NE2 H_NE2 H_NE2 | 1.95 1.95 3.32 |
Information from this table was used to calculate the scoring function presented in Table 2: Ps = (Pn/ΣPn) × 100 % (where Pn is the number of contacts for the metal-specified residue atom in the pdb); infrequently occurring contacts have a negligible score function
C, H, M—one letter code for amino acids
Table 2.
Protein atoms that contact Cu, scoring function and average distances
| Residue-atom | Scoring function Ps | Average distance to metal atom (Å) |
|---|---|---|
| H-NE2 | 27.69 | 2.21 |
| C-SG | 27.00 | 2.24 |
| H-ND1 | 19.46 | 2.15 |
| D-OD1 | 14.00 | 3.13 |
| M-SD | 8.63 | 2.77 |
| D-OD2 | 7.45 | 3.23 |
| M-CG | 7.41 | 3.84 |
| G-O | 1.80 | 3.03 |
| H-CD2 | 1.77 | 3.59 |
| F-CB | 1.02 | 3.84 |
| P-O | 0.81 | 3.77 |
H, C, M, G, D, F, P—one letter code for amino acids
Results
hCTR1 Model Structure Incorporates the Helices of the X-ray Structure
Modeling hCTR1 is a significant challenge: there are no homologous protein structures available where a transmembrane channel is formed by a trimer. hCTR1 was modeled by taking the existing cryoelectron microscopy data [20–21] as a starting point and analyzing possible conformations for the protein using several protein structure prediction methods. Figure 4 shows the structure of the model (ribbon presentation) produced by this procedure. Residues that our analysis suggests are involved in Cu transport are represented in CPK format.
Fig. 4.

Ribbon diagram of hCTR1 with the CPK atom presentation scheme showing the positions of the stacked methionine triads of MET45, MET43, MET154, and MET150. A view from the side showing also the HIS188 and HIS190 residues at the C-terminus; B view from the top
Extracellular Domains of the CTR1 Monomers and Trimer are Stabilized by Hydrophobic Interactions
In the N-terminal extracellular domains of the hCTR1 subunits, the hydrophilic residues are located mostly on the surface of the protein, and a number of histidine residues including HIS3, HIS5-6, HIS22-24, and HIS33, are solvent accessible. As described below, we believe that these residues bind Cu(I) ions as the first step in the transport process. The polar residues ASP2, ASP13, ASP37, LYS52, and GLU55 are also solvent accessible. The abundance of negative residues creates a total negative charge of −9 electrons for the N-terminal extracellular domain of hCTR1, so it will have significant attraction for positively charged ions. The highly hydrophilic surface of the extracellular domain makes it quite soluble in water, but the N-terminal domain also contains a significant number of hydrophobic interactions that provide a physical basis for conformational stability. Residues MET12, PHE47, PHE49, VAL54, LEU56, PHE58, ILE83, the hydrophobic part of TYR11 and MET1 along with the MET42 and LEU57 pair create a very strong intrinsic hydrophobic network. This region has few hydrogen bonds salt bridges or disulfide bonds that might restrict flexibility. As a result, movement of the entire transporter can occur by rotation of the subunits relative to each other. The hydrophobic interactions may provide the flexibility for the proper functioning as an opening and closing transporter in its trimeric form, while also providing sufficient stability to restore the conformation of this domain after it has been perturbed.
It is important to note that the interfaces between the N-terminal extracellular domains of adjoining monomers are also supported exclusively by hydrophobic interactions. If one follows the channel from the first level MET45 triad (Fig. 4b), the first point of hydrophobic interactions exists between methionines 45A, 45B, and 45C. Strong hydrophobic interactions also link the MET45 residues and PRO44 residues of the neighboring subunits. At the second level, the extracellular domain of the trimer is stabilized by hydrophobic interactions involving the MET43 triad. At the third level, hydrophobic interactions involving the MET154 triad and interactions between the MET154 residues with LEU151 of the neighboring subunits contribute to conformational stability. The trimer is also stabilized by hydrophobic interaction within the MET150 triad at the fourth level. Taking into consideration all the hydrophobic interactions that keep the N-terminal parts of CTR1 together leads to the prediction that this transporter would be less stable and consequently less efficient at lower temperatures since hydrophobic interactions lose strength at low temperatures.
The Extracellular Domain of the hCTR1 Trimer Contains Four Methionine Triads that Constitute the Channel
When viewed from the side, one can see that a channel is formed by the four triads of methionines, which align into four stacked rings (Fig. 4a). When viewed from above the membrane (Fig. 4b), it is apparent that all four methionine triads are aligned to form a channel. The ring formed by the lowest triad in the stack, the one closest to the extracellular surface of the membrane, is clearly seen and shows that the structure is in the “closed” state. This is consistent with the observation that the crystal structure determined by cryoelectron microscopy is in closed state. The conformation of the closed state is also supported by MD simulations [35]. The distances between the Cα atoms of the methionine residues in adjacent triad rings varies from 5 to 7 Å, while the distance between sulfur atoms in each adjacent triads varies from ~4 to 6 Å (Fig. 4a, b).
Tail-to-Tail Packing of the C-terminal Domain Creates Specific Cu-Binding Sites
The side chains of histidines 188 and 190 are located close to each other in the C-terminal tail of hCTR1. Rotation of the CYS189 side chains along the CA–CB bond changes the conformation of the two adjacent CYS189 residues so the distance between the S atoms can be suitable for positioning of Cu atoms. It is also noteworthy that the CYS189 residues are located close to MET106 of the neighboring subunits; the distance between their sulfur atoms is<5 Å. Similar pairs of CYS residues participate in Cu ion binding in several other protein structures in the PDB databank, making this region a candidate for a Cu-binding site. Cu bound at this site would stabilize the trimer and C-terminal region of the individual subunits. Positions of Cu atoms near the HIS188, HIS190 residues (violet spheres in Fig. 5) are predicted by our program Metbind.
Fig. 5.

Predicted Cu-binding sites (pink spheres) in the model of hCTR1. Sites are found at each of the four stacked methionine triads, at the lower end of the vestibule on the cytoplasmic side of the protein and at various sites in the N-terminal domain (Color figure online)
Identification of Cu-Binding Sites Within CTR1
Figure 5 shows the predicted Cu-binding sites in hCTR1 that were identified by the most stringent discrimination and filtering criteria of the METBIND program. Each of the sites has a structural homolog in the PDB. Possible binding sites for Cu ions that occur in the extracellular N-terminal region include HIS23-HIS6, HIS6-HIS33, ASP2-HIS3, HIS23-MET7, HIS3-MET4 and at the site between MET12 and the carbonyl oxygen atom of PRO20. All these sites were formed by interactions within the same monomer. Binding of Cu to HIS3, HIS5, and HIS6 has been shown by Cu competition with alanine substituted 1–14 N-terminal hCTR1 peptides [36]. All four stacked rings of methionines are also identified as Cu-binding sites (MET45, MET43, MET154, and MET150). All predicted Cu-binding sites on the cytoplasmic face of the protein were associated with HIS188 and HIS190. HIS–HIS sites have previously been shown to form stable complexes with Cu+1 [37]. It is noteworthy that yeast CTR1 does not have histidines in its extracellular domain. This can be attributed to the fact that yeast must import Cu from acidic environments, where histidine binds Cu poorly. This is in contrast to mammalian systems, where hCTR1 imports Cu from a neutral environment [38]. A possible Cu-binding site may also exist between CYS189 and MET106 of the neighboring subunit; such an interaction could play a role in stabilizing the trimer. These predicted binding sites are organized in a manner expected to provide a clear path from the extracellular domain, through the plasma membrane to the intracellular domain.
Transport of Cu in hCTR and Role of Electrostatic Field
CTR1 is not an ATPase and transport of Cu by hCTR1 is a passive process. We calculated the profile of the electrostatic field of the CTR1 trimer by Poisson–Boltzmann equations. Figure 6 shows the electrostatic profile of hCTR1 with equipotential surfaces of +1.8 (blue) or −1.8 (red) electrons. The model predicts that Cu ions coming from the extracellular environment are attracted to the negative extracellular domain (top of the model structure in Fig. 6), where they are concentrated and directed toward the central MET triads. The central part of the channel is neutral so passage through this section must involve protein-mediated transfer into the intramembranous part of the channel that does not depend on an electrostatic field. A number of possible mechanisms can be proposed for this transfer [39]. An example of such a mechanism for the transfer of Cu from ATOX1 to APT7B has already been reported [40]. The sides of the channel closest to the center of the transporter on the cytoplasmic side are positive and the parts located farther from the center are negative. This unique charge profile at the cytoplasmic side of hCTR1 creates a dipole moment leading metal ions from the exit of the channel toward sites where they are stored before their transfer to the intracellular Cu chaperones.
Fig. 6.
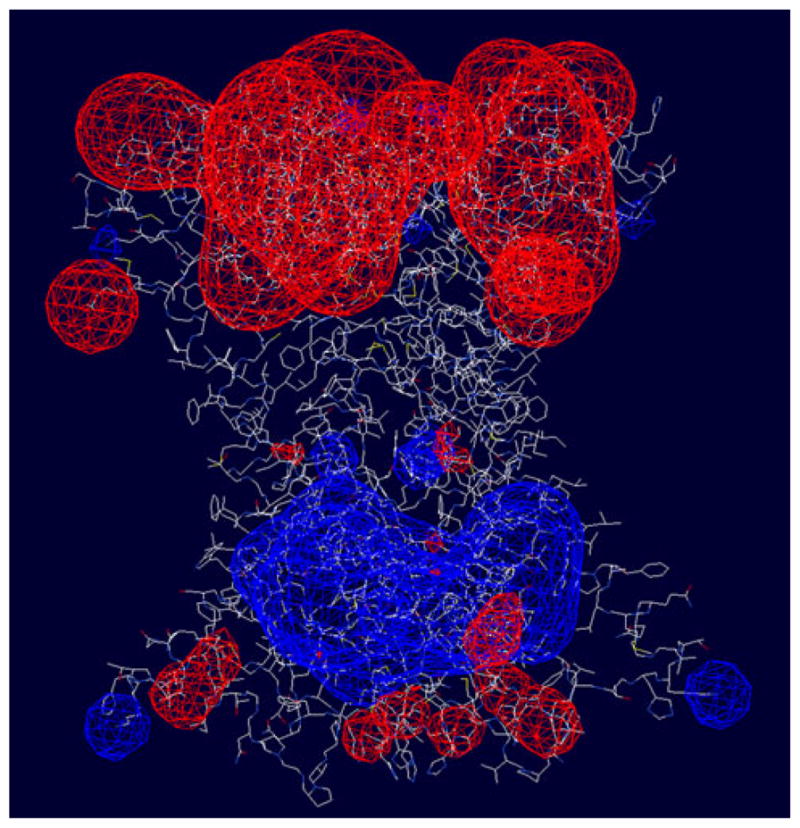
Electrostatic profile of hCTR1 with the values of 1.8 (blue) or −1.8 (red) electrons showing equipotential surfaces (Color figure online)
MDs of CTR1 in the POPC Membrane
The results show that during the simulations the transporter remains reasonably stable (Fig. 7). The regions containing amino acids 67–94 and 126–180, constituting the transmembrane and adjacent residues, are stable with overall RMSDs of <1.5 Å during 20 ns MD simulations (Fig. 8). The N-terminal portion of the transporter has the maximum RMSD during MD, as expected for an extracellular region, and the intracellular loop including residues 95–125 and the C-terminal domain also have significant flexibility (Fig. 8). The behavior of these regions is consistent with the results of cryoelectron microscopy, where the flexibility of these regions was so high that their structures were not resolved. It is important to note that the transporter exists in a large number of possible conformations with finite statistical probability. We expect that our model represents the centroid of these many possible conformers. One of the important features of the model is elucidation of four methionine-triads at residues 43, 45, 150, and 154 of each subunit. Figure 8b shows the evolution of the important distances between the methionine sulfur atoms during MD. One can see that distances remain within a reasonable level of stability between 4 and 6.5 Å.
Fig. 7.

Superimposed conformations of hCTR model at the start and after MD within the POPC membrane. Violet initial; brown 20 ns (Color figure online)
Fig. 8.

Evolution of interatomic distances during MD. a RMSD for different domains of hCTR1. Most stable regions are those containing the transmembrane and adjacent residues 67–94 and 126–180. b Variation in distances between S atoms in the methionine triads
Discussion
Correspondence Between Model Predictions and Mutational Analyses of Cu Transport
Some information is available in the literature on the effect of deletion or mutation of specific motifs and amino acids on the ability of hCTR1 to transport Cu. Table 3 provides a summary of this information. In many cases, the effect of these deletions and mutations is consistent with the all-atom model presented here. The model predicts that transport will be disrupted if any methionine residues in the four triads (MET45, MET43, MET154, and MET150) are converted to an amino acid that is unable to bind Cu. Interestingly, when expressed in Sf9 cells, removing the first two triads formed by MET45 and MET43 (by deletion of the first 53 amino acids) decreased Cu transport only 70 %, whereas removal of the first 69 residues decreased the Cu transport by 90 % [41]. On the other hand, reductions of >10-fold were observed when both MET45 and MET43 were converted to alanine [42], or glutamine [16]. The results suggest that while deletion of METs 43/45 permits efficient diffusion of free Cu ions into the channel, replacing METs 43/45 with non-coordinating residues creates a steric barrier to Cu uptake. In other words, METs43/45 are not required for free Cu ions to enter the channel, but placing a non-coordinating residue at this location creates a physical block to Cu transport. The role of METs43/45 in the carefully regulated physiological movement of Cu from transport proteins to the hCtr1 remains to be evaluated.
Table 3.
Effect of mutations and deletions in CTR1 on Cu transport
| Mutant | Cell type tested | Effect on Cu transport
|
Reference | All-atom model prediction | |
|---|---|---|---|---|---|
| Km/Vmax | Other | ||||
| hCTR1-WT | Sf9 | Km 3.5 | [39] | ||
| hCTR1-WT | Sf9 | Km 10.1 | |||
| hCtr1-N15Q | Sf9 | Km 5.9 | Tryptic digest reduced by Cu | ||
| hCTR1-C161S | Sf9 | Km 8.2 | |||
| hCT1-C189S | Sf9 | Km 11.9 | Trimer can be stabilized with Cu binding to C189 and M106 of the neighboring subunit. Specificity of Cu binding at the exit from the transporter is diminished | ||
| hCTR1-WT | Sf9 |
Km 8.2 Vmax 84.0 |
[40] | ||
| hCTR1-C189S | Sf9 |
Km 6.8 Vmax 66.0 |
Trimer can be stabilized with Cu binding to C189 and M106 of the neighboring subunit. Specificity of Cu binding at the exit from the transporter is diminished | ||
| hCTR1- 40MMMPM45 → 40AAAPA45 | HEK293 | Markedly reduced | [38] | Trimer loses two MET triads created by M43 and M45. These triads are a necessary part of the transport mechanism | |
| hCTR1-M150L | HEK293 | Abolished | Trimer loses a MET triad and the transport residues chain is interrupted | ||
| hCTR1-M154L | HEK293 | Abolished | Trimer loses MET triad and the transport residues chain is interrupted | ||
| hCTR1-WT | Sf9 |
Km 8.9 Vmax 75.7 |
[37] | ||
| hCTR1-MG34 (deleted amino acids 1–34) | Sf9 |
Km 19.8 Vmax 75.8 |
Slightly less uptake than WT | Subunits lose a set of external binding sites for Cu. These sites may work for initial uptake of Cu from transport proteins. Deletion of Cu-binding residues can decrease specificity (increase Km) | |
| hCTR1-MN53 (deleted amino acids 1–53) | Sf9 |
Km 2.6 Vmax 15.8 |
Slightly less uptake than WT | Trimer loses two MET triads created by M43 and M45, as well as a set of external binding sites for Cu. Specificity of Cu transport by the first external MRT triads is lost. This can lead to an increase of Km. The external binding sites may mediate initial acquisition of Cu from the transporting proteins. The resulting parameters are the sum of these two effects | |
| hCTR1-M69 (deleted amino acids 1–69) | Sf9 |
Km 0.9 Vmax 5.7 |
Slightly less uptake than WT | Trimer loses two MET triads created by M43 and M45. Also subunits lose a greater set of external binding sites for Cu. These sites can work for the initial uptake of Cu from the transporting proteins | |
| hCTR1-M150I//M154I | Sf9 |
Km 10.9 Vmax 17.7 |
Significantly less Cu uptake than WT | Trimer loses two MET triads created by M150 and M154 that are essential to transport | |
| hCTR1-C189S | Sf9 | Km 7.0 51.5 | Significantly less Cu uptake than WT | Trimer can be stabilized by Cu binding to C189 and M106 of the neighboring subunit. Specificity of Cu binding at the exit from the transporter is diminished | |
| hCTR1-H139A | Sf9 | Km 11.6 26.7 | Vmax increased | Residue H139 with the neighboring Q142 can serve as an intrachannel binding site for Cu. Changing H139 to alanine can abolish this site and increase Km | |
| hCTR1-H139R | Sf9 |
Km 146.1 533.2 |
Vmax increased | Residue H139 with the neighboring Q142 can serve as an intrachannel binding site for Cu. Changing H139 to the positive argenine makes this site repulsive and Km greater. At the same time it can decrease the length of time that Cu stays in the channel, thus increasing Vmax | |
| hCTR1-WT | SCLC |
Km 15.4 Vmax 87.2 |
[15] | ||
| hCTR1-M43Q | SCLC |
Km 3.5 Vmax 5.2 |
Trimer loses MET triads created by M43. Note that loss of each triad produces comparable results. Supports the model with four layers of triads | ||
| hCTR1-M45Q | SCLC |
Km 4.0 Vmax 6.9 |
Trimer loses MET triads created by M45. Note that loss of each triad produces comparable results. Supports the model with four layers of triads | ||
| hCTR1-M154Q | SCLC |
Km 3.5 Vmax 5.0 |
[15] | Trimer loses MET triads created by M154. Note that loss of each triad produces comparable results. Supports the model with four layers of triads | |
| hCTR1-G167S | SCLC |
Km 3.8 Vmax 6.3 |
G167 of one of the intramembrane helices is very close to A192 of the neighboring helix of the same subunit. Insertion of any side chain (in this case serine) will affect the positions of the helices and may alter the conformation such that transport if less effective | ||
WT wild type
The other two methionine triads that lie more deeply in the intramembranous region (MET150 and MET154) appear to be more important to transport of free Cu. Conversion of MET150 or MET154 to amino acids that are incapable of binding Cu produced almost complete inhibition of Cu transport [16, 41, 42]. Similar observations have been made with yCTR1 [40]. It is noteworthy that when MET150 and MET154 were replaced with either cysteine or histidine (both of which can bind Cu) a functional form of yCTR1 was produced [42]. This provides further evidence for the crucial nature of the interaction of Cu with the methionine triads that line the channel. The all-atom model predicts that the possible dynamic movement of the upper two rings of methionines is substantially greater than that of the lower two rings the movement of which is quite constrained. Flexibility of the N-terminal extracellular part of hCTR1 was reported to be the main reason why the crystal structure determination in this part of the transporter was not successful [24].
The all-atom model of CTR1 predicts that the C-terminal HIS188-CYS189-HIS190 motif of CTR1 serves to bind Cu after it has moved through the channel (Fig. 2). Deletion of either the last 6 or 12 amino acids of CTR1, or conversion of CYS189 to serine slightly reduced Vmax for Cu transport (10–30 %) [41, 43]. The structure of CTR1 presented here predicts that deletion of the last six amino acids of CTR1 will eliminate the Cu-binding site formed by CYS188 and HIS190. Deletion of the last 12 amino acids also eliminates GLU187, which has an electrostatic interaction with ARG102 of the neighboring subunit. The absence of such an interaction would be predicted to alter the conformation of the C-terminals of the hCTR1 trimer and affect Cu transfer to chaperones. The reduction in Vmax is sufficiently small that it does not appear this region influences the rate limiting step for uptake of free Cu ions. Instead, this region may be involved in the transfer of Cu to intracellular transport proteins. Consistent with this notion, the HIS188-CYS-HIS190 motif appears to be required for docking of CTR1 to metallochaperones such as ATOX1 [44], and CYS189 has been reported to serve a gating function as it accepts Cu+l from more proximal portions of the CTR1 channel [24]. How the HIS188-CYS-HIS190 motif functions to transfer Cu(I) to the Cu chaperones has not yet been defined.
The all-atom model provides an explanation for several paradoxical effects on Cu transport observed in mutational studies. For example, conversion of HIS139 to arginine was found to produce a large increase rather than decrease in both Km and Vmax for Cu transport [41]. HIS139 together with GLN142 is an attractive site for transient binding of Cu. Conversion of HIS139 to arginine introduces a repulsive force at this site that would be expected to prevent the positive Cu+1 from binding. These changes would be expected to increase Vmax because the ions would not stop at these sites, but at the same time decrease Cu affinity leading to an increase in Km. Likewise, conversion of GLY167 to serine reduced the Km and markedly decreased the Vmax for Cu transport [16]. This effect can now be explained on the basis of the observation that in the all-atom model this residue is located within one of the transmembrane helices of hCTR1 and is very close to ALA192 of the neighboring helix of the same subunit. Insertion of a side chain of any size would be expected to affect the positions of the helices and reduce the efficiency of transport.
Correspondence Between Model Predictions and Mutational Analysis of CTR1 Degradation
High levels of Cu trigger endocytosis of CTR1, and in some types of cells this is accompanied by degradation [13–15], which may serve to limit accumulation of toxic levels of the metal [17–20]. cDDP triggers CTR1 degradation at much lower concentrations and this limits the amount of cDDP that enters the cell and reduces its cytotoxicity [14, 15]. In yeast, the degradation of CTR1 requires the E3 ubiquitin ligase Rsp5 [20]. In mammalian cells, cDDP induced degradation causes CTR1 becomes ubiquitinated [21]; inhibition of proteasome function can block the cDDP-induced degradation [14] and enhance the uptake and cytotoxicity of the drug [15]. The LYS178/LYS179 motif in the C-terminal domain of CTR1 is a potential site of ubiquitination, as is LYS121; the model predicts these sites are solvent accessible. In the all-atom model, LYS178 and LYS179 are close to the cytoplasmic side of hCTR1 well away from the channel, and thus would not be predicted to have a major effect on Cu transport. However, the model indicates that the side chain of LYS178 is exposed to the solvent and positioned, where it can interact with incoming molecules such as an ubiquitin ligase. The side chain of LYS179 is less exposed to the solvent, but it is positioned close to ARG102 of the neighboring hCTR1 subunit. This combination of two positively charged residues would be expected to create a repulsive force between the subunits that acts to decrease the total attractive forces between the hCTR1 monomers that keep the transporter together. When LYS179 is converted to alanine this repulsion force is deleted and the trimer would be expected to be more stable and less susceptible to degradation. These predictions have been borne out experimentally in mammalian cells. Whereas cDDP induced rapid degradation of wild type hCTR1, it failed to enhance ubiquitination or trigger degradation of a variant in which both LYS178 and 179 had been converted to alanines (Safaei, personal communication, 2011).
Another site of interest in hCTR1 with regard to the induction of degradation is TYR103 that is located in the intracellular loop between the first two transmembrane domains. Tyrosines in the cytosolic domains of proteins are often involved in endocytosis [45]. TYR103 is of particular interest because the TYR–X–X–MET motif within which it resides is a potential phosphorylation site that once phosphorylated this motif would be a candidate for binding to the SH2 domain of the p85 subunit of PI3K, which is involved in the endocytosis of PDGF and many other surface proteins [46]. The all-atom model indicates that TYR103, MET106, and other members of the site TYR-ASN-SER-MET are located in the region that is a part of the outside surface of the transporter.
Predictions Regarding the Mechanism of Cu Movement
One of the interesting features of CTR1 is that it transports Cu+1 but not Cu+2. This transport is unidirectional from outside the cell to its interior. The all-atom model provides some new insights into this behavior. There are distinct differences between the preferences of Cu+2 and Cu+1 ions. The Cu+2 ion prefers a planar or octohedral geometry because of Jahn–Teller distortion of d9 electron configuration, while Cu+1 forms a filled d10 configuration that does not have such stringent preferences and can participate in a number of 2-, 3-, and 4-coordinated sites [47]. This explains why hCTR1 selectively transports only Cu+1. The Cu+1 is stored at the 2- and 3-coordinated sites in the extracellular domain and then shifts to 3- and 4-coordination as it passes through the MET triads. It again returns in Cu+1 form on the intracellular side of the transporter because of a variety of mostly 2-coordinated systems and the organization of 3-coordinated system that mediates transfer to the intracellular Cu chaperones.
Acknowledgments
This work was supported by grants CA152185 and CA095298 from the National Institutes of Health, grant W81XWH-08-1-0135 from the Department of Defense, and a grant from the Clayton Medical Research Foundation. Additional support for core laboratories was from grants P30 NS047101 for the UCSD Neuroscience Microscopy Shared Facility and the UCSD Cancer Center Specialized Support grant P30 CA23100. We would like to thank Dr. Vincenz Unger for helpful discussions, and Dr. Ben-Tal for sharing the results of his simulation studies.
Contributor Information
Igor F. Tsigelny, Email: itsigeln@ucsd.edu, Department of Neurosciences, University of California, San Diego, 9500 Gilman Drive, La Jolla, San Diego, CA 92093-0505, USA. San Diego Supercomputer Center, University of California, San Diego, 9500 Gilman Drive, La Jolla, San Diego, CA 92093-0505, USA
Yuriy Sharikov, Department of Neurosciences, University of California, San Diego, 9500 Gilman Drive, La Jolla, San Diego, CA 92093-0505, USA.
Jerry P. Greenberg, San Diego Supercomputer Center, University of California, San Diego, 9500 Gilman Drive, La Jolla, San Diego, CA 92093-0505, USA
Mark A. Miller, San Diego Supercomputer Center, University of California, San Diego, 9500 Gilman Drive, La Jolla, San Diego, CA 92093-0505, USA
Valentina L. Kouznetsova, San Diego Supercomputer Center, University of California, San Diego, 9500 Gilman Drive, La Jolla, San Diego, CA 92093-0505, USA. Moores Cancer Center, University of California, San Diego, 3855 Health Sciences Drive, La Jolla, CA 92093-0819, USA
Christopher A. Larson, Moores Cancer Center, University of California, San Diego, 3855 Health Sciences Drive, La Jolla, CA 92093-0819, USA
Stephen B. Howell, Moores Cancer Center, University of California, San Diego, 3855 Health Sciences Drive, La Jolla, CA 92093-0819, USA
References
- 1.Puig S, Thiele DJ. Molecular mechanisms of copper uptake and distribution. Current Opinion in Chemical Biology. 2002;6:171–180. doi: 10.1016/s1367-5931(02)00298-3. [DOI] [PubMed] [Google Scholar]
- 2.Linder MC, Hazegh-Azam M. Copper biochemistry and molecular biology. American Journal of Clinical Nutrition. 1996;63:797S–811S. doi: 10.1093/ajcn/63.5.797. [DOI] [PubMed] [Google Scholar]
- 3.Howell SB, Safaei R, Larson CA, Sailor MJ. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Molecular Pharmacology. 2010;77:887–894. doi: 10.1124/mol.109.063172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crider SE, Holbrook RJ, Franz KJ. Coordination of platinum therapeutic agents to met-rich motifs of human copper transport protein1. Metallomics. 2010;2:74–83. doi: 10.1039/b916899k. [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Du X, Li H, Chan DS, Sun H. The effect of the extracellular domain of human copper transporter (hCTR1) on cisplatin activation. Angewandte Chemie (International ed in English) 2011;50:2706–2711. doi: 10.1002/anie.201006739. [DOI] [PubMed] [Google Scholar]
- 6.Lin X, Okuda T, Holzer A, Howell SB. The copper transporter CTR1 regulates cisplatin uptake in Saccharomyces cerevisiae. Molecular Pharmacology. 2002;62:1154–1159. doi: 10.1124/mol.62.5.1154. [DOI] [PubMed] [Google Scholar]
- 7.Ishida S, Lee J, Thiele DJ, Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14298–14302. doi: 10.1073/pnas.162491399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larson CA, Blair BG, Safaei R, Howell SB. The role of the mammalian copper transporter 1 in the cellular accumulation of platinum-based drugs. Molecular Pharmacology. 2009;75:324–330. doi: 10.1124/mol.108.052381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee J, Pena MM, Nose Y, Thiele DJ. Biochemical characterization of the human copper transporter Ctr1. The Journal of Biological Chemistry. 2002;277:4380–4387. doi: 10.1074/jbc.M104728200. [DOI] [PubMed] [Google Scholar]
- 10.Lee J, Petris MJ, Thiele DJ. Characterization of mouse embryonic cells deficient in the Ctr1 high affinity copper transporter. The Journal of Biological Chemistry. 2002;277:40253–40259. doi: 10.1074/jbc.M208002200. [DOI] [PubMed] [Google Scholar]
- 11.Jiang J, Nadas IA, Alison Kim M, Franz KJ. A mets motif peptide found in copper transport proteins selectively binds Cu(I) with methionine-only coordination. Inorganic Chemistry. 2005;44:9787–9794. doi: 10.1021/ic051180m. [DOI] [PubMed] [Google Scholar]
- 12.Wu X, Sinani D, Kim H, Lee J. Copper transport activity of yeast Ctr1 is down regulated via its C-terminus in response to excess copper. The Journal of Biological Chemistry. 2009;284:4112–4122. doi: 10.1074/jbc.M807909200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petris MJ, Smith K, Lee J, Thiele DJ. Copper-stimulated endocytosis and degradation of the human copper transporter, hCtr1. The Journal of Biological Chemistry. 2003;278:9639–9646. doi: 10.1074/jbc.M209455200. [DOI] [PubMed] [Google Scholar]
- 14.Holzer AK, Howell SB. The internalization and degradation of human copper transporter 1 following cisplatin exposure. Cancer Research. 2006;66:10944–10952. doi: 10.1158/0008-5472.CAN-06-1710. [DOI] [PubMed] [Google Scholar]
- 15.Jandial DD, et al. Enhanced delivery of cisplatin to intraperitoneal ovarian carcinomas mediated by the effects of bortezomib on the human copper transporter 1. Clinical Cancer Research. 2009;15:553–560. doi: 10.1158/1078-0432.CCR-08-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang ZD, Stockton D, Savaraj N, Kuo MT. Mechanistic comparison of human copper transporter hCtr1-mediated transports between copper ion and cisplatin. Molecular Pharmacology. 2009;76:843–853. doi: 10.1124/mol.109.056416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molloy SA, Kaplan JH. Copper-dependent recycling of hCTR1, the human high affinity copper transporter. The Journal of Biological Chemistry. 2009;284:29704–29713. doi: 10.1074/jbc.M109.000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holzer AK, Katano K, Klomp LW, Howell SB. Cisplatin rapidly down-regulates its own influx transporter hCTR1 in cultured human ovarian carcinoma cells. Clinical Cancer Research. 2004;10:6744–6749. doi: 10.1158/1078-0432.CCR-04-0748. [DOI] [PubMed] [Google Scholar]
- 19.Guo Y, Smith K, Lee J, Thiele DJ, Petris MJ. Identification of methionine-rich clusters that regulate copper-stimulated endocytosis of the human Ctr1 copper transporter. The Journal of Biological Chemistry. 2004;279:17428–17433. doi: 10.1074/jbc.M401493200. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, Sitaram A, Burd CG. Regulation of copper-dependent endocytosis and vacuolar degradation of the yeast copper transporter, ctr1p, by the rsp5 ubiquitin ligase. Traffic. 2007;8:1375–1384. doi: 10.1111/j.1600-0854.2007.00616.x. [DOI] [PubMed] [Google Scholar]
- 21.Safaei R, Maktabi MH, Blair BG, Larson CA, Howell SB. Effects of the loss of Atox1 on the cellular pharmacology of cisplatin. Journal of Inorganic Biochemistry. 2009;103:333–341. doi: 10.1016/j.jinorgbio.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aller SG, Unger VM. Projection structure of the human copper transporter CTR1 at 6-A resolution reveals a compact trimer with a novel channel-like architecture. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:3627–3632. doi: 10.1073/pnas.0509929103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Feo CJ, Aller SG, Unger VM. A structural perspective on copper uptake in eukaryotes. BioMetals. 2007;20:705–716. doi: 10.1007/s10534-006-9054-7. [DOI] [PubMed] [Google Scholar]
- 24.De Feo CJ, Aller SG, Siluvai GS, Blackburn NJ, Unger VM. Three-dimensional structure of the human copper transporter hCTR1. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4237–4242. doi: 10.1073/pnas.0810286106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia Y, Dewey TG, Shindyalov IN, Bourne PE. A new scoring function and associated statistical significance for structure alignment by CE. Journal of Computational Biology. 2004;11(5):787–799. doi: 10.1089/cmb.2004.11.787. [DOI] [PubMed] [Google Scholar]
- 26.Chivian D, Baker D. Homology modeling using parametric alignment ensemble generation with consensus and energy-based model selection. Nucleic Acids Research. 2006;34:e112. doi: 10.1093/nar/gkl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raman S, et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins. 2009;77(Suppl 9):89–99. doi: 10.1002/prot.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharikov Y, Walker RC, Greenberg J, Kouznetsova V, Nigam SK, Miller MA, Masliah E, Tsigelny IF. MAPAS: A tool for predicting membrane-contacting protein surfaces. Nature Methods. 2008;5:119. doi: 10.1038/nmeth0208-119. [DOI] [PubMed] [Google Scholar]
- 29.Roy A, Kucukural A, Zhang Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nature Protocols. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy A, Xu D, Poisson J, Zhang Y. A protocol for computer-based protein structure and function prediction. Journal of visualized experiments: JoVE. 2011:e3259. doi: 10.3791/3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kale L, et al. NAMD2: Greater scalability for parallel molecular dynamics. Journal of Computational Physics. 1999;151:282–312. [Google Scholar]
- 32.Feller SE, MacKerell AD. An improved empirical potential energy function for molecular simulations of phospholipids. The Journal of Physical Chemistry B. 2000;104:7510–7515. [Google Scholar]
- 33.Tu K, Tobias DJ, Klein ML. Constant pressure and temperature molecular dynamics simulation of a fully hydrated liquid crystal phase dipalmitoylphosphatidylcholine bilayer. Biophysical Journal. 1995;69:2558–2562. doi: 10.1016/S0006-3495(95)80126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feller S, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: the Langevin piston method. Journal of Chemical Physics. 1995;103:4613–4621. [Google Scholar]
- 35.Schushan M, Barkan Y, Haliloglu T, Ben-Tal N. C(alpha)-trace model of the transmembrane domain of human copper transporter 1, motion and functional implications. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10908–10913. doi: 10.1073/pnas.0914717107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haas KL, Putterman AB, White DR, Thiele DJ, Franz KJ. Model peptides provide new insights into the role of histidine residues as potential ligands in human cellular copper acquisition via ctr1. Journal of the American Chemical Society. 2011;133:4427–4437. doi: 10.1021/ja108890c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Himes RA, Park GY, Barry AN, Blackburn NJ, Karlin KD. Synthesis and X-ray absorption spectroscopy structural studies of Cu(I) complexes of histidylhistidine peptides: The predominance of linear 2-coordinate geometry. Journal of the American Chemical Society. 2007;129:5352–5353. doi: 10.1021/ja0708013. [DOI] [PubMed] [Google Scholar]
- 38.Rubino JT, Chenkin MP, Keller M, Riggs-Gelascob P, Franz KJ. A comparison of methionine, histidine and cysteine in copper(I)-binding peptides reveals differences relevant to copper uptake by organisms in diverse environments. Metallomics. 2011;3:61–73. [PubMed] [Google Scholar]
- 39.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nature Chemical Biology. 2008;4:176–185. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 40.Rodriguez-Granillo A, Crespo A, Estrin DA, Wittung-Stafshede P. Copper-transfer mechanism from the human chaperone Atox1 to a metal-binding domain of Wilson disease protein. The Journal of Physical Chemistry. 2010;114:3698–3706. doi: 10.1021/jp911208z. [DOI] [PubMed] [Google Scholar]
- 41.Eisses JF, Kaplan JH. The mechanism of copper uptake mediated by human CTR1: A mutational analysis. Journal of Biological Chemistry. 2005;280:37159–37168. doi: 10.1074/jbc.M508822200. [DOI] [PubMed] [Google Scholar]
- 42.Puig S, Lee J, Lau M, Thiele DJ. Biochemical and genetic analyses of yeast and human high affinity copper transporters suggest a conserved mechanism for copper uptake. Journal of Biological Chemistry. 2002;277:26021–26030. doi: 10.1074/jbc.M202547200. [DOI] [PubMed] [Google Scholar]
- 43.Eisses JF, Kaplan JH. Molecular characterization of hCTR1, the human copper uptake protein. Journal of Biological Chemistry. 2002;277:29162–29171. doi: 10.1074/jbc.M203652200. [DOI] [PubMed] [Google Scholar]
- 44.Xiao Z, Wedd AG. A C-terminal domain of the membrane copper pump Ctr1 exchanges copper(I) with the copper chaperone Atx1. Chemical Communications (Cambridge, England) 2002;2:588–589. doi: 10.1039/b111180a. [DOI] [PubMed] [Google Scholar]
- 45.Esposito DL, Li Y, Cama A, Quon MJ. Tyr(612) and Tyr(632) in human insulin receptor substrate-1 are important for full activation of insulin-stimulated phosphatidylinositol 3-kinase activity and translocation of GLUT4 in adipose cells. Endocrinology. 2001;142:2833–2840. doi: 10.1210/endo.142.7.8283. [DOI] [PubMed] [Google Scholar]
- 46.Wu H, Windmiller DA, Wang L, Backer JM. YXXM motifs in the PDGF-beta receptor serve dual roles as phosphoinositide 3-kinase binding motifs and tyrosine-based endocytic sorting signals. Journal of Biological Chemistry. 2003;278:40425–40428. doi: 10.1074/jbc.C300225200. [DOI] [PubMed] [Google Scholar]
- 47.Haas K. PhD dissertation. Duke University; Durham, NC: 2010. Copper at the interface of chemistry and biology: New insights into hCtr1 function and the role of histidine in human cellular copper acquisition. [Google Scholar]