

Abstract
Cancer stem cells (CSCs) are a small subpopulation of cancer cells that have increased resistance to conventional therapies and are capable of establishing metastasis. However, only a few biomarkers of CSCs have been identified. Here, we report that ganglioside GD2 (a glycosphingolipid) identifies a small fraction of cells in human breast cancer cell lines and patient samples that are capable of forming mammospheres and initiating tumors with as few as 10 GD2+ cells. In addition, the majority of GD2+ cells are also CD44hiCD24lo, the previously established CSC-associated cell surface phenotype. Gene expression analysis revealed that GD3 synthase (GD3S) is highly expressed in GD2+ as well as in CD44hiCD24lo cells and that interference with GD3S expression, either by shRNA or using a pharmacological inhibitor, reduced the CSC population and CSC-associated properties. GD3S knockdown completely abrogated tumor formation in vivo. Also, induction of epithelial-mesenchymal transition (EMT) in transformed human mammary epithelial cells (HMLER cells) dramatically increased GD2 as well as GD3S expression in these cells, suggesting a role of EMT in the origin of GD2+ breast CSCs. In summary, we identified GD2 as a new CSC-specific cell surface marker and GD3S as a potential therapeutic target for CSCs, with the possibility of improving survival and cure rates in patients with breast cancer.
Introduction
In several types of cancer, a distinct subpopulation of cancer cells has a greater capacity to initiate new tumors compared with the bulk of the tumor cells upon transplantation into mice (1, 2). These cancer cells have both long-term self-renewal capacity and the ability to initiate tumors. Since their properties are similar to those of normal stem cells, these cancer cells have been termed cancer stem cells (CSCs) (3, 4). More recently, CSCs were found to be inherently resistant to conventional cancer therapies and capable of establishing metastases (5, 6). Accordingly, much effort is being undertaken to identify clinically relevant biomarkers for better identification and targeting of CSCs.
Al-Hajj and colleagues isolated a subpopulation of lineage-negative cancer cells expressing high levels of membrane CD44 and lacking or displaying a low level of CD24 (CD44hiCD24lo). These cells displayed the requisite features of breast CSCs. They found that the CD44hiCD24lo population had a 10- to 15-fold-increased ability to form tumors in NODΩSCID mice compared with bulk tumor cells (7). In addition, CD44hiCD24lo cells have a greater capacity to propagate as mammospheres, which is an in vitro surrogate assay for self-renewal (8). Moreover, a higher proportion of CD44hiCD24lo cells in breast cancer has been associated with shorter disease-free and overall survival and with a greater incidence of distant metastases (9, 10); the clinical outcome associated with tumors rich in CD44+ cells is significantly inferior, and they are characterized by activation of TGF-β signaling (11).
More recently, the Weinberg group reported that epithelial cells induced to undergo epithelial-mesenchymal transition (EMT) acquired stem cell properties (12). Moreover, induction of EMT, by ectopic expression of Snail or Twist or by treatment with TGF-β, in immortalized human mammary epithelial cells (HMECs, also known as HMLE cells) resulted in the acquisition of the CD44hiCD24lo marker profile as well as significantly greater mammosphere formation and tumor initiation potential. Building on this, we recently found that HMECs in which EMT was induced through ectopic expression of Twist or Snail also acquired functional properties of human bone marrow–derived mesenchymal stem/stromal cells (MSCs), such as the ability to differentiate into mesodermal lineages and home to wounds (13). Based on these findings, we hypothesized that cell surface markers known to be expressed on MSCs may also be expressed on breast CSCs and could be utilized to identify and target breast CSCs.
Ganglioside GD2 (according to Svennerholm’s nomenclature system; ref. 14) is highly expressed on bone marrow–derived MSCs, and therefore, this marker is being used for the prospective isolation of these cells (15). Gangliosides are sialic acid–bearing glycosphingolipids expressed on all vertebrate cells (16). They are anchored to the plasma membrane through their ceramide lipids, with their varied glycans extending into the extracellular space (17). Among gangliosides, GM2, GD2, and GD3 are highly expressed in human tumors of neuro-ectodermal origin, such as melanomas, gliomas, and neuroblastomas, whereas they are absent or weakly expressed in normal tissues (18, 19). GD2 is a b-series ganglioside expressed mostly on the cell membrane. GD2 and GD3 are produced from their precursors GD3 and GM3, respectively, by the activity of enzymes GD2 synthase (GD2S) and GD3 synthase (GD3S), respectively (20).
In this report, we demonstrate that GD2-positive breast cancer cells display molecular and functional properties of CSCs. GD3S expression is critical for the expression of GD2, and suppression of its biosynthesis using shRNA or a small molecule against GD3S hampered mammosphere formation and tumor initiation. Our findings suggest that GD2 is a novel biomarker of breast CSCs and that GD3S, the enzyme involved in GD2 biosynthesis, is essential for breast CSC function.
Results
GD2 enriches for breast CSCs.
We recently reported that, following the induction of EMT, human mammary epithelial cells show functional properties similar to those of human bone marrow–derived MSCs (13). Therefore, we hypothesized that the cell markers expressed on the surface of MSCs could also be expressed on the surface of breast CSCs. To test this hypothesis, we analyzed for the expression of several known MSC cell surface markers (i.e., CD105, CD90, CD106, CD166, CD73, CD271, MSCA-1, and GD2) on HMECs that had been experimentally transformed to become tumorigenic using oncogenic V12-H-Ras (HMLER cells) (21). Absolute expression of most of the markers analyzed could not divide HMLER cells into two distinct subpopulations (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI59735DS1), similar to CD44hiCD24lo cells (12). However, ganglioside GD2, one of the cell surface markers for MSCs, was able to separate HMLER cells into GD2+ (4.5% ± 2.4%) and GD2– (92.7% ± 3.8%) populations (Figure 1A and Supplemental Figure 1). Strikingly, GD2+ HMLER cells isolated using FACS appeared spindle-shaped, with limited cell-cell contacts; conversely, the GD2– cells displayed cobblestone epithelial morphology (Figure 1B). Moreover, the GD2+ HMLER cells proliferated approximately 5-fold slower than the GD2– HMLER cells (Figure 1C).
Figure 1. GD2 identifies CSCs in breast cancer.

(A) HMLER cells were stained with anti-GD2 antibody by indirect staining and analyzed on an LSR II flow cytometer. GD2+/– gates were drawn based on IgG2a isotype control. FSC, forward scatter. (B) GD2+/– HMLER cells were cell sorted and cultured in cell culture dishes for 4 days. Scale bars: 50 μm. (C) 2 × 104 GD2+/– HMLER cells were cultured in 6-well cell culture dishes in triplicate. Total cells were counted on days 2, 4, and 6 using a Vi-CELL (Beckman Coulter) cell counter. (D) HMLER or MDA-MB-231 cells (1 × 103) were sorted into each well of 24-well ultra-low attachment dishes containing mammosphere growth medium using the FACSAria II cell sorter. Cells were cultured for 12 days, and the photos were taken using a light microscope. Scale bars: 100 μm. (E and F) Number of mammospheres formed from GD2+/– HMLER (E) and MDA-MB-231 (F) cells. The experiment was performed in triplicate. P < 0.01 (G) GD2+/– MDA-MB-231 cells were sorted (1 cell or 5 cells/well) into 96-well ultra-low-attachment dishes containing mammosphere growth medium. Cells were cultured for 12 days, and mammospheres were counted using a light microscope. Scale bars: 200 μm. (H) Number of mammospheres formed from single GD2+/– MDA-MB-231 cells. *P < 0.002. (I) Size of mammospheres measured using a hemocytometer. *P < 0.0001.
To further investigate the functional properties of GD2+ and GD2– cells, we sorted HMLER and MDA-MB-231 cells based on GD2 expression and examined them by mammosphere assay. Interestingly, the GD2+ cells from HMLER and MDA-MB-231 cells formed 2-fold more mammospheres compared with GD2– cells (Figure 1, D–F, P < 0.01). Direct sorting of GD2+ and GD2– MDA-MB-231 cells into low-attachment 96-well plates at either 1 or 5 cells per well also resulted in a 2-fold increase in sphere formation by GD2+ cells regardless of the number of cells per well compared with GD2– cells (Figure 1, G and H). In addition, the mammospheres generated by GD2+ cells were 3 times larger than those generated by GD2– cells (Figure 1, G and I), indicating that the GD2+ cells are capable of growing better in suspension cultures.
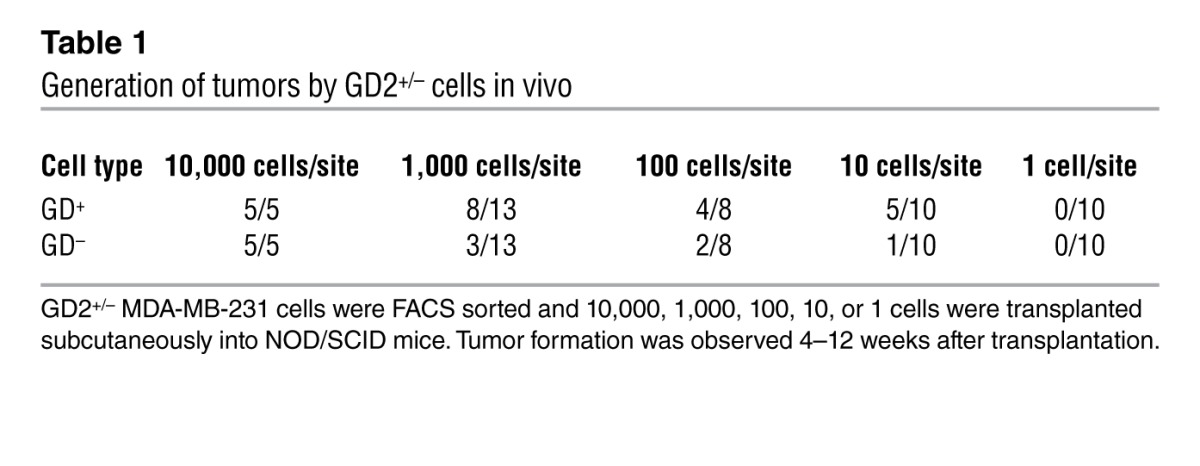
CSCs are known to be more migratory and invasive (1, 3). To examine the migration and invasion potential of GD2+/– cells, we fractionated HMLER cells into GD2+ and GD2– cells and analyzed them for migration and invasion using Boyden chamber Matrigel invasion assays. After 24 hours of incubation, GD2+ HMLER cells migrated to a more than 4-fold greater extent compared with GD2– cells, indicating that GD2+ cells are highly migratory (Supplemental Figure 2). The hallmark of CSCs is their ability to initiate tumor better than their bulk tumor counterparts (1, 2). To determine the tumor-initiating potential of GD2+ cells, we sorted GD2+ and GD2– MDA-MB-231 cells and transplanted them subcutaneously into the flank of NOD/SCID mice at limiting dilutions. At lower cell numbers including 100 or 10 cells/site, the GD2+ cells generated 2- and 5-fold more tumors, respectively, compared with the GD2– fraction (Table 1). However, at higher cell numbers (10,000 or 1,000 cells/site), there were no significant differences in tumor initiation between GD2+ and GD2– cells. These data firmly established that GD2 is a marker of cells capable of initiating tumors at a higher frequency than cells without GD2.
Table 1.
Generation of tumors by GD2+/– cells in vivo
Percentage of GD2+ cells is highest in cell lines with a basal molecular signature.
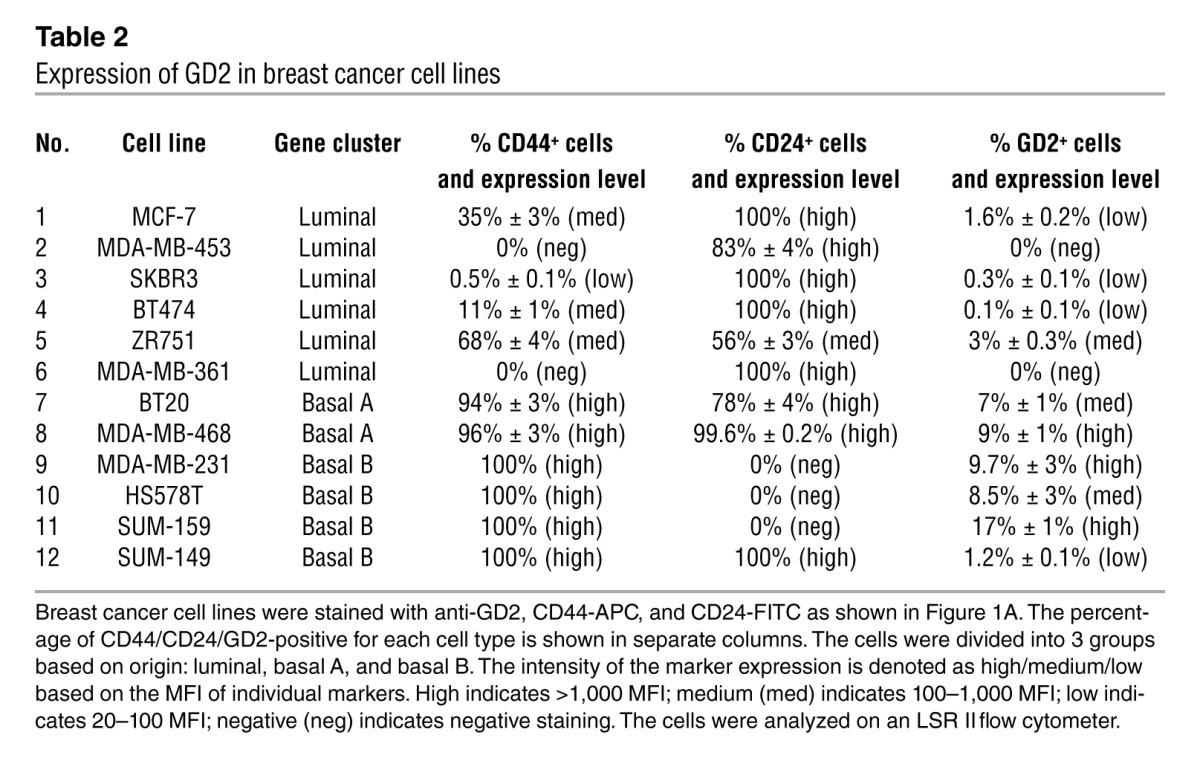
On the basis of gene expression profile (22), breast cancer cell lines have been classified into 3 groups: luminal, basal A, and basal B. We randomly selected 12 breast cancer cell lines representing these 3 subgroups and analyzed them for GD2 expression. Interestingly, the majority of these lines, independent of the subgroup, contained a subpopulation of GD2+ cells at variable levels (Table 2). However, basal cell lines contained a much greater number (mean 9%, range 1.2%–17%, n = 6) of GD2+ cells compared with luminal cell lines (median 0.2%, range 0–3%, n = 6, Table 1, P = 0.00237). Since basal-derived cell lines show greater tumor initiation potential and contain more CSCs based on the previously reported CD44hiCD24lo profiles (23), this finding once again confirms GD2 as a stem cell marker.
Table 2.
Expression of GD2 in breast cancer cell lines
GD2 identifies the CD44hiCD24lo population in breast cancer cell lines and patient samples.
Since we found that GD2, similar to previously reported CD44 and CD24 cell surface markers, is capable of separating cancer cells into two populations with differing tumor-initiating potential (7), we hypothesized that GD2 would be mostly expressed in the CD44hiCD24lo cancer cell fraction. To test this, we initially analyzed the expression of CD44hiCD24lo cells in GD2+ HMLER cells and found that more than 85% (85% ± 3.5%) of GD2+ HMLER cells also displayed a CD44hiCD24lo CSC profile, whereas less than 1% (0.7% ± 0.2%) of GD2– HMLER cells were CD44hiCD24lo (Figure 2A). In addition, through reverse gating analysis of CD44hiCD24lo HMLER cells, we noted that more than 84% (84% ± 2.5%) of CD44hiCD24lo HMLER cells were also positive for GD2 (Figure 2B), whereas less than 5% of CD44loCD24hi HMLER cells were GD2+ (4.3% ± 1.2%). To further determine the correlation between the expression of GD2 and the CD44/CD24 profiles, we sequentially gated HMLER cells into GD2hi, GD2lo, and GD2neg cells. This analysis revealed that GD2 expression levels correlated strongly with the CD44hiCD24lo phenotype (Supplemental Figure 3A). Moreover, by determining the MFI, we found that GD2 expression levels correlated positively with CD44 expression (correlation index, r2 = 0.85; P < 0.0003; Supplemental Figure 3B). To validate the coexpression of GD2 on CD44hiCD24lo cells, we used anti-GD2 antibody from a different source (Abcam, clone 2Q549) to stain HMLER cells in a 4-step staining procedure as explained before (24), along with anti-CD44 and anti-CD24 antibodies. Analysis of GD2+ cells revealed that these cells coexpress CD44hiCD24lo, confirming our initial findings with the 14G2a clone (Supplemental Figure 4).
Figure 2. GD2 identifies CD44hiCD24lo stem cell phenotype in breast cancer cells.

(A) HMLER cells were stained with anti-GD2 antibody and with CD44-APC and CD24-FITC using the 4-step staining protocol described in Methods. Cells were electrically gated on GD2+/– cells and displayed in a pseudocolor dot plot with CD44 on the y axis and CD24 on the x axis using FlowJo data analysis software. (B) In an identical experiment, CD44hi/loCD24lo/hi cells were displayed on a pseudocolor dot plot with GD2 on the y axis and FSC on the x axis. (C) Primary breast tumor samples were processed as described in Methods, and the single cells in suspension were stained with anti-GD2, CD44-APC, CD24-FITC, CD45-FITC, and DAPI using the 4-step staining protocol. Cells were initially gated on DAPI-negative cells to exclude dead cells, and the cells were then gated on CD45– cells to exclude hematopoietic cells. GD2+CD45– cells were displayed on a dot plot, with CD44 on the y axis and CD24 on the x axis. Analysis was perfumed using an LSR II flow cytometer. Data were analyzed using FlowJo software.
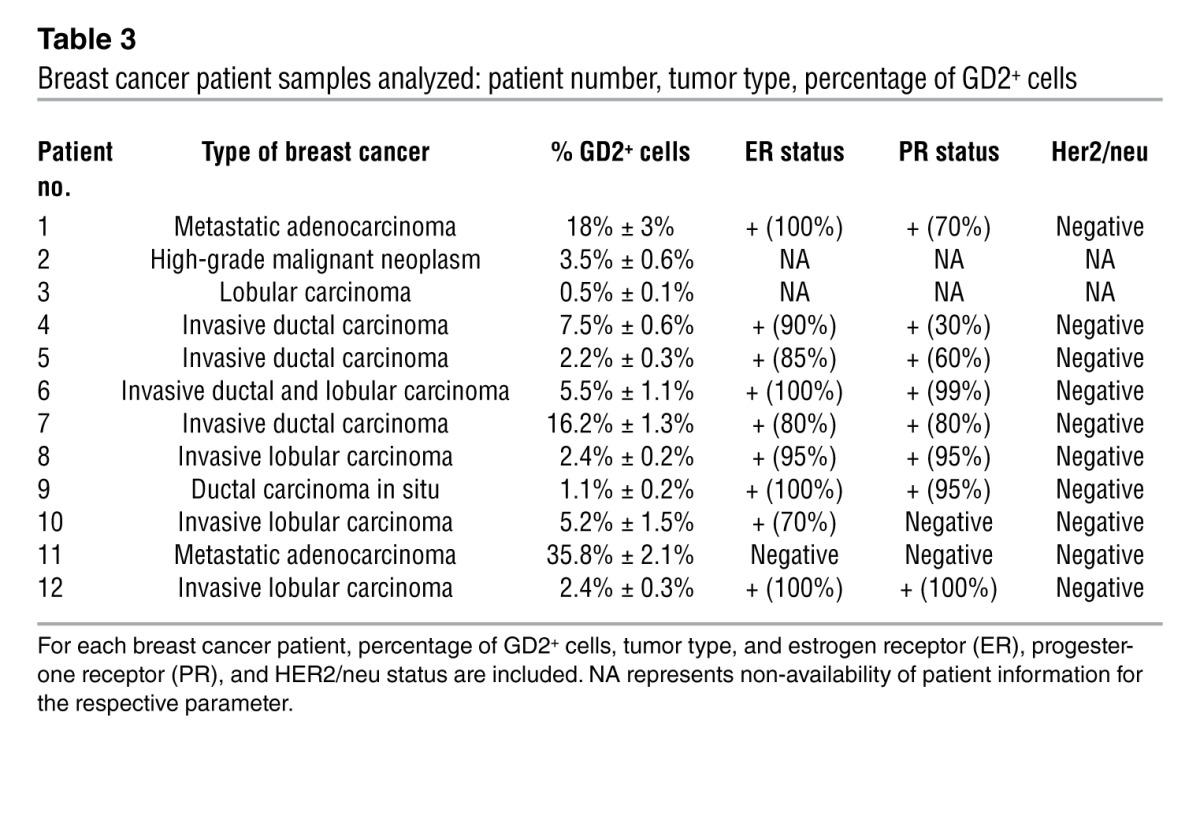
To further investigate the correlation between GD2 expression and the CD44hiCD24lo profile, we also analyzed primary breast tumor samples (n = 12, Table 3). Using multi-parameter flow cytometry, we excluded CD45+ inflammatory and other hematopoietic cells from dissociated tumor samples. The non-hematopoietic CD45– fraction was then analyzed for the expression of GD2, CD44, and CD24. This analysis of the CD45– fraction revealed that GD2 was expressed, at variable levels from 0.5% to 35% (median 4.35%, range 0.5%–35.8%), in tumor samples (Table 3). Importantly, similar to what we observed in cell lines, more than 95.5% (95.5% ± 2.7%) of GD2+CD45– tumor cells also co-segregated with the CD44hiCD24lo phenotype (Figure 2C). In contrast, only 2.4% (2.4% ± 0.4%) of GD2–CD45– cells exhibited the CD44hiCD24lo phenotype (Figure 2C). Together these findings clearly indicated that GD2 is a marker of a subset of cancer cells with stem cell properties. To validate that the identified GD2+ cells are in fact tumor and not MSCs, we stained human breast tumor tissues with anti-GD2 and epithelial-specific anti–pan-cytokeratin antibodies. We found coexpression of GD2 and cytokeratin in some of the breast cancer cells, suggesting that GD2 identifies breast tumors cells (Supplemental Figure 5, A and B).
Table 3.
Breast cancer patient samples analyzed: patient number, tumor type, percentage of GD2+ cells
GD2+ and CD44hiCD24lo cells have similar gene signatures.
Since we found that GD2 is capable of independently enriching for CSCs as a single marker compared with the previously known double marker CD44hiCD24lo, we compared the global gene expression profiles in these two populations isolated from HMLER cells using microarray analysis. We initially compared the GD2+ fraction with the GD2– fraction of cells (GD2 set) and the CD44hiCD24lo with the CD4loCD24hi fraction (CD44 set) and identified gene signatures specific to the GD2+ and CD44hiCD24lo fractions (Figure 3A). Comparison of the top 600 differentially expressed genes in the GD2 set (GEO GSE36643) and the CD44 set (GSE36643) identified 231 genes as being identical in the two sets (Supplemental Table 1). In addition, we applied Pearson’s χ2 test with a Yates continuity correction to assess the association between these two cell types and found that the identified 231 genes correlated (100%) between the two groups described above (Figure 3B). This gene expression analysis along with the cell surface protein analysis shown in Figure 2 indicated that GD2+ cells share not only functional properties but also a gene signature with CD44hiCD24lo cells.
Figure 3. GD2+ and CD44hiCD24lo cells have a similar gene signature.

(A) Heat maps derived from microarray analysis of CD44hi/loCD24lo/hi and GD2+/– populations of HMLER cells. (B) Two hundred thirty-one genes of the top 600 differentially expressed genes were identical in GD2+ versus GD– and CD44hiCD24lo versus CD44loCD24hi groups. These genes were cross-classified in a 2-by-2 table by GD2+ up-/downregulation and CD44hiCD24lo up-/downregulation. Pearson’s χ2 test with a Yates continuity correction was applied to assess the association. Statistical significance was assessed at the 0.05 level. (C) Biosynthesis reaction of GD2. (D and E) To measure the expression of GD2S/GD3S mRNA, CD44hiCD24lo or CD44loCD24hi and GD2+/– cells from HMLER (D) or MDA-MB-231 cells (E) were FACS sorted, and mRNA was analyzed using qRT-PCR. *P < 0.001.
Among the genes differentially expressed between GD2+ and GD2– populations, GD3S, a key enzyme involved in the biosynthesis of GD3 (an intermediate for GD2, Figure 3C), was found to be upregulated approximately 9-fold in GD2+ compared to GD2– cells (Supplemental Table 2). The microarray data were validated by qRT-PCR (Figure 3D). However, expression of the gene encoding GD2S, which is involved in conversion of GD3 to GD2, was not altered (Figure 3D). Expression of a number of genes involved in migration and invasion, including MMPs (MMP2, MMP7, and MMP19), and EMT-associated markers, including N-cadherin and vimentin, were expressed at higher levels, whereas E-cadherin was expressed at low levels in GD2+ cells (Supplemental Table 2). We confirmed these findings by qRT-PCR (Supplemental Figure 6). In addition, CD44 mRNA was upregulated and CD24 mRNA downregulated in GD2+ relative to GD2– cells, which was confirmed by FACS analysis (Figure 2A). In addition, the stem cell marker nestin was also found to be upregulated in GD2+ cells compared with GD2– cells (Supplemental Figure 7). These and other genes that were differentially expressed in GD2+ versus GD2– cells are listed in Supplemental Table 2. Conversely, as in GD2+ versus GD2– cells, GD3S was overexpressed more than 10-fold in CD44hiCD24lo compared with CD4loCD24hi cells (Supplemental Figure 8), but no significant difference was found in the expression of GD2S between CD44hiCD24lo and CD4loCD24hi cells. This again demonstrates that the expression of GD3S and GD2 strongly correlates with the CD44hiCD24lo phenotype. Similar to HMLER cells, GD2+ cells from MDA-MB-231 cells expressed GD3S at a more than 5-fold-higher level than GD– cells, and consistent with our earlier finding, no significant differences in GD2S expression were observed (Figure 3E).
GD2– cells can spontaneously generate GD2+ cells.
Since we observed only a 2-fold difference in mammosphere formation and a 2- to 5-fold difference in tumor initiation between GD2+ and GD2– populations, we investigated whether this was due to the generation of GD2+ cells from GD2– cells. In fact, GD2+ and GD2– cells were sorted from HMLER (Figure 4A) and MDA-MB-231 cells (Figure 4B) and cultured in vitro for 12 days in their respective growth media. Surprisingly, approximately 10% of GD2+ HMLER cells had become GD2–, and 15% of GD2– cells had spontaneously generated GD2+ cells, and this proportion was almost identical to that in the unfractionated original HMLER cells (Figure 4A). Similarly, the GD2+ and GD2– cells from MDA-MB-231 cells also generated 81% (81% ± 2.5%) of GD2– and 12% of GD2+ cells, respectively, again reflecting the percentage of GD2+ cells within the parental MDA-MB-231 cell composition. To investigate the generation of GD2+ cells from GD2– cells and vice versa in vivo, GFP-labeled MDA-MB-231 cells were sorted into GD2+ and GD2– fractions, and 1 × 106 GD2+ and GD2– cells (GD2+/– cells) were subcutaneously transplanted into NOD/SCID mice. Four weeks later the tumors were dissected, and single-cell suspensions were prepared as described in Methods. The cells were then stained with anti-GD2 antibody and analyzed by flow cytometry. Tumors generated by GD2+ cells consisted of nearly 91% ± 4.5% GD2– cells, whereas 2.4% ± 1.1% of cells in GD2– derived tumors were positive for GD2. These findings indicate that GD2+ cells can spontaneously achieve a GD2– phenotype and vice versa in vivo (Supplemental Figure 9, A and B).
Figure 4. GD2-depleted cells are able to generate GD2+ cells in culture: a possible role of EMT.

(A and B) GD2+/– cells from HMLER (A) MDA-MB-231 (B) cells were FACS sorted and cultured in MEGM medium for 10 days. After incubation, the cells were stained with GD2 antibody (BD) and analyzed on an LSR II flow cytometer. Note the regeneration of GD+ cells in a GD2-depleted population. (C) To determine the possible role of EMT, HMLER cells transduced with two known EMT inducers (Twist and Snail) were stained with anti-GD2 antibody and analyzed on an LSR II flow cytometer. Expression of GD2 is shown on the y axis and FSC on the x axis. Lower panels in A and C represent antibody staining controls without primary antibody for each cell line. (D) Graphic representation of percentage of GD2+ vector control or Twist- or Snail-transduced HMLER cells. (E and F) mRNA expression analysis of GD3S (E) and GD2S (F) in vector- or Twist- or Snail-transduced cells was performed by real-time TaqMan qRT-PCR. *P < 0.001.
Induction of EMT generates GD2+ cells.
Since we recently reported that the induction of EMT in HMLER cells results in the acquisition of stem cell properties (12), we also examined the expression of GD2 on HMLER cells induced to undergo EMT by the ectopic expression of either Twist or Snail. Strikingly, we found that the induction of EMT by Snail or Twist resulted in a significant increase in the percentage of GD2+ populations from the initial 18% (control) to 40% in HMLER-Snail cells and 100% in HMLER-Twist cells (Figure 4, C and D). Corroborating our previous data suggesting a correlation between GD3S and CSCs, we also found that the expression of GD3S mRNA increased in the EMT-derived HMLER cells following induction of EMT by 2.5-fold in Snail cells and 8-fold in Twist cells (Figure 4E), which correlates with the total percentage of GD2+ cells in their respective population (40% in Snail and 100% in Twist cells). In contrast, we found no significant difference in the expression of GD2S (Figure 4F), supporting the hypothesis that GD3S is the key regulator in the biosynthesis of GD2.
GD3S is necessary for CSC properties.
To investigate the functional role of GD2 in CSCs, we suppressed the expression of GD3S, the critical enzyme involved in the biosynthesis of GD2, in MDA-MB-231 cells using a lentiviral-based shRNA expression vector and achieved more than 80% knockdown (Figure 5A). As expected, GD3S knockdown reduced the percentage of GD2+ cells from 12.3% (12.3% ± 1.7%) to 5.5% (5.5% ± 0.8%) in MDA-MB-231 cells (Figure 5, B and C). Since GD3S is known to regulate a-series gangliosides including GM3, we tested whether knockdown of GD3S could induce the expression of GM3 in MDA-MB-231 cells. Flow cytometric analysis revealed that expression of GM3 was increased from 0.4% ± 0.3% (control cells) to 15% ± 1.4% (in GD3S knockdown [GD3S-KD] cells), suggesting that knockdown of GD3S was efficient in these cells (Supplemental Figure 10, A and B). In addition, functional analysis revealed that GD3S-KD-MDA-MB-231 (GD3S-KD-MDA231) cells migrated approximately 3-fold less in Transwell Matrigel invasion assays (Figure 5D) and formed 3-fold fewer mammospheres compared with controls (Figure 5E). To further investigate the effects of suppression of GD3S on tumor formation, we subcutaneously injected MDA-MB-231cells expressing either control shRNA or the GD3S shRNA into the flank of NOD/SCID mice. Strikingly, even after 8 weeks, 1 × 106 GD3S shRNA cells had not formed tumors, whereas the control shRNA cells had formed tumors in 4 of 4 mice (Figure 5F). The growth rate (tumor size) was also dramatically altered, as plotted in Figure 5G.
Figure 5. Knockdown of GD3S reduces cell proliferation, mammosphere formation, and tumor initiation in MDA-MB-231 cells.

(A) To measure knockdown of GD3S, vector control, or GD3S-KD-MDA231 cells were analyzed for mRNA expression of GD3S by real-time TaqMan RT-PCR. Relative expression of GD3S is shown. *P < 0.0001. (B) To measure levels of GD2 on the cell surface, vector control or GD3S-KD-MDA231 cells were stained with anti-GD2 antibody and analyzed on an LSR II flow cytometer (BD). GD2 expression is shown on the y axis and FSC on the x axis. (C) Percentage of GD2+ cells in vector control and GD3S-KD-MDA231 cells. *P < 0.01. (D) To measure cell migration, vector control and GD3S-KD-MDA231 cells were cultured in the presence or absence of 30% serum in a Transwell migration chamber. The average number of cells per microscopic field is shown. *P < 0.001. (E) Mammosphere formation assay using either vector control or GD3S-KD-MDA231 cells was performed by seeding 1,000 cells per well in 24-well cell culture dishes containing mammosphere growth medium. After 10 days, the mammospheres were counted under a light microscope. Scale bar: 200 μm. Numbers of mammospheres formed from either vector control or GD3S-KD-MDA231 cells are shown. *P < 0.0001. (F) To examine tumor initiation potential, 1 × 106 vector control or GD3S-KD-MDA231 cells were transplanted subcutaneously into flanks of NOD/SCID mice. At the end of the ninth week, mice were shaved to remove excess hair on the tumors, and photographs were taken. (G) The tumors size was measured between 4 and 9 weeks. P < 0.000001.
Triptolide, a small molecule inhibitor, inhibits GD3S expression and CSC properties.
Triptolide, a small molecule anti-inflammatory drug, has been shown to inhibit GD3S in a melanoma cancer cell line (25). Therefore, we investigated whether triptolide could inhibit GD3S in breast cancer cell lines as well. MDA-MB-231 and SUM-159 cells were treated with different concentrations of triptolide for 24 hours. Triptolide inhibited GD3S mRNA expression in both cell types in a dose-dependent manner, with greater than 95% inhibition at 125 nM (Figure 6, A and B). To test whether inhibition of GD3S by triptolide also inhibited GD2 expression, we treated MDA-MB-231 cells with different concentrations of triptolide for either 24 or 48 hours. Absolute cell counts were measured using flow cytometry. A dose- and time-dependent decrease in GD2+ cells was observed after triptolide treatment, indicating the successful inhibition of GD3S by triptolide (Figure 6C). Of note, a decrease in GD2+ cells was seen under conditions that induced apoptosis in less than 5% of cells.
Figure 6. Triptolide inhibits the expression of GD3S, induces apoptosis in MDA-MB-231 cells, and blocks tumor growth in NOD/SCID mice.

MDA-MB-231 cells (A) or SUM-159 cells (B) (5 × 105/well) of 6-well cell culture dishes were treated with 25, 50, 75, 100, or 125 nM triptolide for 24 hours. –/–, no treatment. Total RNA was extracted, and GD3S expression was measured by qRT-PCR. (C and D) To measure GD2+ cell growth inhibition, 5 × 105 MDA-MB-231 (C) and SUM-159 (D) cells were plated in each well of 6-well cell culture dishes and treated with 25, 50, 75, 100, or 125 nM triptolide for 24 or 48 hours. After incubation, the cells were detached with trypsin and stained with anti-GD2 antibody and Sytox Red (for dead cells; Invitrogen). The stained cells were analyzed on an LSR II flow cytometer. Absolute numbers of live cells were calculated by measuring 1,000 events for Trucount beads as explained in Methods. (E) To determine the inhibition of tumor growth, 1 × 106 MDA-MB-231 cells were subcutaneously transplanted into NOD/SCID mice (n = 8; 4 mice/group). A group of the mice were treated with 0.15 mg/kg/d triptolide, and the control group was treated with PBS every day by i.p. injection. At the end of 8 weeks, mice were sacrificed, and tumors were dissected out and photographed. (F) Tumor sizes from the mice in experiment in E were measured every week after tumor engraftment, and the measurements are shown. P < 0.001, week 3. (G) The survival analysis was based on Kaplan-Meier estimation, and groups were compared by the log-rank test. Control (n = 4, black line) and triptolide (n = 4, blue line) were analyzed for cumulative survival. Survival was defined as the time (in weeks) from transplantation until death. P = 0.015.
To further examine whether triptolide could also inhibit tumor growth in vivo, we introduced 1 × 106 MDA-MB-231 cells subcutaneously into NOD/SCID mice (2 injections per mouse and 4 mice per group). After the tumors reached 50 mm3, we randomly divided the mice into two groups and treated half of the mice with triptolide (0.15 mg/kg/d) and the other half with PBS (control mice) every day by i.p. injection. Interestingly, after 4 weeks, triptolide-treated animals showed a dramatic decrease in tumor growth compared with control mice. Fifty percent of triptolide treated breast tumors were completely tumor free, and there was a more than 8-fold reduction in tumor volume in 25% of mice (Figure 6D). In addition, tumors in triptolide-treated mice were 3-fold smaller in size and 4-fold lighter by weight (Figure 6E and Supplemental Figure 11). Moreover, in a repeat, identical experiment, triptolide significantly prolonged survival of the treated mice (log-rank, control vs. triptolide, P = 0.0015) (Figure 6F). These findings indicate that GD3S plays a major role in regulating GD3S expression and the resulting GD2+ population. Specifically, it affects cell proliferation and tumor initiation of GD2+ breast cancer cells and when inhibited, greatly diminishes tumor growth and increases metastasis-free survival of breast cancer–bearing mice.
Discussion
In this study, we identified the ganglioside GD2 as a marker for breast CSCs. Specifically, GD2+ cells displayed stem cell properties similar to those of the CD44hiCD24lo population. GD2 was found to be expressed predominantly in the CD44hiCD24lo fraction isolated from breast cancer cell lines and primary patient breast cancer cells. In addition, the gene expression signature derived from GD2+ cells correlated strikingly with that obtained from CD44hiCD24lo cells. Moreover, inhibition of GD2 production using either shRNA against GD3S or a small molecule inhibitor of GD3S (triptolide) resulted in significant reduction in the number of total GD2+ cells as well as in CSC-associated properties. Collectively, our results indicate that GD2 can be used in place of the CD44hiCD24lo phenotype for the purification of CSCs and, unlike that phenotype, can be targeted with small molecule or shRNA inhibitors of its regulator GD3S.
Of note, most of the basal breast cancer cell lines used in this study displayed a higher percentage of GD2+ cells than the luminal-derived breast cancer cell lines. This observation is in line with the clinical observation of the more aggressive phenotype of basal-like versus luminal breast cancer (23). CD44, apart from being a mesenchymal marker, also serves as a prognostic marker for metastatic breast cancer (26, 27). Microarray analysis of GD2+ cells showed that mesenchymal markers including N-cadherin, vimentin, and fibronectin and the stem cell marker nestin were highly expressed in these cells; in addition, epithelial markers including E-cadherin and CD24 were downregulated, supporting the mesenchymal nature of these cells. In addition, the observation that GD2 expression correlated with CD44 expression, that GD2+ cells appeared mesenchymal, and that GD3S expression was induced during EMT indicates that the GD2/GD3S pathway could be used to diagnose and treat tumors that are CSC enriched and contain cells that have undergone EMT.
The identification of GD2 as a marker for breast CSCs is based on our recent work linking EMT-derived breast epithelial cells with bone marrow–derived multipotent MSCs (13). In support of this notion, Weissman’s group recently demonstrated that CD271, another bone marrow–derived MSC marker, is also highly expressed on melanoma-initiating cells (28), suggesting that a number of cell surface markers expressed on MSCs may also be shared by CSCs from different tumor types. Very recently, Chang et al. reported that glycol-sphingolipids including Globo-H and SSEA-3 were highly expressed in breast CSCs (29), suggesting the predominant expression of glyco-sphingolipids in breast CSCs.
Induction of EMT in HMLER cells using either Snail or Twist resulted in mesenchymal appearance as well as in increased expression of GD2. Conversely, GD2+ cells isolated from HMLER cells displayed mesenchymal morphology. This suggests that both EMT-derived and preexisting CSCs express GD2 and that the inhibition of GD2 production by shRNA against GD3S or by triptolide may inhibit CSCs independent of the mechanism of this inhibition. While we focused on the properties of GD2 function, we have not explored the role of GD2 during EMT. For example, it is not clear, whether inhibiting GD2 production will also inhibit EMT.
Tumor cells, when cultured in vitro, tend to maintain a small percentage of GD2+ cells. Moreover, fractionated GD2– cells generate GD2+ cells when cultured in vitro. This once again indicates that cancer cells acquire stem cell capacity through dedifferentiation from differentiated cancer cells probably via EMT. Recently, Weinberg’s group reported that FACS-sorted CD44loCD24hi transformed HMLE cells could generate CD44hiCD24lo cells and vice versa, suggesting a spontaneous generation of stem-like cells from more differentiated cells (30). The finding that spontaneous generation of GD2+ cells from GD2– cells of HMLER or MDA-MB-231 cells is in line with recent reports that cancer cells acquire stem cell capacity through dedifferentiation from differentiated cancer cells and vice versa (31).
We noted that GD3S, but not GD2S, correlates with GD2 expression even though GD2S is the immediate enzyme responsible for the conversion of GD3 to GD2. Further analysis indicates that only GD3S and not GD2S mRNA expression was regulated during EMT. Moreover, Dae et al. found that ZEB1, a known EMT-inducing transcription factor, directly binds to the GD3S promoter and induces transcription of GD3S in glioblastoma cells (32). These findings suggest that the EMT-inducing factors such as ZEB1 might bind to the GD3S promoter and upregulate its expression in CSCs as well as in EMT-derived cancer cells, which then generate GD3, due to the high basal expression of GD2S in these cells, which probably immediately converts to GD2 by ubiquitously expressed GD2S.
The functional role of GD2 in CSCs is not clear. Current evidence suggests that gangliosides are widely expressed on tumor cells (19, 33), and these gangliosides may help tumor cells escape from immune cells within the tumor microenvironment (34). This idea has been supported by several recent reports that gangliosides including GD1a and GD1b, GD3, and GM3 help tumors evade immune attack. It has been shown that gangliosides induce apoptosis in immune cells, including T and NK cells (35, 36). Siglec-7 (sialic acid–binding immunoglobulin-like lectin), an inhibitory receptor, on NK cells was specifically shown to bind to the b-series of gangliosides including GD3 and GD2 and that cells that were engineered to overexpress the b-series of gangliosides suppress NK cell–mediated cytotoxicity in a Siglec-7–dependent manner (37, 38). Considering the fact that metastatic cancer cells have to travel long distances to find a suitable microenvironment to initiate secondary tumors and that these cells have to overcome a complex immune system in order to reach their destination, the immunosuppressive function of GD2 may be critical for their metastatic ability. We are in the process of further investigating the role of GD2 in immune evasion.
GD2 is expressed not only in breast cancer but also in other cancers including melanomas, gliomas, and neuroblastomas (18, 19, 39). Therapeutic effects of anti-GD2 monoclonal antibodies have been reported in neuroblastomas (40, 41). Therefore, GD2 could constitute a potential marker of CSCs in other tumor types, in addition to breast cancer, and may be further developed as a therapeutic target. Collectively, our findings suggest that GD2 and GD3S could identify CSCs in breast cancer, and that inhibition of GD2 biosynthesis by targeting GD3S may serve as a novel means to inhibit CSC-associated tumor growth, chemotherapy resistance and tumor metastasis.
Methods
Antibodies.
The following antibodies were used: anti-GD2 (clone 14G2a, isotype IgG2a), anti-GD3 (clone R24, isotype IgG3), as well as isotype control antibodies for IgG2a and IgG3 from BD. Anti-GD2 antibody (clone 2Q549) was purchased from Abcam. Anti-GM3 (isotype IgMk) antibody was purchased from Cosmo Bio USA. Antibody conjugates anti-CD44 (allophycocyanin [APC]), anti-CD105 (phycoerythrin [PE]), anti-CD106–PE, and anti-CD117–PE were from eBiosciences; anti-CD166–PE, anti-CD73–PE, anti-CD140b–PE, anti-CD90 (FITC), and anti-CD24–FITC were from BD. Anti-CD271–APC antibody was from Miltenyi Biotec.
Cell culture.
HMECs (also known as HMLE cells) expressing H-Ras oncogene (HMLER) were cultured as described previously (13). In brief, HMECs obtained from Clonetics were immortalized with the catalytic subunit of human telomerase and SV40 large T antigen. These cells were transduced with pBabe-puro retroviral vectors expressing Twist, Snail, or empty vector. Human breast cancer cell lines MDA-MB-231, MCF7, MDA-MB-453, SKBR3, BT474, ZR751, MDA-MB-361, B-20, MDA-MB-468, HS578T, SUM-159, and SUM-149 were cultured according to ATCC recommendations.
Patient samples.
Primary breast tumors were obtained from patients undergoing mammectomy. Following institutional protocols and guidelines, tumors were transferred to the laboratory immediately after surgery, where they were cut into approximately 2-mm3 pieces. The tumor tissues were washed once with PBS and then incubated with a combination of collagenase III (Sigma-Aldrich) and hyaluronidase (Sigma-Aldrich) for 8–10 hours with constant shaking at 37°C. After incubation, the cells were washed once with PBS containing 10% serum to stop any residual enzyme activity. The resulting cell pellet was resuspended in PBS and processed with Ficoll density gradient centrifugation to eliminate red blood cells and undigested tissue compartments. The resulting buffy coat was then collected and used for antibody staining.
Flow cytometry.
Single-color staining of HMLER cells was performed as described previously (24). Briefly, cells were washed twice with PBS containing 1% FBS (FACS buffer), incubated with the indicated primary antibodies for 15 minutes on ice, washed in FACS buffer, and incubated with the F(ab)2 fragment of goat anti-mouse secondary antibody conjugated with R-PE (Dako) for 15 minutes. After washing, the cells were analyzed with an LSR II flow cytometer (BD) using FCS-Express (De Novo Software) or FlowJo software (Tree Star Inc.). For double or triple fluorescence staining of HMLER and primary breast tumors, cells were first stained with GD2, GD3, GM3, IgG2a, IgG3, and IgM isotype control antibodies (BD) as described above. Next, free binding sites of the secondary antibody were blocked with mouse IgG polyclonal antibody (SouthernBiotech), and cells were diluted 1:20 with PBS or FACS buffer for 25 minutes on ice, stained with the indicated antibody conjugates, and (after washing) analyzed with an LSR II flow cytometer. For flow cytometric analysis of cell lines and primary tumor cells, approximately 1 × 106 cells were stained with primary antibodies or antibody conjugates as described above. At least 1 × 104 events were measured in cell lines and 1 × 105 cells from primary breast tumors. Sorting of single cells or bulk populations was performed on a FACSAria II cell sorter (BD).
Immunohistochemistry.
After deparaffinization, the tissue sections (4 μm) were retrieved by being boiled in retrieval buffer (Dako) for 20 minutes. After washing, tissue sections were incubated in protein blocking buffer for 15 minutes (Protein Block Serum-Free, Dako) at room temperature and then incubated with anti-GD2 antibody (BD, IgG2a, 1:100 dilution, v/v) and anti–pan-cytokeratin antibody (ab6401, IgG1, Abcam, 1:300 dilution, v/v) at 4°C overnight in a humidified chamber. After washing, sections were incubated with fluorochrome-conjugated secondary antibodies (anti-mouse IgG2a conjugated with Alexa Fluor 488 and anti-mouse IgG1 conjugated with Alexa Fluor 594, both from Invitrogen). The nuclei were stained with DAPI. IgG2a, k antibody (1:100 dilution, BD Biosciences — Pharmingen) was used for isotype control. Following staining, the sections were photographed using an Olympus FV1000 laser confocal microscope.
Viral transduction.
Lentiviral plasmids containing GD3S shRNA (PLKO.1,RHS3979-9603453) or an empty vector were purchased from Open Biosystems. Viral supernatants were generated as described before (42) using a viral packaging system that includes psPAX2 and pM2DG plasmids (provided by Boyko S. Atanassov, MD Anderson Cancer Center). Two days after transfections, the viral supernatants were collected and used for viral transduction.
Mammosphere assay.
GD2+/– cells from HMLER or MDA-MB-231 cells were FACS sorted after staining with anti-GD2 antibody (BD). To measure the mammosphere formation potential of the sorted cells (GD2+/– of HMLER or MDA-MB-231), 1 × 103 cells were plated in ultra-low-attachment 24-well dishes containing mammosphere growth medium (MEGM with all required growth factors as described above for the growth of HMLER cells plus B-27 supplements, without bovine pituitary extract (BPE]) from Lonza. After 12 days of culture at 37°C, the resulting mammospheres were counted. To generate mammospheres from single cells, GD2+/– cells from MDA-MB-231 cells were directly FACS sorted into a low-attachment 96-well cell plate containing mammosphere growth medium. After 12 days of culture at 37°C, the resulting mammospheres were counted.
Microarray analysis.
GD2+/– cells of HMLER cells were cell sorted, and total RNA was extracted using the QIAGEN Miniprep kit. Total RNA was analyzed on an Affymetrix human genome chip at the MD Anderson DNA core facility. The experiment was performed in triplicate. Statistical analysis of the Affymetrix CEL data was performed using the Bioconductor (43) package in the R (2009 release) statistical software environment. To assess differential expression, the intensity values in the CEL files were transformed to robust multichip average (RMA) expression measures (44). The RMA measure is reported in a log2 scale. Using the Limma R package (45), we fit linear models with the lmFit function to assess differential expression between two RNA sources: GD2+ versus GD2– and CD44hiCD24lo versus CD44loCD24hi cells. To obtain empirical Bayes test statistics for each gene, including moderated t statistics and log-odds of differential expression, the fitted model object was further analyzed using the eBayes function. Adjustment for multiple testing was based on controlling the false discovery rate (the expected proportion of false discoveries among the rejected hypotheses) by the method of Benjamini and Hochberg (46). Results are reported for the top 600 differentially expressed genes for each of the comparisons.
To assess the degree of agreement between GD2+ and CD44hiCD24lo groups with respect to their differential expression regulation (GD2+ vs. GD– and CD44hiCD24lo vs. CD44loCD24hi), we identified 231 genes between the two sets of top 600 differentially expressed genes. These genes were cross-classified in a 2-by-2 table by GD2+ up-/downregulation and CD44hiCD24lo up-/downregulation. Pearson’s χ2 test with a Yates continuity correction was applied to assess the association. Statistical significance was assessed at the 0.05 level.
In vivo tumor initiation assay.
GD2+/– cells from MDA-MB-231 cells were FACS sorted using FACSAria II cell sorter (BD). GD2+/– cells were transplanted subcutaneously into NOD/SCID mice at concentrations of 10,000, 1,000, and 100 cells per site. Two injection sites were used per mouse, and 3–7 mice were used in each group. After 10–12 weeks the tumors were identified by palpation. In a similar experiment, GD3S-knockdown or empty vector control MDA-MB-231 cells were subcutaneously transplanted (1 × 106 per site) into NOD/SCID mice to for analysis of the tumor initiation potential. Four mice were used in this experiment, with GD3S-KD-MDA231 cells transplanted on the dorsal right and vector control cells on the dorsal left. Mice were killed after tumors reached a diameter of 1.5 cm, in accordance with institutional guidelines. To determine the generation of GD– cells from GD2+ cells and vice versa in vivo, 1 × 106 GD2+ (Supplemental Figure 6A) or GD2– (Supplemental Figure 6B) GFP+ MDA-MB-231 cells were transplanted subcutaneously into NOD/SCID mice. The tumors were dissected 4 weeks after transplantation, minced, and digested into single-cell suspensions using collagenase III and hyaluronidase (both from Sigma-Aldrich). The cell suspension was analyzed using an LSR II flow cytometer.
qRT-PCR using TaqMan assays.
To analyze the mRNA expression of GD3S and GD2S, total RNA was extracted from CD44hi/loCD24lo/hi HMLER cells and GD2+/– HMLER cells. Real-time RT-PCR was performed using TaqMan gene expression assays from Applied Biosystems as described previously (47). The assay for GD3S was Hs00268157; the assay for GD2S was B4GALNT1; and the assay for nestin was Hs00707120_s1. qRT-PCR for E-cadherin, N-cadherin, vimentin, Snail, and Twist was performed as described before (13).
In vitro Matrigel invasion assay.
An in vitro Matrigel invasion assay was performed using 24-well Biocoat Matrigel Invasion Chambers containing BD Falcon cell culture inserts with a PET membrane (8-μm-diameter pore size) that had been treated with Matrigel Matrix (BD Biosciences) as described previously (13). Briefly, MDA-MB-231 cells that were knocked down for GD3S or empty vector control cells were serum starved for 24 hours before the assay to avoid any receptor-blocking factors from serum. Cell suspensions of MDA231-vector control or MDA231-GD3S-KD were adjusted to a concentration of 15 × 104 cells/ml, and 200 μl of the adjusted cell suspension (3 × 104 cells/insert) was immediately placed in the Matrigel-coated upper chamber. After incubation at 37°C for 24 hours in a 5% CO2 incubator, the residual cells on the upper surface of the filter were completely removed with cotton swabs. The membranes were then stained, and cell counting was performed as described before (13). Each assay was performed in triplicate in 3 separate experiments.
Triptolide treatments.
For mRNA analysis 5 × 105 MDA-MB-231 or SUM-159 breast cancer cells were incubated with triptolide (Alexis Biochemicals) at concentrations of 25, 50, 75, 100, and 125 nM for 24 hours at 37°C in 6-well cell culture dishes. For the growth inhibition studies, 3 × 105 MDA-MB-231 cells cultured in 6-well tissue culture dishes were treated with triptolide at specified concentrations for either 24 or 48 hours. After incubation the cells were detached using trypsin and stained with anti-GD2 antibody (by indirect staining protocol) and Pacific blue–conjugated Annexin V (Invitrogen) following the manufacturer’s instructions. The cells were analyzed in an LSR II flow cytometer. The absolute number of cells was determined by measuring Trucount counting beads (Invitrogen) as per the manufacturer’s instructions. To examine tumor growth inhibition, survival of murine tumor xenografts was determined by subcutaneous administration of 1 × 106 MDA-MB-231 cells into 8- to 10-week-old NOD/SCID mice. After growing for 2 weeks, tumor xenografts reached a size of approximately 50 mm3. Thereafter, triptolide (0.15 mg/kg/d) was administered i.p. into the mice on a daily basis. The mice were sacrificed when the control tumors reached 2 cm or larger according to institutional guidelines.
Statistics.
Unless otherwise indicated, data are mean ± SEM. Statistical significance of tumor growth was determined by 2-way ANOVA for repeated measures. All other group differences were evaluated by 2-tailed unpaired Student’s t test. Survival data were analyzed using Kaplan-Meier log-rank tests. P values less than 0.05 were considered significant.
Study approval.
Animal protocols were approved by the Animal Care and Use Committee of the MD Anderson Cancer Center. Patient tumors were obtained following written informed consent in accordance with tissue procurement protocols approved by the Institution Review Board of the MD Anderson Cancer Center.
Supplementary Material
Acknowledgments
We thank the Breast Tumor Bank at MD Anderson Cancer Center for providing tumor samples and Naoto T. Ueno (MD Anderson Cancer Center) for providing some of the breast cancer cell lines used. S.A. Mani’s laboratory is supported by an MD Anderson Research Trust Fellow Award. The work was supported in part by grants from the NIH (CA-55164, CA-16672, and CA116199) and the Paul and Mary Haas Chair in Genetics (to M. Andreeff).
Footnotes
Conflict of interest: The authors are inventors of a patent application based on the work described here.
Citation for this article:J Clin Invest. 2012;122(6):2066–2078. doi:10.1172/JCI59735.
References
- 1.Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324(5935):1670–1673. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schatton T, Frank NY, Frank MH. Identification and targeting of cancer stem cells. Bioessays. 2009;31(10):1038–1049. doi: 10.1002/bies.200900058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 4.Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007;317(5836):337. doi: 10.1126/science.1142596. [DOI] [PubMed] [Google Scholar]
- 5.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10(2):R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertolini G, et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A. 2009;106(38):16281–16286. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponti D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65(13):5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 9.Liu R, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356(3):217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 10.Sheridan C, et al. CD44+/CD24– breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006;8(5):R59. doi: 10.1186/bcr1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bourguignon LY, Singleton PA, Zhu H, Zhou B. Hyaluronan promotes signaling interaction between CD44 and the transforming growth factor beta receptor I in metastatic breast tumor cells. J Biol Chem. 2002;277(42):39703–39712. doi: 10.1074/jbc.M204320200. [DOI] [PubMed] [Google Scholar]
- 12.Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Battula VL, et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells. 2010;28(8):1435–1445. doi: 10.1002/stem.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svennerholm L. Chromatographic separation of human brain gangliosides. J Neurochem. 1963;10:613–623. doi: 10.1111/j.1471-4159.1963.tb08933.x. [DOI] [PubMed] [Google Scholar]
- 15.Martinez C, Hofmann TJ, Marino R, Dominici M, Horwitz EM. Human bone marrow mesenchymal stromal cells express the neural ganglioside GD2: a novel surface marker for the identification of MSCs. Blood. 2007;109(10):4245–4248. doi: 10.1182/blood-2006-08-039347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Posse de Chaves E, Sipione S. Sphingolipids and gangliosides of the nervous system in membrane function and dysfunction. FEBS Lett. 2009;584(9):1748–1759. doi: 10.1016/j.febslet.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 17.Sonnino S, Prinetti A. Gangliosides as regulators of cell membrane organization and functions. Adv Exp Med Biol. 2010;688:165–184. doi: 10.1007/978-1-4419-6741-1_12. [DOI] [PubMed] [Google Scholar]
- 18.Lloyd KO, Old LJ. Human monoclonal antibodies to glycolipids and other carbohydrate antigens: dissection of the humoral immune response in cancer patients. Cancer Res. 1989;49(13):3445–3451. [PubMed] [Google Scholar]
- 19.Hakomori S. Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res. 1996;56(23):5309–5318. [PubMed] [Google Scholar]
- 20.Fishman PH, Brady RO. Biosynthesis and function of gangliosides. Science. 1976;194(4268):906–915. doi: 10.1126/science.185697. [DOI] [PubMed] [Google Scholar]
- 21.Elenbaas B, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15(1):50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neve RM, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10(6):515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorlie T, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Battula VL, et al. Isolation of functionally distinct mesenchymal stem cell subsets using antibodies against CD56, CD271, and mesenchymal stem cell antigen-1. Haematologica. 2009;94(2):173–184. doi: 10.3324/haematol.13740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kwon HY, et al. Triptolide downregulates human GD3 synthase (hST8Sia I) gene expression in SK-MEL-2 human melanoma cells. Exp Mol Med. 2010;42(12):849–855. doi: 10.3858/emm.2010.42.12.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orian-Rousseau V. CD44, a therapeutic target for metastasising tumours. Eur J Cancer. 2010;46(7):1271–1277. doi: 10.1016/j.ejca.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 27.Seiter S, et al. Prevention of tumor metastasis formation by anti-variant CD44. J Exp Med. 1993;177(2):443–455. doi: 10.1084/jem.177.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boiko AD, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466(7302):133–137. doi: 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang WW, et al. Expression of Globo H and SSEA3 in breast cancer stem cells and the involvement of fucosyl transferases 1 and 2 in Globo H synthesis. Proc Natl Acad Sci U S A. 2008;105(33):11667–11672. doi: 10.1073/pnas.0804979105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaffer CL, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108(19):7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quintana E, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010;18(5):510–523. doi: 10.1016/j.ccr.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dae HM, et al. Isolation and functional analysis of the human glioblastoma-specific promoter region of the human GD3 synthase (hST8Sia I) gene. Acta Biochim Biophys Sin (Shanghai). 2009;41(3):237–245. doi: 10.1093/abbs/gmp007. [DOI] [PubMed] [Google Scholar]
- 33.Hakomori S. Tumor-associated carbohydrate antigens defining tumor malignancy: basis for development of anti-cancer vaccines. Adv Exp Med Biol. 2001;491:369–402. doi: 10.1007/978-1-4615-1267-7_24. [DOI] [PubMed] [Google Scholar]
- 34.Potapenko M, Shurin GV, de Leon J. Gangliosides as immunomodulators. Adv Exp Med Biol. 2007;601:195–203. doi: 10.1007/978-0-387-72005-0_20. [DOI] [PubMed] [Google Scholar]
- 35.Biswas K, et al. GM2 expression in renal cell carcinoma: potential role in tumor-induced T-cell dysfunction. Cancer Res. 2006;66(13):6816–6825. doi: 10.1158/0008-5472.CAN-06-0250. [DOI] [PubMed] [Google Scholar]
- 36.Sa G, et al. GD3, an overexpressed tumor-derived ganglioside, mediates the apoptosis of activated but not resting T cells. Cancer Res. 2009;69(7):3095–3104. doi: 10.1158/0008-5472.CAN-08-3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Avril T, Floyd H, Lopez F, Vivier E, Crocker PR. The membrane-proximal immunoreceptor tyrosine-based inhibitory motif is critical for the inhibitory signaling mediated by Siglecs-7 and -9, CD33-related Siglecs expressed on human monocytes and NK cells. J Immunol. 2004;173(11):6841–6849. doi: 10.4049/jimmunol.173.11.6841. [DOI] [PubMed] [Google Scholar]
- 38.Ikehara Y, Ikehara SK, Paulson JC. Negative regulation of T cell receptor signaling by Siglec-7 (p70/AIRM) and Siglec-9. J Biol Chem. 2004;279(41):43117–43125. doi: 10.1074/jbc.M403538200. [DOI] [PubMed] [Google Scholar]
- 39.Schulz G, Cheresh DA, Varki NM, Yu A, Staffileno LK, Reisfeld RA. Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res. 1984;44(12 pt 1):5914–5920. [PubMed] [Google Scholar]
- 40.Cheung NK, et al. Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. J Clin Oncol. 1987;5(9):1430–1440. doi: 10.1200/JCO.1987.5.9.1430. [DOI] [PubMed] [Google Scholar]
- 41.Kushner BH, Kramer K, Modak S, Cheung NK. Successful multifold dose escalation of anti-GD2 monoclonal antibody 3F8 in patients with neuroblastoma: a phase I study. J Clin Oncol. 2011;29(9):1168–1174. doi: 10.1200/JCO.2010.28.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rey O, Young SH, Jacamo R, Moyer MP, Rozengurt E. Extracellular calcium sensing receptor stimulation in human colonic epithelial cells induces intracellular calcium oscillations and proliferation inhibition. J Cell Physiol. 2010;225(1):73–83. doi: 10.1002/jcp.22198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Irizarry RA, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 45.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 46.Klipper-Aurbach Y, et al. Mathematical formulae for the prediction of the residual beta cell function during the first two years of disease in children and adolescents with insulin-dependent diabetes mellitus. Med Hypotheses. 1995;45(5):486–490. doi: 10.1016/0306-9877(95)90228-7. [DOI] [PubMed] [Google Scholar]
- 47.Konopleva M, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10(5):375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.