

Abstract
Purpose. DLC-1 is a tumor suppressor gene frequently silenced in human cancers. However, the pathogenicity of DLC-1 epigenetic silencing in the mucosa-adenoma-carcinoma transformation process of colorectal cancer (CRC) has not been studied. Methods. Promoter methylation status of DLC-1 was evaluated in 4 human CRC cell lines, 48 normal mucosa, 57 adenomas, and 80 CRC tissues with methylation-sensitive high-resolution melting analysis (MS-HRMA), while the mRNA expression was examined by qPCR. HRMA was utilized to detect the KRAS codon 12, 13 and BRAF V600Emutations. Results. Partial (1%–10%) and extensive (10%–100%) DLC-1 promoter methylations were observed in 10% and 0% of normal mucosa, 46% and 14% of adenomas, and 60% and 36% of CRCs, respectively. The promoter methylation of DLC-1 was related with the reduction of gene expression and the advanced Duke's stages (Stage C and D). DLC-1 promoter methylation and KRAS mutations are common concurrent pathological alternations. Conclusions. Epigenetic alternation plays a key role in the transcriptional silencing of DLC-1. It is also an independent risk factor related to the carcinogenesis of colorectal tumors and spans over its pathogenesis process. Therefore, DLC-1 promoter methylation quantitation may have a promising significance in the evaluation and management of CRC patients.
1. Introduction
Colorectal cancer (CRC) arises as a consequence of genetic and epigenetic alterations. Somatic mutations and epigenetic alterations have been frequently observed in CRC and are considered to be the driver factors of colorectal tumorigenesis [1]. The neoplastic progression of CRCs mostly follows a mucosa-adenoma-carcinoma step-by-step pattern, which is accompanied by an accumulation of successive genetic alterations (e.g., APC, KRAS/BRAF, and TP53) [2–5]. Widespread promoter CpG island methylation, also referred to as the CpG island methylator phenotype (CIMP), has been extensively studied in CRC. CIMP-high CRC significantly correlates to microsatellite instability, BRAF mutation and low TP53 mutation rate, while CIMP-low tumors present a higher rate of KRAS mutation [6]. Despite these extensive researches, the molecular mechanisms underlying the colorectal mucosa-adenoma-carcinoma transformation process and the progression of CRC are still not fully understood.
Deleted in liver cancer-1, DLC-1 gene is a recently identified tumor suppressor gene. It is located on chromosome 8p21-8p22 and affected in multiple cancers. DLC-1 is a regulator of the Rho family of small GTPases [7–12]. The principal function of DCL-1 is to catalyze the conversion of active guanosine triphosphate- (GTP-) bound RhoA to the inactive guanosine biphosphate- (GDP-) bound form. DLC-1 is recurrently downregulated or inactivated by epigenetic mechanisms in the initiation and progression of cancers [13].
Ullmannova and Popescu [14] first reported the downregulation of DLC-1 mRNA expression in CRCs utilizing cancer-profiling arrays. Moreover, aberrant methylation of CpG islands in the promoter region of DLC-1 is a common mechanism leading to the transcriptional silencing [15–18], suggesting DLC-1 methylation is associated with the downregulation of this gene in CRC and DLC-1 may be a potential tumor suppressor gene. However, methylation and mRNA expression status of DLC-1 and its role in the adenoma-carcinoma progression have not been defined.
To study the role of DLC-1 promoter methylation in the colorectal carcinogenesis routing down the adenoma-carcinoma process, here we quantified the methylation status and mRNA expression of DLC-1 and assessed its relation to various clinicopathological parameters and molecular features, especially the mutation status of KRAS and BRAF in 185 colorectal tissue samples from different disease stages.
2. Materials and Methods
2.1. Patients
Eighty colorectal carcinomas, 57 adenomas, and 48 samples of adjacent histologically normal mucosa were collected from the Department of Gastroenterology at Shanghai Changning District Cental Hospital and Huashan Hospital. Tissue samples were frozen within 2 hours of removal and then stored at −80°C. All the tumors contained more than 80% tumor cells as confirmed by histological examination of sequential sections. The patient's gender, age, Duke's stage, tumor differentiation, and tumor size and location were obtained from surgical and pathological records. In compliance with the Helsinki Declaration of 1975 as revised in 1996, this study was approved by the Institutional Review Board of Shanghai Changning District Center Hospital.
2.2. Cell Lines
Four human colorectal carcinoma cell lines (SW480, LoVo, LS 174T, and COLO 320) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) cell bank. The SW480 and LS 174T cells were cultured in RPMI 1640 medium; the LoVo and COLO 320 cells were cultured in F12K medium (Gibco, Grand Island, NY, USA). Both the mediums were supplemented with 10% fetal bovine serum (Sigma, St. Louis, MO, USA) and incubated in 5% CO2 at 37°C.
2.3. Methylation-Sensitive High-Resolution Melting Analysis (MS-HRMA) of DLC-1
Genomic DNA was extracted from each tumor tissue sample and cell line using QiaAmp NDA extraction kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. Genomic DNA was then modified by sodium bisulfite using EZ DNA Methylation kit (Zymo Research, Irvine, CA, USA) according to the manufacturer's instructions. A methylated reference sample, the CpGenome Universal methylated human male genomic DNA (Chemicon, Billerica, MA, USA), and genomic DNA isolated from the peripheral blood mononuclear cells of a healthy male individual were subjected to the bisulfite modification procedure and used as control standards. The methylated reference was then diluted with the unmethylated control in 0%, 1%, 10%, 30%, 50%, 80%, and 100% ratios to carry out the standard curve for the MS-HRMA. The PCR amplification and HRMA were then performed using the methylation specified primers (MS-HRM-F, MS-HRM-R, Table 1). The 20 μL volume master mix contained 10 μL Premix Taq Hot Start Version (2X) (TaKaRa BIO, Shiga, Japan), 1 μL forward primer (5 μM), 1 μL reverse primer (5 μM), 1 μL SYTO 9 dye (30 μM) (Invitrogen, Carlsbad, CA, USA), 6 μL deionized distilled water, and 1 μL DNA template (15–25 ng/μL). The mix was subjected to PCR on a Rotor-Gene Q real-time platform (Qiagen, Valencia, CA, USA). The samples were denaturated at 95°C for 2 min, followed by 40 cycles of 95°C for 30 sec, 58°C for 30 sec, and 72°C for 30 sec. After PCR, the products were again denaturated at 95°C for 2 min and cooled down to 40°C for 2 min to form the heteroduplex. HRMA was then performed at 0.2°C/s from 50°C to 95°C. Each sample was tested in duplicate. The HRMA and data interpretation were performed using Rotor-Gene Q 1.7 software. A differential profile was then evaluated for each sample by comparing fluorescence at the melting point with the fluorescence of the unmethylated control. There was a linear correlation between the differential fluorescence and the dilution of methylated DNA.
Table 1.
Primers for MS-HRMA, HRMA, and real-time PCR.
| Usage | Primer | Sequence (5′–3′) | Amplicon (bp) |
|---|---|---|---|
| MS-HRMA | MS-HRM-F | TCGTTACGGTTTTAGAAAGAAA | 134 |
| MS-HRM-R | TTCGCTCCCAACCAAAACATAA | ||
|
| |||
| HRMA | KRAS-F | CTGAATATAAACTTGTGGTAGTTGGA | 59 |
| KRAS-R | TATCGTCAAGGCACTCTTGC | ||
| BRAF-F | GGTGATTTTGGTCTAGCTACAG | 147 | |
| BRAF-R | AGTAACTCAGCAGCATCTCAGG | ||
|
| |||
| Real-time PCR | GAPDH-F | GAAGGTGAAGGTCGGAGTCA | 226 |
| GAPDH-R | GAAGATGGTGATGGGATTTC | ||
| DLC-1-F | ACCTGATCACGCAACAGTGAAACA | 191 | |
| DLC-1-R | AGACGCCTGCATAGAGCCTCA | ||
2.4. Bisulfite DNA Sequencing
PCR primers were designed as previously described by Guan et al. [19] to amplify a 292 bp region of the DLC-1 promoter that encompasses 35 CpGs (Figure 2(a)). The PCR product was then subcloned into the pMD19-T expression vector by using a TA Cloning Kit (TaKaRa BIO, Shiga, Japan). We selected four clones from each plate and sent the recombinant plasmids to MAP Biotech (Shanghai, China) to complete the bisulfite DNA sequencing procedure.
Figure 2.
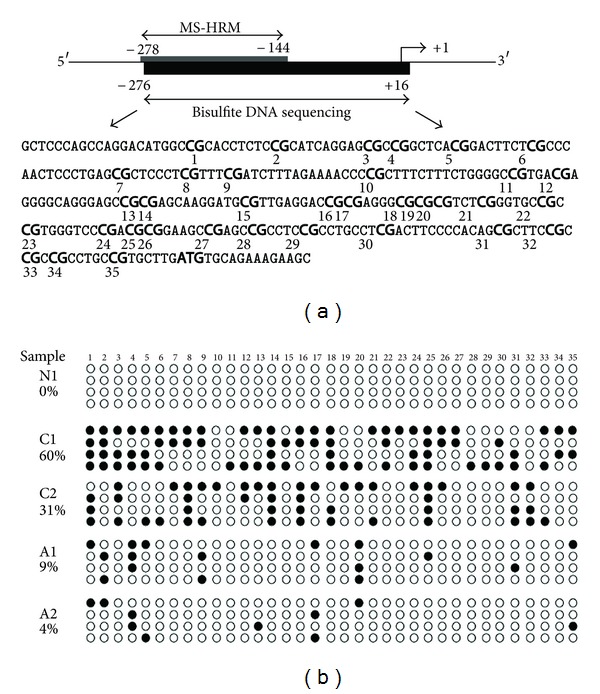
Bisulfite sequencing of the CpG island in DLC-1 promoter region. (a) Schematic depiction of the DLC-1 promoter-associated CpG island, which spans the region from −278 to +16 (the transcription start site ATG as +1). Regions analyzed by MS-HRM and bisulfite genomic sequencing (Bis-DLC-1) are shown. The Bis-DLC-1 region encompassed 292 bp and contained 35 CpG dinucleotides. (b) Methylation patterns of the Bis-DLC-1 region of the DLC-1 CpG island in a normal mucosa tissue (N1), two adenomas (A1 and A2), and two CRCs (C1 and C2) samples that were identified by MS-HRM as methylated. Methylated and unmethylated CpG sites are shown as solid circles and open circles, respectively.
2.5. Quantitative Reverse Transcription PCR
Total RNA was extracted using Trizol (Invitriogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Reverse transcription reactions were carried out on 1 ug total RNA with the PrimeScript RT reagent kit (TaKaRa BIO, Shiga, Japan) using the random hexamer primers. The real-time PCR was then performed using SYBR Premix DimerEraser kit (TaKaRa, Shiga, Japan). The 2−ΔΔCt method was used to calculate the relative fold difference of DLC-1 mRNA expression in all the cancerous tissue samples compared to the average ratio of normal samples. A twofold decreased expression was considered significant. All the reactions were carried out on the ABI PRISM 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). The primers used (DLC-1F and DLC-1R; GAPDH-F and GAPDH-R) were listed in Table 1. The experiments were of strict compliance with the MIQE (Minimum Information about Quantitative Real-Time PCR Experiments) guidelines.
2.6. HRMA Analysis of KRAS Codon 12, 13 and BRAF V600E Mutations
Genomic DNA was extracted from each tumor tissue sample and cell line using QiaAmp DNA extraction kit (Qiagen, Valencia, CA, USA) according to the manufacturer's instructions. AsPC-1 (KRAS condon 12, 13 homozygous mutation), HT29 (BRAF V600E heterozygous mutation), and SW480 cells (BRAF V600E wild-type and KRAS wild-type, homozygous) were used as positive controls for KRAS codon 12, 13 mutation, BRAF V600E mutation, and negative controls, respectively. The DNA was then amplified by the specific primers (KRAS-F and KRAS-R; BRAF-F and BRAF-R, Table 1) [20, 21], producing small amplicons for the HRMA. The PCR mix preparation and the HRMA procedure were carried out with the identical methodology as described in the MS-HRMA of DLC-1 section. For each sample, the normalized melting curves were evaluated and then compared with the mutant and wild-type controls in a deduced difference plot. The samples with distinct melting curves compared with the wild-type allele were recorded as positive.
2.7. Statistical Analysis
The association of DLC-1 methylation and mRNA expression with patients' clinical and genetic variables was analyzed with the χ 2 test. P < 0.05 was considered statistically significant.
3. Results
3.1. MS-HRMA Analysis of DLC-1 Methylation
MS-HRMA was designed to examine the methylation status in promoter regions of DLC-1 in four colon cancer cell lines and various tumor and adjacent normal tissues (Figure 1). When methylation level higher than >1% was considered methylation positive, DLC-1 methylation was observed in 5/48 (10%) of normal mucosa, 26/57 (46%) of adenomas, and 48/80 (60%) of CRCs. However, extensive methylation (methylation level of 10–100%) was observed in only 8/57 (14%) of adenomas, 29/80 (36%) of CRCs, and 0% of the normal mucosa (Table 2). Three of four colon (75%) carcinoma cell lines were methylated in the DLC-1 promoter region at various levels (SW480: 76.9%, LoVo: 62.5%, and LS174T: 44%), whereas no CpG site was methylated in COLO 320 cells.
Figure 1.
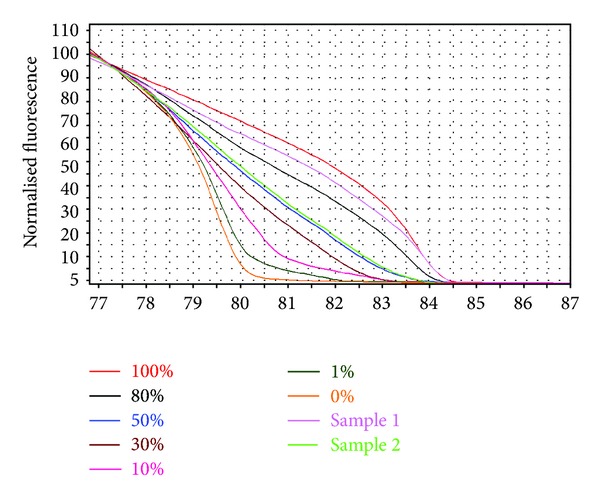
MS-HRMA was used for quantification of DLC-1 methylation status. Profile of fluorescence obtained at the melting temperature for serial dilutions of methylated DNA (from 100 to 0%) and two samples.
Table 2.
Patients' basic clinical characteristics and molecular alternation information.
| Normal mucosa | Adenomas | Carcinomas | P value | |
|---|---|---|---|---|
| Age (y) | ||||
| <60 | 19 | 19 | 33 | 0.631 |
| ≥60 | 29 | 38 | 47 | |
| Gender | ||||
| Female | 18 | 28 | 45 | 0.121 |
| Male | 30 | 29 | 35 | |
| DLC-1 methylation | ||||
| Nonmethylation | 43 | 31 | 32 | <0.001 |
| Partial methylation | 5 | 18 | 19 | |
| Extensive methylation | 0 | 8 | 29 | |
| DLC-1 mRNA expression | ||||
| Abundant | 41 | 27 | 17 | <0.001 |
| Reduction | 7 | 30 | 63 | |
| KRAS | ||||
| Wild-type | 44 | 47 | 48 | <0.001 |
| Mutation | 4 | 10 | 32 | |
| BRAF | ||||
| Wild-type | 47 | 55 | 78 | 0.894 |
| Mutation | 1 | 2 | 2 |
3.2. Bisulfite DNA Sequencing
Bisulfite DNA sequencing was employed to determine the comprehensive methylation pattern of the 5′-CpG islands in the DLC-1 promoter. Five CRCs and five adenoma samples that were identified by MS-HRMA as extensive methylation and partial methylation, respectively, and two normal mucosa tissue samples were amplified with a 292 bp fragment in the DLC-1 promoter, covering 35 CpG sites. Bisulfite DNA sequencing confirmed the CpG islands in the normal sample unmethylated; all five CRC samples were frequently methylated. The tumor samples shared some common methylation sites, while the overall methylation patterns were distinct. Consistent with the MS-HRMA data, the CpG island exhibited that more CpG sites were methylated in clones obtained from CRCs than those from adenomas. We found the methylation of DLC-1 promoter more frequently methylated in the CRC samples (C1, 60% and C2, 31%) than in the adenomas (A1, 9% and A2, 4%) (Figure 2(b)).
3.3. Correlation of DLC-1 Methylation with Clinical and Pathological Features
The correlations between the methylation status of DLC-1 and clinical features in the CRC and adenoma patients were presented in Table 3. DLC-1 methylation was significantly higher in the CRC tissues with advanced Duke's stage (Stage C, versus Stage A + B, P = 0.025; Stage D, versus Stage A + B, P = 0.002). However, no significant difference in the promoter methylation level was observed between Stages C and D (P = 0.902). Other than this, there were no significant associations between DLC-1 methylation status and other clinical factors. Among adenomas, there were no significant associations between DLC-1 methylation status and clinical parameters.
Table 3.
Methylation status, pathological features, and molecular alternations in CRCs and adenomas.
| CRCs | Adenomas | |||||||
|---|---|---|---|---|---|---|---|---|
| Non methylation | Partial methylation | Extensive methylation | P value | Non methylation | Partial methylation | Extensive methylation | P value | |
| Age (y) | ||||||||
| <60 | 14 | 4 | 15 | 0.098 | 8 | 8 | 3 | 0.397 |
| ≥60 | 18 | 15 | 14 | 23 | 10 | 5 | ||
| Gender | ||||||||
| Female | 14 | 11 | 20 | 0.139 | 13 | 11 | 4 | 0.433 |
| Male | 18 | 8 | 9 | 18 | 7 | 4 | ||
| Location | ||||||||
| Distal | 17 | 15 | 16 | 0.151 | 23 | 15 | 6 | 0.829 |
| Proximal | 15 | 4 | 13 | 8 | 3 | 2 | ||
| Dukes' stage | ||||||||
| A + B | 23 | 7 | 7 | 0.004 | ||||
| C | 4 | 5 | 8 | |||||
| D | 5 | 7 | 14 | |||||
| Differentiation | ||||||||
| Well and moderately | 19 | 9 | 11 | 0.259 | ||||
| Poorly | 13 | 10 | 18 | |||||
| Size | ||||||||
| <1 cm | 14 | 7 | 13 | 0.88 | 14 | 6 | 2 | 0.598 |
| ≥1 cm | 18 | 12 | 16 | 17 | 12 | 6 | ||
| KRAS mutation status | ||||||||
| Wild-type | 24 | 13 | 11 | 0.01 | 26 | 14 | 7 | 0.889 |
| Mutation | 8 | 6 | 18 | 5 | 4 | 1 | ||
3.4. Correlation of DLC-1 Methylation with DLC-1 mRNA Expression
We determined the mRNA expression of DLC-1 gene in 4 colon cancer cell lines, normal mucosa, adenomas, and CRC tissues. GAPDH mRNA was used as a housekeeper for cDNA integrity. DLC-1 mRNA expression was detected in Colo320 cells but not in SW480, LoVo, and LS 174T cell lines which harbor methylation in the promoter region of DLC-1. DLC-1 mRNA expression was observed in 41/48 (85%) of normal mucosa specimens. Of the 57 adenomas and 80 CRC tissues, DLC-1 mRNA was downregulated in 30/57 (52%) and 63/80 (79%) samples, respectively (Table 2). Since very low level of methylation could not lead to significant downregulation of gene expression, we considered 10% methylation of DLC-1 promoter region as the cutoff. There was a correlation between DLC-1 mRNA expression reduction and extensive promoter methylation status (Table 4, P = 0.022). Our results suggest that the reduction or loss of DLC-1 mRNA expression was related to the extensive methylation in DLC-1 promoter.
Table 4.
Correlation between DLC-1 mRNA expression and promoter methylation status in adenomas and CRCs.
| mRNA expression | Unmethylation and Partial methylation (<10%) | Extensive methylation (≥10%) | P value |
|---|---|---|---|
| Reduced | 62 | 31 | 0.022 |
| Abundant | 38 | 6 |
3.5. Correlation of DLC-1 Methylation with KRAS and BRAF Mutations
We found KRAS mutations in 4/48 (8%) normal mucosa, 10/57 (18%) adenomas, and 32/80 (40%) CRCs (Table 2). When we examined the association of KRAS mutations to DLC-1 methylation, a statistically significant correlation was observed only in the CRCs (P = 0.010), but not in adenomas (P = 0.889). Hereafter, we investigated the association between extensive methylation of DLC-1 promoter and mutations in KRAS codon12, 13. In terms of CRCs, 62% (18/29) of tumors with extensive DLC-1 methylation showed KRAS mutations, while KRAS mutation alterations were present in only 32% (6/19) of tumors with partial methylation and 25% (8/32) of tumors with unmethylated DLC-1 promoter. These data demonstrated that KRAS mutations significantly correlated with extensive DLC-1 promoter methylation in CRCs. However, we did no further investigation in the correlation between BRAF mutation and DLC-1 methylation because only 4 of 185 tissues turned out to be BRAF V600E positive from the HRMA, limiting the statistical power of the data.
4. Discussion
The identification of genes contributing to the development of colon cancer is critical to the understanding of molecular mechanisms of carcinogenesis and may provide new strategies for clinical therapy. A new candidate tumor suppressor gene, DLC-1, was first identified as a rat p122RhoGAP homolog [10]. This gene is diminished or silenced in various types of human cancers as well as in metastatic cells compared to nonmetastatic cells [7–9, 11, 12]. DLC-1 has not been extensively studied in CRC. Moreover, the transcriptional regulation of DLC-1 gene expression through epigenetic mechanisms has not been investigated in the normal adenoma-carcinoma sequence. The relationships between DLC-1 methylation status and clinic pathological variables in CRC remain to be elucidated.
In this study, we used MS-HRMA to detect DLC-1 promoter methylation in 48 normal mucosa, 57 adenomas, 80 CRC tissue samples, and 4 CRC cell lines. The methylation status of DLC-1 did not show a strong correlation with the widely accepted risk factors of CRC including age and sex (estrogen) [22]. We found the mRNA expression of DLC-1 was decreased when DLC-1 promoter was methylated in the cancerous tissues. Moreover, partial methylation was frequently observed in adenomas as well as CRC. Extensive methylation was primarily observed in CRCs but less prevalent in adenomas. In addition, by quantifying the methylation status in DLC-1 promoter, we also found an accumulation of aberrant methylation following the adenoma-carcinoma sequence. During this stepwise progression, the methylation level of the DLC-1 promoter region increased gradually and the existence of cytosine methylation expanded widely. None of the normal adjacent mucosa specimens showed extensive methylation in the DLC-1 promoter region. Extensive methylation is a characteristic of a more advanced CRC while partial methylation is the feature of an earlier stage of CRC. These results indicate that the methylation of DLC-1 promoter runs through the whole course of colorectal tumorigenesis.
These pieces of evidence indicate that the epigenetic mechanism is a driving factor of DLC-1 transcriptional silencing and may be involved in the tumorigenesis of CRC as an independent risk factor.
DLC-1 methylation levels were also found to be significantly associated with Duke's stage. The methylation levels were significantly higher in advanced stage (Stages C and D) tumors, which indicated the role of DLC-1 in the CRC progression. This result is consistent with previous findings, which showed the methylation status of DLC-1 was related to TNM stages [23]. Jin et al. [24] reported that the knocking down of DLC-1 transcriptional expression by RNAi resulted in the promotion of LoVo CRC cell proliferation, migration, and cell cycle progression, that is critical for tumor growth and metastasis. Thus, our results further imply that methylation of DLC-1 promoter is a potentially biomarker for prognosis evaluation of CRC.
KRAS oncogene is a guanine nucleotide-binding protein with GTPase activity that is involved in signal transduction. In this study, we confirmed that the extensive DLC-1 methylation was associated with the KRAS mutations in CRCs but not in adenomas. Our previous study showed that DLC-1 methylation significantly correlated with PIK3CA mutations in Paget's disease [25]. These findings highlighted the interaction between genetic and epigenetic alterations in CRC, although the mechanism underlying this phenomenon requires further study. On the other hand, overactivation of certain oncogenic pathways is known to affect the activity of methyltransferases and the regulation of gene transcription, thus possibly affecting components of the MAPK pathway, such as RAS, RAF, MEK, and ERK [26, 27]. Data from KRAS transformation studies suggest that activated KRAS promotes P16 methylation [28]. Stable transformation of colon cancer cells with KRAS increased DNA methyltransferase activity and P16 gene methylation [29]. Collectively, the results from our study and previous work by others suggest epigenetic alterations of DLC-1 might occur as a consequence of overactivation of the oncogenic pathway in cancer.
In conclusion, the current data suggested that methylation-induced epigenetic silencing of DLC-1 is involved in the colorectal tumorigenesis and has a strong correlation with the Duke's stage, and with KRAS mutation. Quantitative detection of DLC-1 promoter methylation may have a promising clinical significance in the evaluation of CRC patients and in the management of the disease.
Author's Contribution
Both Haixia Peng and Feng Long contributed equally to this study.
Conflict of Interests
The authors declare that they have no conflict of interests.
Acknowledgments
This study was supported by a fund from the Science and Technology Commission of Shanghai (114119b0600), and Shanghai Municipal Health Bureau Hundred Talents Program (XBR2011044). The authors thank Dr. Bing Su and his colleague from Roswell Park Cancer Institute of USA for language editing.
References
- 1.Markowitz SD, Bertagnolli MM. Molecular basis of colorectal cancer. New England Journal of Medicine. 2009;361(25):2404–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker SJ, Preisinger AC, Jessup JM, et al. p53 Gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Research. 1990;50(23):7717–7722. [PubMed] [Google Scholar]
- 3.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 4.Forrester K, Almoguera C, Han K, Grizzle WE, Perucho M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature. 1987;327(6120):298–303. doi: 10.1038/327298a0. [DOI] [PubMed] [Google Scholar]
- 5.Goss KH, Groden J. Biology of the adenomatous polyposis coli tumor suppressor. Journal of Clinical Oncology. 2000;18(9):1967–1979. doi: 10.1200/JCO.2000.18.9.1967. [DOI] [PubMed] [Google Scholar]
- 6.Barault L, Charon-Barra C, Jooste V, et al. Hypermethylator phenotype in sporadic colon cancer: study on a population-based series of 582 cases. Cancer Research. 2008;68(20):8541–8546. doi: 10.1158/0008-5472.CAN-08-1171. [DOI] [PubMed] [Google Scholar]
- 7.Goodison S, Yuan J, Sloan D, et al. The RhoGAP protein DLC-1 functions as a metastasis suppressor in breast cancer cells. Cancer Research. 2005;65(14):6042–6053. doi: 10.1158/0008-5472.CAN-04-3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Healy KD, Hodgson L, Kim TY, et al. DLC-1 suppresses non-small cell lung cancer growth and invasion by RhoGAP-dependent and independent mechanisms. Molecular Carcinogenesis. 2008;47(5):326–337. doi: 10.1002/mc.20389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liao YC, Lo SH. Deleted in liver cancer-1 (DLC-1): a tumor suppressor not just for liver. International Journal of Biochemistry and Cell Biology. 2008;40(5):843–847. doi: 10.1016/j.biocel.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yuan BZ, Miller MJ, Keck CL, Zimonjic DB, Thorgeirsson SS, Popescu NC. Cloning, characterization, and chromosomal localization of a gene frequently deleted in human liver cancer (DLC-1) homologous to rat RhoGAP. Cancer Research. 1998;58(10):2196–2199. [PubMed] [Google Scholar]
- 11.Yuan BZ, Zhou X, Durkin ME, et al. DLC-1 gene inhibits human breast cancer cell growth and in vivo tumorigenicity. Oncogene. 2003;22(3):445–450. doi: 10.1038/sj.onc.1206064. [DOI] [PubMed] [Google Scholar]
- 12.Zhou X, Thorgeirsson SS, Popescu NC. Restoration of DLC-1 gene expression induces apoptosis and inhibits both cell growth and tumorigenicity in human hepatocellular carcinoma cells. Oncogene. 2004;23(6):1308–1313. doi: 10.1038/sj.onc.1207246. [DOI] [PubMed] [Google Scholar]
- 13.Durkin ME, Yuan BZ, Zhou X, et al. DLC-1: a Rho GTPase-activating protein and tumour suppressor. Journal of Cellular and Molecular Medicine. 2007;11(5):1185–1207. doi: 10.1111/j.1582-4934.2007.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ullmannova V, Popescu NC. Expression profile of the tumor suppressor genes DLC-1 and DLC-2 in solid tumors. International Journal of Oncology. 2006;29(5):1127–1132. [PubMed] [Google Scholar]
- 15.Kim TY, Jong HS, Song SH, et al. Transcriptional silencing of the DLC-1 tumor suppressor gene by epigenetic mechanism in gastric cancer cells. Oncogene. 2003;22(25):3943–3951. doi: 10.1038/sj.onc.1206573. [DOI] [PubMed] [Google Scholar]
- 16.Wong CM, Lee JMF, Ching YP, Jin DY, Ng IOL. Genetic and epigenetic alterations of DLC-1 gene in hepatocellular carcinoma. Cancer Research. 2003;63(22):7646–7651. [PubMed] [Google Scholar]
- 17.Yuan BZ, Durkin ME, Popescu NC. Promoter hypermethylation of DLC-1, a candidate tumor suppressor gene, in several common human cancers. Cancer Genetics and Cytogenetics. 2003;140(2):113–117. doi: 10.1016/s0165-4608(02)00674-x. [DOI] [PubMed] [Google Scholar]
- 18.Yuan BZ, Jefferson AM, Baldwin KT, Thorgeirsson SS, Popescu NC, Reynolds SH. DLC-1 operates as a tumor suppressor gene in human non-small cell lung carcinomas. Oncogene. 2004;23(7):1405–1411. doi: 10.1038/sj.onc.1207291. [DOI] [PubMed] [Google Scholar]
- 19.Guan M, Zhou X, Soulitzis N, Spandidos DA, Popescu NC. Aberrant methylation and deacetylation of deleted in liver cancer-1 gene in prostate cancer: potential clinical applications. Clinical Cancer Research. 2006;12(5):1412–1419. doi: 10.1158/1078-0432.CCR-05-1906. [DOI] [PubMed] [Google Scholar]
- 20.Pichler M, Balic M, Stadelmeyer E, et al. Evaluation of high-resolution melting analysis as a diagnostic tool to detect the BRAF V600E mutation in colorectal tumors. Journal of Molecular Diagnostics. 2009;11(2):140–147. doi: 10.2353/jmoldx.2009.080100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang C, Guo W, Wu J, et al. Differential high-resolution melting analysis for the detection of K-ras codons 12 and 13 mutations in pancreatic cancer. Pancreas. 2011;40(8):p. 1283. doi: 10.1097/MPA.0b013e318220af91. [DOI] [PubMed] [Google Scholar]
- 22.Chlebowski RT, Wactawski-Wende J, Ritenbaugh C, et al. Estrogen plus progestin and colorectal cancer in postmenopausal women. New England Journal of Medicine. 2004;350(10):991–1004. doi: 10.1056/NEJMoa032071. [DOI] [PubMed] [Google Scholar]
- 23.Liu JB, Zhang SQ, Shi MX, Shao BF, Zhang YX. Relationship between methylation status of DLC-1 gene and metastasis of hepatocellular carcinoma. World Chinese Journal of Digestology. 2008;16(11):1237–1240. [Google Scholar]
- 24.Jin Y, Tian X, Shang Y, Huang P. Inhibition of DLC-1 gene expression by RNA interference in the colon cancer Lo Vo cell line. Oncology Reports. 2008;19(3):669–674. [PubMed] [Google Scholar]
- 25.Kang Z, Xu F, Zhang QA, et al. Correlation of DLC1 gene methylation with oncogenic PIK3CA mutations in extramammary Paget's disease. Modern Pathology. 2012;25(8):1160–1168. doi: 10.1038/modpathol.2012.65. [DOI] [PubMed] [Google Scholar]
- 26.Carragher LAS, Snell KR, Giblett SM, et al. V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO Molecular Medicine. 2010;2(11):458–471. doi: 10.1002/emmm.201000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwon O, Jeong SJ, Kim SO, et al. Modulation of E-cadherin expression by K-Ras; involvement of DNA methyltransferase-3b. Carcinogenesis. 2010;31(7):1194–1201. doi: 10.1093/carcin/bgq071. [DOI] [PubMed] [Google Scholar]
- 28.Tannapfel A, Benicke M, Katalinic A, et al. Frequency of p16(INK4A) alterations and k-ras mutations in intrahepatic cholangiocarcinoma of the liver. Gut. 2000;47(5):721–727. doi: 10.1136/gut.47.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guan RJ, Fu Y, Holt PR, Pardee AB. Association of K-ras mutations with p16 methylation in human colon cancer. Gastroenterology. 1999;116(5):1063–1071. doi: 10.1016/s0016-5085(99)70009-0. [DOI] [PubMed] [Google Scholar]