

Abstract
Hepatocellular carcinoma (HCC) is one of the most common and aggressive human cancers worldwide. HCC is an example of inflammation-related cancer and represents a paradigm of the relation occurring between tumor microenvironment and tumor development. Tumor-associated macrophages (TAMs) are a major component of leukocyte infiltrate of tumors and play a pivotal role in tumor progression of inflammation-related cancer, including HCC. Several studies indicate that, in the tumor microenvironment, TAMs acquire an M2-polarized phenotype and promote angiogenesis, metastasis, and suppression of adaptive immunity through the expression of cytokines, chemokines, growth factors, and matrix metalloproteases. Indeed, an established M2 macrophage population has been associated with poor prognosis in HCC. The molecular links that connect cancer cells and TAMs are not completely known, but recent studies have demonstrated that NF-κB, STAT-3, and HIF-1 signaling pathways play key roles in this crosstalk. In this paper, we discuss the current knowledge about the role of TAMs in HCC development, highlighting the role of TAM-derived cytokines, chemokines, and growth factors in the initiation and progression of liver cancer and outlining the signaling pathways involved in the interplay between cancer cells and TAMs.
1. Introduction
Hepatocellular carcinoma (HCC) is one of the most aggressive human cancers and the third leading cause of death worldwide [1]. Despite the recent advance in diagnosis and treatment of HCC, it remains a highly lethal disease due to recurrence of metastasis [2].
HCC is an example of inflammation-related cancer, as the chronic inflammatory state appears to be necessary for the initiation and development of liver cancer. Several studies have shown that chronic infections with hepatitis viruses (hepatitis B virus, HBV and hepatitis C virus, HCV) are major risk factors for HCC development. Other risk factors to liver carcinogenesis include chronic alcohol abuse, biliary disease, metabolic disorders, drugs, toxins, and genetic conditions, such as hereditary hemochromatosis and 1-antitrypsin deficiency [3].
The chronic inflammation is characterized by the continued expression of cytokines and recruitment of immune cells to the liver. Activated inflammatory cells release free radicals, such as reactive oxygen species (ROS) and nitric oxide (NO) reactive species, which in turn can cause DNA damage and lead to gene mutations, thus fostering neoplastic transformation. In fact, hepatic oxidative stress is also strongly associated with increased risk for HCC in patients with chronic HBV and HCV infections [3, 4]. The chronic inflammation also affects many cellular pathways, leading to fibrosis and cirrhosis and finally hepatocarcinogenesis. Liver injury induces tissue repair and liver regeneration, which involve deregulated growth and death of hepatocytes. High cell turnover induces several critical alterations for malignant transformation, including structural and/or functional modifications of proteins involved in cell-cycle control, apoptosis, oxidative stress, lipid peroxidation and DNA repair damage [5, 6]. Moreover, Tumor-Necrosis-Factor-alpha (TNF-α)-induced Nuclear Factor kappa B (NF-κB) activation plays a key role in hepatocarcinogenesis [7, 8].
It is therefore clear that the crosstalk between tumor cells and their surrounding microenvironment is required for HCC development. The tumor microenvironment plays critical roles in modulating the process of liver fibrosis, hepatocarcinogenesis, epithelial-mesenchymal transition (EMT), tumor invasion, and metastasis. HCC microenvironment consists of (a) stromal cells, such as carcinoma-associated fibroblasts (CAFs), hepatic stellate cells (HSCs), endothelial cells and immune cells, (b) growth factors and inflammatory cytokines, and (c) extracellular matrix proteins [1]. Tumor-associated macrophages (TAMs) are a major component of leukocyte infiltrate and play a crucial role in this scenario, by producing signal molecules, which promote and sustain tumor progression (Figure 1). This review focuses on the activities that TAMs exert in HCC, paying close attention to cytokines, chemokines, and growth factors secreted by TAMs, as well as to intracellular signaling pathways that play pivotal roles in regulating TAM functions and TAM-cancer cell crosstalk. A better understanding of the molecular events underlining the relationship between cancer cells and TAMs may be useful for the discovery of novel therapeutic targets.
Figure 1.
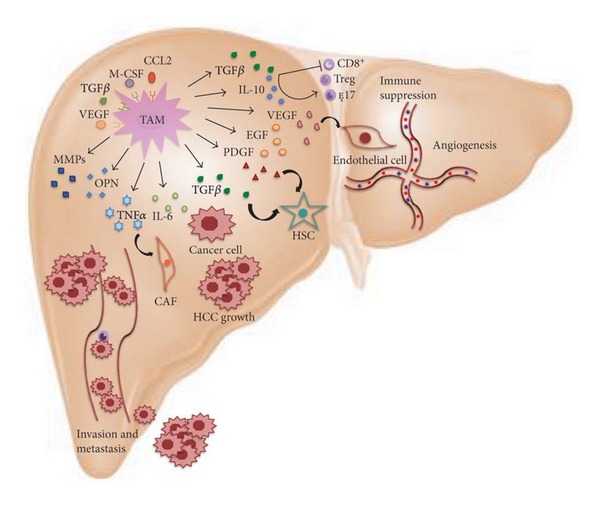
The roles of tumor-associated macrophages (TAMs) in liver cancer. TAMs promote hepatocellular carcinoma (HCC) growth, angiogenesis, invasion, and metastasis, as well as the suppression of antitumor immune response by interacting with both stromal and cancer cells within the tumor microenvironment. TAMs are recruited in HCC milieu by M-CSF, CCL2, VEGF, and TGFβ, and they, in turn, release many cytokines, chemokines and growth factors, which are implicated in such crosstalk. In particular, IL-6 and TGFβ favor tumor growth, whereas TNFα, OPN, MMPs, and IL-6 are involved in invasion and metastasis; TGFβ, in concert with IL-10, promotes the suppression of antitumor immune response. Finally, angiogenesis is induced by several molecules, including VEGF, EGF, PDGF, and TGFβ. Refer to the text for abbreviations.
2. TAMs and HCC
Among the immune cell types present within the HCC, TAMs play a leading role in the setting of the crosstalk between tumor and stromal cells [9]. Indeed, TAMs are key actors of cancer-related inflammation, representing the main type of inflammatory cells infiltrating tumors [10]. TAMs originate from circulating monocytic precursors, which are recruited within the tumor microenvironment by tumor-derived signals, including Chemokine (C-C motif) Ligand 2 (CCL2) and Macrophage Colony-Stimulating Factor (M-CSF), and differentiate into mature macrophages. Thanks to their plasticity, macrophages are able to exert both anti- and protumor activities through the expression of different functional programs in response to distinct microenvironmental signals [11]. Accordingly with the phenotypic polarization, macrophage state of activation can be classified as M1 (or classical activated) or M2 (or alternatively activated) (Figure 2) [12, 13]. Macrophages mount M1 phenotype in response to microbial stimuli and Interferon gamma (INF-γ): classical activation of macrophages is characterized by high capability to present antigen and high expression of Interleukin 12 (IL-12) and other proinflammatory cytokines, thus being able to trigger T helper 1 (Th1) immune response [14, 15]. They have cytotoxic activity towards ingested microorganisms and cancer cells, by producing high amounts of toxic intermediates, such as NO and ROS [14, 15]. On the other hand, when monocytes are exposed to Interleukin 4 (IL-4), Interleukin 13 (IL-13), Interleukin 10 (IL-10), glucocorticoids, and immune complexes/Toll-like Receptor (TLR) ligands, they polarize towards M2 phenotype, characterized by poor antigen presenting capability, expression of a distinctive set of cytokines and chemokines, such as IL-10, Transforming Growth Factor β (TGF-β) Chemokine (C-C motif) Ligand 17 (CCL17), Chemokine (C-C motif) Ligand 22 (CCL22), and Chemokine (C-C motif) Ligand 24 (CCL24). M2 macrophages activate T helper 2 (Th2) immune response and promote angiogenesis, tissue remodeling and repair [10]. Further, M2 macrophages express specific changes in some metabolic pathways: arginine metabolism is orientated toward the production of ornithine and polyamine by arginase I and II instead of citrulline and NO by inducible nitric oxide synthase (iNOS) [16, 17]. However, it is clear from previous studies that the different functional patterns mounted by macrophages in response to distinct microenvironmental signals do not display a precise dichotomy between M1 and M2 responses [15, 18]. In this regard, Stout et al. proposed a model of functional adaptivity, suggesting not only that macrophages can adapt to microenvironmental signals by mounting different functional pattern, but also that they can progressively change their functional phenotype in response to progressive variation of these signals [18].
Figure 2.
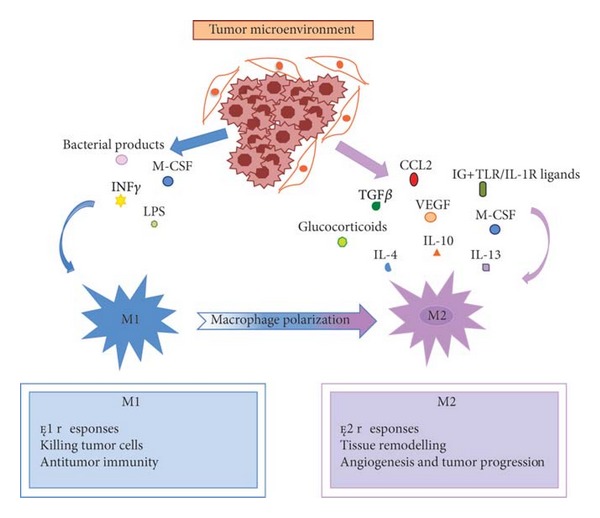
The phenotypic polarization of macrophages in the tumor microenvironment. Macrophages can be schematically classified into two main classes depending on their phenotypic polarization: macrophages mount M1 phenotype in response to M-CSF, INFγ, LPS and other microbial products, whereas they differentiate into M2 in the presence of TGFβ, VEGF, CCL2, M-CSF, IL-4, IL-10, IL-13, glucocorticoids and immune complexes/TLR ligands. M1 and M2 display different functions. M1 macrophages are able to trigger Th1 immune response and exert cytotoxic activity towards ingested microorganisms and cancer cells. M2 macrophages activate Th2 immune response and promote angiogenesis, tissue remodeling, and tumor progression. Refer to the text for abbreviations.
Within the tumor microenvironment, TAMs are mainly polarized towards M2 phenotype. In fact, TAMs were reported to express high levels of IL-10 and arginase I and low levels of proinflammatory cytokines, NO and ROS. Furthermore, TAMs are poor antigen-presenting cells [10, 13]. Notably, arginase expression by TAMs was previously suggested to be an adaptor mechanism to avoid formation of cytotoxic NO concentrations in tumors [19]. Mouse macrophages overexpressing arginase promoted proliferation of tumor cells in vivo [20] and macrophage-dependent tumor vascularization required polyamine synthesis [21].
Several clinical studies have shown that increased number of TAMs frequently correlate with angiogenesis, metastasis, and poor prognosis [14, 22, 23]. Nevertheless, there is a consensus view that macrophage polarization is strongly related to tumor stage, suggesting that a dynamic switching from M1 phenotype during the early phases of chronic inflammation to M2-like one in established tumors might occur. Moreover, several studies observed a “mixed” phenotype-expressing TAM population in different established murine and human tumors [24–29]. For instance, Sugai et al. observed increased levels of both IL-12 and IL-10 in monocytes from progressed gastric cancer patients with respect to healthy donors [27]. The hallmark of plasticity is also highlighted by the fact that the phenotype of macrophages also differs from tumor to tumor or within different areas of the same tumor. Soluble mediators secreted by cancer cells can alter the physiological development of macrophages, triggering transient early activation of monocytes in peritumoral stroma, and inducing immunosuppressive macrophages in cancer nests [30]. According to this model, tumor cells reeducate macrophages to adopt specific phenotype depending on the tumor area in which they localize: monocytes in peritumoral stroma are rapidly activated after their first exposure to tumor microenvironment, while the same cells become exhausted when they are in close proximity of tumor cells, thus failing to trigger an effective antitumor immune response [31]. In human HCC, most of the macrophages localized in the peritumoral stromal region mount an activated phenotype, with high expression of HLA-DR (Human Leukocyte Antigen), Interleukin 1 beta (IL-1β), Interleukin 6 (IL-6), and Interleukin 23 (IL-23), whereas they exhibit a HLA-DRlowIL-10low phenotype in the cancer nests [30, 32].
The importance of analyzing macrophage activation state is due to the fact that the type of macrophage polarization at the tumor site represents a prognostic factor. In HCC, tumor cells have been demonstrated to recruit and activate TAMs by secretion of Vascular Endothelial Growth Factor (VEGF), Platelet-derived Growth Factor (PDGF), TGF-β, CCL2, or M-CSF [33–35]. The expression of glypican-3 on the surface of liver cancer cells seems to be also implicated in TAM recruitment [36]. Recent studies have reported high expression of M-CSF in peritumoral liver tissue, which was associated with macrophage density, intrahepatic metastasis, and poor survival after hepatectomy [34, 37]. Moreover, a higher mRNA level of CCL2 is found in human HCC and the inhibition of Akt/NF-κB-induced CCL2 production resulted in a reduced migration and invasion of HCC cell lines [38, 39].
The upregulation of M2-associated genes in HCC-infiltrating macrophages, such as CD163, Fc fragment of IgG, and C-type lectin domain, has been recently demonstrated by examining gene expression profile [40]. Moreover, an established M2 macrophage population has been associated with poor prognosis in HCC [41]. The association between TAM density and unfortunate prognosis has been confirmed by several works [34, 42–44]. However, Li et al. observed that increased TAM infiltration was associated with improved overall survival after tumor resection, suggesting that TAMs could protect HCC patients from recurrence and metastasis [45]. Similarly, Chew et al. have shown that high levels of proinflammatory molecules derived from tumor-infiltrating cells were associated with a better survival in HCC patients [46]. Despite this latter experimental evidence, the recruitment and activation of TAMs are considered key events in HCC progression due to their secretion of soluble mediators, which in turn sustain tumor cell survival, proliferation and dissemination. In fact, Kuang et al. demonstrated that the inhibition of monocytes/macrophages inflammation in hepatoma-bearing mice through macrophage depletion experiments markedly reduced tumor growth [32]. Moreover, macrophages play also a role in the implantation of metastatic niches at distant sites of primary tumor; these niches seems to be a reservoir of myeloid derived cells, where monocytes would be rapidly mobilized to differentiate into macrophages in response to tumor signals, thus amplifying metastatic seeding and growth [13]. In this regard, Wyckoff et al. demonstrated that genetic reduction of TAMs resulted in a decrease of circulating tumor cells in a mouse model of breast cancer [47].
3. TAM-Derived Factors in HCC
3.1. TAM-Derived Cytokines and Chemokines
Inflammatory milieu has been established as a crucial component of HCC development and the crosstalk between tumor and stromal cells plays crucial roles in the regulation of HCC progression [1]. TAMs are involved in a complex interrelationship with cancer cells, and TAM-released cytokines and chemokines play an important role in the initiation and progression of liver cancer, regulating tumor growth, invasion, and metastasis. Inflammatory cytokines profile has been recognized to have a prognostic value. In fact, the expression of inflammation-associated genes, such as IL-6 and TNF-α, in peritumoral liver tissue was reported to predict late HCC recurrence [48, 49].
3.1.1. IL-6
IL-6 is a pleiotropic cytokine with a wide range of biological functions in immune regulation, inflammation, and oncogenesis. IL-6 plays a crucial role in the pathogenesis of HCC. In a chemically-induced HCC mouse model, diethylnitrosamine (DEN) exposure promotes IL-6 production by Kupffer cells in response to Interleukin 1 alpha (IL-1α) release from damaged hepatocytes). In turn, IL-6 promotes abnormal compensatory proliferation of surviving hepatocytes, thus stimulating the initiation and development of HCC. IL-6 exerts its oncogenic activity by triggering downstream Signal Transducer and Activator of Transcription 3 (STAT-3) and Extracellular-signal-Regulated Kinases (ERK) pathways, which in turn control target genes involved in both cell proliferation and survival [50]. In this model, IL-6−/− mice developed fewer tumors than wild type controls [51]. Estrogens suppress IL-6 production by Kupffer cells, partially explaining the gender bias in liver cancer [51]. Moreover, high IL-6 expression and the activation of IL-6 signaling pathway lead to HCC development in obese mice, representing a link between obesity and HCC [52]. Increased serum levels of IL-6 have been associated with high risk to develop HCC in patients with chronic hepatitis B and C; accordingly, high serum IL-6 has been frequently observed in patients with HCC and it was associated with a poor prognosis [53–55]. Furthermore, IL-6 showed prometastatic properties, and an early study demonstrated that exogenously administered IL-6 affected the metastatic potential of rat hepatocellular carcinoma cells [56]. Increased IL-6 serum levels could also be used to differentiate primary or metastatic HCC from benign lesions [57]. TAM-derived IL-6 seems also to favor the epithelial-mesenchymal transition of HCC, which has an important role in tumor progression [58, 59]. Indeed, IL-6 is believed to have immunosuppressive properties and to affect, in concert with other cytokines, T cell subset differentiation [60]. Kuang et al. showed that TAM-released proinflammatory cytokines IL-6 and IL-1β promoted T helper 17 (Th17) cell expansion in HCC, whereas Zhang et al. reported that high levels of Th17 cells correlated with microvessel density and poor survival in HCC patients [32, 61].
3.1.2. TNF-α
TNF-α is mainly produced by macrophages and is fundamental for liver regeneration following liver injury or partial hepatectomy [62]. TNF-α is strongly involved in the pathogenesis of HCC, promoting invasion, angiogenesis, and metastasis [23]. In fact, TNF-α has been shown to promote HCC in a genetic model of inflammation-induced carcinogenesis, in which mice lacking the P-glycoprotein Mdr2 developed cholestatic inflammation followed by HCC. In this model, in vivo administration of TNF-α-neutralizing antibody impaired HCC development [7]. Moreover, TNF-α plays a crucial role in DEN-induced HCC mouse model. TNF-α, in concert with IL-6, promoted hepatosteatosis and steatohepatitis, thus favoring obesity-enhanced HCC. In fact, the ablation of TNF-α signaling abolished HCC development in these mice [52]. TNF-α is associated with cell-cycle progression, tumor growth, and oxidative stress, stimulating the expression of Transforming Growth Factor alpha (TGF-α) in mouse hepatocytes and the formation of 8-oxodeoxyguanosine, a critical biomarker of oxidative stress and carcinogenesis [63–65]. Furthermore, TNF-α significantly induced phosphorylation of p38 Mitogen-Activated Protein Kinase (MAPK), ERK, Akt, and production of Interleukin 8 (IL-8) from HCC cells [66]. TAM-derived TNF-α also stimulated the activation of CAF, which are dominant elements in tumor inflammatory milieu, and was involved in promoting Th17 cell expansion [32, 67]. Furthermore, TNF-α and IL-1β supported tumor immune escape in vitro, by inducing the expression of TNF-related Apoptosis-inducing Ligand (TRAIL) on the surface of liver cancer cells, which in turn promoted apoptosis of activated T cells [68]. TNF-α also stimulated the expression of the negative costimulatory molecule B7 homolog 1 (B7-H1) (or Programmed Cell Death 1 Ligand 1, PDL-1) on macrophage surface, thus suppressing CD8+ T-cell antitumor immune response [43, 69]. The downstream principal mediator of protumoral activity of TNF-α is NF-κB, whose target genes are involved in cell proliferation and survival [3]. Of note, TNF-α is also induced by NF-κB in a positive feedback loop. TNF-α levels in HCC patients have also been investigated, but controversial data have been reported, whereas TNF-α (308) single nucleotide polymorphism was associated with cancer susceptibility [70].
3.1.3. IL-10
IL-10 is one of the most important immunosuppressive cytokines. Several studies have reported high IL-10 levels in HCC patients, as reviewed by Budhu and Xin [5]. IL-10, along with TNF-α, autocrinally stimulated the expression of B7-H1 on macrophage surface, impairing CD8+ T cell activity and supporting tumor immune escape [43, 69]. Moreover, B7-H1 expression by liver cancer cells correlated with TAM infiltration in HCC tissue and was dependent on IL-10-induced NF-κB an STAT-3 signaling pathways [71]. IL-10 is also involved in the induction of FOXP3+ (Forkhead Box P3) regulatory T cell (Treg) differentiation. Tregs strongly suppressed the activity of effector T cells and are associated with HCC aggressive clinicopathological features and poor survival [42, 72–74]. Moreover, high prevalence of Treg cells along with high levels of IL-10 has been observed in HCC patients [75]. In addition to play an immunosuppressive role, an increased expression of IL-10 is also correlated with high angiogenic activity in a HCC mouse model [76].
3.1.4. IL-17
The proinflammatory cytokine Interleukin 17 (IL-17) has been demonstrated to foster tumor immune escape in HCC [77]. IL-17 stimulated macrophages to express inflammatory cytokines, such as IL-1β, IL-10 and TNF-α, that autocrinally upregulated B7-H1 expression; thereafter, B7-H1-bearing macrophages in the peritumoral stroma suppressed cytotoxic T cell response via B7-H1 signaling [77]. Several reports demonstrated an association between high infiltration of IL-17-producing cells in the peritumoral stroma and the progression of HCC. Therefore, IL-17-mediated immunosuppression might represent a further mechanism by which this cytokine exerts its protumoral activity in liver cancer [32, 78].
3.1.5. Chemokines
In HCC progression, chemokines and their receptors play an intricate role. Chemokines could affect the recruitment of immunosuppressive Th17 cells, as reported by Kuang et al., which detected an increase of Th17 cell population in response to high local levels of CCL22 and Chemokine (C-C motif) Ligand 20 (CCL20) [78]. Furthermore, Zhang et al. observed higher levels of CCL20 in tumor tissue compared with nontumor one in HCC patients [61]. Uchida et al. reported that the incidence of intrahepatic metastasis was higher in patients with increased expression of the CCL20 receptor (CC-chemokine receptor 6, CCR6) than in patients with low expression of this molecule. The authors suggested that CCR6 might be associated with the intrahepatic metastasis of HCC and may be used as a prognostic factor after hepatic resection for HCC [79]. In addition, Shih et al. identified CCR6 as a marker of endothelial progenitor cells in tumor [80]. Indeed, M2-like TAMs produce several chemokines such as CCL17, CCL22, and CCL24, that interact with their receptors expressed mainly by Th2 and Treg cells, promoting the recruitment of these ineffective T cell subsets [16]. The Chemokine (C-X-C motif) Ligand 12 (CXCL12) is involved in Treg recruitment. In fact, Shen et al. reported that CXCL12 was responsible for the increased recruitment of Treg cells to tumor sites in HCC patients by the activation of CXCL12/CXC-Chemokine Receptor 4 (CXCR4) signaling [75]. The CXCL12-CXCR4 axis has been demonstrated to promote growth, invasion and metastasis of HCC cell lines [81, 82]. CXCL12-CXCR4 system is also involved in the secretion of Matrix Metalloprotease 9 (MMP-9) and Matrix Metalloprotease 2 (MMP-2), thus favoring metastasis [83]. Schimanski et al. also reported that strong expression of CXCR4 was significantly associated with progressed HCC, correlating with distant dissemination of lymphatic metastasis and a reduced 3-year-survival rate [84]. Recently, up-regulation of IL-8 receptor CXC-Chemokine Receptor 2 (CXCR2) was found in HCC patients and correlated with intrahepatic metastasis [85].
3.2. TAM-Derived Growth Factors
The deregulated expression of growth factors and the activation of their signaling pathways are hallmarks of chronic inflammatory liver diseases and HCC [3]. Aberrant growth factors expression contribute to neoplastic transformation and to the maintenance of acquired tumoral phenotype of HCC cells [86]. Moreover, TAM-derived growth factors strongly affect angiogenesis, as confirmed by Peng et al., which showed that TAM count was correlated to microvessels density [87]. Furthermore, marginal macrophage density is associated with angiogenesis and poor prognosis [44]. In addition to hepatocytes, extracellular matrix-producing cells and inflammatory cells such as macrophages have been demonstrated to produce TGF-β1, PDGF, VEGF, and the Epidermal Growth Factor Receptor (EGFR) ligands (TGF-α, Epidermal Growth Factor (EGF) and Amphiregulin) [1, 6, 88].
3.2.1. TGF-β
TGF-β exerts a dual role in liver hepatocarcinogenesis, showing both antitumoral and protumoral activities depending on HCC stage. In precancerous state, TGF-β1 acts as tumor suppressor, mediating anti-proliferative and proapoptotic signals, whereas in the established tumors TGF-β1 promotes tumorigenesis through several mechanisms [89]. First, TGF-β1, like IL-10, is an immunosuppressive molecule. TGF-β1 has been demonstrated to suppress INF-γ production by CD8+ T cells and to promote, in concert with IL-6, IL-1β, and IL-10, Treg generation, and Th17 differentiation, thus favoring tumor growth and progression [75, 90–92]. Moreover, TGF-β1 is one of the soluble factors able to activate HSCs, another crucial population within HCC microenvironment [1]. TGF-β1 expression is also associated with migration, invasion, angiogenesis, and metastasis. In fact, TGF-β induces α3β1 integrin, which is a marker of invasiveness [93, 94], as well as the secretion of VEGF [95]. In a recent preclinical study, targeting TGF-β has been demonstrated to affect several kinases involved in HCC cell migration control, such as SMAD-2 and focal adhesion kinase (FAK). Blocking TGF-β resulted in an overall inhibition of tumor growth and metastasis [96]. In HCC, TGF-β1 also promotes EMT through the downregulation of E-cadherin, a major component of epithelial adherent junctions, and the upregulation of the E-cadherin repressor snail and PDGF intracellular signaling [97, 98]. In accordance to the pivotal role of TGF-β1 in EMT, a recent work demonstrated that coupled with E-cadherin downregulation, TGF-β1 can induce N-cadherin, vimentin, and the HCC-associated antigen CD147 [99]. Experimental evidence in the literature reported that TGF-β also regulates several microRNAs involved in HCC pathogenesis, such as miR-23a, 27a, 24 and 181b [100, 101]. In particular, miR-181b has been demonstrated to up-regulate MMP-2 and MMP-9, favoring invasion and metastasis [100]. Early studies reported an increase of TGF-β levels in both plasma and tissue of HCC patients [102, 103]. Furthermore, TGF-β overexpression was associated with short survival in HCC patients [104]. Nevertheless, a recent study by Mamiya et al. reported that a decreased expression of TGF-β receptor type II was associated with intrahepatic metastasis, in accordance with the controversial role of TGF-β in HCC [105].
3.2.2. PDGF-β
As mentioned above, PDGF signaling is also important in HCC. In addition to its involvement in EMT, PDGF is considered an angiogenic factor, due to its capability to stabilize blood vessels [106]. PDGF also promotes the activation of CAFs and HSCs. Moreover, PDGF induces HSC differentiation into proliferating and ECM-producing myofibroblasts, which results in increased liver fibrosis and subsequent development of HCC [107]. Moreover, in this cell population, PDGF can also induce the up-regulation of amphiregulin, in turn involved in the activation of the EGFR signaling [108].
3.2.3. EGFR Ligands
EGFR overexpression has been observed in HCC tissue and seems to be correlated to a poor patients survival. In the same way, overexpression of its ligands has been reported in HCC tumor samples [109]. The role of EGFR pathway in HCC pathogenesis was well clarified by the use of genetically modified mouse models and confirmed by the experimental evidence that EGFR inhibition prevented chemically induced HCC in rats [3, 110]. Activation of EGFR signaling is strongly associated with angiogenesis because this pathway induces VEGF production [111].
3.2.4. VEGF
VEGF is a critical player in liver cancer angiogenesis. VEGF exerts its effect on the proliferation of both endothelial and VEGF-A receptor-expressing cancer cells [112]. High levels of this growth factor and its receptors VEGFR1, VEGFR2, and VEGFR3 have been reported in HCC cell lines, tissue, and in the blood circulation of HCC patients. Activation of VEGF signaling has been associated with vascular invasion, HCC grade, and poor outcome and survival (as reviewed in [113]).
3.3. Other TAM-Derived Mediators
In HCC, TAMs sustain invasion, angiogenesis, and metastasis through the expression of other several mediators different from cytokines, chemokines, or growth factors, including MMPs, osteopontin (OPN), and cyclooxygenase 2 (COX-2) [114].
With regard to MMPs, previous studies have reported an elevated expression of MMP-9 in HCC that was associated with growth and invasiveness [115–118]. A recent study by Roderferl et al. reported that MMP-9-expressing macrophages were involved in matrix remodeling and degradation at the invasive front of murine HCC [119]. Thanks to their proteolytic activity, MMP-9 and MMP-2 promote ECM-stored growth factors mobilization, including VEGF, thus favoring angiogenesis in HCC [120, 121]. Moreover, MMPs activated TGF-β during the EMT in HCC [122]. MMP-9 has also been demonstrated to induce HCC invasion and metastasis by cleaving and consequently activating the OPN precursor [123].
OPN is a phosphorylated acidic glycoprotein which was found to be expressed in macrophages after liver injury, taking part in the host response [124]. OPN is involved in the control of inflammation and tumor progression and contributes to HCC invasion and metastasis interacting with integrins [125]. OPN plasma levels were found increased in HCC patients and were associated with reduced liver function and worse prognosis [126]. Neutralizing OPN by anti-OPN antibodies resulted in strong inhibition of invasion and metastasis of HCC cells in vitro and in vivo [127].
COX-2 is involved in the synthesis of lipid inflammatory mediators from arachidonic acid and its overexpression has been observed in HCC patients [128, 129]. Furthermore, Cervello et al. reported that in primary HCC COX-2 expression was significantly correlated to the presence of inflammatory cells, including macrophages. The authors suggested that COX-2 expressing inflammatory cells were involved in the early stages of hepatocarcinogenesis [129].
In addition, TAM-derived urokinase-type plasminogen activator has been correlated with invasion and metastasis of HCC [130].
4. Signaling Pathways Linking TAMs and HCC
Although many studies have shown significant alterations in the expression and activity of different cytokines and their signaling systems in liver cirrhosis and HCC, the critical components linking inflammation and liver cancer are only beginning to be unmasked. Experimental evidence gathered in genetic mouse models over the past few years identified the transcription factor NF-κB, hypoxia inducible factor 1α (HIF-1α), and STAT-3 as the major molecular players linking inflammation and cancer [131] (Figure 3).
Figure 3.
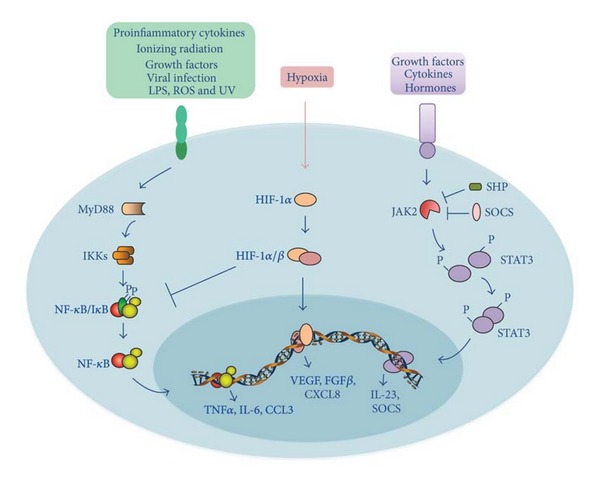
Schematic representation of NF-κB, HIF-1, and STAT-3 signaling pathways linking inflammation and liver cancer. Proinflammatory signals bind to their corresponding receptors, leading to the recruitment of receptor-associated proteins, such as MyD-88. In turn, these associated proteins trigger a phosphorylation cascade that leads to activation of the IKK complex, which is responsible for the phosphorylation of the IκB protein. Phosphorylated IκB is degraded by proteasome, thereby allowing free NF-κB dimers to translocate to the nucleus and transactivate target genes, such as TNFα, IL-6, and CCL3. HIF-1α is the central regulator of the hypoxic response. HIF-1α is activated by hypoxia and its activity progressively increases with a decrease in O2 gradient. Heterodimerization of HIF-1α with HIF-1β allows for DNA binding to hypoxia response elements (HRE) and transactivation of its target genes, such as VEGF, FGFβ, and CXCL8. Cytokine, hormone and growth factor stimulation activates JAK2, which in turn phosphorylates STAT3, allowing its dimerization, nuclear translocation, and transactivation of target genes, such as IL-23. STAT-3 signaling is turned off by protein inhibitors, such as SHP and SOCS, which are induced by STAT-3 in a negative feedback loop. Refer to the text for abbreviations.
4.1. NF-κB
The transcription factor NF-κB is a key orchestrator of innate immunity and inflammation, and recent evidence suggests that it represents a molecular link between inflammation and cancer [132]. NF-κB transcription factor family consists of five members: NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB, and c-Rel, which can form homo- or heterodimers [133]. NF-κB is retained in the cytoplasm of resting cells by binding with Inhibitor of NF-κB (IκB) proteins and can be rapidly activated upon stimulation by IκB phosphorylation. IκB proteins are phosphorylated by IκB kinase complex (IKK), which consist of two catalytic subunits (IKKα and IKKβ) and a regulatory subunit (IKKγ or NEMO). Recent studies have demonstrated that NF-κB can be activated in two different ways, the canonical pathway and the alternative pathway. Microbial products, such as Lipopolysaccharide (LPS), and proinflammatory cytokines, such as TNF-α and IL-1β, activate the classical pathway, leading to IKKβ-dependent phosphorylation of IκBα and RelA-p50 complexes activation (Figure 3). The alternative pathway is activated by lymphotoxin β (LTβ), CD40 ligand (CD40L), B-cell activating factor (BAFF), and RANK ligand (RANKL) and results in activation of RelB-p52 by IKKα-mediated phosphorylation and processing of p100 [133].
Genetic studies targeting NF-κB activation in liver epithelial cells and in liver macrophages have demonstrated that this factor plays a key role in HCC development. Mice with hepatocyte-specific ablation of IKKβ [50], NEMO [134], or with overexpression of an IκBα superrepressor [7] showed increased cell death, enhanced compensatory proliferation, predisposition to malignancy, and increased tumor susceptibility. Thus, NF-κB has a tumor suppressor function in liver parenchymal cells. On the contrary, NF-κB activation in liver macrophages has a protumoral significance. In fact, either genetic deletion of NF-κB or IL-6 [50, 51], or the inhibition of inflammatory cytokines, such as TNF-α [7], determined a significant reduction in HCC tumor load. Thus, detection of dying cells by Kupffer cells induces them to release more NF-κB-regulated inflammatory cytokines, which are necessary for growth and survival of malignant hepatocytes. The mechanism by which necrotic hepatocytes activate NF-κB in Kupffer cells was found to depend on the release of IL-1α by dying cells, which activates a Myeloid-Differentiation-Factor-88 (MyD-88)-dependent signaling pathway upon binding to IL-1 receptor (IL1R) on Kupffer cells [135].
These studies in HCC mouse models suggested that NF-κB plays a proinflammatory role in macrophages during early stages of the tumor growth. The roles of NF-κB in TAMs from established HCC cancers have not been yet elucidated, but recent works have dissected its function in TAMs from established tumors in a model of murine fibrosarcoma [29, 136], mouse mammary carcinoma [137], and mouse ovarian carcinoma [138]. Defective NF-κB activation was identified as signature of tumor-promoting, M2-like TAMs in fibrosarcoma. In fact, several NF-κB-regulated genes such as IL-12p40, TNF-α, Chemokine (C-C motif) Ligand 3 (CCL3), and IL-6 were downregulated in TAMs upon LPS treatment in vitro [29], and Saccani et al. showed that this phenotype was dependent on high levels of transcriptional inactive p50/p50 homodimers [136]. p50/p50 homodimers can compete with p50/p65 transcriptional active complex for the binding to promoter regions of proinflammatory genes. In this work, the authors showed that p50−/− TAMs have a proinflammatory, anti-tumor M1 phenotype, resulting in reduced tumor growth [136]. Indeed, Colombo et al. showed that activation of NF-κB is associated with mouse mammary tumor regression [137]. By contrast, a recent work by Hagemann et al. has demonstrated that inhibition of IKKβ (and therefore of NF-κB) in TAMs promotes an M1-like phenotype, whereas functional IKKβ/NF-κB activation maintains these cells in an alternative, tumor-promoting M2 phenotype [138]. These apparent discrepancy between Sica et al. studies on p50−/− TAMs and Hagemann et al. work on IKKβ −/− TAMs can be due to the different tumor types and experimental approaches utilized, as reviewed in [139]. In fact, it is well known that macrophage phenotype can differ markedly between different cancers [140]. Moreover, macrophages are very plastic cells, able to modify quickly their phenotype depending on the tumor stage [141].
4.2. STAT-3
STAT-3 is a member of the signal transducer and activator of transcription (STAT) and has been identified as one of the master regulator of macrophage transcriptional programs [142]. STAT-3 pathway is rapidly activated by several cytokines (i.e., IL-6), hormones, and growth factors (i.e., EGF and VEGF). The interaction of these ligands with their receptors triggers Janus Kinases (JAK) activation, especially JAK2, which, in turn, phosphorylates STAT-3 on the critical Tyr75 residue. The phosphorylation of STAT-3 at Tyr75 mediates STAT-3 dimerization, nucleus translocation, and DNA-binding of target genes involved in proliferation, survival, angiogenesis, and metastasis [143] (Figure 3). COX-2 has also been demonstrated to activate STAT-3 signaling pathway [144]. In normal condition STAT-3 activation is a transient and tightly regulated event. In fact, STAT-3 signaling is turned off by protein inhibitors, including SH2-containing phosphatases (SHP) and the suppressors of cytokine signaling (SOCS), which are activated by STAT-3 in a negative feedback loop [143] (Figure 3). The constitutive activation of STAT-3 signaling pathway was associated with M2 phenotype and it has been observed in both cancer and tumor-infiltrating inflammatory cells, including TAMs [3, 14, 145]. STAT-3 blocks IL-12p35 expression by dendritic cells and promotes the expression of the protumoral Interleukin 23 (IL-23) cytokine by TAMs [146]. STAT-3 activation in immune cells also exerts an immunosuppressive effect, thus counteracting anti-tumor immune response [147]. High STAT-3 levels have been detected in a large number of HCC samples and have been associated with invasiveness and poor prognosis [148–150]. Although the exact molecular events that lead to STAT-3 activation in human HCC are not well understood, NF-κB-regulated IL-6 released by TAMs seems to be the major STAT-3 activator. Accordingly, IL-6−/− mice showed a reduced STAT-3 activation and were less susceptible to develop DEN-induced HCC [51]. Moreover, a sixfold reduction in DEN-induced HCC load was observed in hepatocyte-specific STAT-3 deficient mice (Stat3∆hep) [151]. Furthermore, Stat3∆hep mice developed smaller tumors than control mice, suggesting a role for STAT-3 in hepatocyte proliferation and survival [151]. The pivotal role of JAK/STAT-3 pathway in inflammation-related liver cancer was confirmed by SOCS knocking out studies [152–154]. In fact, Socs3−/− mice were more susceptible to DEN-induced HCC and developed tumors increased in number and size [153, 154]. Furthermore, the inhibition of STAT-3 and NF-κB signaling pathways blocks the TAM-induced upregulation of B7-H1 on HCC cells [71].
Targeting STAT-3 signaling pathway might be a hopeful approach in the treatment of HCC. Liu et al. demonstrated that blocking STAT-3 phosphorylation with specific small molecule inhibitors causes apoptosis in HCC cell lines, whereas Stat3 antisense oligonucleotide strongly inhibited growth and metastasis of HCC in vivo [155, 156].
4.3. HIF-1
Hypoxic areas are often found in many solid tumors, including HCC. In addition, TAMs have been shown to accumulate in these poorly vascularized regions [157, 158]. Under low oxygen conditions, both tumor cells and macrophages mount a proangiogenic program mediated by Hypoxia Inducible Factor 1 (HIF-1). HIF-1 is a transcriptional activator complex constituted with two types of subunits, an inducible alpha subunit (HIF-1α, HIF-2α, or HIF-3α), and one constitutively expressed HIF-1β subunit. Hypoxia stabilizes HIF-1α, preventing its posttranslational hydroxylation and consequently proteasome-mediated degradation. In addition, hypoxia promotes HIF-1α association with HIF-1β, as well as cofactor recruitment [159] (Figure 3). HIF-1α is also transcriptionally regulated by NF-κB, as demonstrated by Rius et al. [160]. The authors reported that bone marrow-derived macrophages upregulated NF-κB, which in turn induced HIF-1α following short-term exposure to hypoxia. In addition, Maeda et al. observed the downregulation of HIF-1α in mice lacking IKKβ in both hepatocytes and Kupffer cells [134]. TAM adaptation to hypoxia is mediated by the induction of HIF-1 and HIF-2-regulated genes, including VEGF, Fibroblast Growth Factor beta (FGF-β), and IL-8, as well as glycolytic enzymes [161, 162]. Furthermore, HIF-1 pathway has been demonstrated to play a role in TAM recruitment and activation. In fact, hypoxia affected the localization of both tumor and stromal cells by up-regulating their expression of CXCR4 receptor, CXCR4 ligand, and CXCL12 [163, 164]. The pivotal role of HIF-1 in sustaining oncogenic activities of TAMs has been demonstrated by HIF1α and HIF2α ablation experiments [165–167]. The lack of HIF-1α impaired macrophage motility and migration through extracellular matrix, as well as their suppressive abilities [165, 166]. The loss of HIF-2α ablation in macrophages affected TAM recruitment in a mouse model of HCC and it was associated with reduced tumor cell proliferation. In the same work, the authors demonstrated that HIF-2α regulated proinflammatory cytokine expression, including IL-6, by binding their promoter regions [167]. Thus, the ablation of both HIF-1α and HIF-2α resulted in reduced tumor volume and progression in different cancer models, suggesting that HIF-1α and HIF-2α regulate overlapping pathways. As suggested for NF-κB and STAT-3, HIF-1 might be a potential target in HCC therapy. In fact, two different works reported that antisense HIF-1 therapy was effective in inhibiting HCC cell proliferation as well as in enhancing chemotherapy antitumor efficacy in a xenograft model of liver cancer [168, 169].
5. Conclusions and Perspectives
The crucial role of the tumor microenvironment in HCC pathogenesis is now widely accepted. TAMs represent one of the main tumor-infiltrating immune cells that sustain tumor progression. In fact, several experimental evidence showed that TAMs promote cancer cell growth, invasion, and metastasis by stimulating proliferation, survival, and EMT transition of cancer cells, as well as, by inducing angiogenesis, ECM remodeling and suppression of antitumor immunity. Thus, targeting tumor-stroma interactions is believed to be an attractive therapeutic strategy in the management of HCC. To date, most of the therapeutics have been designed to block receptors and downstream signaling pathways, so inhibiting stroma-derived protumoral signals, such as the kinase inhibitor drugs. Sorafenib, an oral inhibitor of VEGFR-2/3, PDGFR, and Raf kinases, has been demonstrated to be effective and safe in Phase III clinical trials and it is the standard therapeutic agent for advanced HCC [170, 171]. Moreover, a recent study by Zhang et al. showed that sorafenib is more effective when used in combination with zoledronic acid and clodronate-encapsulated liposomes, which deplete macrophages population [172]. A phase II clinical study of sorafenib and zoledronic acid to treat advanced HCC is currently recruiting participants. Similarly, targeting TGF-β signaling has been demonstrated to be a potential therapy in HCC, because TGF-β receptor I kinase inhibitor LY2109761 has been reported to inhibit HCC migration in vitro and tumor growth, intravasation, and metastasis in vivo [96]. Other drugs belonging to kinase inhibitors class, such as brivanib (which targets VEGFR2 and FGFR1), linifanib (which targets PDGFR and VEGFR), sunitinib (which targets PDGFR, VEGFR, c-Kit, and Flt-3), erlotinib (which targets EGFR), and PI-88 (which targets heparanase and sulfatases) are currently being investigated for efficacy and safety in liver cancer-phase III clinical trials. Similarly, efficacy and safety of recombinant monoclonal antibodies against VEGF (bevacizumab), VEGFR2 (ramucirumab), and EGFR (cetuximab) are now evaluated in phase II-III clinical trials for the treatment of HCC (as reviewed in [1]). Moreover, on the basis of experimental evidence, targeting signaling pathways involved in the crosstalk between stromal and tumor cells might also be an effective strategy against HCC.
Thus, clarifying the molecular mechanisms underlying the crosstalk between tumor and stromal cells within HCC microenvironment will be beneficial to identify new targets for liver cancer therapy.
Authors' Contribution
F. Zazzeroni and E. Alesse contributed equally to this work.
Acknowledgments
This work was supported in part by MIUR FIRB Grant no. RBAP10A9H9 to E. Alesse, by Associazione Italiana per la Ricerca sul Cancro (AIRC) grant to F. Zazzeroni and by MIUR PRIN Grant no. 2009EWAW4M_003 to F. Zazzeroni.
References
- 1.Yang JD, Nakamura I, Roberts LR. The tumor microenvironment in hepatocellular carcinoma: current status and therapeutic targets. Seminars in Cancer Biology. 2011;21(1):35–43. doi: 10.1016/j.semcancer.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruix J, Boix L, Sala M, Llovet JM. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5(3):215–219. doi: 10.1016/s1535-6108(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 3.Berasain C, Castillo J, Perugorria MJ, Latasa MU, Prieto J, Avila MA. Inflammation and liver cancer: new molecular links. Annals of the New York Academy of Sciences. 2009;1155:206–221. doi: 10.1111/j.1749-6632.2009.03704.x. [DOI] [PubMed] [Google Scholar]
- 4.Porta C, Riboldi E, Sica A. Mechanisms linking pathogens-associated inflammation and cancer. Cancer Letters. 2011;305(2):250–262. doi: 10.1016/j.canlet.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 5.Budhu A, Xin WW. The role of cytokines in hepatocellular carcinoma. Journal of Leukocyte Biology. 2006;80(6):1197–1213. doi: 10.1189/jlb.0506297. [DOI] [PubMed] [Google Scholar]
- 6.Leonardi GC, Candido S, Cervello M, et al. The tumor microenvironment in hepatocellular carcinoma. International Journal of Oncology. 2012;40(6):1733–1747. doi: 10.3892/ijo.2012.1408. [DOI] [PubMed] [Google Scholar]
- 7.Pikarsky E, Porat RM, Stein I, et al. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431(7007):461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 8.Luedde T, Schwabe RF. NF-κB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nature Reviews Gastroenterology and Hepatology. 2011;8(2):108–118. doi: 10.1038/nrgastro.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mantovani A, Germano G, Marchesi F, Locatelli M, Biswas SK. Cancer-promoting tumor-associated macrophages: new vistas and open questions. European Journal of Immunology. 2011;41(9):2522–2525. doi: 10.1002/eji.201141894. [DOI] [PubMed] [Google Scholar]
- 10.Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. Journal of Leukocyte Biology. 2009;86(5):1065–1073. doi: 10.1189/jlb.0609385. [DOI] [PubMed] [Google Scholar]
- 11.Porta C, Larghi P, Rimoldi M, et al. Cellular and molecular pathways linking inflammation and cancer. Immunobiology. 2009;214(9-10):761–777. doi: 10.1016/j.imbio.2009.06.014. [DOI] [PubMed] [Google Scholar]
- 12.Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Current Opinion in Immunology. 2010;22(2):231–237. doi: 10.1016/j.coi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in Immunology. 2002;23(11):549–555. doi: 10.1016/s1471-4906(02)02302-5. [DOI] [PubMed] [Google Scholar]
- 15.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nature Reviews Immunology. 2005;5(12):953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 16.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nature Immunology. 2010;11(10):889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 17.Movahedi K, Laoui D, Gysemans C, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Research. 2010;70(14):5728–5739. doi: 10.1158/0008-5472.CAN-09-4672. [DOI] [PubMed] [Google Scholar]
- 18.Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. The Journal of Immunology. 2005;175(1):342–349. doi: 10.4049/jimmunol.175.1.342. [DOI] [PubMed] [Google Scholar]
- 19.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature Reviews Immunology. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Research. 2001;61(3):1100–1106. [PubMed] [Google Scholar]
- 21.Davel LE, Jasnis MA, De la Torre E, et al. Arginine metabolic pathways involved in the modulation of tumor-induced angiogenesis by macrophages. FEBS Letters. 2002;532(1-2):216–220. doi: 10.1016/s0014-5793(02)03682-7. [DOI] [PubMed] [Google Scholar]
- 22.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nature Reviews Cancer. 2004;4(1):71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 23.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Research. 2006;66(2):605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 24.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. The Journal of Clinical Investigation. 2007;117(5):1155–1166. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. The Journal of Immunology. 2005;174(8):4880–4891. doi: 10.4049/jimmunol.174.8.4880. [DOI] [PubMed] [Google Scholar]
- 26.Tsai CS, Chen FH, Wang CC, et al. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. International Journal of Radiation Oncology Biology Physics. 2007;68(2):499–507. doi: 10.1016/j.ijrobp.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 27.Sugai H, Kono K, Takahashi A, et al. Characteristic alteration of monocytes with increased intracellular IL-10 and IL-12 in patients with advanced-stage gastric cancer. Journal of Surgical Research. 2004;116(2):277–287. doi: 10.1016/j.jss.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 28.Hagemann T, Wilson J, Burke F, et al. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. The Journal of Immunology. 2006;176(8):5023–5032. doi: 10.4049/jimmunol.176.8.5023. [DOI] [PubMed] [Google Scholar]
- 29.Biswas SK, Gangi L, Paul S, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-κB and enhanced IRF-3/STAT1 activation) Blood. 2006;107(5):2112–2122. doi: 10.1182/blood-2005-01-0428. [DOI] [PubMed] [Google Scholar]
- 30.Kuang DM, Wu Y, Chen N, Cheng J, Zhuang SM, Zheng L. Tumor-derived hyaluronan induces formation of immunosuppressive macrophages through transient early activation of monocytes. Blood. 2007;110(2):587–595. doi: 10.1182/blood-2007-01-068031. [DOI] [PubMed] [Google Scholar]
- 31.Wu Y, Zheng L. Dynamic education of macrophages in different areas of human tumors. doi: 10.1007/s12307-012-0113-z. Cancer Microenvironment. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuang DM, Peng C, Zhao Q, Wu Y, Chen MS, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology. 2010;51(1):154–164. doi: 10.1002/hep.23291. [DOI] [PubMed] [Google Scholar]
- 33.Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Medicine. 2008;5(1, article e19) doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu XD, Zhang JB, Zhuang PY, et al. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. Journal of Clinical Oncology. 2008;26(16):2707–2716. doi: 10.1200/JCO.2007.15.6521. [DOI] [PubMed] [Google Scholar]
- 35.Benetti A, Berenzi A, Gambarotti M, et al. Transforming growth factor-β1 and CD105 promote the migration of hepatocellular carcinoma-derived endothelium. Cancer Research. 2008;68(20):8626–8634. doi: 10.1158/0008-5472.CAN-08-1218. [DOI] [PubMed] [Google Scholar]
- 36.Takai H, Kato A, Kato C, et al. The expression profile of glypican-3 and its relation to macrophage population in human hepatocellular carcinoma. Liver International. 2009;29(7):1056–1064. doi: 10.1111/j.1478-3231.2008.01968.x. [DOI] [PubMed] [Google Scholar]
- 37.Jia JB, Wang WQ, Sun HC, et al. High expression of macrophage colony-stimulating factor-1 receptor in peritumoral liver tissue is associated with poor outcome in hepatocellular carcinoma after curative resection. Oncologist. 2010;15(7):732–743. doi: 10.1634/theoncologist.2009-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haybaeck J, Zeller N, Wolf MJ, et al. A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer Cell. 2009;16(4):295–308. doi: 10.1016/j.ccr.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen TA, Wang JL, Hung SW, Chu CL, Cheng YC, Liang SM. Recombinant VP1, an akt inhibitor, suppresses progression of hepatocellular carcinoma by inducing apoptosis and modulation of CCL2 production. PLoS ONE. 2011;6(8) doi: 10.1371/journal.pone.0023317.e23317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakai Y, Honda M, Fujinaga H, et al. Common transcriptional signature of tumor-infiltrating mononuclear inflammatory cells and peripheral blood mononuclear cells in hepatocellular carcinoma patients. Cancer Research. 2008;68(24):10267–10279. doi: 10.1158/0008-5472.CAN-08-0911. [DOI] [PubMed] [Google Scholar]
- 41.Budhu A, Forgues M, Ye QH, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell. 2006;10(2):99–111. doi: 10.1016/j.ccr.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 42.Zhou J, Ding T, Pan W, Zhu LY, Li A, Zheng L. Increased intratumoral regulatory T cells are related to intratumoral macrophages and poor prognosis in hepatocellular carcinoma patients. International Journal of Cancer. 2009;125(7):1640–1648. doi: 10.1002/ijc.24556. [DOI] [PubMed] [Google Scholar]
- 43.Kuang DM, Zhao Q, Peng C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. The Journal of Experimental Medicine. 2009;206(6):1327–1337. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ding T, Xu J, Wang F, et al. High tumor-infiltrating macrophage density predicts poor prognosis in patients with primary hepatocellular carcinoma after resection. Human Pathology. 2009;40(3):381–389. doi: 10.1016/j.humpath.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 45.Li YW, Qiu SJ, Fan J, et al. Tumor-infiltrating macrophages can predict favorable prognosis in hepatocellular carcinoma after resection. Journal of Cancer Research and Clinical Oncology. 2009;135(3):439–449. doi: 10.1007/s00432-008-0469-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chew V, Tow C, Teo M, et al. Inflammatory tumour microenvironment is associated with superior survival in hepatocellular carcinoma patients. Journal of Hepatology. 2010;52(3):370–379. doi: 10.1016/j.jhep.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 47.Wyckoff JB, Wang Y, Lin EY, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Research. 2007;67(6):2649–2656. doi: 10.1158/0008-5472.CAN-06-1823. [DOI] [PubMed] [Google Scholar]
- 48.Lee JS, Heo J, Libbrecht L, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nature Medicine. 2006;12(4):410–416. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 49.Hoshida Y, Villanueva A, Kobayashi M, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. The New England Journal of Medicine. 2008;359(19):1995–2004. doi: 10.1056/NEJMoa0804525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121(7):977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 51.Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–124. doi: 10.1126/science.1140485. [DOI] [PubMed] [Google Scholar]
- 52.Park EJ, Lee JH, Yu GY, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wong VWS, Yu J, Cheng ASL, et al. High serum interleukin-6 level predicts future hepatocellular carcinoma development in patients with chronic hepatitis B. International Journal of Cancer. 2009;124(12):2766–2770. doi: 10.1002/ijc.24281. [DOI] [PubMed] [Google Scholar]
- 54.Nakagawa H, Maeda S, Yoshida H, et al. Serum IL-6 levels and the risk for hepatocarcinogenesis in chronic hepatitis C patients: an analysis based on gender differences. International Journal of Cancer. 2009;125(10):2264–2269. doi: 10.1002/ijc.24720. [DOI] [PubMed] [Google Scholar]
- 55.Hsia CY, Huo TI, Chiang SY, et al. Evaluation of interleukin-6, interleukin-10 and human hepatocyte growth factor as tumor markers for hepatocellular carcinoma. European Journal of Surgical Oncology. 2007;33(2):208–212. doi: 10.1016/j.ejso.2006.10.036. [DOI] [PubMed] [Google Scholar]
- 56.Reichner JS, Mulligan JA, Spisni R, Sotomayor EA, Albina JE, Bland KI. Effect of IL-6 overexpression on the metastatic potential of rat hepatocellular carcinoma cells. Annals of Surgical Oncology. 1998;5(3):279–286. doi: 10.1007/BF02303786. [DOI] [PubMed] [Google Scholar]
- 57.Coskun U, Bukan N, Sancak B, et al. Serum hepatocyte growth factor and interleukin-6 levels can distinguish patients with primary or metastatic liver tumors from those with benign liver lesions. Neoplasma. 2004;51(3):209–213. [PubMed] [Google Scholar]
- 58.Tang Y, Kitisin K, Jogunoori W, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-β and IL-6 signaling. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(7):2445–2450. doi: 10.1073/pnas.0705395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Werno C, Menrad H, Weigert A, et al. Knockout of HIF-1α in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis. 2010;31(10):1863–1872. doi: 10.1093/carcin/bgq088. [DOI] [PubMed] [Google Scholar]
- 60.Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 2010;140(6):845–858. doi: 10.1016/j.cell.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 61.Zhang JP, Yan J, Xu J, et al. Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. Journal of Hepatology. 2009;50(5):980–989. doi: 10.1016/j.jhep.2008.12.033. [DOI] [PubMed] [Google Scholar]
- 62.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43(1):S45–S53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 63.Gallucci RM, Simeonova PP, Toriumi W, Luster MI. TNF-α regulates transforming growth factor-α expression in regenerating murine liver and isolated hepatocytes. The Journal of Immunology. 2000;164(2):872–878. doi: 10.4049/jimmunol.164.2.872. [DOI] [PubMed] [Google Scholar]
- 64.Cosgrove BD, Cheng C, Pritchard JR, Stolz DB, Lauffenburger DA, Griffith LG. An inducible autocrine cascade regulates rat hepatocyte proliferation and apoptosis responses to tumor necrosis factor-α . Hepatology. 2008;48(1):276–288. doi: 10.1002/hep.22335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wheelhouse NM, Chan YS, Gillies SE, et al. TNF-alpha induced DNA damage in primary murine hepatocytes. International Journal of Molecular Medicine. 2003;12(6):889–894. [PubMed] [Google Scholar]
- 66.Wang Y, Wang W, Wang L, Wang X, Xia J. Regulatory mechanisms of interleukin-8 production induced by tumour necrosis factor-α in human hepatocellular carcinoma cells. Journal of Cellular and Molecular Medicine. 2012;16(3):496–506. doi: 10.1111/j.1582-4934.2011.01337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mueller L, Goumas FA, Affeldt M, et al. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. American Journal of Pathology. 2007;171(5):1608–1618. doi: 10.2353/ajpath.2007.060661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shiraki K, Yamanaka T, Inoue H, et al. Expression of TNF-related apoptosis-inducing ligand in human hepatocellular carcinoma. International Journal of Oncology. 2005;26(5):1273–1281. [PubMed] [Google Scholar]
- 69.Ke W, Kryczek I, Chen L, Zou W, Welling TH. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Research. 2009;69(20):8067–8075. doi: 10.1158/0008-5472.CAN-09-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Talaat RM, Esmail AA, Elwakil R, Gurgis AA, Nasr MI. Tumor necrosis factor-alpha -308G/A polymorphism and risk of hepatocellular carcinoma in hepatitis C virus-infected patients. Chinese Journal of Cancer. 2012;31(1):29–35. doi: 10.5732/cjc.011.10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen J, Li G, Meng H, et al. Upregulation of B7-H1 expression is associated with macrophage infiltration in hepatocellular carcinomas. Cancer Immunology, Immunotherapy. 2012;61(1):101–108. doi: 10.1007/s00262-011-1094-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ju MJ, Qiu SJ, Fan J, et al. Peritumoral activated hepatic stellate cells predict poor clinical outcome in hepatocellular carcinoma after curative resection. American Journal of Clinical Pathology. 2009;131(4):498–510. doi: 10.1309/AJCP86PPBNGOHNNL. [DOI] [PubMed] [Google Scholar]
- 73.Gao Q, Qiu SJ, Fan J, et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. Journal of Clinical Oncology. 2007;25(18):2586–2593. doi: 10.1200/JCO.2006.09.4565. [DOI] [PubMed] [Google Scholar]
- 74.Fu J, Xu D, Liu Z, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132(7):2328–2339. doi: 10.1053/j.gastro.2007.03.102. [DOI] [PubMed] [Google Scholar]
- 75.Shen X, Li N, Li H, Zhang T, Wang F, Li Q. Increased prevalence of regulatory T cells in the tumor microenvironment and its correlation with TNM stage of hepatocellular carcinoma. Journal of Cancer Research and Clinical Oncology. 2010;136(11):1745–1754. doi: 10.1007/s00432-010-0833-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ryschich E, Lizdenis P, Ittrich C, et al. Molecular fingerprinting and autocrine growth regulation of endothelial cells in a murine model of hepatocellular carcinoma. Cancer Research. 2006;66(1):198–211. doi: 10.1158/0008-5472.CAN-05-1636. [DOI] [PubMed] [Google Scholar]
- 77.Zhao Q, Xiao X, Wu Y, et al. Interleukin-17-educated monocytes suppress cytotoxic T-cell function through B7-H1 in hepatocellular carcinoma patients. European Journal of Immunology. 2011;41(8):2314–2322. doi: 10.1002/eji.201041282. [DOI] [PubMed] [Google Scholar]
- 78.Kuang DM, Peng C, Zhao Q, et al. Tumor-activated monocytes promote expansion of IL-17-producing CD8+ T cells in hepatocellular carcinoma patients. The Journal of Immunology. 2010;185(3):1544–1549. doi: 10.4049/jimmunol.0904094. [DOI] [PubMed] [Google Scholar]
- 79.Uchida H, Iwashita Y, Sasaki A, et al. Chemokine receptor CCR6 as a prognostic factor after hepatic resection for hepatocellular carcinoma. Journal of Gastroenterology and Hepatology. 2006;21(1):161–168. doi: 10.1111/j.1440-1746.2005.04157.x. [DOI] [PubMed] [Google Scholar]
- 80.Shih Y-T, Wang M-C, Peng H-H, et al. Modulation of chemotactic and pro-inflammatory activities of endothelial progenitor cells by hepatocellular carcinoma. Cellular Signalling. 2012;24(3):779–793. doi: 10.1016/j.cellsig.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 81.Sutton A, Friand V, Brulé-Donneger S, et al. Stromal cell-derived factor-1/chemokine (C-X-C motif) ligand 12 stimulates human hepatoma cell growth, migration, and invasion. Molecular Cancer Research. 2007;5(1):21–33. doi: 10.1158/1541-7786.MCR-06-0103. [DOI] [PubMed] [Google Scholar]
- 82.Liu H, Pan Z, Li A, et al. Roles of chemokine receptor 4 (CXCR4) and chemokine ligand 12 (CXCL12) in metastasis of hepatocellular carcinoma cells. Cellular and Molecular Immunology. 2008;5(5):373–378. doi: 10.1038/cmi.2008.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chu H, Zhou H, Liu Y, Liu X, Hu Y, Zhang J. Functional expression of CXC chemokine recepter-4 mediates the secretion of matrix metalloproteinases from mouse hepatocarcinoma cell lines with different lymphatic metastasis ability. International Journal of Biochemistry and Cell Biology. 2007;39(1):197–205. doi: 10.1016/j.biocel.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 84.Schimanski CC, Bahre R, Gockel I, et al. Dissemination of hepatocellular carcinoma is mediated via chemokine receptor CXCR4. British Journal of Cancer. 2006;95(2):210–217. doi: 10.1038/sj.bjc.6603251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu Z, Yang L, Xu J, Zhang X, Wang B. Enhanced expression and clinical significance of chemokine receptor CXCR2 in hepatocellular carcinoma. Journal of Surgical Research. 2011;166(2):241–246. doi: 10.1016/j.jss.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 86.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25(27):3787–3800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 87.Peng SH, Deng H, Yang JF, et al. Significance and relationship between infiltrating inflammatory cell and tumor angiogenesis in hepatocellular carcinoma tissues. World Journal of Gastroenterology. 2005;11(41):6521–6524. doi: 10.3748/wjg.v11.i41.6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Drucker C, Parzefall W, Teufelhofer O, et al. Non-parenchymal liver cells support the growth advantage in the first stages of hepatocarcinogenesis. Carcinogenesis. 2006;27(1):152–161. doi: 10.1093/carcin/bgi202. [DOI] [PubMed] [Google Scholar]
- 89.Giannelli G, Mazzocca A, Fransvea E, Lahn M, Antonaci S. Inhibiting TGF-β signaling in hepatocellular carcinoma. Biochimica et Biophysica Acta. 2011;1815(2):214–223. doi: 10.1016/j.bbcan.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 90.Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. International Journal of Biological Sciences. 2011;7(5):651–658. doi: 10.7150/ijbs.7.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Volpe E, Servant N, Zollinger R, et al. A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human TH-17 responses. Nature Immunology. 2008;9(6):650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 92.Manel N, Unutmaz D, Littman DR. The differentiation of human TH-17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγt. Nature Immunology. 2008;9(6):641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fransvea E, Mazzocca A, Antonaci S, Giannelli G. Targeting transforming growth factor (TGF)-βRI inhibits activation of β1 integrin and blocks vascular invasion in hepatocellular carcinoma. Hepatology. 2009;49(3):839–850. doi: 10.1002/hep.22731. [DOI] [PubMed] [Google Scholar]
- 94.Giannelli G, Fransvea E, Marinosci F, et al. Transforming growth factor-β1 triggers hepatocellular carcinoma invasiveness via α3β1 integrin. American Journal of Pathology. 2002;161(1):183–193. doi: 10.1016/s0002-9440(10)64170-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mazzocca A, Fransvea E, Lavezzari G, Antonaci S, Giannelli G. Inhibition of transforming growth factor β receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology. 2009;50(4):1140–1151. doi: 10.1002/hep.23118. [DOI] [PubMed] [Google Scholar]
- 96.Fransvea E, Mazzocca A, Santamato A, Azzariti A, Antonaci S, Giannelli G. Kinase activation profile associated with TGF-β-dependent migration of HCC cells: a preclinical study. Cancer Chemotherapy and Pharmacology. 2011;68(1):79–86. doi: 10.1007/s00280-010-1459-x. [DOI] [PubMed] [Google Scholar]
- 97.Giannelli G, Bergamini C, Fransvea E, Sgarra C, Antonaci S. Laminin-5 with transforming growth factor-β1 induces epithelial to mesenchymal transition in hepatocellular carcinoma. Gastroenterology. 2005;129(5):1375–1383. doi: 10.1053/j.gastro.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 98.Van Zijl F, Mair M, Csiszar A, et al. Hepatic tumor-stroma crosstalk guides epithelial to mesenchymal transition at the tumor edge. Oncogene. 2009;28(45):4022–4033. doi: 10.1038/onc.2009.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu J, Ru NY, Zhang Y, et al. HAb18G/CD147 promotes epithelial-mesenchymal transition through TGF-β signaling and is transcriptionally regulated by Slug. Oncogene. 2011;30(43):4410–4427. doi: 10.1038/onc.2011.149. [DOI] [PubMed] [Google Scholar]
- 100.Wang B, Hsu SH, Majumder S, et al. TGFβ-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene. 2010;29(12):1787–1797. doi: 10.1038/onc.2009.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Huang S, He X, Ding J, et al. Upregulation of miR-23a∼27a∼24 decreases transforming growth factor-beta-induced tumor-suppressive activities in human hepatocellular carcinoma cells. International Journal of Cancer. 2008;123(4):972–978. doi: 10.1002/ijc.23580. [DOI] [PubMed] [Google Scholar]
- 102.Bedossa P, Peltier E, Terris B, Franco D, Poynard T. Transforming growth factor-beta 1 (TGF-β1) and TGF-β1 receptors in normal, cirrhotic, and neoplastic human livers. Hepatology. 1995;21(3):760–766. [PubMed] [Google Scholar]
- 103.Shirai Y, Kawata S, Tamura S, et al. Plasma transforming growth factor-β1 in patients with hepatocellular carcinoma: comparison with chronic liver diseases. Cancer. 1994;73(9):2275–2279. doi: 10.1002/1097-0142(19940501)73:9<2275::aid-cncr2820730907>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 104.Okumoto K, Hattori E, Tamura K, et al. Possible contribution of circulating transforming growth factor-β1 to immunity and prognosis in unresectable hepatocellular carcinoma. Liver International. 2004;24(1):21–28. doi: 10.1111/j.1478-3231.2004.00882.x. [DOI] [PubMed] [Google Scholar]
- 105.Mamiya T, Yamazaki K, Masugi Y, et al. Reduced transforming growth factor-β receptor II expression in hepatocellular carcinoma correlates with intrahepatic metastasis. Laboratory Investigation. 2010;90(9):1339–1345. doi: 10.1038/labinvest.2010.105. [DOI] [PubMed] [Google Scholar]
- 106.Hirschi KK, D’Amore PA. Control of angiogenesis by the pericyte: molecular mechanisms and significance. EXS. 1997;79:419–428. doi: 10.1007/978-3-0348-9006-9_18. [DOI] [PubMed] [Google Scholar]
- 107.Campbell JS, Hughes SD, Gilbertson DG, et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(9):3389–3394. doi: 10.1073/pnas.0409722102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Perugorria MJ, Latasa MU, Nicou A, et al. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology. 2008;48(4):1251–1261. doi: 10.1002/hep.22437. [DOI] [PubMed] [Google Scholar]
- 109.Berasain C, Castillo J, Prieto J, Avila MA. New molecular targets for hepatocellular carcinoma: the ErbB1 signaling system. Liver International. 2007;27(2):174–185. doi: 10.1111/j.1478-3231.2006.01424.x. [DOI] [PubMed] [Google Scholar]
- 110.Schiffer E, Housset C, Cacheux W, et al. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology. 2005;41(2):307–314. doi: 10.1002/hep.20538. [DOI] [PubMed] [Google Scholar]
- 111.Tabernero J. The role of VEGF and EGFR inhibition: implications for combining Anti-VEGF and Anti-EGFR Agents. Molecular Cancer Research. 2007;5(3):203–220. doi: 10.1158/1541-7786.MCR-06-0404. [DOI] [PubMed] [Google Scholar]
- 112.Yang JC, Teng CF, Wu HC, et al. Enhanced expression of vascular endothelial growth factor-A in ground glass hepatocytes and its implication in hepatitis B virus hepatocarcinogenesis. Hepatology. 2009;49(6):1962–1971. doi: 10.1002/hep.22889. [DOI] [PubMed] [Google Scholar]
- 113.Zhu AX, Duda DG, Ancukiewicz M, et al. Exploratory analysis of early toxicity of sunitinib in advanced hepatocellular carcinoma patients: kinetics and potential biomarker value. Clinical Cancer Research. 2011;17(4):918–927. doi: 10.1158/1078-0432.CCR-10-0515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu S-D, Ma Y-S, Fang Y, Liu L-L, Fu D, Shen X-Z. Role of the microenvironment in hepatocellular carcinoma development and progression. Cancer Treatment Reviews. 2012;38(3):218–225. doi: 10.1016/j.ctrv.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 115.Arii S, Mise M, Harada T, et al. Overexpression of matrix metalloproteinase 9 gene in hepatocellular carcinoma with invasive potential. Hepatology. 1996;24(2):316–322. doi: 10.1053/jhep.1996.v24.pm0008690399. [DOI] [PubMed] [Google Scholar]
- 116.Maatta M, Soini Y, Liakka A, Autio-Harmainen H. Differential expression of matrix metalloproteinase (MMP)-2, MMP-9, and membrane type 1-MMP in hepatocellular and pancreatic adenocarcinoma: implications for tumor progression and clinical prognosis. Clinical Cancer Research. 2000;6(7):2726–2734. [PubMed] [Google Scholar]
- 117.Cheng JCH, Chou CH, Kuo ML, Hsieh CY. Radiation-enhanced hepatocellular carcinoma cell invasion with MMP-9 expression through PI3K/Akt/NF-κB signal transduction pathway. Oncogene. 2006;25(53):7009–7018. doi: 10.1038/sj.onc.1209706. [DOI] [PubMed] [Google Scholar]
- 118.Sakamoto Y, Mafune K, Mori M, et al. Overexpression of MMP-9 correlates with growth of small hepatocellular carcinoma. International Journal of Oncology. 2000;17(2):237–243. doi: 10.3892/ijo.17.2.237. [DOI] [PubMed] [Google Scholar]
- 119.Roderfeld M, Rath T, Lammert F, Dierkes C, Graf J, Roeb E. Innovative immunohistochemistry identifies MMP-9 expressing macrophages at the invasive front of murine HCC. World Journal of Hepatology. 2010;2(5):175–179. doi: 10.4254/wjh.v2.i5.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kohga K, Tatsumi T, Takehara T, et al. Expression of CD133 confers malignant potential by regulating metalloproteinases in human hepatocellular carcinoma. Journal of Hepatology. 2010;52(6):872–879. doi: 10.1016/j.jhep.2009.12.030. [DOI] [PubMed] [Google Scholar]
- 121.Littlepage LE, Sternlicht MD, Rougier N, et al. Matrix metalloproteinases contribute distinct roles in neuroendocrine prostate carcinogenesis, metastasis, and angiogenesis progression. Cancer Research. 2010;70(6):2224–2234. doi: 10.1158/0008-5472.CAN-09-3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yu Q, Stamenkovic I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes and Development. 1999;13(1):35–48. doi: 10.1101/gad.13.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Takafuji V, Forgues M, Unsworth E, Goldsmith P, Wang XW. An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene. 2007;26(44):6361–6371. doi: 10.1038/sj.onc.1210463. [DOI] [PubMed] [Google Scholar]
- 124.Zhang H, Ye QH, Ren N, et al. The prognostic significance of preoperative plasma levels of osteopontin in patients with hepatocellular carcinoma. Journal of Cancer Research and Clinical Oncology. 2006;132(11):709–717. doi: 10.1007/s00432-006-0119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ramaiah SK, Rittling S. Reviews: pathophysiological role of osteopontin in hepatic inflammation, toxicity, and cancer. Toxicological Sciences. 2008;103(1):4–13. doi: 10.1093/toxsci/kfm246. [DOI] [PubMed] [Google Scholar]
- 126.Kim J, Ki SS, Lee SD, et al. Elevated plasma osteopontin levels in patients with hepatocellular carcinoma. American Journal of Gastroenterology. 2006;101(9):2051–2059. doi: 10.1111/j.1572-0241.2006.00679.x. [DOI] [PubMed] [Google Scholar]
- 127.Ye QH, Qin LX, Forgues M, et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nature Medicine. 2003;9(4):416–423. doi: 10.1038/nm843. [DOI] [PubMed] [Google Scholar]
- 128.Leng J, Han C, Demetris AJ, Michalopoulos GK, Wu T. Cyclooxygenase-2 promotes hepatocellular carcinoma cell growth through Akt activation: evidence for Akt inhibition in celecoxib-induced apoptosis. Hepatology. 2003;38(3):756–768. doi: 10.1053/jhep.2003.50380. [DOI] [PubMed] [Google Scholar]
- 129.Cervello M, Foderà D, Florena AM, et al. Correlation between expression of cyclooxygenase-2 and the presence of inflammatory cells in human primary hepatocellular carcinoma: possible role in tumor promotion and angiogenesis. World Journal of Gastroenterology. 2005;11(30):4638–4643. doi: 10.3748/wjg.v11.i30.4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zheng Q, Tang ZY, Xue Q, Shi DR, Song HY, Tang HB. Invasion and metastasis of hepatocellular carcinoma in relation to urokinase-type plasminogen activator, its receptor and inhibitor. Journal of Cancer Research and Clinical Oncology. 2000;126(11):641–646. doi: 10.1007/s004320000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 132.Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441(7092):431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 133.Bonizzi G, Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends in Immunology. 2004;25(6):280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 134.Luedde T, Beraza N, Kotsikoris V, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11(2):119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 135.Sakurai T, He G, Matsuzawa A, et al. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14(2):156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Saccani A, Schioppa T, Porta C, et al. p50 nuclear factor-κB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Research. 2006;66(23):11432–11440. doi: 10.1158/0008-5472.CAN-06-1867. [DOI] [PubMed] [Google Scholar]
- 137.Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Research. 2005;65(8):3437–3446. doi: 10.1158/0008-5472.CAN-04-4262. [DOI] [PubMed] [Google Scholar]
- 138.Hagemann T, Lawrence T, McNeish I, et al. “Re-educating” tumor-associated macrophages by targeting NF-κB. The Journal of Experimental Medicine. 2008;205(6):1261–1268. doi: 10.1084/jem.20080108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hagemann T, Biswas SK, Lawrence T, Sica A, Lewis CE. Regulation of macrophage function in tumors: the multifaceted role of NF-κB. Blood. 2009;113(14):3139–3146. doi: 10.1182/blood-2008-12-172825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? The Lancet. 2001;357(9255):539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 141.Mantovani A, Sica A, Locati M. New vistas on macrophage differentiation and activation. European Journal of Immunology. 2007;37(1):14–16. doi: 10.1002/eji.200636910. [DOI] [PubMed] [Google Scholar]
- 142.Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Letters. 2008;267(2):204–215. doi: 10.1016/j.canlet.2008.03.028. [DOI] [PubMed] [Google Scholar]
- 143.Calvisi DF. Dr. Jekyll and Mr. Hyde: a paradoxical oncogenic and tumor suppressive role of signal transducer and activator of transcription 3 in liver cancer. Hepatology. 2011;54(1):9–12. doi: 10.1002/hep.24435. [DOI] [PubMed] [Google Scholar]
- 144.Dalwadi H, Krysan K, Heuze-Vourc’h N, et al. Cyclooxygenase-2-dependent activation of signal transducer and activator of transcription 3 by interleukin-6 in non-small cell lung cancer. Clinical Cancer Research. 2005;11(21):7674–7682. doi: 10.1158/1078-0432.CCR-05-1205. [DOI] [PubMed] [Google Scholar]
- 145.Kortylewski M, Kujawski M, Wang T, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nature Medicine. 2005;11(12):1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 146.Kortylewski M, Xin H, Kujawski M, et al. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell. 2009;15(2):114–123. doi: 10.1016/j.ccr.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nature Reviews Immunology. 2007;7(1):41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 148.He G, Karin M. NF-κB and STAT3- key players in liver inflammation and cancer. Cell Research. 2011;21(1):159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Calvisi DF, Ladu S, Gorden A, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130(4):1117–1128. doi: 10.1053/j.gastro.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 150.Wurmbach E, Chen YB, Khitrov G, et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 2007;45(4):938–947. doi: 10.1002/hep.21622. [DOI] [PubMed] [Google Scholar]
- 151.He G, Yu GY, Temkin V, et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17(3):286–297. doi: 10.1016/j.ccr.2009.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yoshida T, Ogata H, Kamio M, et al. SOCS1 is a suppressor of liver fibrosis and hepatitis-induced carcinogenesis. The Journal of Experimental Medicine. 2004;199(12):1701–1707. doi: 10.1084/jem.20031675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Riehle KJ, Campbell JS, McMahan RS, et al. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. The Journal of Experimental Medicine. 2008;205(1):91–103. doi: 10.1084/jem.20070820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ogata H, Kobayashi T, Chinen T, et al. Deletion of the SOCS3 gene in liver parenchymal cells promotes hepatitis-induced hepatocarcinogenesis. Gastroenterology. 2006;131(1):179–193. doi: 10.1053/j.gastro.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 155.Liu Y, Liu A, Xu Z, et al. XZH-5 inhibits STAT3 phosphorylation and causes apoptosis in human hepatocellular carcinoma cells. Apoptosis. 2011;16(5):502–510. doi: 10.1007/s10495-011-0578-0. [DOI] [PubMed] [Google Scholar]
- 156.Li WC, Ye SL, Sun RX, et al. Inhibition of growth and metastasis of human hepatocellular carcinoma by antisense oligonucleotide targeting signal transducer and activator of transcription 3. Clinical Cancer Research. 2006;12(23):7140–7148. doi: 10.1158/1078-0432.CCR-06-0484. [DOI] [PubMed] [Google Scholar]
- 157.Yan W, Chang Y, Liang X, et al. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology. 2012;55(6):1863–1875. doi: 10.1002/hep.25572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nature Reviews Cancer. 2008;8(8):618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 159.Weigert A, Brüne B. Nitric oxide, apoptosis and macrophage polarization during tumor progression. Nitric Oxide—Biology and Chemistry. 2008;19(2):95–102. doi: 10.1016/j.niox.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 160.Rius J, Guma M, Schachtrup C, et al. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α . Nature. 2008;453(7196):807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104(8):2224–2234. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- 162.Knowles HJ, Mole DR, Ratcliffe PJ, Harris AL. Normoxic stabilization of hypoxia-inducible factor-1α by modulation of the labile iron pool in differentiating U937 macrophages: effect of natural resistance-associated macrophage protein 1. Cancer Research. 2006;66(5):2600–2607. doi: 10.1158/0008-5472.CAN-05-2351. [DOI] [PubMed] [Google Scholar]
- 163.Schioppa T, Uranchimeg B, Saccani A, et al. Regulation of the chemokine receptor CXCR4 by hypoxia. The Journal of Experimental Medicine. 2003;198(9):1391–1402. doi: 10.1084/jem.20030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nature Medicine. 2004;10(8):858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 165.Cramer T, Yamanishi Y, Clausen BE, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Doedens AL, Stockmann C, Rubinstein MP, et al. Macrophage expression of hypoxia-inducible factor-1α suppresses T-cell function and promotes tumor progression. Cancer Research. 2010;70(19):7465–7475. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Imtiyaz HZ, Williams EP, Hickey MM, et al. Hypoxia-inducible factor 2α regulates macrophage function in mouse models of acute and tumor inflammation. The Journal of Clinical Investigation. 2010;120(8):2699–2714. doi: 10.1172/JCI39506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.WeiXing C, Tiantian H, Qun N, Chaohui Y, Ping X. Inhibitory effect of Hypoxia inducible factor-1 antisense oligonucleotide on growth of human hepatocellular carcinoma cells. Medical Oncology. 2008;25(1):88–92. doi: 10.1007/s12032-007-0050-8. [DOI] [PubMed] [Google Scholar]
- 169.Liu F, Wang P, Jiang X, et al. Antisense hypoxia-inducible factor 1α gene therapy enhances the therapeutic efficacy of doxorubicin to combat hepatocellular carcinoma. Cancer Science. 2008;99(10):2055–2061. doi: 10.1111/j.1349-7006.2008.00905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. The Lancet Oncology. 2009;10(1):25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 171.Cheng A-L, Guan Z, Chen Z, et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma according to baseline status: Subset analyses of the phase III Sorafenib Asia-Pacific trial. European Journal of Cancer. 2012;48(10):1452–1465. doi: 10.1016/j.ejca.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 172.Zhang W, Zhu XD, Sun HC, et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clinical Cancer Research. 2010;16(13):3420–3430. doi: 10.1158/1078-0432.CCR-09-2904. [DOI] [PubMed] [Google Scholar]