

Abstract
Aims/hypothesis
Although skeletal muscle insulin resistance has been associated with activation of c-Jun N-terminal kinase (JNK), whether increased JNK activity causes insulin resistance in this organ is not clear. In this study we examined the metabolic consequences of isolated JNK phosphorylation in muscle tissue.
Methods
Plasmids containing genes encoding a wild-type JNK1 (WT-JNK) or a JNK1/JNKK2 fusion protein (rendering JNK constitutively active; CA-Jnk) were electroporated into one tibialis anterior (TA) muscle of C57Bl/6 mice, with the contralateral TA injected with an empty vector (CON) to serve as a within-animal control.
Results
Overproduction of WT-JNK resulted in a modest (~25%) increase in phosphorylation (Thr183/Tyr185) of JNK, but no differences were observed in Ser307 phosphorylation of insulin receptor substrate 1 (IRS-1) or total IRS-1 protein, nor in insulin-stimulated glucose clearance into the TA muscle when comparing WT-JNK with CON. By contrast, overexpression of CA-Jnk, which markedly increased the phosphorylation of CA-JNK, also increased serine phosphorylation of IRS-1, markedly decreased total IRS-1 protein, and decreased insulin-stimulated phosphorylation of the insulin receptor (Tyr1361) and phosphorylation of Akt at (Ser473 and Thr308) compared with CON. Moreover, overexpression of CA-Jnk decreased insulin-stimulated glucose clearance into the TA muscle compared with CON and these effects were observed without changes in intramuscular lipid species.
Conclusions/interpretation
Constitutive activation of JNK in skeletal muscle impairs insulin signalling at the level of IRS-1 and Akt, a process which results in the disruption of normal glucose clearance into the muscle.
Keywords: Inflammation, Insulin resistance, JNK, Obesity, Serine threonine kinases, Skeletal muscle
Introduction
Although obesity is closely associated with insulin resistance, the exact mechanisms by which increased adiposity contributes to this disorder are not fully resolved. Insulin resistance is, however, associated with excess ectopic lipid deposition in liver and skeletal muscle [1]. Moreover, the accumulation of deleterious lipid species such as diacylglycerol (DAG) [2] and ceramide [3] are known to dysregulate insulin action, either directly or indirectly, via the activation of important serine threonine kinases such as c-Jun N-terminal kinase (JNK), IκB (inhibitor of nuclear factor κB) kinase β (IKK-β) or protein kinase C. Both JNK and IKK-β are central to inflammatory signalling and have been shown to play a critical role in the development of insulin resistance, since genetic ablation of either JNK or IKK-β protects against diet-induced or genetic obesity [4, 5]. However, since activation of both JNK and IKK-β is thought to be elevated in insulin-resistant skeletal muscle, complete or partial ablation of the activation of these kinases may not be the most appropriate physiological model to examine their cause and effect relationship with respect to insulin action.
As discussed, global JNK1-deficient animals are protected from insulin resistance induced by a high-fat diet [4], but the role of selective ablation of JNK1 in skeletal muscle is unclear, since it either improves [6] or does not affect [7] insulin action. Furthermore, the potential compensatory interactions between JNK isoforms are also likely to complicate the tissue-specific deletion models [8]. Of note, pharmacological and/or physiological interventions that reduce lipid-induced activation of JNK in skeletal muscle also reduce insulin resistance [9, 10]. One mechanism by which activation of JNK is thought to impair insulin action is via enhanced serine phosphorylation of insulin receptor substrate 1 (IRS-1) [11], although it has recently been hypothesised that JNK may play a role in modulating intramyocellular lipid accumulation via altered activity of lipoprotein lipase (LPL) [6].
Skeletal muscle accounts for ~80% of insulin-stimulated whole-body glucose disposal, and in states of insulin resistance skeletal muscle is by far the most affected organ with respect to impaired insulin-stimulated glucose disposal [12]. It is somewhat surprising, therefore, that the effect of increased JNK activation on insulin sensitivity in this tissue has not been extensively studied. Accordingly, in the current study we used in vivo electroporation to deliver plasmids containing genes encoding either wild-type (WT-JNK) or constitutively active (CA-JNK) Jnk constructs specifically in skeletal muscle and examined insulin-stimulated glucose clearance and insulin signal transduction. While a modest, short-term increase in phosphorylation of JNK was insufficient to lead to impaired insulin action, constitutive activation of this inflammatory protein increases Ser307 phosphorylation of IRS-1 and decreases IRS-1 levels and phosphorylation of the insulin receptor and Akt, impairing insulin action.
Methods
Animals
Male WT C57Bl/6 mice (AMREP AS, Melbourne, VIC, Australia) were used for all experiments. All experiments were approved by the Alfred Medical Research Education Precinct Animal Ethics Committee and commenced when mice were 8 weeks of age. Mice were fed a chow diet containing 5% of total energy from fat (Specialty Feeds, Glen Forrest, WA, Australia). Animals were administered food and water ad libitum and housed in a controlled environment (22.0±0.5°C) with a 12 h light–dark cycle.
Plasmid construction
JNKK2-JNK1 fusion protein
The CA-JNK fusion protein was generated by enzyme substrate fusion as previously described [13]. Briefly, Jnk1 (also known as Mapk8) was fused in frame with its specific activator Jnkk2 (also known as Map2k7) with DNA encoding a decapeptide linker (Gly-Glu)5 inserted between the coding sequences and inserted into the pSRα3HA vector. Fusion of JNK1 to its upstream activator JNKK2 results in its constitutive activation [13].
WT-JNK
As previously described [14], the WT-JNK plasmid contains human JNK1 cDNA with an HA-tag at the 5′ region cloned by blunt-end ligation at the Xho I site in the pCAGGS vector [15]. Overproduced WT-JNK is functional in skeletal muscle in vivo [14].
Green fluorescent protein
The green fluorescent protein (gfp)-expressing plasmid (pCDNA3 GFP) was constructed by obtaining the pCDNA3 vector from Invitrogen (Carlsbad, CA, USA), and the gfp cDNA was cloned into the vector. Plasmid DNA was prepared using a Plasmid Purification Megaprep kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s specifications. The DNA concentration was quantified using a Nanodrop ND-1000 Spectrophotometer (Biolab, Scoresby, VIC, Australia) and the DNA dissolved in saline (154 mmol/l NaCl) to a final concentration of 4 μg/μl.
In vivo electrotransfer
The purified plasmids were directly injected into mouse tibialis anterior (TA) muscle as previously described [16, 17]. The TA muscle was chosen because it can be electroporated easily without the requirement for surgery, and the fibre composition is representative of the entire musculature of the mouse. Briefly, mice were anaesthetised with isoflurane, their hindlimbs shaved and TA muscles injected with 30 μl 0.5 U/μl hyaluronidase. After 2 h, mice were again anaesthetised with isoflurane and 100 μg WT-JNK, CA-Jnk or pcDNA3 GFP (in 25 μl saline) injected into the right TA muscle using a 29-gauge needle. The left TA muscle was injected with 25 μl empty vector which served as a control (CON). Stainless steel electrodes were placed on the muscle and square-wave electrical pulses (200 V/cm) were applied eight times with an electrical pulse generator at a rate of 1 pulse/s, with each pulse being of 20 ms duration. Insulin-stimulated glucose clearance experiments (subsequently described) were performed on animals 7 days after electroporation, a time point when we have shown peak overexpression of constructs (Fig. 1). The muscles were rapidly frozen and stored at −80°C.
Fig. 1.

GFP in TA muscles following gene transfer by electroporation. pCDNA3 GFP expression vector (100 μg) or saline was injected into the TA muscles of mice, followed by electroporation under anaesthesia. (a) Immunoblot of GFP from two animals (b) Control image under fluorescence where muscle had been electroporated with saline only. (c) TA muscle cross-section under normal light. (d) GFP staining in a TA muscle electroporated with gfp
In vivo insulin-stimulated muscle glucose clearance
Insulin-stimulated glucose clearance into the TA muscle was examined as previously described [18]. Briefly, 5 h fasted mice were anaesthetised with sodium pentobarbitone (50 mg/kg i.p.) and injected intravenously with 0.5 U/kg insulin (Actrapid; Novo Nordisk, Copenhagen, Denmark) and 370,000 Bq 2-deoxy[3H]glucose (GE Healthcare Life Sciences, Little Chalfont, UK). Samples of blood were taken from the tail 2, 5, 10, 15, 20 and 30 min after injection and mixed with 100 μl 10 U/ml heparin/saline. Blood glucose was simultaneously measured using an Accu-Chek Advantage meter (Roche Diagnostics, NSW, Australia). Blood radioactivity was determined at each time point by liquid scintillation beta counting. After 30 min, mice were killed and the TA muscles removed and frozen in liquid nitrogen. To analyse the clearance of 2-deoxy[3H]glucose, the muscle was homogenised, centrifuged at 15,700×g for 10 min and the supernatant fraction collected. To determine the total counts, 200 μl supernatant fraction was analysed by liquid scintillation counting. A 400 μl sample of the supernatant fraction was passed through an anion exchange column (AS 1-X8 resin; Bio-Rad, Hercules, CA, USA) for determination of the free counts. The rate of tissue metabolism of 2-deoxy[3H]glucose (K) was calculated using the single-injection model [19]. The tissue content of 2-deoxy[3H]-glucose-6-phosphate (C; dpm/g) was used to calculate the amount of 2-deoxy[3H]glucose metabolised in the TA muscle. The initial blood 2-deoxy[3H]glucose concentration (Cpo; dpm/ml) and the rate of disappearance of 2-deoxy[3H]glucose from the blood (Kp) were derived from a single exponential fit of the 2-deoxy[3H]glucose concentration in the blood at the time points studied for each mouse.
Analysis of insulin signalling
Mice were fasted for 5 h and anaesthetised by injection of 50 mg/kg i.p. sodium pentobarbitone. The peritoneal cavity was then opened and insulin (1 U/kg) was injected into the hepatic portal vein. After 10 min, TA muscles were dissected and immediately frozen in liquid nitrogen. TA muscles were harvested from a separate group of mice that were not insulin stimulated. These tissues were used for analysis of insulin signalling in the basal state.
Western blotting
Muscle samples were lysed and protein concentration determined. Lysates were solubilised and 40 μg protein was loaded and resolved by SDS-PAGE on polyacrylamide gels, transferred to membranes and blocked with 5% milk. Immunoblotting was performed using the following primary antibodies: LPL (Abcam, Cambridge, MA, USA); α-tubulin (Sigma, St Louis, MO, USA); GFP, phosphorylated Akt (Ser473), phosphorylated Akt (Thr308), Akt, phosphorylated JNK (Thr183/Tyr185), JNK, phosphorylated IRS-1 (Ser307), phosphorylated IRS-1 (Tyr612), total insulin receptor, phosphorylated insulin receptor-β (Tyr1361) and β-actin (all from Cell Signaling Technology, Danvers, MA, USA). After incubation with appropriate secondary antibody, the immunoreactive proteins were detected with enhanced chemiluminescence and quantified by densitometry.
Sectioning and imaging of GFP
The TA muscle sample was dissected and placed in embedding medium (Tissue-Tek; ProSciTech, QLD, Australia) so that the muscle fibres were vertical, frozen in precooled isopentane and stored at −80°C. Samples were cut in cross-sections in a cryostat (Leica CM1900; Leica Microsystems, Germany) at a thickness of 10 μm and picked up using a Polysine 25×75×1.0 mm precleaned slide (Lomb Scientific, Sydney, NSW, Australia). Slides were viewed under an Olympus CKX41 microscope (Mt Waverley, VIC, Australia) under normal light to orientate the sections and then with fluorescence to identify GFP (×10 magnification). Three sections from each sample were analysed and averaged for GFP content and expressed as a percentage of total cells in the field of view.
Lipid analysis
Lipidomic analyses were performed on TA muscles that were homogenised in 300 μl PBS buffer, pH 7.47, according to previous methods [20] with modifications. Briefly, lipids were extracted from 50 μg protein in 96-well plates, whereby there was no partitioning between the aqueous and organic phases, recovering all lipids in a single phase suitable for liquid chromatography–mass spectrometry analysis. Lipid analysis was performed by liquid chromatography, electrospray ionisation–tandem mass spectrometry using an HP 1200 liquid chromatography system combined with a PE Sciex API 4000 Q/TRAP mass spectrometer with a turbo-ionspray source (350°C) and Analyst 1.5 data system as previously described [21].
Statistics
Data were analysed using paired t test analysis. All data are presented as mean±standard error of the mean (SEM). Statistical significance was set at p<0.05.
Results
Delivery of GFP into skeletal muscle
To determine the in vivo electroporation efficiency of our system, we injected 100 μg pCDNA3 GFP vector or saline only into TA muscles. Muscles were then electrically stimulated to facilitate gene transfer into the muscle cells and harvested 7 days later. Figure 1a depicts a representative western blot probed for GFP, where GFP abundance was observed in the test leg and not in the control leg. Fluorescence microscope visualisation and analysis revealed that the percentage of fibres that produced GFP as a percentage of total muscle cells was 53.9±3.9% (Fig. 1b–d).
Overexpression of WT-JNK into TA modestly increases JNK phosphorylation but does not affect insulin signalling or action
Phosphorylation (Thr183/Tyr185) and total JNK were measured by western blotting 7 days after electroporation of the WT-JNK plasmid into TA muscle. Electroporation of WT-JNK increased JNK protein levels approximately sixfold (Fig. 2a) but resulted in a relatively modest (~25%), though nonetheless significant, increase in JNK phosphorylation (Fig. 2b). Interestingly, and somewhat unexpectedly, we consistently observed that the WT-JNK overexpression resulted in an increase predominately in the 46 kDa band for total JNK and the 54 kDa band for phosphorylated JNK.
Fig. 2.

Representative western blots and quantification of total (a) and phosphorylated (Thr183/Tyr185) (b) WT-JNK in TA muscle from C57Bl/6 mice 7 days following electroporation of an empty vector (C; CON) or WT-JNK (T; Test) plasmid. The 46 and 54 kDa bands can be seen. Data are expressed as mean±SEM; *p<0.05, **p<0.01; n=6–9 per group; p, phosphorylated
We next examined whether such increases in total and phosphorylated JNK affected insulin signalling and/or action. Initially, we performed experiments on both individual basal and insulin-stimulated groups of mice using Akt measured by western blotting. Both test leg (WT-JNK) and CON muscle had similar increases in magnitude of phosphorylation of Akt at two critical residues (Ser473 and Thr308) in response to insulin compared with the basal group (Electronic supplementary material [ESM] Fig. 1a). Following this, we next performed experiments whereby all animals were insulin stimulated so that a bone fide within-animal comparison could be evaluated. No differences were observed in Ser307 phosphorylation of IRS-1, insulin-stimulated Tyr612 phosphorylation or total IRS-1 when comparing the test (WT-JNK) with the CON muscle (Fig. 3a). In addition, while there was a small, but significant, reduction in insulin-stimulated threonine (Thr308) phosphorylation of Akt when comparing the test with the control muscle, no differences were observed in insulin-stimulated Ser473 phosphorylation of Akt when comparing muscles (Fig. 3b). Moreover, and most importantly, we observed no differences in insulin-stimulated glucose clearance when comparing muscles from these mice (Fig. 3c), suggesting that the small decrease observed in the phosphorylation of Akt Thr308 was not sufficient in itself to impact upon glucose clearance.
Fig. 3.
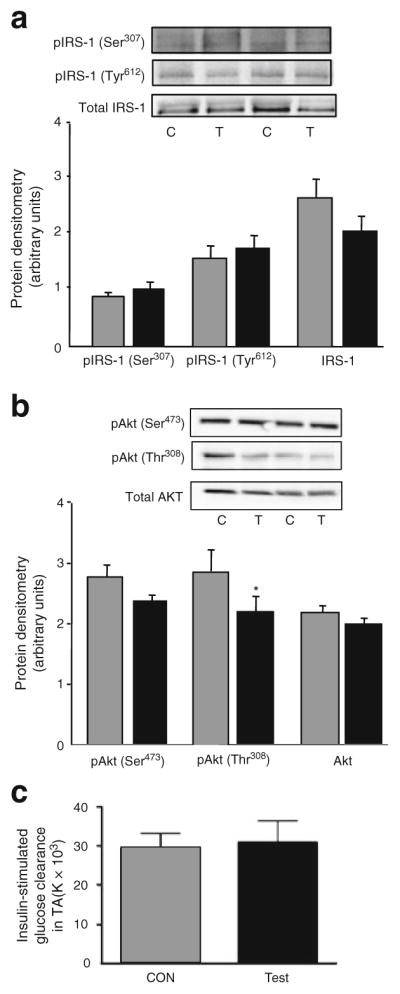
Representative western blots and quantification of Ser307 and Tyr612 IRS-1 phosphorylation and total IRS-1 (a), Akt phosphorylation (Ser473 and Thr308) and total Akt (b) and insulin-stimulated glucose clearance (c) in TA muscle from C57Bl/6 mice 7 days following electroporation of an empty vector (C; CON; grey bars) or WT-JNK (T; Test; black bars) plasmid. Ser307 phosphorylation measures were made in the absence of insulin stimulation; all other measures were insulin stimulated. For signalling, insulin (1 U/kg) was injected into the hepatic portal vein and tissue was excised after 10 min. For glucose clearance, 0.5 U/kg insulin and 370,000 Bq 2-deoxy[3H]glucose were injected. Data are expressed as mean±SEM; *p<0.05; n=6–13 per group; p, phosphorylated
Overexpression of CA-Jnk into TA increases CA-JNK phosphorylation, which impairs insulin signalling and action
We next electroporated the CA-Jnk construct into the TA muscle and measured total and phosphorylated (Thr183/Tyr185) JNK. Electroporation of the CA-Jnk plasmid increased both total and phosphorylated (Fig. 4a) CA-JNK (>80 kDa band). However, such an intervention did not affect either total (Fig. 4a, b) or phosphorylated (Fig. 4a, c) endogenous JNK (54/46 kDa bands).
Fig. 4.
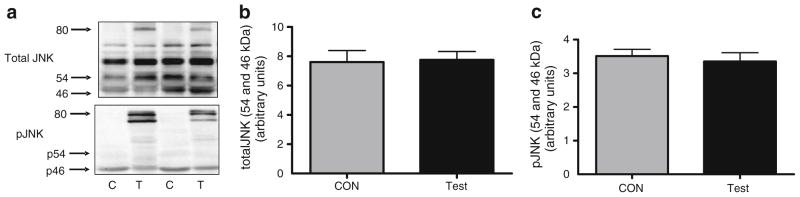
Representative western blots of total and phosphorylated (Thr183/Tyr185) CA-JNK (>80 kDa) (a), showing >80, 46 and 54 kDa bands, and quantification of total (b) and phosphorylated (Thr183/ Tyr185) (c) endogenous JNK in TA muscle from C57Bl/6 mice 7 days following electroporation of an empty vector (C; CON) or CA-Jnk (T; Test) plasmid. Data are expressed as mean±SEM; n=6–7 per group; p, phosphorylated
We next examined whether electroporation of the CA-Jnk construct had any effect on insulin signalling and/or action. As described earlier, we initially performed experiments on separate cohorts of mice that were either stimulated with (insulin) or without (basal) insulin (ESM Fig. 1b). Although Akt was phosphorylated in response to insulin in both treatments, the degree of magnitude of phosphorylation at both sites was less in the leg with CA-Jnk overexpressed (ESM Fig. 1b). Further cohorts were subsequently all insulin stimulated to enable accurate within-animal analysis to determine differences in the insulin signalling cascade between CA-JNK and control muscle. Electroporation of the CA-Jnk plasmid significantly decreased phosphorylation of the insulin receptor-β, while total levels of insulin receptor were not different (Fig. 5a). Further, the test leg had an increased Ser307 phosphorylation of IRS-1 (Fig. 5b) and a marked decrease in total IRS-1 expression (Fig. 5b), while insulin-stimulated Tyr612 phosphorylation of IRS-1 tended (non-significantly) to be decreased with CA-JNK overexpression (Fig. 5b). In addition, insulin-stimulated Akt phosphorylation at two important residues (Thr308 and Ser473) was markedly decreased when comparing the test with the control muscle (Fig. 5c). Moreover, from a functional perspective, insulin-stimulated glucose clearance was impaired when comparing the test with the control muscle (Fig. 5d).
Fig. 5.

Representative western blots and quantification of insulin receptor-β (IR-β) Tyr1361 phosphorylation and total insulin receptor (a), Ser307 and Tyr612 IRS-1 phosphorylation and total IRS-1 (b), phosphorylation (Ser473 and Thr308) and total Akt (c), and insulin-stimulated glucose clearance (d) in TA muscle from C57Bl/6 mice 7 days following electroporation of an empty vector (C; CON; grey bars) or CA-Jnk (T; Test; black bars) plasmid. Ser307 phosphorylation measurements were made in the absence of insulin stimulation; all other measurements were insulin stimulated. For signalling, insulin (1 U/kg) was injected into the hepatic portal vein and tissue was excised after 10 min. For glucose uptake, 0.5 U/kg insulin and 370,000 Bq3 2-deoxy[3H]glucose were injected. Data are expressed as mean±SEM; *p<0.05, **p<0.01; n=8–11 per group (n=4 for insulin receptor measurements); p, phosphorylated; tub, tubulin
Overexpression of CA-Jnk into TA does not affect LPL abundance or intramuscular lipid accumulation
While activation of JNK can impair insulin signalling via phosphorylation of IRS-1 [22], it has recently been suggested that JNK may also play a role in intramuscular lipid accumulation via modulation of LPL [6]. Accordingly, we measured the protein levels of LPL and performed a comprehensive lipidomic analysis in skeletal muscle overexpressing CA-Jnk. No differences were observed in LPL protein levels when comparing the test with the control muscle (ESM Fig. 2). In addition, no differences were observed in triacylglycerol (TAG) (Fig. 6a, ESM Table 1), DAG (Fig. 6b, ESM Table 1) or ceramide (Fig. 6c, ESM Table 1) accumulation when comparing the test with the control muscle.
Fig. 6.

TAG (a), DAG (b) and ceramide (c) species in TA muscle from C57Bl/6 mice 7 days following electroporation of an empty vector (CON, grey bars) or CA-Jnk (black bars) plasmid. Data are expressed as mean± SEM; n=9 per group
Discussion
While several studies have examined either whole-body [4, 7] or tissue-specific [6, 23–27] ablation of JNK and its involvement in insulin resistance, relatively few studies have attempted to determine whether elevated JNK in metabolically relevant tissues is sufficient to disrupt metabolic homeostasis. One previous study has overproduced JNK in skeletal muscle, but in that study insulin action was not examined [14]. We clearly show that electroporation of the CA-Jnk construct in skeletal muscle impairs insulin action, decreases phosphorylation of the insulin receptor and Akt, increases serine phosphorylation of IRS-1 and decreases IRS-1 levels, suggesting that increased JNK activation in this tissue may contribute to the pathogenesis of insulin resistance.
We observed a different response for insulin-stimulated glucose disposal and insulin signalling depending on whether we used the WT-JNK plasmid or the CA-Jnk plasmid. As both plasmids were shown to phosphorylate JNK in skeletal muscle, it is possible that the different effects of the WT-JNK, compared with the CA-JNK, were related to the magnitude of the increase in JNK phosphorylation, since the WT-JNK electroporation resulted in an approximate 25% increase in JNK phosphorylation compared with a marked increase in phosphorylation of the >80 kDa band for the CA-Jnk electroporation (Fig. 4a). We recently demonstrated that treadmill exercise increases JNK phosphorylation in the skeletal muscle of mice by approximately 25% [28], and it is well known that insulin sensitivity is enhanced in the immediate postexercise period. Together, these recent data, coupled with the data presented here, suggest that small increases in the phosphorylation of JNK in skeletal muscle do not have a deleterious effect on insulin signalling or action. However, caution should be applied to these direct comparisons, as exercise or exercise training most likely results in cyclic or transient changes in JNK production, and in the exercise model the increase in phosphorylated JNK occurs in conjunction with numerous other biochemical and metabolic alterations that cannot easily be separated out. As the current study was short term (7 days), it is possible that small but chronic increases in JNK could potentially negate insulin signalling/action.
We [9] and others [29] have demonstrated that, similar to mouse models, JNK phosphorylation is also elevated in skeletal muscle in human obesity, insulin resistance and/or type 2 diabetes by 50% in the former study [9] and threefold in the latter study [29]. The electroporation technique is advantageous in that it allows for each mouse to act as its own control in the absence of differing hormones and/or substrates in the circulation which may make results difficult to interpret. However, one major limitation of this technique is that it only allows for transient increases in protein abundance and, consequently, the TA muscles were only exposed to the WT-JNK and CA-JNK for 7 days. Consequently, whether the elevated JNK phosphorylation seen in skeletal muscle in human obesity, insulin resistance and/or type 2 diabetes is sufficient in itself to contribute to the pathogenesis of disease cannot be determined from the present study. However, since the magnitude of the increase in the phosphorylation of JNK at ~80 kDa with overexpression of CA-Jnk was somewhat comparable to that observed in the skeletal muscle of at least one human study [29], our data certainly suggest that increased JNK activation in skeletal muscle may contribute in part to the pathogenesis of insulin resistance.
To our knowledge, this is the first study to show that increased JNK activation can lead to decreased IRS-1 protein levels, presumably via IRS-1 ubiquitination mediated via serine phosphorylation of this protein. We show that CA-Jnk overexpression increases Ser307 phosphorylation of IRS and decreases IRS-1 protein abundance by more than 50% (Fig. 5b). Consistent with our observation, it has recently been shown that the E3 ligase CUL7 can target IRS-1 for degradation via IRS-1 serine phosphorylation [30]. However, in the latter study, isolated Ser307 phosphorylation, in itself, was not sufficient to result in IRS-1 degradation, suggesting that in our system several serine residues on IRS-1 were most likely phosphorylated together with Ser307. In fact, while IRS-1 Ser307 phosphorylation generally serves as a reliable surrogate, JNK activation is likely to cause IRS-1 serine phosphorylation at multiple residues, collectively resulting in impaired insulin signalling. This is also supported by the recent observation that isolated prevention of IRS-1 Ser307 phosphorylation in a genetic model does not result in enhanced insulin signalling [31]. Indeed, more than 100 potential serine phosphorylation sites exist in IRS-1 and as the IRS proteins contain a binding site for the docking of JNK [32] there are a plethora of prospective residues that may be working in unison with the Ser307 site. Likewise, there are multiple tyrosine phosphorylation sites on IRS-1 and although there was a tendency for phosphorylation of IRS-1 at Tyr612 to be reduced with electroporation of CA-Jnk this slight reduction was not statistically significant (Fig. 5b). Nevertheless, over-expression of CA-Jnk caused a marked decrease in Akt phosphorylation at two critical residues (Thr308 and Ser473), and it is likely this is the cause of the reduced muscle glucose clearance in response to insulin stimulation. Further studies are warranted to investigate the precise roles of serine and tyrosine residues in this system.
The insulin receptor is a transmembrane receptor and tyrosine phosphorylation is one of the earliest cellular responses to insulin stimulation. As we observed a decrease in phosphorylation of insulin receptor-β in the test leg injected with CA-Jnk, it would suggest that this proximal step in the pathway is a target for JNK-induced insulin signalling defects in skeletal muscle. It is unclear whether such alterations are sufficient to induce all the changes to downstream signalling events we observed and eventual impaired glucose clearance. Certainly, considering our observed changes in total IRS-1 levels, our data would suggest that the effects of CA-Jnk overexpression are multifaceted.
While it is likely that the reduction in insulin signalling and action observed with CA-JNK phosphorylation was mediated by a combination of reduced insulin receptor phosphorylation and serine phosphorylation, and subsequent ubiquitination of IRS-1 and alteration in Akt phosphorylation status, recent studies have suggested that JNK may also play a role in modulating intramyocellular lipid accumulation via altered activity of LPL [6]. In addition, the bioactive lipid species ceramide has been implicated in insulin resistance [3] and it has been suggested that ceramides may be regulated via inflammatory signalling pathways that also lead to JNK activation [33]. Accordingly, we examined the abundance of LPL and the accumulation of bioactive lipid species in the CA-JNK and CON muscles. We demonstrated that LPL levels were unaltered by CA-Jnk overexpression (ESM Fig. 2). Moreover, we show that neither the molecular species (Fig. 6) nor the sum of (ESM Table 1) TAG, DAG and ceramide species was affected by overexpression of CA-Jnk; therefore, the decreased insulin signalling and action observed in the CA-Jnk electroporated muscle were entirely independent of changes in bioactive lipid accumulation.
An interesting point to come from the WT-JNK component of the study was that a small but significant decrease in phosphorylation of Akt at Thr308 was observed without this impacting on glucose clearance rates into the skeletal muscle. Either this decrease was too small to exert any further downstream effects or these data question the relative contribution of the two different phosphorylation sites of Akt on glucose clearance. While often insulin resistance or disrupted glucose uptake is associated with impairments in both Akt phosphorylation sites following insulin stimulation, this is not always the case. For instance, muscle-specific rictor knockout mice exhibit glucose intolerance and decreased skeletal muscle insulin-stimulated glucose uptake and phosphorylation of Akt at Ser473, yet the phosphorylation of Akt at Thr308 is normal [34]. Together with our data in the current study, findings like this indicate that alterations in the phosphorylation status of Akt at Thr308 in skeletal muscle are not inextricably linked to functional differences in glucose clearance at the muscle site.
The WT-JNK plasmid increased JNK phosphorylation at the 54 kDa band and although as mentioned above this was associated with decreased insulin-induced Akt threonine phosphorylation it did not result in impairment in insulin-stimulated glucose clearance. It is known from work in JNK1 and JNK2-deficient mouse embryonic fibroblasts that Jnk1 and Jnk2 express both 46 and 54 kDa protein versions of JNK [35]. Therefore, it is difficult to separate the actions of both the 54 and 46 kDa versions of JNK, since the Jnk1 gene encodes both proteins and the Jnk2 gene plays a role in encoding both proteins. Whole-body genetic studies investigating the role of JNK1 and JNK2 indicate that it is likely that both loci contribute to metabolic regulation [8]. Of note, the only other study to date that has overexpressed WT-JNK in skeletal muscle did not show a significant elevation in Ser307 phosphorylation of IRS-1, but these authors did not examine insulin action [14].
In summary, we demonstrate that a marked increase in JNK phosphorylation, mediated by skeletal muscle over-expression of a constitutively active Jnk plasmid is sufficient in itself to result in reduced insulin-stimulated glucose clearance via a mechanism that probably involves alteration of the insulin signalling cascade. These data suggest that the increased skeletal muscle JNK activation seen in patients with insulin resistance may be a contributing factor to the pathology of this disorder.
Supplementary Material
Acknowledgments
We wish to acknowledge D. Ramsay and her team at Alfred Medical Research and Education Precinct Animal Services for their assistance with the animal studies. We acknowledge J. I. Miyazaki (University of Tokyo) for the original donation of the pCAGGS expression vector. We thank G. Hotamisligil and C. Görgün (Harvard School of Public Health) for supplying the CA-Jnk plasmid and J. McMullen (Baker IDI Heart and Diabetes Institute) for her assistance with the project.
Funding This study was supported by a Ramaciotti Establishment grant to CRB as well as grants from the National Health and Medical Research Council of Australia (NHMRC Project Grants 472650, 526619, 586636 to MAF and GIL) and the Victorian Government Operational Infrastructure Support Program. CRB is supported by a Career Development Award of the NHMRC. MAF is a Senior Principal Research Fellow of the NHMRC. PJM is a Senior Research Fellow of the NHMRC. DCH is supported by a National Heart Foundation (Australia) Postdoctoral Fellowship (PF 10M 5347). LJG is supported by a grant from the National Institutes of Health, USA (AR42238).
Abbreviations
- CA-JNK
Constitutively active JNK
- CA-Jnk
Plasmid containing constitutively active mouse Jnk gene
- CON
Empty vector control
- DAG
Diacylglycerol
- GFP
Green fluorescent protein
- IKK-β
IκB (inhibitor of nuclear factor κB) kinase β
- IRS-1
Insulin receptor substrate 1
- JNK
c-Jun N-terminal kinase
- LPL
Lipoprotein lipase
- TA
Tibialis anterior
- TAG
Triacylglycerol
- WT-JNK
Wild-type JNK1protein
- WT-JNK
Plasmid containing the wild-type human JNK1 gene
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00125-012-2652-8) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Duality of interest The authors declare that there is no duality of interest associated with the manuscript.
Contribution statement DCH and CRB contributed equally to this study. DCH, CRB, CPP, GIL, TLA, EE, TG and JMW performed experiments and acquired, analysed and interpreted data. KSSL, AX, NF, PJM, LJG and MAF facilitated experiments through the provision of reagents, analysed data and interpreted the data. CRB and MAF designed the study and supervised the project. DCH, CRB and MAF wrote the manuscript. All authors contributed to discussions and critically edited, reviewed and gave final approval of this version of the manuscript to be published.
Contributor Information
D. C. Henstridge, Cellular and Molecular Metabolism Laboratory, Baker IDI Heart and Diabetes Institute, 75 Commercial Rd, Melbourne, VIC 3004, Australia
C. R. Bruce, Cellular and Molecular Metabolism Laboratory, Baker IDI Heart and Diabetes Institute, 75 Commercial Rd, Melbourne, VIC 3004, Australia
C. P. Pang, Cellular and Molecular Metabolism Laboratory, Baker IDI Heart and Diabetes Institute, 75 Commercial Rd, Melbourne, VIC 3004, Australia
G. I. Lancaster, Cellular and Molecular Metabolism Laboratory, Baker IDI Heart and Diabetes Institute, 75 Commercial Rd, Melbourne, VIC 3004, Australia
T. L. Allen, Cellular and Molecular Metabolism Laboratory, Baker IDI Heart and Diabetes Institute, 75 Commercial Rd, Melbourne, VIC 3004, Australia
E. Estevez, Cellular and Molecular Metabolism Laboratory, Baker IDI Heart and Diabetes Institute, 75 Commercial Rd, Melbourne, VIC 3004, Australia
T. Gardner, Cellular and Molecular Metabolism Laboratory, Baker IDI Heart and Diabetes Institute, 75 Commercial Rd, Melbourne, VIC 3004, Australia
J. M. Weir, Metabolomics Laboratory, Baker IDI Heart and Diabetes Institute, Melbourne, VIC, Australia
P. J. Meikle, Metabolomics Laboratory, Baker IDI Heart and Diabetes Institute, Melbourne, VIC, Australia
K. S. L. Lam, Department of Medicine and Research Center for Heart, Brain, Hormones, and Healthy Aging, University of Hong Kong, Hong Kong, SAR, People’s Republic of China
A. Xu, Department of Medicine and Research Center for Heart, Brain, Hormones, and Healthy Aging, University of Hong Kong, Hong Kong, SAR, People’s Republic of China
N. Fujii, Department of Health Promotion Science, Tokyo Metropolitan University, Hachioji, Tokyo, Japan
L. J. Goodyear, Joslin Diabetes Center, Boston, MA, USA
M. A. Febbraio, Email: mark.febbraio@bakeridi.edu.au, Cellular and Molecular Metabolism Laboratory, Baker IDI Heart and Diabetes Institute, 75 Commercial Rd, Melbourne, VIC 3004, Australia
References
- 1.Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87:507–520. doi: 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chibalin AV, Leng Y, Vieira E, et al. Downregulation of diacylglycerol kinase delta contributes to hyperglycemia-induced insulin resistance. Cell. 2008;132:375–386. doi: 10.1016/j.cell.2007.12.035. [DOI] [PubMed] [Google Scholar]
- 3.Holland WL, Brozinick JT, Wang LP, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 5.Yuan M, Konstantopoulos N, Lee J, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 6.Sabio G, Kennedy NJ, Cavanagh-Kyros J, et al. Role of muscle c-Jun NH2-terminal kinase 1 in obesity-induced insulin resistance. Mol Cell Biol. 2010;30:106–115. doi: 10.1128/MCB.01162-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Witczak CA, Hirshman MF, Jessen N, et al. JNK1 deficiency does not enhance muscle glucose metabolism in lean mice. Biochem Biophys Res Commun. 2006;350:1063–1068. doi: 10.1016/j.bbrc.2006.09.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci U S A. 2006;103:10741–10746. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung J, Nguyen AK, Henstridge DC, et al. HSP72 protects against obesity-induced insulin resistance. Proc Natl Acad Sci U S A. 2008;105:1739–1744. doi: 10.1073/pnas.0705799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schenk S, Horowitz JF. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J Clin Invest. 2007;117:1690–1698. doi: 10.1172/JCI30566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 12.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 13.Zheng C, Xiang J, Hunter T, Lin A. The JNKK2-JNK1 fusion protein acts as a constitutively active c-Jun kinase that stimulates c-Jun transcription activity. J Biol Chem. 1999;274:28966–28971. doi: 10.1074/jbc.274.41.28966. [DOI] [PubMed] [Google Scholar]
- 14.Fujii N, Boppart MD, Dufresne SD, et al. Overexpression or ablation of JNK in skeletal muscle has no effect on glycogen synthase activity. Am J Physiol Cell Physiol. 2004;287:C200–C208. doi: 10.1152/ajpcell.00415.2003. [DOI] [PubMed] [Google Scholar]
- 15.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 16.Bruce CR, Brolin C, Turner N, et al. Overexpression of carnitine palmitoyltransferase I in skeletal muscle in vivo increases fatty acid oxidation and reduces triacylglycerol esterification. Am J Physiol Endocrinol Metab. 2007;292:E1231–E1237. doi: 10.1152/ajpendo.00561.2006. [DOI] [PubMed] [Google Scholar]
- 17.Bruce CR, Hoy AJ, Turner N, et al. Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat diet-induced insulin resistance. Diabetes. 2009;58:550–558. doi: 10.2337/db08-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooney GJ, Astbury LD, Williams PF, Caterson ID. Insulin response in individual tissues of control and gold thioglucose-obese mice in vivo with [1-14C]2-deoxyglucose. Diabetes. 1987;36:152–158. doi: 10.2337/diab.36.2.152. [DOI] [PubMed] [Google Scholar]
- 19.Hom FG, Goodner CJ, Berrie MA. A [3H]2-deoxyglucose method for comparing rates of glucose metabolism and insulin responses among rat tissues in vivo. Validation of the model and the absence of an insulin effect on brain. Diabetes. 1984;33:141–152. doi: 10.2337/diab.33.2.141. [DOI] [PubMed] [Google Scholar]
- 20.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 21.Matthews VB, Allen TL, Risis S, et al. Interleukin-6-deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia. 2010;53:2431–2441. doi: 10.1007/s00125-010-1865-y. [DOI] [PubMed] [Google Scholar]
- 22.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 23.Sabio G, Cavanagh-Kyros J, Barrett T, et al. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes Dev. 2010;24:256–264. doi: 10.1101/gad.1878510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabio G, Cavanagh-Kyros J, Ko HJ, et al. Prevention of steatosis by hepatic JNK1. Cell Metab. 2009;10:491–498. doi: 10.1016/j.cmet.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabio G, Das M, Mora A, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. 2008;322:1539–1543. doi: 10.1126/science.1160794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solinas G, Vilcu C, Neels JG, et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007;6:386–397. doi: 10.1016/j.cmet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 27.Vallerie SN, Furuhashi M, Fucho R, Hotamisligil GS. A predominant role for parenchymal c-Jun amino terminal kinase (JNK) in the regulation of systemic insulin sensitivity. PLoS One. 2008;3:e3151. doi: 10.1371/journal.pone.0003151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitham M, Chan MH, Pal M, et al. Contraction-induced IL-6 gene transcription in skeletal muscle is regulated by c-jun terminal kinase/activator protein-1. J Biol Chem. 2012;287:10771–10779. doi: 10.1074/jbc.M111.310581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased p85/55/50 expression and decreased phosphotidylinositol 3-kinase activity in insulin-resistant human skeletal muscle. Diabetes. 2005;54:2351–2359. doi: 10.2337/diabetes.54.8.2351. [DOI] [PubMed] [Google Scholar]
- 30.Xu X, Sarikas A, Dias-Santagata DC, et al. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol Cell. 2008;30:403–414. doi: 10.1016/j.molcel.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab. 2010;11:84–92. doi: 10.1016/j.cmet.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- 33.Holland WL, Knotts TA, Chavez JA, Wang LP, Hoehn KL, Summers SA. Lipid mediators of insulin resistance. Nutr Rev. 2007;65:S39–S46. doi: 10.1111/j.1753-4887.2007.tb00327.x. [DOI] [PubMed] [Google Scholar]
- 34.Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC., Jr Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances basal glycogen synthase activity. Mol Cell Biol. 2008;28:61–70. doi: 10.1128/MCB.01405-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaeschke A, Karasarides M, Ventura JJ, et al. JNK2 is a positive regulator of the cJun transcription factor. Mol Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.