

Abstract
Multiple sclerosis (MS) is a chronic inflammatory disorder of the central nervous system, traditionally considered to be an autoimmune, demyelinating disease. Based on this understanding, initial therapeutic strategies were directed at immune modulation and inflammation control. At present, there are five licensed first-line disease-modifying drugs for MS in Europe, and two second-line treatments. Currently available MS therapies have shown significant efficacy throughout many trials, but they produce different side effects. Despite disease-modifying drugs being well known and safe, they require regular and frequent parenteral administration and are associated with limited long-term treatment adherence. Therefore, the development of new therapeutic strategies is warranted. Several oral compounds are in late stages of development for treating MS. fingolimod is an oral sphingosine-1-phosphate receptor modulator that has demonstrated superior efficacy compared with placebo and interferon β-1a in phase III studies. It has already been approved in the treatment of MS. This review focuses on advances in current and novel oral treatment approaches in MS. We summarily review the oral compounds in this study, focusing on the recent development, approval, and the clinical experience with fingolimod.
Keywords: multiple sclerosis, oral compounds, fingolimod, sphingosine-1-phosphate, patient satisfaction, adherence
Introduction
Multiple sclerosis (MS) is a chronic inflammatory immune-mediated disease of the central nervous system (CNS). The worldwide incidence of MS is rising and is currently estimated to be about 3.6/100,000 person-years in women and 2.0/100,000 person-years in men. The female preponderance, common to many autoimmune diseases, is increasing, from an estimated 1.4 in 1955 to 2.3 in 2000.1
Although there is this wide incidence variability, MS is still the most common nontraumatic cause of neurological disability and impairment in young patients in Western Europe and North America.2 Most patients (80%) present with a relapsing and remitting course (RRMS), which is characterized by recurring attacks of acute focal neurological deficits or exacerbations of existing deficits (relapses), followed gradually by partial or full recovery (remission).3 The multifocal nature of the disease manifests clinically as a range of sensorimotor, cerebellar, visual, sphincteric, brain stem, cognitive, and neuropsychiatric symptoms. After 10–20 years, approximately half of these patients convert to the secondary progressive (SPMS) phase of the disease, in which there is acceleration of disability accumulating irreversible neurologic deficits in the absence of clinical relapses.4 The remaining 20% with progressive clinical deterioration from the onset of the disease have primary progressive MS (PPMS). Even though the immunopathogenesis of MS is complex and still unclear, it has been supposed that RRMS is characterized by strong inflammation activity, and PPMS and SPMS are thought to be dominated by axonal degeneration in the absence of overt inflammation, which is most likely a result of oxidative damage and/or increased susceptibility to injury caused by the process of loosening of the myelin sheath.5 Many different immune cells are involved in the pathogenesis of MS, including T cells, B cells, and macrophages.6 Given the prominence of inflammatory changes in acute MS lesions, therapy for the disease has focused particularly, for the past three decades, on anti-inflammatory strategies. Thus, eight compounds have nowaday licenses for treating MS, and they especially target the inflammatory component of the disease. Treatment of RRMS typically consists of direct symptom management, brief corticosteroid administration for acute exacerbations, and the regular use of disease modifying drugs (DMDs). Currently approved immumomodulator treatments for RRMS include glatiramer acetate (GA) (Copaxone) and recombinant beta interferons (IFNβs) (IFNβ-1a, Avonex; IFNβ-1a, Rebif; IFNβ-1b, Betaseron, Extavia) that represent the gold standard in modifying the course of MS. Natalizumaab (Tysabri), mitoxantrone (Novantrone), and fingolimod (Gilenya) are also available for treatment of MS as second-line therapy in more severe disease. Since MS is a currently incurable, chronic disease, long-term DMD therapy is required, necessitating commitment from patients to continue their treatment indefinitely. At present, most approved MS treatments are injected (subcutaneously or intramuscularly) or are given by intravenous infusion, which can be associated with reduced convenience, compliance, and with injection- or infusion-related adverse reactions.7 Moreover, in clinical practice, DMDs or immunosuppressive treatments are frequently associated with suboptimal response in terms of efficacy. The existing medications are only partially effective in halting MS relapses and in particular disability progression. Given the limitations of current interventions, management of MS could be significantly improved by new treatments that influence not only the immune system but also the pathologic changes in the CNS while also being amenable to oral administration, possibly avoiding the drawbacks of parenteral administration. Thus, there is an important need for new therapeutic strategies, not only those that may offer greater patient satisfaction, such as oral medications and monoclonal antibodies, but also agents intended to promote neuroprotection and neurorepair. fingolimod (FTY720), the first oral drug approved, could combine both these aspects. Here, we briefly review compounds that have successfully completed first phase III clinical trials and then focus on FTY720 and how its recent approval will change the treatment paradigm.
Oral MS therapies in development
A number of potential therapies for MS are now in late-stage development. New and novel therapeutic agents are being trialed in MS centers worldwide. These include not only oral agents for relapsing and progressive forms of the disease but also monoclonal antibodies. To improve patient adherence to AMD and aespecially to make their therapeutic compliance and quality of life better, clearly oral agents – if at least as effective as or even more effective than the currently available injectable therapies – would be a welcome advance. Several oral therapies are under evaluation in clinical trials. Currently, the most promising agents are listed in Table 1. Among these compounds, two agents have already been considered by regulatory agencies (fingolimod, cladribine), with different outcomes. Regulatory applications for fingolimod were submitted in December 2009, and the drug received approval from the FDA on September 21, 2010.8 On January 20, 2011, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorization for fingolimod, intended for the treatment of adult patients with RRMS with high disease activity. At present, fingolimod is available in Russia, the US, and Europe. Applications for cladribine as an oral therapy for MS were submitted to the European Medicines Agency (EMA) and the FDA in 2009. Although additional information was given, cladribine received a negative European recommendation from the CHMP on September 23, 2010 (http://www.merckserono.com/corp.merckserono_2011/en/images/20100924_en_tcm1494_59896.pdf?Version=).9
Table 1.
Summary of oral therapies in late-stage development (phase III) for the treatment of multiple sclerosis
| Compound | Company | Structure | Side effects | Stage of development | Mechanism of action | Comments |
|---|---|---|---|---|---|---|
| Fumaric acid (BG-12) | Biogen Idec | HO2CCH = CHC02H (E)-butenedioic acid | Skin flushing, pruritis, gastrointestinal disturbance, myalgia, dizziness, headache | Phase III trial completed | Switching T-helper (Th)l cells into an interleukin-4-dominated Th2 phenotype; induction of the expression of phase II detoxifying enzymes; impairing cell traffic | Available as white solid tablets; long experience with psoriasis Three times a day, every day |
| Teriflunomide (HMR I726) | Sanofi-Aventis | C12H9F3N202(2Z)-2-cyano-3-hydroxy-N-(4-[trifluoromethyl]phenyl) but-2-enamide | Nasopharyngitis, alopecia, nausea, alanine aminotransferase increase, paresthesia, back and limb pain, diarrhea, and arthralgia | Phase III trial completed | Blocks de novo pyrimidine synthesis by inhibiting dihydroorotate dehydrogenase | Teriflunomide is the active metabolite of leflunomide, a well-known drug approved for the treatment of rheumatoid arthritis Once daily, every day |
| Cladribrine | Merck Serono | C10H12CIN5O3 5-(6-amino-2-chloro-purin-9-yl)-2-(hydroxymethyl) oxolan-3-ol | Dose-dependent myelosuppression, opportunistic infections | Phase III trial ongoing in MS. Approved in Russia and Australia* | Cladribine is a chlorinated purine analogue, resistant to the action of adenosine deaminase. In cells, cladribine is phosphorylate to the active triphosphate form, which impairs the deoxyribonucleic acid synthesis and the cellular metabolism | Cladribine twice received a negative European recommendation from the CHMP. Following feedback from regulatory authorities, the company decided no longer to pursue the global process for approvalOnce daily for 2–4 weeks per year |
| Fingolimod (FTY720) | Novartis | C19H33NO2 2-amino-2-(2-[4-octylphenyl]ethyl) propane-1,3-diol | Transient reduction in the heart rate within hours after the first dose, increased mean arterial blood pressure, and airway obstruction | Phase IlIa trial completed Approved in US and Russia | Agonist at the G protein-coupled sphingosine 1-phosphate receptor-1 (S1P1) on lymphocytes: downmodulating the receptor, the cells become unresponsive to S1P, required to egress from the limphonodes into blood | EMA adopted a positive opinion, recommending the granting of a marketing authorization for fingolimod, intended for the treatment of adult RRMS with high disease activityOnce daily, every day |
| Laquinimod (ABR-215062) | Teva pharmaceutical industries | C19H17CIN2O35-Chloro-N-ethyl-4-hydroxy-1 -methyl-2-oxo-N-phenyl-1,2-dihydroquinoline-3-carboxamide | Potential hepatotoxicity; possible proinflammatory effect. Reported pleuritis, Budd-Chiari syndrome, pituitary adenoma with hemorrhage and possible Crohn’s disease. Pharyngolaryngeal pain, dyspepsia | Phase III trial completed | Modulates of the balance of the T-helper cells 1 and 2 induction of transforming growth factor-β inhibit infiltration of CD4+ T cells and macrophages into the central nervous system | Potent therapeutic efficacy both in MS-like acute and chronic experimental autoimmune encephalomyelitis models Once daily, every day |
Note: *Merck Serono intends to withdraw the product from the market.
Abbreviations: RRMS, relapsing-remitting multiple sclerosis; CHMP, Committee for Medicinal Products for Human Use; EMA, European Medicines Agency.
Cladribine, a synthetic deoxyadenosine analogue, is an oral immunomodulatory agent that produces targeted, sustained reduction of T and B lymphocytes. Short-course therapy with cladribine tablets was investigated for RRMS in the phase III CLARITY (Cladribine Tablets Treating Multiple Sclerosis Orally) study. The cladribine tablets’ dosing regimen consisted of two or four short courses per year (cumulative doses of 3.5 and 5.35 mg/kg over the 96-week study, respectively). Treatment with 3.5 and 5.25 mg/kg cladribine tablets significantly reduced the annualized relapse rate (relative reduction 57.6% and 54.5% vs placebo, respectively, both P < 0.001) and resulted in significantly more relapse-free patients (79.7% and 78.9% vs 60.9%, respectively; odds ratios 2.53 and 2.43; both P < 0.001).10 Three measures were detected on magnetic resonance imaging (MRI) activity: T1 gadolinium-enhancing (Gd+) lesions, active T2 lesions, and combined unique lesions (all P < 0.001 vs placebo). Lymphopenia occurred more frequently in patients treated with cladribine tablets at both dosages than with placebo.11
Fumaric acid is an unsaturated dicarboxylic acid, isomeric to maleic acid, which acts as an intermediate in the Krebs cycle. Its exact mechanism of action is still unclear, but it appears to induce depletion of peripheral blood leukocytes, owing mainly to a reduction in the number of T cells. In vitro studies indicate that fumaric acid esters induce a shift from T-helper (Th)1 to Th2 cytokines as part of their treatment effect. Current evidence suggests that BG-12 is a potent activator of Nrf2, a transcription factor of a major cytoprotective (neuroprotective) and anti-inflammatory pathway.12 In a phase IIb study, 257 patients with multiple sclerosis were enrolled to receive three different doses of BG-12 or placebo.13 Compared with placebo, BG-12 at 240 mg three times a day (TID) reduced the number of GdE lesions from week 12 to 24 by 69% (P < 0.0001). The numbers of new or enlarging T2-hyperintense and new T1-hypointense lesions were also reduced (P = 0.0006 and P = 0.014, respectively).
A phase III program with multicenter, 2-year, randomized, double-blind, placebo-controlled, dose-comparison studies was undertaken in Europe and in North America to determine the long-term safety and efficacy of BG-12. This included the DEFINE (Efficacy and Safety of Oral BG00012 in Relapsing–Remitting Multiple Sclerosis) and CONFIRM (Efficacy and Safety Study of Oral BG00012 with Active Reference in Relapsing–Remitting Multiple Sclerosis) studies; the CONFIRM study also contains a GA group to compare BG-12 with an established therapy. Both trials have been completed and results recently presented. The DEFINE study showed how BG-12 reduced the risk of relapse by 49% in the twice a day (BID) group (hazard ratio 0.51, 95% confidence interval 0.40–0.66; P < 0.0001) and by 50% in the TID, group (hazard ratio 0.50, 95% confidence interval 0.39–0.65; P < 0.0001). Moreover, BG-12 BID reduced the annualized relapse rate (ARR) by 53%, while BG-12 TID reduced the ARR by 48% (P < 0.0001 for both).14 MRI scans were performed at baseline, 24 weeks, 1 year, and 2 years to determine the number of T2-hyperintense lesions, Gd+ lesions, and T1-hypointense lesions (a tertiary end point). At 2 years, results demonstrated that RRMS patients receiving BG-12 experienced significant reductions in the number of brain lesions compared to patients on placebo.15 CONFIRM was a multicenter, randomized, placebo-controlled, reference comparator study that evaluated the efficacy and safety of BG-12 over 2 years in RRMS patients. BG-12 BID and TID significantly reduced the ARR by 44% (P < 0.0001) and 51% (P < 0.0001), respectively, versus placebo at 2 years. GA reduced the ARR by 29% (P = 0.0128). BG-12 BID and TID and GA reduced the risk of relapse by 34% (P = 0.0020), 45% (P < 0.0001), and 29% (P = 0.0097), respectively. Reduction in 12-week confirmed disability progression was 21% for BG-12 BID, 24% for TID, and 7% for GA (nonsignificant for all) versus placebo at 2 years. The incidence of adverse events (AEs) and serious AEs (SAEs) was similar across all groups.16
Teriflunomide is the active metabolite of leflunomide. Leflunomide blocks de novo pyrimidine synthesis by inhibiting dihydroorotate dehydrogenase in T cells and other rapidly dividing cell populations, leading to a decrease in DNA synthesis.17 leflunomide is licensed for the treatment of rheumatoid arthritis and is also effective in experimental autoimmune neuritis and rat experimental autoimmune encephalomyelitis (EAE).1819 A 36-week, randomized, double-blind, placebo-controlled, parallel-group trial assessed the safety and efficacy of this new drug on MS: 179 patients (157 with RRMS, 29 with SPMS) were treated with placebo (n = 61), teriflunomide 7 mg/day (n = 61), and teriflunomide 14 mg/day (n = 57) for 36 weeks. Treatment with teriflunomide 7 or 14 mg/day resulted in significant suppression of >61% of MRI activity relative to placebo (P < 0.03 at 7 mg/day and P < 0.001 at 14 mg/day). The annualized relapse rate between placebo and teriflunomide 14 mg/day was similar to that reported for IFNβ and GA (32%). The higher dose of teriflunomide (1 4 mg/day) appears to be more effective than the 7 mg/day dose in terms of relapse rate, although no dose effect was observed on primary end points such as Gd+ lesions and new/enlarging T2 lesions.20 These results have been recently confirmed in an open-label, long-term extension of this study. Disability progression, ARRs, and MRI activity remained low throughout the course of the extension, providing evidence that the previously reported beneficial effects of teriflunomide on clinical and MRI end points are maintained over the long term, for up to 8.5 years.21
A large, randomized, double-blind, placebo-controlled phase III parallel-group study has been completed (TEMSO). Patient with RRMS or progressive relapsing MS (n = 1088) were randomized (1:1:1) to receive placebo, teriflunomide 7 mg/day or 1 4 mg/day for 1 08 weeks. teriflunomide reduced the ARR (0.54 for placebo vs 0.37 for teriflunomide at either 7 or 14 mg), with relative-risk reductions of 31.2% and 31.5%, respectively (P < 0.001 for both comparisons with placebo). The proportion of patients with confirmed disability progression was 27.3% with placebo, 21.7% with teriflunomide at 7 mg (P = 0.08), and 20.2% with teriflunomide at 14 mg (P = 0.03). Both teriflunomide doses were superior to placebo on a range of end points measured by MRI. Diarrhea, nausea, and hair thinning were more common with teriflunomide than with placebo. The incidence of elevated alanine aminotransferase levels (≥ 1 times the upper limit of the normal range) was higher with teriflunomide at 7 mg and 14 mg (54.0% and 57.3%, respectively) than with placebo (35.9%); the incidence of levels that were at least three times the upper limit of the normal range was similar in the lower-and higher-dose teriflunomide groups and the placebo group (6.3%, 6.7%, and 6.7%, respectively). Serious infections were reported in 1.6%, 2.5%, and 2.2% of patients in the three groups, respectively22 Trials investigating teriflunomide in clinically isolated syndrome are under way as combination studies of teriflunomide with IFNβ or GA. The last mentioned study seems to be really meaningful, exploring the possibility of using teriflunomide as an add-on therapy.
Laquinimod is a novel synthetic compound with oral bioavailability that is in development as an oral formulation for the treatment of MS. Laquinimod was effective in a rat model of EAE, in which its efficacy was ascribed to modulation of the balance of Th cells’ 1 and 2 induction of transforming growth factor β.23 However, the exact mechanism of action in MS patients is still ambiguous. The efficacy of laquinimod was studied in two phase II studies. The first phase II clinical trials showed inconclusive results on the effect of a 0.3 mg dose and led to further exploration of the therapeutic dose in an additional phase IIb study. The second study was a phase IIb, multinational, randomized double-blind parallel-group, placebo-controlled 36-week study evaluating the efficacy, tolerability, and safety of two daily oral doses (0.3 and 0.6 mg) of laquinimod (as compared to placebo) in subjects with RRMS. The statistical analysis on the intention-to-treat population for the primary end point demonstrated a statistically significant treatment effect of laquinimod 0.6 mg compared to placebo (P = 0.0048), with a reduction of 40% in the cumulative number of Gd+ lesions at weeks 24, 28, 32, and 36. A nonsignificant treatment effect of 8% was observed with laquinimod 0.3 mg (P = 0.6740).24 Given these results, the sponsor moved forward with phase III clinical trials of laquinimod at the 0.6 mg/day dose, which have just been completed. Overall, the results from the phase III studies indicate that while laquinimod is relatively effective in reducing disease activity as measured by relapse rate, it is very effective on some MRI markers of neurodegeneration, indicating a possible role as neuroprotective treatment in MS. Treatment with laquinimod as compared with placebo was associated with a modest reduction in the mean (±standard error) ARR (0.30 ± 0.02 vs 0.39 ± 0.03, P = 0.002) and with a reduction in the risk of confirmed disability progression (11.1% vs 15.7%, hazard ratio 0.64, 95% confidence interval 0.45-0.91; P = 0.01). The mean cumulative numbers of Gd+ lesions and new or enlarging lesions on T2-weighted images were lower for patients receiving laquinimod than for those receiving placebo (1.33 ± 0.14 vs 2.12 ± 0.22 and 5.03 ± 0.08 vs 7.14 ± 0.07, respectively; P < 0.001 for both comparisons).25 The most significant side effects appeared to be urinary tract infections and a temporary abnormality in liver-function tests. Based on a thorough review of all laquinimod clinical data available to date, the primary safety concerns include potential hepatotoxicity and a possible proinflammatory effect. Four SAEs were reported: pleuritis, Budd-Chiari syndrome, pituitary adenoma with hemorrhage, and a possible diagnosis of Crohn’s disease. Pharyngolaryngeal pain, dyspepsia, and ankle edema were also observed. Recently, pooled analyses assessing the effect of laquinimod on relapse, disability, and brain-atrophy measures from the completed phase III trials ALLEGRO and BRAVO have been presented. Pooled analyses on ARR found that laquinimod reduced relapse rate by 21.4% (0.30 ARR for laquinimod vs 0.38 for placebo patients, P = 0.0005). Pooled analyses on the risk to confirmed disability progression sustained for 3 months found laquinimod significantly reduced the risk (34.2%, P = 0.0017) compared to placebo. Using a more stringent criterion of sustained disability for 6 months, there was a significant reduction by laquinimod (46%, P < 0.0001). Percent change in brain volume was reduced by 30% (−0.835 vs −1.188% for laquinimod and placebo patients, P < 0.001).26
Introduction to the compound
Fingolimod (FTY720) is an oral drug with a novel mechanism of action and unique immunological and neurobiological properties. Fingolimod is a structural analogue of sphingosine that does not impair T- and B-cell activation, proliferation, and effective function, but interferes with cell traffic between lymphoid organs and blood.27 Moreover, fingolimod is a lipophilic compound that can cross the blood-brain barrier, and research suggests that its neuroprotective properties might be a reflection of its ability to directly interact with specific receptors on neurons and glia.28 Following preclinical studies in animal models of organ transplantation, fingolimod was first evaluated for the prevention of allograft rejection in renal transplant patients, but phase III trials failed to show benefit over standard treatments.29 Encouraging results obtained from animal models of EAE provided the rationale for clinical evaluation in humans, and one phase II proof-of-concept study confirmed the favorable effect of fingolimod oral therapy on standardized measures of disease activity in patients with RRMS.
Chemistry, pharmacodynamics, and pharmacokinetics
Fingolimod (2-amino-2-[2-{4-octylphenyl}ethyl]-1,3-propanediol) is an immunosuppressive synthetic compound derived from myriocin, a metabolite isolated from the fungus Isaria sinclairii, used as a drug in Oriental medicine30 (Figure 1). This compound is the sphingosine-1-phosphate (S1P) receptor modulator. S1P is produced by phosphorylation of sphingosine by ubiquitously expressed sphingosine kinases, a breakdown product of the cell-membrane constituent sphingomyelin. It is present at concentrations of 100–1000 nmol/L in blood, and as with most small lipids is preferentially bound to albumin and other plasma proteins.31 S1P could act both as second messenger and as an extracellular ligand for a family of plasma-membrane G protein-coupled receptors.32 There are five known S1P receptor subtypes – S1P1–5 – and these are expressed on a wide range of cell types, including lymphocytes and neural cells (Table 2). Each S1P receptor subtype is associated with at least one subclass of G protein, which activate different intracellular signaling pathways.33 The receptors S1P1–3 are widely expressed by a variety of tissues, whereas S1P4 is exclusively found on lymphoid and hematopoietic tissues and S1P5 is mainly expressed in the CNS.34 The ubiquitous expression of S1P receptors and their coupling to different G proteins explains the varied biological effects of the S1P/S1P-receptor system.35 In particular, S1P1 expressed on lymphocytes regulates the normal egress of lymphocytes from lymphoid tissues,36 whereas S1P receptors expressed in the CNS have been shown to modulate several functions, such as neurogenesis, neural function, and migration.34 Thus, this explains the possible clinical and adverse effects that agents like fingolimod targeting the S1P receptor system can potentially induce. As a structural analogue of natural sphingosine, fingolimod after ingestion can undergo rapid phosphorylation in vivo, especially in the liver, by sphingosine kinase-2 to produce the phosphorylated form of the compound. The latter can bind four of the five receptor subtypes with high affinity: S1P1–S1P3–S1P4–S1P5.37 Following engagement through the agonist, the S1P1 receptor is internalized and can no longer bind to its natural circulating ligand, S1P, inducing a long-lasting internalization, ubiquitination, and intracellular degradation of the receptors, rendering the cells unresponsive to endogenous S1P.38 The effects of fingolimod reflect the ubiquitous expression of the S1P receptor and are shown on different levels, principally regarding the immune system and the CNS. Following administration, blood concentration increases slowly to reach peak values at 8–36 hours postdosing. The elimination half-life of fingolimod averages 8.8 days, so a once-daily dosing is permitted. Clearance of fingolimod depends mainly on hepatic oxidative metabolism, and none of the identified metabolites possess immunomodulatory activity.39 The main pharmacodynamic effect of fingolimod consistently observed in human studies has been temporary reversible lymphopenia.39,40
Figure 1.
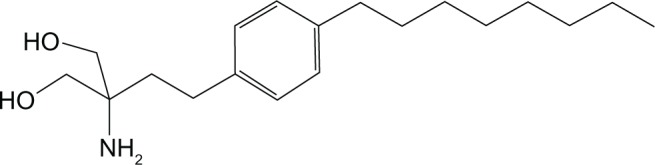
Fingolimod chemical structure.
Table 2.
Distribution and functions of S1P receptors
| Receptors | Cellular distribution | Fingolimod binding | Key functions |
|---|---|---|---|
| S1P1 (EDG1) | • Lymphocytes, mast cells, eosinophils • Neurons, astrocytes, oligodendrocytes, microglia • Atrial myocytes, endothelium smooth-muscle cells, Schwann cells |
Yes | • Lymphocyte egress from secondary lymphoid organs • Neural cell migration/function • Embryonic development of cardiovascular and nervous systems • Blood-vessel formation • Endothelial barrier function |
| S1P2 (EDG5) | • Neurons, microglia, astrocytes • Smooth-muscle cells, Schwann cells |
No | • Vascular tone • Endothelial barrier function • Inner-ear maintenance affecting hearing and balance • Nerve conduction |
| S1P3 (EDG3) |
• Neurons, astrocytes, microglia • Atrial myocytes, endothelium, smooth-muscle cells • Lung • Kidney • Intestine • Cartilage • Schwann cells |
Yes | • Endothelial barrier function • Neural cell migration/function |
| S1P4 (EDG6) | • Leukocytes • Schwann cells |
Yes | Unknown |
| S1P5 (EDG8) | • Oligodendrocytes, microglia, astrocytes | Yes | • Oligodendrocyte function • NK-cell migration |
Abbreviations: EDG, endothelial differentiation sphingolipid G protein-coupled receptor; NK, natural killer; S1P, sphingosine 1-phospate.
Clinical efficacy
On the basis of the results demonstrating the potency of fingolimod in human organ transplantation,40 preclinical studies in various EAE models were designed.41 These studies demonstrated the capability of fingolimod in preventing and treating EAE. Delivery of fingolimod from the day of immunization or at a presymptomatic disease stage prevents the development of neurological signs in both monophasic and relapsing forms of EAE.41,42 fingolimod is currently being assessed in one of the largest phase III MS study programs ever undertaken, having shown promise in a phase II, 6-month, placebo-controlled study in patients with relapsing MS in which oral fingolimod compared with placebo significantly reduced the ARR and inflammatory activity according to MRI scans (Table 3).
Table 3.
Summary of fingolimod trials
| Study | Study design | Treatment in study | Primary end points | Eligibility criteria | Main results |
|---|---|---|---|---|---|
| Kappos et al43 | Phase II, 6-month, doubleblind, parallel-group, placebo-controlled, multicenter | fingolimod 5 mg orally, daily fingolimod1.25 mg orally, daily/placebo | Total no of Gd+ lesions on T1-w MRI at month 6 | RRMS, SPMS 18–60 years EDSS 0–6, no evidence of relapse in the last 30 days | Patients free from Gd+ lesions: 82%* |
| Cohen et al46 (TRANSFORMS) | Phase III, 12-month, doubleblind, double-dummy, parallel-group, active-controlled, multicenter | fingolimod 1.25 mg orally, daily fingolimod0.5 mg orally, daily IFNβ-1a 30 μg intramuscularly, weekly | ARR over 12 months | RRMS 18–55 years EDSS 0–5.5, recent history of relapse | ARR: 0.20*; 0.16* Patients free from relapses: 79.8%*; 82.6%* |
| Kappos et al47 (FREEDOMS) | Phase III, 24-month, doubleblind, parallel-group, placebo-controlled, multicenter | fingolimod 1.25 mg orally, daily fingolimod 0.5 mg orally, daily/placebo | ARR over 24 months | RRMS 18–55 years EDSS 0–5.5, recent history of clinical relapse | ARR: 0.16*; 0.18* Patients free from relapses: 74.7% ± 2.2%* 70.4% ± 2.3%* |
Note: *P < 0.001.
Abbreviations: ARR, annualized relapse rate for confirmed relapses; EDSS, expanded disability status scale; Gd+, gadolinium-enhanced; IFNβ-1a, interferon beta-1a; MRI, magnetic resonance imaging; RRMS, relapsing–remitting multiple sclerosis; SPMS, secondary progressive multiple sclerosis; T1.-w: T1-weighted.
Phase II studies
The first study evaluating the activity of fingolimod in patients with RRMS was a randomized, double-blind, placebo-controlled, 6-month study.43 The study was designed to explore safety and tolerability and the treatment effect on MRI lesion parameters but not on clinical outcomes. In the core study, patients were randomly assigned, in a 1:1:1 ratio, to 1.25 mg of fingolimod, 5.0 mg of fingolimod, or a matching placebo given once daily as oral capsules. The intention-to-treat population comprised 277 patients, of which 255 (92%) completed the study. The primary efficacy end point of the study was the total number of Gd+ lesions per patient recorded on T1 -weighted MRI at monthly intervals for 6 months. Secondary MRI variables included the total volume of Gd+ lesions per patient, the proportion of patients with Gd+ lesions, the total number of new lesions per patient on T2-weighted images, changes in lesion volume on T2-weighted images, and brain volume from baseline to month 6. Clinical end points included the number of patients remainingfree of relapse, the ARR, and the time to the first relapse.
The total cumulative numbers of lesions per patient on postbaseline, monthly Gd+T1 -weighted MRI scans were lower in both fingolimod groups than in the placebo group (P < 0.001 for the 1.25 mg dose and P = 0.006 for the 5.0 mg dose). At month 6, the proportion of patients who were free of Gd+ lesions was greater in both fingolimod groups than in the placebo group (P < 0.001 for both comparisons). Regarding the clinical end point, significant improvements over placebo were observed in the fingolimod groups, including a relative reduction in the ARR (by 53% in the 5.0 mg group and by 55% in the 1.25 mg group).
After the core-study completion, patients could enter a dose, open-label, uncontrolled, active-drug study extension. Placebo recipients were rerandomized to one of the FTY720 doses; those already receiving FTY720 continued at the same dose.44,45 During the month 15–24 study visits, patients receiving FTY720 5.0 mg were switched to 1.25 mg because a benefit–risk assessment indicated that the higher dose offered no efficacy advantage and possibly a less favorable safety profile.
Of the 250 patients who entered the extension, 189 (75.6%) completed to month 24 and 173 (69%) to month 36. In the group of patients who switched from placebo to fingolimod, the number of Gd+ lesions decreased markedly following initiation of fingolimod treatment to 0.4 at month 12 and remained low (0.1) at month 36, and relapse rate to a level comparable with that of patients treated with fingolimod in the core study. The proportion of patients free from Gd+ lesions in the placebo/fingolimod group was 50% at baseline, 47% at month 6, and 89% at month 36. The proportions of patients free from Gd+ lesions in the fingolimod 1.25 mg and 5.0/1.25 mg groups at baseline were 52% and 48%, respectively; at month 36, the corresponding proportions were 88% and 89%, respectively. In the continuous-fingolimod groups, the mean number of Gd+ lesions was 3.2 at baseline, 1.4 at month 6, and 0.2 at month 36 in the 1.25 mg group, and 2.6 at baseline, 0.4 at month 6, and 0.3 at month 36 in the 5.0/1.25 mg group. The mean number of new T2 lesions acquired across all treatment groups was 0.6 at month 24 (since month 12) and 0.7 at month 36 (since month 24). Considering clinical outcomes at month 36, the ARR was 0.20 and 0.21 in the fingolimod 1.25 mg and 5.0/1.25 mg groups, respectively, and 0.31 in the placebo/fingolimod group. The proportions of patients relapse-free at month 36 were 68% in the fingolimod 1.25 mg group and 73% in the fingolimod 5.0/1.25 mg group rather than estimated 51% of patients in the placebo/fingolimod group at month 36.
Phase III studies
On the basis of the results detected from phase II studies, two multicenter, randomized, double-blind phase III trials have been designed.4647 As these results suggest that the therapeutic benefits attributed to fingolimod might exceed those expected from approved disease-modifying injectable therapies such as IFNβ or glatiramer acetate, the Trial Assessing Injectable Interferon versus FTY720 Oral in Relapsing-Remitting Multiple Sclerosis (TRANSFORMS)46 tested directly this hypothesis. In this 12-month, double-blind double-dummy study, they randomly assigned 1292 patients between 18 and 55 years of age with RRMS, according to the McDonald criteria,48 who had had a recent history of at least one relapse in the last year before screening to receive either oral fingolimod at a daily dose of either 1.25 or 0.5 mg or intramuscular IFNβ-1a at a weekly dose of 30 μg. The primary efficacy end point was the ARR. Key secondary end points were the number of new or enlarged lesions on T2-weighted MRI scans at 12 months and the time to confirmed disability progression meant a progression of disability that was sustained for at least 3 months. In all, 1153 patients (89%) completed the study, and 1123 (87%) continued to receive the assigned study drug. There was a significantly greater reduction in the ARRs in both fingolimod groups than in the IFN group. The ARR was lower with fingolimod (0.5 mg, 0.16; 1.25 mg, 0.20) than with IFNβ-1a (0.33, P < 0.001). More patients treated with fingolimod remained free from relapses (80%–83%) compared with IFNβ-1a (69%, P < 0.001). The proportion of patients with confirmed disability progression was low and similar in all groups (6%–8%). Over the treatment period of 12 months, patients in both fingolimod groups had fewer new or enlarged hyperintense lesions on T2-weighted images than those receiving IFNβ-1a (mean number, 0.5 mg, 1.7; 1.25 mg, 1.5; IFNβ-1a, 2.6; P < 0.001 and P = 0.004). Patients free from Gd+ lesions were also lower with fingolimod (0.5 mg, 90.1%; 1.25 mg, 91.2%) compared with those taking IFNβ-1a (80.8%, P < 0.001 for both groups).47
The second phase III study investigating fingolimod is the FTY720 Research Evaluating Effects of Daily Oral Therapy in Multiple Sclerosis (FREEDOMS) trial,47 a 24-month, double-blind placebo-controlled randomized study. A total of 1272 patients between 18 and 55 years of age with RRMS defined by the McDonald criteria who had a score of 0-5.5 on the expanded disability status scale and had had one or more relapses in the previous year or two or more in the previous 2 years were enrolled. Patients received oral fingolimod at a dose of 0.5 mg or 1.25 mg daily or placebo. As with the TRANSFORMS study, the primary end point was the ARR and the secondary end point was represented by the time to disability progression. MRI measures of inflammation, burden of disease, and tissue destruction in patients were also analyzed. A total of 1033 patients (81.2%) completed the 24-month study. All clinical and MRI-related efficacy end points significantly favored both doses of fingolimod over placebo, and there were no significant differences in efficacy between the two fingolimod doses: the aggregate ARR was lower with fingolimod at a dose of 0.5 mg (0.18) and fingolimod at a dose of 1.25 mg (0.16) than with placebo (0.40), representing relative reductions of 54% and 60%, respectively, in the aggregate ARR (P < 0.001 for both groups). In the fingolimod groups compared with the placebo group, the time to a first relapse was longer, the risk of relapse was reduced, and proportionately more patients remained free of relapse during the 24-month period. fingolimod reduced the risk of disability progression, confirmed after 3 months, over the 24-month study period (hazard ratios 0.68 for the 1.25 mg dose and 0.70 for the 0.5 mg dose, P < 0.02).47 The cumulative probability of disability progression (confirmed after 3 months) was 17.7% for 0.5 mg of fingolimod, 16.6% for 1.25 mg of fingolimod, and 24.1% for placebo. At month 24, patients receiving fingolimod had fewer new or enlarged hyperintense lesions on T2-weighted images than those in the placebo group (means for 0.5 and 1.25 mg, 2.5; placebo, 9.8; P < 0.001). fingolimod was also superior to placebo with regard to other MRI measures of disease activity as Gd+ lesions.47 Moreover, and really interestingly, in patients treated with fingolimod, the brain reduction was smaller than in patients treated with placebo.
Safety and tolerability
The safety profile of fingolimod has been extensively studied, based on acquired experience, and the available data indicate that the drug has a relatively good safety profile. It is clearly known that the incidence of AEs is dose-dependent and increases with rising doses of fingolimod.43,46,47 The phase II study showed a higher incidence of AEs in the group of patients treated with a high dose of fingolimod (5.0 mg; 96% against 82% in the placebo group and 84% in the fingolimod 1.25 mg group). In phase III trials, the incidence of AEs was approximately the same across the study groups. AEs related to fingolimod included infections, particularly respiratory, urinary tract, and herpes virus infections, increased levels of alanine aminotransferase, bradycardia, and atrioventricular block at the time of treatment initiation, hypertension, and macular edema. The incidence of SAEs was comparable among the study groups, with the exception of the TRANSFORMS trial, in which SAEs were more frequent in patients assigned to a higher dose (1.25 mg) of fingolimod (11%) than in those receiving 0.5 mg (7%).
Even though the pathophysiology of macular edema in patients treated with fingolimod is still unclear, it was confirmed in 13 patients receiving fingolimod in phase III trials, of which eleven were treated with 1.25 mg. Most cases occurred within the first 3–4 months and resolved after treatment discontinuation.46,47 No cases were detected during phase II studies in MS.43
As expected, the drug induced a decrease of peripheral blood lymphocyte counts to 20%–30% of baseline values. Lymphocyte counts remained stable throughout the treatment period, and returned to baseline values within weeks after treatment discontinuation. As a consequence of the effects of fingolimod on circulating lymphocyte, the drug apparently increases the risk of infections. In phase III trials, the overall incidence of infections was similar across the study groups, in the range between 51% and 72%. Mild and moderate upper and lower respiratory tract infections occurred more frequently among patients receiving fingolimod. Herpes zoster occurred in six patients during months 0–36 in phase II studies.43 Herpes virus infections were diagnosed in 2%–9% of patients. In the FREEDOMS study, herpes virus infections were reported in similar proportions across study groups (1.25 mg, 5.8%; 0.5 mg, 8.7%; placebo, 7.9%). On the contrary, in the TRANSFORMS study, these were more common in the 1.25 mg group (5.5% of patients) than in the 0.5 mg and IFNβ-1a groups (2.1% and 2.8%, respectively). Most herpes virus infections were mild, but a total of six SAEs were reported, including one case of fatal disseminated varicella zoster virus infection and one case of fatal herpes simplex virus type 1 encephalitis, both in patients assigned to fingolimod 1.25 mg. Both patients were undergoing concomitant corticosteroid treatment when the infection occurred.46,47 Thus, a possible increase in the risk of reactivation of latent herpes should be deeply investigated when considering fingolimod treatment. Besides lymphopenia, asymptomatic elevation of liver enzyme levels was the most frequent laboratory abnormality and occurred in a dose- dependent manner within the whole range of doses investigated. In clinical studies, alanine aminotransferase levels returned to normal values after treatment discontinuation. In none of these cases were other signs or symptoms of significant hepatocellular injury observed.46,47 Moreover, fingolimod induced a small, dose-dependent increase in airway resistance upon treatment initiation, with no evidence for further progression with continuous dosing.43,46,47
As was expected based on previous findings, though rarely symptomatic, a dose-dependent reduction in heart rate occurred within 6 hours after administration of the first dose. This drop in heart rate was already evident within 1 hour postdosing, reached a maximum mean reduction of approximately 10 beats/minute at 4–5 hours, began to attenuate at 6 hours, and returned towards the baseline values.46,47
In phase III trials, symptomatic bradycardia after the first dose of fingolimod, mainly dizziness, chest discomfort, or palpitations, was observed in <1% of patients. No cases of syncope have been observed. Most events were mild to moderate in severity and resolved within 24 hours without requiring pharmacological interventions. There were no episodes of symptomatic bradycardia occurring beyond 24 hours, and no clinically significant effect on heart rate was observed with sustained administration of the drug.46,47 Although during clinical trials, pharmacological treatment has never been required to treat bradycardia, it has been suggested that intravenous atropine can ameliorate the negative effect of fingolimod on cardiac rhythm.49 In addition to transient changes in cardiac rhythm, fingolimod induced cardiac conduction abnormalities, including first- and second-degree atrioventricular block. In the phase II trial, transient Mobitz type 1 second-degree atrioventricular block occurred on the first day postdosing in eight of 65 patients who received fingolimod.43 In phase III trials, first- and second-degree atrioventricular blocks were infrequently reported (0.4%–1.4% of patients), and these were not symptomatic. However, in the FREEDOMS study, electrocardiography performed on day 1 postdosing revealed first- and second-degree atrioventricular block in seven and 0.6% of patients respectively. No effect on atrioventricular conduction was observed with continued treatment beyond 24 hours.46,47 Initial administration of fngolimod was also associated with a mild reduction in mean arterial blood pressure within 4–5 hours postdosing. This transient reduction in mean arterial pressure was followed by a small and sustained increase (2–3 mmHg over the baseline values) during the first 6 months of treatment, with no further changes in the subsequent months. In phase III trials, hypertension was reported in 4%–6% of participants.46,47
Malignant neoplasms were reported in patients undergoing therapy with fingolimod in phase III trials, including localized skin cancer (Bowen’s disease, one case; basal cell carcinoma, ten cases; malignant melanoma, four cases), all of which were successfully excised, and breast cancer (five cases). One woman died from metastatic breast cancer 10 months after discontinuing fingolimod.46 Both skin and breast cancer were also reported in the control groups (six and three cases in total, respectively). The number of events was not enough to establish a statistical association between fingolimod and the risk of cancer in clinical trials. However, further long-term observation is needed before definitive conclusions can be reached.
Besides more frequent SAEs being observed, two cases of particular interest were experienced during the clinical trials. In the phase II trial, one case of posterior reversible encephalopathy syndrome occurred in a woman with no evident predisposing factors after 10 weeks of treatment with 5 mg of fingolimod. The symptoms and MRI abnormalities improved 72 hours after discontinuation of medication, leaving residual neurological deficits.43
A case of temporo-occipital hemorrhagic and centrally necrotic focal encephalitis of unknown etiology was reported in a woman after 7 months of treatment with 1.25 mg of fingolimod. Although bacterial and viral causes were excluded, antimicrobial treatment was administered. The patient recovered with sequelae.50
Future perspectives
The positive results gained from the studies described above was followed by authorization in Russia and the US. Then fingolimod (Gilenya, Novartis, Basel, Switzerland) was approved in Europe in June 2011 to treat adults with highly active MS. As the FDA listed several recommendations for clinicians using Gilenya, the EMA made all European countries aware of different conditions or restrictions with regard to the safe and effective use of the compound.51 Nonetheless, at present several phase III trial extension studies are ongoing to collect long-term data on the safety and effectiveness of fingolimod, especially exploring the incidence of selected safety-related outcomes of fingolimod treatment during follow-up. Results from an extension study have been recently presented. Newer agents that are more selective for the different types of S1P receptors are currently in development to minimize the AEs observed with fingolimod.52 In order to focus on cardiac safety after the first dose of fingolimod 0.5 mg, the FIRST study has included relapsing MS patients both without cardiac risk factors and with cardiac conditions not included in previous fingolimod clinical trials: overall incidence of atrioventricular blocks on Holter electrocardiography was low, irrespective of on- or off-site first-dose administration or the presence of potential cardiac risk factors. These results are similar to those observed in fingolimod trials and confirm the benign nature of the cardiac effects of treatment initiation with fingolimod.53 In addition, a fundamental purpose is continuously to monitor, evaluate, and assess for major and minor teratogenic effects in the offspring of women exposed to fingolimod before (up to 8 weeks before last menstrual period) and during pregnancy in routine clinical practice. The overall aim is to collect and evaluate data on maternal, fetal, and infant outcomes and compare it with reference populations through the Multi-National Gilenya Pregnancy Exposure Registry in Multiple Sclerosis (MS).54
Conclusion
In the last few years, impressive research and development programs for drug treatments in MS have been developed. The five oral therapies discussed (cladribine, fingolimod, laquinimod, BG-12, and teriflunomide) have just completed phase III studies, and some have just been licensed. Maintaining adherence to DMDs is a daily challenge to those who manage and coordinate care for MS patients. It is well known that patients who have more positive feelings towards prescribed therapy adhere more closely to their therapeutic regimen. The development of drugs with easier administration, such as oral agents discussed above, would further promote adherence and could increase the number of patients with MS in treatment, reducing discomfort and inconvenience, making it easier for patients to adhere to their treatment regimen and thus improving efficacy. The most challenging aspect for physicians will be making their patients understand the need of balancing for each case, the pros and cons of new oral treatment, where serious side effects may outweigh benefits in certain individuals. This is particularly important given the lack of long-term safety data with these new oral treatments versus currently approved disease-modifying therapies. Neurologists are starting to use more powerful but potentially dangerous drugs in the treatment of MS. Moreover, the potential combination of oral drugs and IFNβ or GA needs to be tested. More information regarding the mechanism of action and safety of oral drugs would assist in the design of such combination studies. Safety is likely to become the most important factor in the future development of MS drugs. New oral drugs for MS need to be placed within this evolving marketplace, where ease of delivery together with efficacy and side effects need to be balanced against the known issues but also the known longterm safety of standard injectables.
Fingolimod is the first of a new class of immunosuppressants with a unique mode of action. The preventive and therapeutic effects of oral fingolimod in various models of EAE and its ability to penetrate the blood–brain barrier make it an attractive candidate for use in the treatment of MS. However, safety concerns include the known pharmacodynamic effect of the drug on heart rate and rhythm. In the last few months, the safety profile of the compound has been extremely focused since the EMA decided to review Gilenya following cases of death and serious cardiovascular events in patients who had recently started treatment with the medicine. While the review was ongoing, the CHMP advised health-care professionals to intensify monitoring of patients after the first dose.55 During the last few months, the reviewing process has been concluded, and CHMP stated that the benefits of Gilenya continue to outweigh its risks but recommended changes to the product information to strengthen the warnings and ensure close monitoring of all patients following the first dose.56 Moreover, previous reports of macular edema in the fingolimod transplant program, and experience of seven cases of melanoma, squamous cell, and basal cell carcinomas should require careful safety monitoring.
In conclusion, while the availability of oral therapies has been much anticipated by physicians and patients, in clinical practice neurologists will need to be cautious in selecting such therapies, which may appear to have efficacy and convenience advantages versus current therapies, but may also carry novel safety and tolerability concerns. The decision to use these new therapies will most likely be based on an overall assessment of efficacy, safety, tolerability, adherence, potential need for monitoring, and cost effectiveness. Although better patient compliance is expected with the oral agents compared with the injectables, the safety profiles of these new oral drugs will have to be watched carefully. The safety of drug combinations will be crucial to future therapeutic decision-making, and more research is needed to compare escalation and induction treatment strategies.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Alonso A, Hernán MA. Temporal trends in the incidence of multiple sclerosis: a systematic review. Neurology. 2008;71:129–135. doi: 10.1212/01.wnl.0000316802.35974.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray TJ. Diagnosis and treatment of multiple sclerosis. BMJ. 2006;332:525–527. doi: 10.1136/bmj.332.7540.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 4.Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain. 2006;129:606–616. doi: 10.1093/brain/awl007. [DOI] [PubMed] [Google Scholar]
- 5.Lassmann H, Bruck W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007;17:210–218. doi: 10.1111/j.1750-3639.2007.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frohman EM, Racke MK, Raine CS. Multiple sclerosis – the plaque and its pathogenesis. N Engl J Med. 2006;354:942–955. doi: 10.1056/NEJMra052130. [DOI] [PubMed] [Google Scholar]
- 7.Pozzilli C, Schweikert B, Ecari U, BetaPlus Study group Supportive strategies to improve adherence to IFN beta-1b in multiple sclerosis – results of the βPlus observational cohort study. J Neurol Sci. 2011;307:120–126. doi: 10.1016/j.jns.2011.04.026. [DOI] [PubMed] [Google Scholar]
- 8.US Food and Drug Administration FDA approves first oral drug to reduce MS relapses [press release] Silver Spring MD, USA: FDA; September222010Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm226755.htmAccessed November 19, 2012 [Google Scholar]
- 9.Therapeutic Goods Administration Australian Public Assessment Report for Cladribine Tablets Woden, ACT, Australia: TGA; 2011Available from: http://www.tga.gov.au/pdf/auspar/auspar-movectro.pdfAccessed November 19, 2012 [Google Scholar]
- 10.Giovannoni G, Comi G, Cook S, et al. A placebo controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362:416–426. doi: 10.1056/NEJMoa0902533. [DOI] [PubMed] [Google Scholar]
- 11.Cook S, Vermersch P, Comi G, et al. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (Cladribine Tablets Treating Multiple Sclerosis Orally) study. Mult Scler. 2011;17:578–593. doi: 10.1177/1352458510391344. [DOI] [PubMed] [Google Scholar]
- 12.Lukashev M, Zeng W, Ryan S, et al. Activation of Nrf 2 and modulation of disease progression in EAE models by BG00012 (dimethyl fumarate) suggests a novel mechanism of action combining anti-inflammatory and neuroprotective modalities. Mult Scler. 2007;13:S149. [Google Scholar]
- 13.Kappos L, Gold R, Miller DH, et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: a multicenter, randomized, double-blind, placebo controlled phase IIb study. Lancet. 2008;372:1463–1472. doi: 10.1016/S0140-6736(08)61619-0. [DOI] [PubMed] [Google Scholar]
- 14.Gold R, Kappos L, Bar-Or A, et al. Clinical efficacy of BG-12, an oral therapy, in relapsing–remitting multiple sclerosis: data from the phase 3 DEFINE trial. Mult Scler J. 2011;17:95. [Google Scholar]
- 15.Arnold D, Gold R, Bar-Or A, et al. Efficacy on MRI endpoints of BG-12, an oral therapy, in relapsing–remitting multiple sclerosis: data from the phase 3 DEFINE trial. Mult Scler J. 2011;17:P813. [Google Scholar]
- 16.Fox R, Miller D, Phillips JT, et al. Clinical efficacy of BG-12 in relapsing-remitting multiple sclerosis (RRMS): data from the phase 3 CONFIRM study. Neurology. 2012;78:S01.00. [Google Scholar]
- 17.Warnke C, Meyer zu Hörste G, Hartung HP, Stüve O, Kieseier BC. Review of teriflunomide and its potential in the treatment of multiple sclerosis. Neuropsychiatr Dis Treat. 2009;5:333–340. [PMC free article] [PubMed] [Google Scholar]
- 18.Korn T, Toyka K, Hartung HP, Jung S. Suppression of experimental autoimmune neuritis by leflunomide. Brain. 2001;124:1791–1802. doi: 10.1093/brain/124.9.1791. [DOI] [PubMed] [Google Scholar]
- 19.Merril JE, Hanak S, Pu SF, et al. teriflunomide reduces behavioral, electrophysiological, and histopathological deficits in the Dark Agouti rat model of experimental autoimmune encephalomyelitis. J Neurol. 2009;256:89–103. doi: 10.1007/s00415-009-0075-3. [DOI] [PubMed] [Google Scholar]
- 20.O’Connor PW, Li D, Freedmann MS, et al. A phase II study of the safety and efficacy of teriflunomide in multiple sclerosis with relapses. Neurology. 2006;66:894–900. doi: 10.1212/01.wnl.0000203121.04509.31. [DOI] [PubMed] [Google Scholar]
- 21.Confavreux C, Li DK, Freedman MS, et al. Long-term follow-up of a phase 2 study of oral teriflunomide in relapsing multiple sclerosis: safety and efficacy results up to 8.5 years. Mult Scler. 2012;18:1278–1289. doi: 10.1177/1352458512436594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365:1293–1303. doi: 10.1056/NEJMoa1014656. [DOI] [PubMed] [Google Scholar]
- 23.Yang JS, Xu LY, Xiao BG, Hedlund G, Link H. Laquinimod (ABR-215062) suppresses the development of experimental autoimmune encephalomyelitis, the Th1/Th2 balance and induces the Th3 cytokine TGFβ in Lewis rats. J Neuroimmunol. 2004;156:3–9. doi: 10.1016/j.jneuroim.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 24.Comi G, Pulizzi A, Rovaris M, et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;371:2085–2092. doi: 10.1016/S0140-6736(08)60918-6. [DOI] [PubMed] [Google Scholar]
- 25.Comi G, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366:1000–1009. doi: 10.1056/NEJMoa1104318. [DOI] [PubMed] [Google Scholar]
- 26.Vollmer T, Comi G, Sorensen PS, et al. Clinical efficacy of laquinimod for the treatment of multiple sclerosis; pooled analyses from the ALLEGRO and BRAVO phase III trials. Neurology. 2012;78:S01.007. [Google Scholar]
- 27.Brinkmann V. FTY720 (fingolimod) in multiple sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol. 2009;158:1173–1182. doi: 10.1111/j.1476-5381.2009.00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foster CA, Howard LM, Schweitzer A, et al. Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encepha-lomyelitis: consequences for mode of action in multiple sclerosis. J Pharmacol Exp Ther. 2007;323:469–475. doi: 10.1124/jpet.107.127183. [DOI] [PubMed] [Google Scholar]
- 29.Salvadori M, Budde K, Charpentier B, et al. FTY720 versus MMF with cyclosporine in de novo renal transplantation: a 1-year, randomized controlled trial in Europe and Australasia. Am J Transplant. 2006;6:2912–2921. doi: 10.1111/j.1600-6143.2006.01552.x. [DOI] [PubMed] [Google Scholar]
- 30.Fujita T, Inoue K, Yamamoto S, et al. Fungal metabolites. Part 11. A potent immunosuppressive activity found in Isaria sinclairii metabolite. J Antibiot (Tokyo) 1994;47:208–215. doi: 10.7164/antibiotics.47.208. [DOI] [PubMed] [Google Scholar]
- 31.Chun J, Hartung H P. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 1994;33:91–101. doi: 10.1097/WNF.0b013e3181cbf825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez T, Hla T. Structural and functional characteristics of S1P receptors. J Cell Biochem. 2004;92:913–922. doi: 10.1002/jcb.20127. [DOI] [PubMed] [Google Scholar]
- 34.Dev KK, Mullershausen F, Mattes H, et al. Brain sphingosine-1-phosphate receptors: implication for FTY720 in the treatment of multiple sclerosis. Pharmacol Ther. 2008;117:77–93. doi: 10.1016/j.pharmthera.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 35.Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115:84–105. doi: 10.1016/j.pharmthera.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 36.Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8:1295–1301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- 37.Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annu Rev Biochem. 2004;73:321–354. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 38.Oo ML, Thangada S, Wu MT, et al. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem. 2007;282:9082–9089. doi: 10.1074/jbc.M610318200. [DOI] [PubMed] [Google Scholar]
- 39.Kovarik JM, Schmouder R, Barilla D, Wang Y, Kraus G. Multiple-dose FTY720: tolerability, pharmacokinetics, and lymphocyte responses in healthy subjects. J Clin Pharmacol. 2004;44:532–537. doi: 10.1177/0091270004264165. [DOI] [PubMed] [Google Scholar]
- 40.Budde K, Schmouder RL, Brunkhorst R, et al. First human trial of FTY720, a novel immunomodulator, in stable renal transplant patients. J Am Soc Nephrol. 2002;13:1073–1083. doi: 10.1681/ASN.V1341073. [DOI] [PubMed] [Google Scholar]
- 41.Kataoka H, Sugahara K, Shimano K, et al. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol Immunol. 2005;2:439–448. [PubMed] [Google Scholar]
- 42.Fujino M, Funeshima N, Kitazawa Y, et al. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J Pharmacol Exp Ther. 2003;305:70–77. doi: 10.1124/jpet.102.045658. [DOI] [PubMed] [Google Scholar]
- 43.Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;14(355):1124–1140. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 44.O’Connor P, Comi G, Montalban X, et al. Oral fingolimod (FTY720) in multiple sclerosis: two-year results of a phase II extension study. Neurology. 2009;6(72):73–79. doi: 10.1212/01.wnl.0000338569.32367.3d. [DOI] [PubMed] [Google Scholar]
- 45.Comi G, O’Connor P, Montalban X, et al. Phase II study of oral fingolimod (FTY720) in multiple sclerosis: 3-year results. Mult Scler. 2010;16:197–207. doi: 10.1177/1352458509357065. [DOI] [PubMed] [Google Scholar]
- 46.Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 47.Kappos L, Radue E W, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 48.Polman CH, Reingold SC, Edan G, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the “McDonald Criteria.”. Ann Neurol. 2005;58:840–846. doi: 10.1002/ana.20703. [DOI] [PubMed] [Google Scholar]
- 49.Kovarik JM, Slade A, Riviere GJ, et al. The ability of atropine to prevent and reverse the negative chronotropic effect of fingolimod in healthy subjects. Br J Clin Pharmacol. 2008;66:199–206. doi: 10.1111/j.1365-2125.2008.03199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leypoldt F, Münchau A, Moeller F, Bester M, Gerloff C, Heesen C. Hemorrhaging focal encephalitis under fingolimod (FTY720) treatment: a case report. Neurology. 2009;17(72):1022–1024. doi: 10.1212/01.wnl.0000344567.51394.e3. [DOI] [PubMed] [Google Scholar]
- 51.Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR__Conditions_imposed_on_member_states_for_safe_and_effective_use/human/002202/WC500104527.pdf
- 52.Kasper LH, Due BR et ONO-4641 Study Group . 26th Congress of the European Committee for the Treatment and Research in Multiple Sclerosis. Gothenburg, Sweden: Oct 13–16, 2010. A phase I study to evaluate safety, tolerability and pharmacological properties of a selective sphingosine-1-phosphate (S1P) receptor agonist ONO-4641 in patients with multiple sclerosis [poster no P991] [Google Scholar]
- 53.Comi G, Kappos L, Palace J, et al. Cardiac safety of fingolimod 0.5 mg during the first dose observation in 4-month, open-label, multi-center FIRST study in patients with relapsing MS. Neurology. 2012;78:S41.003. [Google Scholar]
- 54.Geissbühler Y, Butzkueven H, Hernández-Diaz S, et al. Multinational Gilenya (fingolimod) pregnancy exposure registry in multiple sclerosis: study design. Neurology. 2012;78:P06.189. [Google Scholar]
- 55.European Medicines Agency European Medicines Agency starts review of Gilenya (fingolimod) 2012Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/public_health_alerts/2012/01/human_pha_detail_000050.jsp&mid=WC0b01ac058001d126Accessed November 19, 2012
- 56.European Medicines Agency Questions and answers on the review of Gilenya 2012Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Medicine_QA/2012/04/WC500125689.pdfAccessed November 19, 2012