

Abstract
Pemphigus vulgaris (PV) is a mucocutaneous blistering disease characterized by IgG autoantibodies against the stratified squamous epithelium. Current understanding of PV pathophysiology does not explain the mechanism of acantholysis in patients lacking desmoglein antibodies, which justifies a search for novel targets of pemphigus autoimmunity. We tested 264 pemphigus and 138 normal control sera on the multiplexed protein array platform containing 701 human genes encompassing many known keratinocyte cell-surface molecules and members of protein families targeted by organ-non-specific PV antibodies. The top 10 antigens recognized by the majority of test patients’ sera were proteins encoded by the DSC1, DSC3, ATP2C1, PKP3, CHRM3, COL21A1, ANXA8L1, CD88 and CHRNE genes. The most common combinations of target antigens included at least one of the adhesion molecules DSC1, DSC3 or PKP3 and/or the acetylcholine receptor CHRM3 or CHRNE with or without the MHC class II antigen DRA. To identify the PV antibodies most specific to the disease process, we sorted the data based on the ratio of patient to control frequencies of antigen recognition. The frequency of antigen recognition by patients that exceeded that of control by 10 and more times were the molecules encoded by the CD33, GP1BA, CHRND, SLC36A4, CD1B, CD32, CDH8, CDH9, PMP22 and HLA-E genes as well as mitochondrial proteins encoded by the NDUFS1, CYB5B, SOD2, PDHA1 and FH genes. The highest specificity to PV showed combinations of autoantibodies to the calcium pump encoded by ATP2C1 with C5a receptor plus DSC1 or DSC3 or HLA-DRA. The results identified new targets of pemphigus autoimmunity. Novel autoantibody signatures may help explain individual variations in disease severity and treatment response, and serve as sensitive and specific biomarkers for new diagnostic assays in PV patients.
Introduction
Pemphigus vulgaris (PV) is a mucocutaneous blistering disease characterized by IgG autoantibodies against stratified squamous epithelium. PV antibodies demonstrate epithelial cell-surface staining by indirect immunofluorescence (IIF), and, because this staining appears between cells, initially the antibodies were described as “intercellular” antibodies [1], [2]. Although the incidence of PV is only 1 to 16 per million population per year [3], [4], this disease represents a significant burden to health care professionals, and the health care system. Systemic administration of glucocorticosteroid hormones is essential to establish control of disease during the acute stage [5]. While glucocorticosteroid treatment is life saving, it may cause severe side effects, including death [6], [7]. The development of non-steroidal treatment has been hampered by a lack of clear understanding of the mechanisms leading to keratinocyte detachment in PV.
During the last decade, the studies of autoimmune responses in PV have been supplemented and, to some extent, replaced by analyzing the levels of antibodies to desmoglein (Dsg) 3 by enzyme linked immunosorbent assay (ELISA) representing a hallmark and a diagnostic criterion of PV [8]. However, Dsg 3 antibody levels do not always correspond to the presence of cell-surface antibodies by IIF or correlate with disease activity [9], [10], [11] or predict relapse of the disease [12]. Furthermore, anti-Dsg antibodies can be absent in the active stage of disease but present in PV patients during remission [13], [14], [15], [16], [17], [18], patients with unrelated medical conditions, and healthy subjects, including relatives of PV patients [17], [19], [20], [21], [22], [23], [24], [25], [26]. For example, 16 PV patients positive for cell-surface antibodies by IIF had normal Dsg 3 antibody levels [27].
Identification of proteins targeted by autoantibodies in PV is a subject of intense research. The first evidence that keratinocyte antigens other than Dsg 1 and Dsg 3 are pathophysiologically relevant was provided by experiments showing the ability to induce suprabasal acantholysis and gross skin blisters in Dsg3−/− neonatal mice by passive transfer of PV antibodies [28]. In this model, murine epidermis lacks Dsg 3 and the passively transferred PV IgG lacks Dsg 1 antibody. Hence, the injected PV antibodies cause blisters by targeting non-Dsg 1 and Dsg 3 keratinocyte antigens. Current understanding, however, does not adequately explain the mechanism of acantholysis in patients lacking Dsg 1 and 3 antibodies. Furthermore, results of a recent study indicate that autoreactivity in PV relies on somatic mutations generated in response to an antigen unrelated to Dsg 3 [29]. Taken together, these facts justify a search for novel targets of pemphigus autoimmunity.
In general, autoimmune diseases are characterized by the presence of multiple types of autoantibodies mediating a coordinated immunological attack against a fraction of the tissue proteome. For example, 116 autoantibodies were described in patients with systemic lupus erythematous [30]. The number of targeted self- antigens varies dramatically from patient to patient. Therefore, multiplex analysis of autoantibody responses against a spectrum of candidate antigens represents a powerful screening tool to delineate biomarker signatures in autoimmunity, allowing elucidation of the overall autoimmune process rather than individual components [31]. The availability of multiplex technologies has made possible the simultaneous detection of several different autoantibodies overcoming some of the limitations of conventional methods [32]. For instance, antigen arrays proved to be 4- to 8-fold more sensitive than conventional ELISA analyses for detection of autoantibodies specific for some autoantigens [33]. Thus, autoantibody profiling may serve purposes including classification of individual patients and subsets of patients based on their “autoantibody fingerprint,” examination of epitope spreading and antibody isotype usage, discovery and characterization of candidate autoantigens, and tailoring antigen-specific therapy [34], [35].
Historically, studies of autoimmune responses had been conducted by analyzing the presence and/or concentration of single antibodies in biological fluids using conventional immunoassays, such as ELISA, radioimmunoassay, immunoblot, and others. More recently, antigen microarrays have been constructed and validated for over a dozen autoimmune diseases, including connective-tissue diseases, primary biliary cirrhosis, experimental autoimmune encephalomyelitis, multiple sclerosis, rheumatoid arthritis, diabetes, Crohn’s disease and sclerosing cholangitis [36]. Previous reports demonstrated feasibility of the protein microarray as a laboratory tool allowing parallel analysis against hundreds of different antigens in minimal serum quantities (less than 2 µL). We pioneered the use of multiplexed protein array platforms to evaluate PV autoantibody profiles [37]. In our previous study, the sera from acute PV patients and healthy donors were probed using the microarray containing self-antigens characteristic of the organ-non-specific autoimmune disorders, such as rheumatoid arthritis, lupus erythematosus, scleroderma, diabetes and some other autoimmune disorders [37]. The results identified the presence of several non-organ specific antibodies, but the relatively small sample size did not allow determination of their prevalence in PV patients. Most recently, some of these new autoantibodies were validated in an independent proteomic study [38], indicating reliability of identifying novel disease-specific autoantibodies in PV through multiplexed parallel testing.
To elucidate the immunopathological mechanisms underlying keratinocyte detachment in PV, we designed a multiplexed protein array platform encompassing most of known keratinocyte cell-surface molecules as well as members of protein families targeted by organ-non-specific PV antibodies. In the present study, we utilized this specialized microarray to test a large number of PV patient and control sera to define the proteome targeted by PV autoimmunity. As predicted by the multiple hit hypothesis [39], an in-depth analysis of PV sera revealed new self-antigens and identified specific patterns of differentially reacting autoantibodies. Such novel autoantibody signatures may help explain individual variations of disease severity and response to treatment in PV patients, and serve as sensitive and specific biomarkers for new diagnostic assays.
Materials and Methods
Test Sera
We tested 264 PV patient and 138 normal serum specimens. Patient specimens were selected based on IIF results showing the presence of cell-surface antibodies staining stratified squamous epithelial substrate (human skin and/or monkey esophagus purchased from California National Primate Research Center, Davis, CA; http://www.cnprc.ucdavis.edu/) in the characteristic pemphigus pattern at a serum titer of 1∶40 concentration and higher. The Dsg3 antibody level was measured using the MESACUP Dsg3 ELISA test system (MBL International Corp., Nagoya, Japan). Patient specimens were de-identified prior to testing. As controls, we used sera collected from healthy donors at University of California Irvine and normal human sera purchased from Bioreclamation, Inc. (Westbury, NY). This research has been approved by Institutional Review Boards (IRB) at University of California Irvine. Participants provided their written informed consent on the IRB-apprroved consent forms to participate in this study.
Microarray Design and Printing
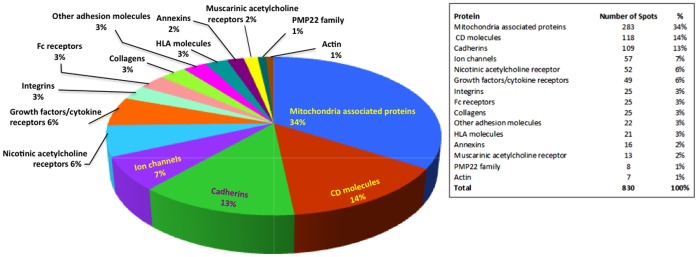
Protein microarray construction consisted of four-steps: (i) PCR amplification of each complete or partial open reading frame (ORF); (ii) in vivo recombination cloning; (iii) in vitro transcription/translation; and (iv) microarray chip printing. ORFs were obtained from the National Institutes of Health Mammalian Gene Collection clones (Invitrogen, Carlsbad, CA and MCLAB, San Francisco, CA), and amplified using custom polymerase chain reaction (PCR) primers comprising 20 base pairs of gene-specific sequences with 15 base pairs of “adapter” sequences. The size of amplified genes ranged from 246 to 6786 base pair. ORFs >4,000 base pairs were cloned as 3 overlapping segments. The adapter sequences, which became incorporated into the termini flanking the amplified gene, were homologous to the cloning site of the linearized T7 expression vector pXT7 allowing the PCR products to be cloned by in vivo homologous recombination in competent DH5α cells [40], [41]. The resulting fusion protein also incorporated a 5′ polyhistidine epitope, an ATG translation start codon, and a 3′ hemagglutinin epitope and T7 terminator. Plasmids were expressed in 5 h in vitro transcription/translation reactions (rapid translation system, RTS, 100 E. coli HY kits; Roche, Switzerland) according to the manufacturer’s instructions. Protein expression was monitored by microarray using monoclonal anti-polyhistidine (clone His-1; Sigma Chemical Co., St Louis, MO) and anti-hemagglutinin (clone 3F10; Roche). Microarrays were printed onto Whatman nitrocellulose coated glass FAST slides using an Omni Grid 100 microarray printer (Genomic Solutions, UK). Each microarray chip consisted of proteins representing 701 unique human genes and their transcript variants, and peptides of the Dsg 1 and Dsg 3 extracellular domains 1–5 (Table S1; Fig. 1 ). The microarrays also included the following controls: (i) the “no DNA” negative control, in which an empty plasmid vector was placed in the RTS reaction; (ii) serially diluted human IgG (a positive control); and (iii) serially diluted EBNA-1 (another positive control, given the high prevalence of latent Epstein Barr viral infection).
Figure 1. Composition of the multiplexed protein array platform.
Antibody Assays and Reading of Microarrays
Prior to array staining, experimental and control serum samples were diluted to 1/25 in Whatman’s Protein Array Blocking Buffer containing Escherichia coli lysate at a final concentration of 30% and incubated at room temperature for 1 h with constant mixing. The arrays were rehydrated in the blocking buffer for 30 min that was replaced with preabsorbed test sera and incubated overnight at 4°C with constant agitation. The slides were washed five times in 10 mM Tris (pH 8.0)-150 mM NaCl containing 0.05% Tween 20 buffer, and bound antibodies were detected after 1 h incubation in Biotin-SP conjugated affinity purified human goat secondary antibody to IgG Fc (Fc-γ fragment specific) (Jackson Immunoresearch, West Grove, PA) diluted 1/200 in blocking buffer. The slides were washed three times, and bound antibodies detected by 1 h incubation with StreptAvidin conjugated with the dye PBXL-3 as tertiary antibody (streptavidin-conjugated SureLight® P-3) (Columbia Biosciences Corporation, Columbia, MD) diluted 1/200 in blocking buffer. After being washed three times, the slides were air-dried under brief centrifugation and stored at 18°C in a desiccator. The arrays were examined with a Perkin Elmer ScanArray Express HT apparatus at a wavelength of 670 nm and intensities were quantified using ProScanArray Express software (Perkin Elmer, Waltham, MA). All signal intensities were corrected for spot-specific background.
Data Analysis
Signal intensity was obtained from ScanArray and background signal was corrected by subtracting “no DNA” negative control plus 2 standard deviations (SD) from the signals in each antigen. This standardized signal was used to determine positives in response to antigen on array. The positives were defined as the signal intensity higher than the average signal intensity of control group plus 2 SD. A two-tailed unpaired Student’s t-test was used to verify the significance of differences between the groups, and p values less than 0.05 were considered significant. The microarray’s ability to detect disease-related antibodies was evaluated based on the ratio of positive patient vs. control sera. JMP Software (SAS Institute Inc., Cary, NC) was used to perform the principal component analysis as detailed elsewhere [42].
Results
Self-antigens Targeted by Pemphigus Antibodies
Analyses of the data revealed positive reactions of both PV patient and control sera with a large number of peptides included in the microarray (Table S1). The specificity was determined by the frequency of antigen recognition by PV sera. To identify the PV antibodies most specific to the disease process, we re-sorted the data based on the ratio of patient to control frequencies of antigen recognition in the microarray. The unsupervised principal component analysis demonstrated that the top 30 antigens recognized by PV antibodies with the highest sensitivity or top 15 antigens recognized with the highest specificity were clearly distinct from controls ( Fig. 2 ). These results indicated that PV features a unique serological profile that distinguishes pemphigus autoimmunity from normal immune response.
Figure 2. Principal component analysis of top antigens.

Unsupervised principal component analysis of the signal intensity for samples from PV patients and healthy controls revealed that these two groups could be segregated on the basis of top 30 antigens with the highest sensitivity (A) and 15 antigens with the highest specificity (B) for PV.
The sensitivity analysis ( Fig. 3 ) demonstrated that the majority of patients’ sera targeted the DR α chain of the class II major histocompatibility complex (MHC) encoded by the human leukocyte antigen (HLA)-DRA gene (45% PV patients), desmocollin 1 and desmocollin 3 (DSC1 and DSC3, respectively; 44% each), ATPase, Ca++ transporting, type 2C, member 1 (ATP2C1; 43%), plakophilin 3 (PKP3; 43%), M3 subtype of muscarinic acetylcholine receptor (AChR) (CHRM3; 42%), collagen α1, type XXI, (COL21A1; 42%), annexin A8-like 1 molecule (ANXA8L1; 42%), complement component 5a receptor 1 (CD88; 42%) and ε subunit of nicotinic AChR (CHRNE; 41%).
Figure 3. Sensitivity of the microarray in detecting disease-related autoantibodies.
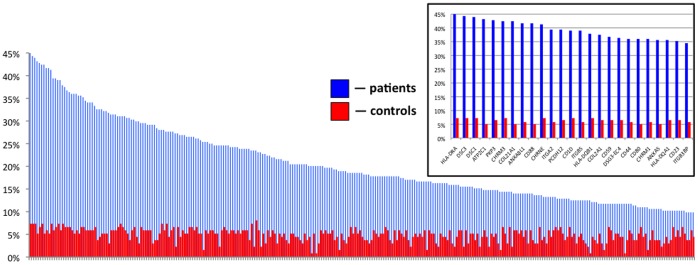
Sorted by percent of positive samples in the group of antigens recognized by 10 percent and more of PV patients. Inset: 25 top antigens.
The specificity analysis ( Fig. 4 ) demonstrated that self-antigens recognized by PV antibodies with the frequency that exceeded that of control by 10 and more times included the cell-surface molecules sialic acid-binding immunoglobulin-like lectin 3 (CD33; ratio = 27.7) and glycoprotein Ibα (GP1BA; 27.7), δ subunit of nicotinic AChR (CHRND; 17.6), proton-coupled amino acid transporter 4 (SLC36A4; 17.3), the antigen-presenting protein CD1B (13.1), Fc-fragment of IgG (CD32; 12.5), cadherins 8 (CDH8; 11.3) and 9 (CDH9; 11.5), peripheral myelin protein 22 (PMP22; 11.0), the MHC class I molecule E (HLA-E; 10.8) and the mitochondrial proteins NADH-ubiquinone oxidoreductase (NDUFS1; 16.2), cytochrome b5 outer mitochondrial membrane isoform precursor (CYB5B; 13.1), superoxide dismutase (SOD2; 10.6), α subunit of pyruvate dehydrogenase E1 component (PDHA1; 10.3) and fumarate hydratase (FH; 10.1).
Figure 4. Specificity of the microarray in revealing disease-specific PV antibodies.

Sorted by ratio of positive patient/control samples in the group of antigens recognized by two and more time frequently by PV patient than control sera. Inset: 25 top antigens.
Non-Dsg 3 Antibodies Produced by the Dsg 3 Antibody-positive PV Patients
Although sera from all 264 patients demonstrated a pemphigus cell-surface staining pattern by IIF, only 183 (69%) of them recognized at least one Dsg 3 peptide in the microarray (Table S1). This was not surprising, because Amagai’s group reported that only 60 to 75 percent of PV patients develop autoantibodies recognizing recombinant Dsg 3 proteins produced in a bacterial expression system [43]. In our study, the Dsg 3 antibody-negative patient sera likely were from PV patients who did not have detectable levels of Dsg 3 antibody, which is not uncommon [16], and/or pemphigus foliaceus (PF) patients, since both PV and PF antibodies demonstrate an indistinguishable IIF staining pattern [21], [44], [45]. Therefore, to extrapolate our results specifically to the immunopathology of PV, we separately determined antigen reaction patterns of the Dsg 3 antibody-containing patient sera (Table S2). The antigen reactivity pattern of the Dsg 3 antibody-positive sera was similar to that of the entire collection of patients’ sera (Table S1). Most importantly, the top 10 most frequently recognized self-antigens were identical, but displayed a slightly different order within the group ( Table 1 ). Therefore, the microarray results of the entire serum collection were representative of PV autoimmunity.
Table 1. Top 10 antigens most frequently recognized by PV antibodies.
| Symbol | Name | % of all patient samples | % of Dsg3-antibody positive samples |
| HLA-DRA | major histocompatibility complex, class II, DR α chain | 45 | 53 |
| DSC3 | desmocollin 3 isoform Dsc3a preproprotein | 44 | 54 |
| DSC1 | desmocollin 1 isoform Dsc1a preproprotein | 44 | 54 |
| ATP2C1 | ATPase, Ca++ transporting, type 2C, member 1 | 43 | 50 |
| PKP3 | plakophilin 3 | 43 | 53 |
| CHRM3 | cholinergic muscarinic receptor type 3 | 42 | 51 |
| COL21A1 | collagen, type XXI, α1 | 42 | 50 |
| ANXA8L1 | annexin A8-like 1 | 42 | 49 |
| CD88 | complement component 5a receptor 1 | 42 | 50 |
| CHRNE | nicotinic acetylcholine receptor ε subunit | 41 | 50 |
Combinations of most Common Individual Antigens Targeted by Antibodies in PV Patients
The top 10 most commonly recognized antigens ( Table 1 ) were subjected to an association analysis. We combined two and more from the top 10 most commonly recognized antigens and calculated the number of patient and control sera that contained antibodies against those combined antigens, then computed the ratio of patient over control sera positivity. The most common combinations of target antigens included at least one of the adhesion molecules DSC1, DSC3 and PKP3 and/or the AChR CHRM3 or CHRNE with or without the MHC class II antigen DRA ( Fig. 5A ). The highest specificity to PV showed combinations of antibodies to the calcium pump encoded by ATP2C1 with CD88 (C5a receptor) plus DSC1 or DSC3 or HLA-DRA ( Fig. 5B ).
Figure 5. Combinations of top 10 most common individual antigens targeted by PV antibodies.

A, Top 25 combinations sorted by percent of positive samples. B, Top 25 antigen sorted by the patient/control ratio.
Discussion
In this study, we used sera from a large cohort of pemphigus patients to identify novel targets for PV antibodies. We applied a protein microarray approach to characterize the autoantibody response profile associated with the disease process. This approach has been employed broadly to identify differentially reactive, serodiagnostic antigens against infectious agents, and is now also being used in autoimmune diseases. In the past, our group has extensively characterized disease-associated antibody profiles in large cohorts of healthy and infected individuals (e.g., [42], [46]), and also initiated the proteomic analysis of pemphigus autoimmunity [37]. The pilot study of PV sera demonstrated that the reactivities of novel PV antibodies correlate closely with the Dsg 3 antibody levels [37]. In the present study, we used polypeptides representing different regions of Dsg 3 because, at variance with early studies [47], [48], it has been recently demonstrated that the Dsg 3 autoantibody response in PV is polyclonal [29]. We also tested autoantibody reactivities with other proteins implicated in PV pathogenesis, such as AChRs [39].
We sought to reconcile the knowledge about important role of Dsg 3 antibody in PV with the evidence that Dsg 3 is not essential for keratinocyte adhesion and Dsg 3 antibody is not requisite for acantholysis (reviewed in [49]). The results revealed new targets of pemphigus autoimmunity, and demonstrated variability of the autoantibody profile among different PV patients. We identified both the most common and the most specific autoantibodies directed against members of the cell adhesion molecule and the AChR families, which are known targets in pemphigus, as well as a number of new organ-specific and non-specific targets from previously unsuspected protein families. Based on these data, it is apparent that the immunopathology of PV is complex and variable, inducing alterations in vital keratinocyte functions due to a simultaneous hits by an array of autoantibodies of different antigenic specificities. Targeting of non-Dsg molecules potentially exacerbates the pathogenic effects of Dsg 3 antibody. Our new PV autoantibody database should provide a roadmap to navigate pemphigus research toward new disease pathways and treatment approaches.
An important role of the conformational epitope of self-antigens in PV has been vividly demonstrated in the studies of the recombinant Dsg 3 species that were raised in different expression systems and had various posttranslational modifications [43], [50], [51], [52]. An inherent drawback of the high-throughput approach applied here is that not all proteins on the array likely are folded in the same way as in human cells, and/or that they are not post-translationally modified in the same way. Consequently, they may not display all possible antigenic epitopes. This potential limitation, however, did not prevent us from reaching the main objective of our study, which was identification of new pathogenic PV autoantibodies that may work together with Dsg 3 antibodies to disrupt keratinocyte adhesion. Hundreds of autoantigens were found to be significantly more reactive with PV than control sera. Importantly, the top antigenic targets significantly separated PV patients from healthy individuals, demonstrating strong sensitivity and specificity. This observation indicated that the two approaches to microarray data analysis can be used together. The proteomic approach, therefore, serves as a starting point to “rule in,” but not necessarily “rule out,” individual self-antigens or their combinations that contribute to the autoimmune response.
The results indicate that acantholysis in PV likely derives from simultaneous inactivation of several physiological pathways maintaining keratinocyte adhesion, as predicted by the “multiple hit” hypothesis of pemphigus pathophysiology [39]. In addition to Dsg 3, other keratinocyte adhesion molecules are presumably destroyed or inactivated by PV antibodies. Notably, previous results showing that chimeric proteins containing the extracellular epitope of Dsg 1 or Dsg 3 combined with the Fc portion of human IgG absorbed out all disease-causing pemphigus IgGs [50], [53], [54] should be interpreted with caution, because both this and previous [37] proteomics studies demonstrated that PV patients produce autoantibodies against Fc-IgG.
Our results are consistent with previous reports that desmocollins [55], [56] and classical cadherins [57], [58] also are targeted by pemphigus antibodies. The significance of the autoantibody targeting DSC3 in PV is underscored by the evidence that: (i) the DSC3 loss of function mouse model exhibits phenotypic similarity to PV [59], (ii) adsorption with DSC3 can eliminate acantholytic activity of PV IgG [60], and (iii) DSC3 monoclonal antibody causes intraepidermal blistering in in vitro model of human skin and loss of cell-cell adhesion in keratinocyte cultures [61]. Indeed, production of certain autoantibodies against cell adhesion molecules and structural proteins, such as the intracellular desmosomal plaque protein, plakophilin 3, and the collagen α1, type XXI, may be secondary to acantholysis, as discussed in detail elsewhere [62].
The high percentage of patients’ sera reacting with AChRs of the muscarinic and/or the nicotinic classes is in keeping with previous reports that downstream signaling from these receptors regulates keratinocyte cell-cell adhesion through physiological control of phosphorylation/dephosphorylation of desmosomal and classical cadherins [63]. Blocking AChRs expressed on keratinocytes leads to disassembly of desmosomal and adherence junctions due to phosphorylation of key adhesion molecules. Our early reports that PV and PF patients develop antibodies to keratinocyte AChRs [28], [64] and that cholinomimetic drugs can ameliorate pemphigus [65] have been recently corroborated by new clinical and laboratory data. AChR antibodies in PV patients correlate with disease extent at the time of diagnosis and during follow-up [66], and, in a patient with bipolar disorder, cholinolytic drugs worsen PF [67].
The high reactivity of PV sera with the annexin A8-like molecule was not surprising either, because it had been reported that PV patients develop antibodies to different annexins [68]. Probing of keratinocyte λgt11 cDNA library with the PV IgG eluted from a 75 kD band that stained epidermis in a pemphigus-like cell-surface pattern and caused acantholysis in keratinocyte monolayers revealed a novel type of AChRs, termed pemphaxin (a.k.a. annexin 9) [69]. Annexin A8 is specifically expressed in adult stratified epithelia [70], where it may participate in the organization of certain actin associated membrane domains and regulate late endosome organization and function [71]. By analogy to annexins 1, 2, 3 and 9 that act as non-professional AChRs [69], [72], the annexin A8-like molecule also may mediate acetylcholine signaling and, thus, be involved in regulation of keratinocyte shape and adhesion. This possibility needs further investigation.
The microarray results from this study demonstrate that mitochondrial antibodies are highly specific to the PV sera. The presence of antibodies against mitochondrial proteins in PV and PF patients had been reported [73], [74]. Mitochondrial antibodies are apparently pathogenic because their absorption abolishes the ability of PV IgG to cause acantholysis both in vitro and in vivo [74]. Based on the known functions of targeted proteins, the following mitochondrial pathways may be subject to dysfunction: tricarboxylic acid cycle, oxidative phosphorylation, O2 respiration, and production/inactivation of reactive oxygen species (ROS). The mitochondrial dysfunction in PV has been directly or indirectly suggested by an increase of lipid peroxidation, reflecting an increased production of ROS [75], [76], and the peroxidant-antioxidant balance, measuring oxidative stress [77], and activation of the mitochondria-dependent intrinsic apoptotoc pathway in keratinocytes exposed to PV IgG [74], [78], [79]. At this point, however, it remains unclear how PV mitochondrial antibodies enter keratinocytes. A cell-surface protein may act as a surrogate for the nominal mitochondrial antigen. For example, since annexins can relocate to the cytosol reaching mitochondria [80], mitochondrial antibodies may enter keratinocytes bound to annexins. Additionally or alternatively, instead of binding to an antigen on the plasma membrane, an antibody may be internalized in a complex with Fc receptors expressed on keratinocytes [81], [82].
Of particular interest is a discovery in PV of autoimmunity against members of the PMP (peripheral myelin protein)-22/gas3 family termed PMP-22 and PERP (p53 apoptosis effector related to PMP-22). The high specificity of PMP22 antibody demonstrated in this study confirms our previous microarray results [37]. A relatively high prevalence of autoantibody against the structurally related PERP (31% positive patients, 5% controls), a tetraspan membrane protein originally identified as an apoptosis-associated target of the p53 tumor suppressor, was not surprising either. The relevance of anti-PERP autoimmunity to the pathophysiology of PV is underscored by the fact that PERP knockout mice display a phenotypic similarity to PV [83]. Perp−/− mice die within the first week of life as a result of severe adhesion defects and blistering of the skin and oral mucosa. Further, they exhibit highly abnormal desmosomes by electron microscopy. Dissolution of desmosomes and PV-like intraepidermal split in Perp−/− mice may result from aberrant inside-out signaling along the altered cell death pathways, because PERP is involved in the extrinsic apoptotic pathway, playing a “death receptor” role [84].
An interesting and unexpected discovery from this study is the PV autoantibody targeting the human secretory pathway Ca2+/Mn2+-ATPase, or hSPCA1, encoded by the ATP2C1 gene located on chromosome 3. One copy of this gene is mutated in patients with Hailey–Hailey disease (a.k.a. familial benign chronic pemphigus) exhibiting the clinical-and-pathological features resembling very closely PV [85], [86]. Both Hailey-Hailey disease keratinocytes [87] and normal keratinocytes treated with PV antibodies [88] exhibit altered intracellular calcium metabolism that can lead to abnormal cell-cell adhesion [89].
This microarray study also revealed previously unrecognized autoantibodies to a number of proteins known to subserve various vital functions in cell types other than keratinocytes. The findings suggest that these cells contribute to the pathophysiology of PV and/or that the targeted proteins also function in keratinocytes. The highest specificities to PV demonstrated autoantibodies to the cell-surface molecules, sialic acid-binding immunoglobulin-like lectin 3 and glycoprotein Ibα, both of which are known to be involved in cell adhesion [90], [91].
The complement component 5a receptor 1 (a.k.a. C5a receptor) is a G protein-coupled transmembrane protein [92]. Although contribution of the complement system to the pathophysiology of pemphigus is controversial [93], [94], clinical evidence suggests that it plays a pathogenic role [95]. Hence, if autoantibodies against C5a receptor inhibit the complement cascade, they may play a protective role. Alternatively, complement activation by anti-C5a receptor may facilitate acantholysis.
Autoantibodies against the antigen-presenting protein CD1B, and MHC class I and II molecules may interfere with normal functioning of the skin immune system [38]. They may also directly affect keratinocyte shape and adhesion. Incubation of skin explant culture with HLA-A, -B and -C alloantibodies has been reported to produce keratinocyte detachment and structural disorganization similar to that found in cultures treated with PV antibodies [96].
Another protein targeted by disease-specific PV antibodies, proton-coupled amino acid transporter 4, coded by the gene SLC36A4, is a widely distributed member of the solute carrier family. It is a high-affinity transporter for proline and tryptophan [97]. In the past, we have reported that PV antibody targets a novel antigen similar to the taurine transporter that controls cellular size and water content [98], but we have much to learn about transporter proteins in keratinocyte biology and pemphigus pathophysiology.
We believe that a simultaneous and, perhaps, synchronized inactivation of the physiological pathways regulating and mediating keratinocyte adhesive function is required to disrupt the most important phylogenetic function of tegumental cells, such as integrity of epidermal barrier. To further explore the “polypathogenic” nature of pemphigus autoimmunity, we looked for the most common combinations of individual autoantibodies associated with Dsg 3 autoimmunity in PV. The results demonstrated that synergy may stem from functional cooperation of antibodies to distinct proteins mediating the same biologic function, such as heterophilic trans-interactions of desmogleins and desmocollins within the desmosome [99]. Simultaneous blockade of both desmosomal protein partners would distort cell-cell attachment in epidermis more efficiently, compared to interference with cis-interactions of single-type desmosomal proteins suggested by the “monopathogenic” theory of pemphigus pathophysiology [8]. Individual variations within the constellations of pathogenic antibodies targeting molecules that mediate and regulate keratinocyte adhesion likely determine the magnitude of the “multiple hit” attack required to disrupt the integrity of epidermis in a particular PV patient and explain the clinical and immunopathological variability of PV.
In conclusion, considerable progress in elucidation of the immunopathology of pemphigus can be achieved through the use of proteomic technology enabling a large-scale characterization of immune responses against self-antigens that may be involved in development and progression of the disease process in PV. Thus, autoantibody profiling using antigen arrays is well-positioned to become an anchor technology for the development of multiplex autoantibody-based biomarker assays for use in management of pemphigus patients.
Supporting Information
Reactivities of patient and control sera on protein micoarrays.
(PDF)
Reactivities of Dsg3-positive patients’ sera on protein micoarrays.
(PDF)
Acknowledgments
The late J-C Bystryn, M.D., contributed to this study by providing 50 patient serum specimens that were stored at Beutner Laboratories (3580 Harlem Road, Buffalo, NY 1421) and subsequently shipped to Dr. Grando’s laboratory.
Funding Statement
The authors have no support or funding to report.
References
- 1. Beutner EH, Lever WF, Witebsky E, Jordon R, Chertock B (1965) Autoantibodies in Pemphigus Vulgaris: Response to an Intercellular Substance of Epidermis. JAMA 192: 682–688. [DOI] [PubMed] [Google Scholar]
- 2. Anderson HJ, Newcomer VD, Landau JW, Rosenthal LH (1970) Pemphigus and other diseases. Results of indirect intercellular immunofluorescence. Arch Dermatol 101: 538–546. [PubMed] [Google Scholar]
- 3. Pisanti S, Sharav Y, Kaufman E, Posner LN (1974) Pemphigus vulgaris: incidence in Jews of different ethnic groups, according to age, sex, and initial lesion. Oral Surg Oral Med Oral Pathol 38: 382–387. [DOI] [PubMed] [Google Scholar]
- 4. Chams-Davatchi C, Valikhani M, Daneshpazhooh M, Esmaili N, Balighi K, et al. (2005) Pemphigus: analysis of 1209 cases. International Journal of Dermatology 44: 470–476. [DOI] [PubMed] [Google Scholar]
- 5. Carson PJ, Hameed A, Ahmed AR (1996) Influence of treatment on the clinical course of pemphigus vulgaris. Journal of the American Academy of Dermatology 34: 645–652. [DOI] [PubMed] [Google Scholar]
- 6. Ahmed AR, Moy R (1982) Death in pemphigus. Journal of the American Academy of Dermatology 7: 221–228. [DOI] [PubMed] [Google Scholar]
- 7. Rosenberg FR, Sanders S, Nelson CT (1976) Pemphigus: a 20-year review of 107 patients treated with corticosteroids. Archives of Dermatology 112: 962–970. [DOI] [PubMed] [Google Scholar]
- 8. Amagai M, Stanley JR (2012) Desmoglein as a target in skin disease and beyond. J Invest Dermatol 132: 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lambert LL, Spriet E, Vandewiele A, Naeyaert J (2006) Desmoglein 1 and 3 IgG auto-antibody titers do not correlate with pemphigus disease activity in a prospective study. J Invest Dermatol 126 Suppl. 1 11 (Abstract 65)..16417211 [Google Scholar]
- 10. Abasq C, Mouquet H, Gilbert D, Tron F, Grassi V, et al. (2009) ELISA testing of anti-desmoglein 1 and 3 antibodies in the management of pemphigus. Arch Dermatol 145: 529–535. [DOI] [PubMed] [Google Scholar]
- 11. Kamiya K, Aoyama Y, Shirafuji Y, Hamada T, Morizane S, et al. (2012) Detection of antibodies against the non-calcium-dependent epitopes of desmoglein 3 in pemphigus vulgaris and their pathogenic significance. British Journal of Dermatology 167: 252–261. [DOI] [PubMed] [Google Scholar]
- 12. Akman A, Uzun S, Alpsoy E (2010) Immunopathologic features of pemphigus in the east Mediterranean region of Turkey: a prospective study. Skinmed 8: 12–16. [PubMed] [Google Scholar]
- 13. Arin MJ, Engert A, Krieg T, Hunzelmann N (2005) Anti-CD20 monoclonal antibody (rituximab) in the treatment of pemphigus. Br J Dermatol 153: 620–625. [DOI] [PubMed] [Google Scholar]
- 14. Kwon EJ, Yamagami J, Nishikawa T, Amagai M (2008) Anti-desmoglein IgG autoantibodies in patients with pemphigus in remission. J Eur Acad Dermatol Venereol 22: 1070–1075. [DOI] [PubMed] [Google Scholar]
- 15. Sharma VK, Prasad HR, Khandpur S, Kumar A (2006) Evaluation of desmoglein enzyme-linked immunosorbent assay (ELISA) in Indian patients with pemphigus vulgaris. Int J Dermatol 45: 518–522. [DOI] [PubMed] [Google Scholar]
- 16. Belloni-Fortina A, Faggion D, Pigozzi B, Peserico A, Bordignon M, et al. (2009) Detection of autoantibodies against recombinant desmoglein 1 and 3 molecules in patients with pemphigus vulgaris: correlation with disease extent at the time of diagnosis and during follow-up. Clin Dev Immunol 2009: 187864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Khandpur S, Sharma VK, Sharma A, Pathria G, Satyam A (2010) Comparison of enzyme-linked immunosorbent assay test with immunoblot assay in the diagnosis of pemphigus in Indian patients. Indian J Dermatol Venereol Leprol 76: 27–32. [DOI] [PubMed] [Google Scholar]
- 18. Horvath B, Huizinga J, Pas HH, Mulder AB, Jonkman MF (2012) Low-dose rituximab is effective in pemphigus. British Journal of Dermatology 166: 405–412. [DOI] [PubMed] [Google Scholar]
- 19. Brandsen R, Frusic-Zlotkin M, Lyubimov H, Yunes F, Michel B, et al. (1997) Circulating pemphigus IgG in families of patients with pemphigus: comparison of indirect immunofluorescence, direct immunofluorescence, and immunoblotting. Journal of the American Academy of Dermatology 36: 44–52. [DOI] [PubMed] [Google Scholar]
- 20. Kricheli D, David M, Frusic-Zlotkin M, Goldsmith D, Rabinov M, et al. (2000) The distribution of pemphigus vulgaris-IgG subclasses and their reactivity with desmoglein 3 and 1 in pemphigus patients and their first-degree relatives. Br J Dermatol 143: 337–342. [DOI] [PubMed] [Google Scholar]
- 21. Torzecka JD, Narbutt J, Sysa-Jedrzejowska A, Waszczykowska E, Lukamowicz J, et al. (2003) Detection of pemphigus autoantibodies by IIF and ELISA tests in patients with pemphigus vulgaris and foliaceus and in healthy relatives. Med Sci Monit 9: CR528–533. [PubMed] [Google Scholar]
- 22. Torzecka JD, Wozniak K, Kowalewski C, Waszczykowska E, Sysa-Jedrzejowska A, et al. (2007) Circulating pemphigus autoantibodies in healthy relatives of pemphigus patients: coincidental phenomenon with a risk of disease development? Archives for Dermatological Research Archiv fur Dermatologische Forschung 299: 239–243. [DOI] [PubMed] [Google Scholar]
- 23. Hilario-Vargas J, Dasher DA, Li N, Aoki V, Hans-Filho G, et al. (2006) Prevalence of anti-desmoglein-3 antibodies in endemic regions of Fogo Selvagem in Brazil. J Invest Dermatol 126: 2044–2048. [DOI] [PubMed] [Google Scholar]
- 24. Yoshimura T, Seishima M, Nakashima K, Yasuhara Y, Adachi S, et al. (2001) Increased antibody levels to desmogleins 1 and 3 after administration of carbamazepine. Clin Exp Dermatol 26: 441–445. [DOI] [PubMed] [Google Scholar]
- 25. Gallo R, Massone C, Parodi A, Guarrera M (2002) Allergic contact dermatitis from thiurams with pemphigus-like autoantibodies. Contact Dermatitis 46: 364–365. [DOI] [PubMed] [Google Scholar]
- 26.Cozzani E, Rosa GM, Drosera M, Intra C, Barsotti A, et al.. (2010) ACE inhibitors can induce circulating antibodies directed to antigens of the superficial epidermal cells. Archives for Dermatological Research Archiv fur Dermatologische Forschung. [DOI] [PubMed]
- 27. Zagorodniuk I, Weltfriend S, Shtruminger L, Sprecher E, Kogan O, et al. (2005) A comparison of anti-desmoglein antibodies and indirect immunofluorescence in the serodiagnosis of pemphigus vulgaris. Int J Dermatol 44: 541–544. [DOI] [PubMed] [Google Scholar]
- 28. Nguyen VT, Lee TX, Ndoye A, Shultz LD, Pittelkow MR, et al. (1998) The pathophysiological significance of non-desmoglein targets of pemphigus autoimmunity. Pemphigus vulgaris and foliaceus patients develop antibodies against keratinocyte cholinergic receptors. Archives of Dermatology 134: 971–980. [DOI] [PubMed] [Google Scholar]
- 29.Di Zenzo G (2012) Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Invest doi:101172/JCI64413. [DOI] [PMC free article] [PubMed]
- 30. Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y (2004) Autoantibody explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin Arthritis Rheum 34: 501–537. [DOI] [PubMed] [Google Scholar]
- 31. Hueber W, Robinson WH (2006) Proteomic biomarkers for autoimmune disease. Proteomics 6: 4100–4105. [DOI] [PubMed] [Google Scholar]
- 32. Tozzoli R (2008) The diagnostic role of autoantibodies in the prediction of organ-specific autoimmune diseases. Clin Chem Lab Med 46: 577–587. [DOI] [PubMed] [Google Scholar]
- 33. Robinson WH, DiGennaro C, Hueber W, Haab BB, Kamachi M, et al. (2002) Autoantigen microarrays for multiplex characterization of autoantibody responses. Nat Med 8: 295–301. [DOI] [PubMed] [Google Scholar]
- 34. Hueber W, Utz PJ, Steinman L, Robinson WH (2002) Autoantibody profiling for the study and treatment of autoimmune disease. Arthritis Res 4: 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Plebani M, Pittoni M, Celadin M, Bernardi D, Mion MM (2009) Recent advances in diagnostic technologies for autoimmune diseases. Autoimmun Rev 8: 238–243. [DOI] [PubMed] [Google Scholar]
- 36. Fathman CG, Soares L, Chan SM, Utz PJ (2005) An array of possibilities for the study of autoimmunity. Nature 435: 605–611. [DOI] [PubMed] [Google Scholar]
- 37. Kalantari-Dehaghi M, Molina DM, Farhadieh M, John Morrow W, Liang X, et al. (2011) New targets of pemphigus vulgaris antibodies identified by protein array technology. Experimental Dermatology 20: 154–156. [DOI] [PubMed] [Google Scholar]
- 38. Sinha AA (2012) Constructing immunoprofiles to deconstruct disease complexity in pemphigus. Autoimmunity 45: 36–43. [DOI] [PubMed] [Google Scholar]
- 39. Grando SA (2000) Autoimmunity to keratinocyte acetylcholine receptors in pemphigus. Dermatology (Basel) 201: 290–295. [DOI] [PubMed] [Google Scholar]
- 40. Davies DH, Liang X, Hernandez JE, Randall A, Hirst S, et al. (2005) Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc Natl Acad Sci U S A 102: 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luevano M, Bernard HU, Barrera-Saldana HA, Trevino V, Garcia-Carranca A, et al.. (2010) High-throughput profiling of the humoral immune responses against thirteen human papillomavirus types by proteome microarrays. Virology. [DOI] [PMC free article] [PubMed]
- 42. Liang L, Tan X, Juarez S, Villaverde H, Pablo J, et al. (2011) Systems biology approach predicts antibody signature associated with Brucella melitensis infection in humans. J Proteome Res 10: 4813–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Amagai M, Karpati S, Prussick R, Klaus-Kovtun V, Stanley JR (1992) Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. Journal of Clinical Investigation 90: 919–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Judd KP, Mescon H (1979) Comparison of different epithelial substrates useful for indirect immunofluorescence testing of sera from patients with active pemphigus. Journal of Investigative Dermatology 72: 314–316. [DOI] [PubMed] [Google Scholar]
- 45. Jiao D, Bystryn JC (1997) Sensitivity of indirect immunofluorescence, substrate specificity, and immunoblotting in the diagnosis of pemphigus. J Am Acad Dermatol 37: 211–216. [DOI] [PubMed] [Google Scholar]
- 46. Vigil A, Davies DH, Felgner PL (2010) Defining the humoral immune response to infectious agents using high-density protein microarrays. Future Microbiology 5: 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Payne AS, Ishii K, Kacir S, Lin C, Li H, et al. (2005) Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest 115: 888–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yamagami J, Takahashi H, Ota T, Amagai M (2008) Genetic characterization of human Dsg3-specific B cells isolated by flow cytometry from the peripheral blood of patients with pemphigus vulgaris. J Dermatol Sci 52: 98–107. [DOI] [PubMed] [Google Scholar]
- 49. Grando SA (2012) Pemphigus autoimmunity: hypotheses and realities. Autoimmunity 45: 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Amagai M, Hashimoto T, Shimizu N, Nishikawa T (1994) Absorption of pathogenic autoantibodies by the extracellular domain of pemphigus vulgaris antigen (Dsg3) produced by baculovirus. Journal of Clinical Investigation 94: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Amagai M, Ishii K, Hashimoto T, Gamou S, Shimizu N, et al. (1995) Conformational epitopes of pemphigus antigens (Dsg1 and Dsg3) are calcium dependent and glycosylation independent. Journal of Investigative Dermatology 105: 243–247. [DOI] [PubMed] [Google Scholar]
- 52. Amagai M, Ishii K, Takayanagi A, Nishikawa T, Shimizu N (1996) Transport to endoplasmic reticulum by signal peptide, but not proteolytic processing, is required for formation of conformational epitopes of pemphigus vulgaris antigen (Dsg3). Journal of Investigative Dermatology 107: 539–542. [DOI] [PubMed] [Google Scholar]
- 53. Amagai M, Hashimoto T, Green KJ, Shimizu N, Nishikawa T (1995) Antigen-specific immunoadsorption of pathogenic autoantibodies in pemphigus foliaceus. Journal of Investigative Dermatology 104: 895–901. [DOI] [PubMed] [Google Scholar]
- 54. Amagai M, Nishikawa T, Nousari HC, Anhalt GJ, Hashimoto T (1998) Antibodies against desmoglein 3 (pemphigus vulgaris antigen) are present in sera from patients with paraneoplastic pemphigus and cause acantholysis in vivo in neonatal mice. Journal of Clinical Investigation 102: 775–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dmochowski M, Hashimoto T, Garrod DR, Nishikawa T (1993) Desmocollins I and II are recognized by certain sera from patients with various types of pemphigus particularly Brazilian pemphigus foliaceus. Journal of Investigative Dermatology 100: 380–384. [DOI] [PubMed] [Google Scholar]
- 56. Dmochowski M, Hashimoto T, Chidgey MAJ, Yue KKM, Wilkinson RW, et al. (1995) Demonstration of antibodies to bovine desmocollin isoforms in certain pemphigus sera. British Journal of Dermatology 133: 519–525. [DOI] [PubMed] [Google Scholar]
- 57. Evangelista F, Dasher DA, Diaz LA, Prisayanh PS, Li N (2008) E-cadherin is an additional immunological target for pemphigus autoantibodies. J Invest Dermatol 128: 1710–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flores G, Culton DA, Prisayanh P, Qaqish BF, James K, et al.. (2012) IgG Autoantibody Response against Keratinocyte Cadherins in Endemic Pemphigus Foliaceus (Fogo Selvagem). J Invest Dermatol. [DOI] [PMC free article] [PubMed]
- 59. Chen J, Den Z, Koch PJ (2008) Loss of desmocollin 3 in mice leads to epidermal blistering. Journal of Cell Science 121: 2844–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, et al. (2010) Autoimmunity to desmocollin 3 in pemphigus vulgaris. American Journal of Pathology 177: 2724–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Spindler V, Heupel WM, Efthymiadis A, Schmidt E, Eming R, et al. (2009) Desmocollin 3-mediated binding is crucial for keratinocyte cohesion and is impaired in pemphigus. Journal of Biological Chemistry 284: 30556–30564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Amagai M, Ahmed AR, Kitajima Y, Bystryn JC, Milner Y, et al. (2006) Are desmoglein autoantibodies essential for the immunopathogenesis of pemphigus vulgaris, or just ‘witnesses of disease’? Exp Dermatol 15: 815–831. [DOI] [PubMed] [Google Scholar]
- 63. Chernyavsky AI, Arredondo J, Piser T, Karlsson E, Grando SA (2008) Differential coupling of M1 muscarinic and α7 nicotinic receptors to inhibition of pemphigus acantholysis. J Biol Chem 283: 3401–3408. [DOI] [PubMed] [Google Scholar]
- 64. Nguyen VT, Ndoye A, Grando SA (2000) Novel human α9 acetylcholine receptor regulating keratinocyte adhesion is targeted by pemphigus vulgaris autoimmunity. Am J Pathol 157: 1377–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nguyen VT, Arredondo J, Chernyavsky AI, Pittelkow MR, Kitajima Y, et al. (2004) Pemphigus vulgaris acantholysis ameliorated by cholinergic agonists. Arch Dermatol 140: 327–334. [DOI] [PubMed] [Google Scholar]
- 66. Tirado-Sanchez A, Vazquez-Gonzalez D, Ponce-Olivera RM, Lopez-Lozano HE (2012) Acetylcholine receptor antibodies in patients with pemphigus vulgaris: correlation with disease extent at the time of diagnosis and during follow-up. Dermatol Online J 18: 14. [PubMed] [Google Scholar]
- 67. Botelho-Nogueira L, Quarantini L, Miranda-Scippa A (2011) Use of anticholinergic drugs and worsening of pemphigus foliaceus in a patient with bipolar disorder. Rev Bras Psiquiatr 33: 412–413. [DOI] [PubMed] [Google Scholar]
- 68. Bastian BC, Nuss B, Romisch J, Kraus M, Brocker EB (1994) Autoantibodies to annexins: a diagnostic marker for cutaneous disorders? Journal of Dermatological Science 8: 194–202. [DOI] [PubMed] [Google Scholar]
- 69. Nguyen VT, Ndoye A, Grando SA (2000) Pemphigus vulgaris antibody identifies pemphaxin–a novel keratinocyte annexin-like molecule binding acetylcholine. J Biol Chem 275: 29466–29476. [DOI] [PubMed] [Google Scholar]
- 70. Runkel F, Michels M, Franken S, Franz T (2006) Specific expression of annexin A8 in adult murine stratified epithelia. J Mol Histol 37: 353–359. [DOI] [PubMed] [Google Scholar]
- 71. Goebeler V, Poeter M, Zeuschner D, Gerke V, Rescher U (2008) Annexin A8 regulates late endosome organization and function. Molecular Biology of the Cell 19: 5267–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zimmerman UJ, Hennigan BB, Liu L, Campbell CH, Fisher AB (1995) Annexins I, II and III are specific choline binding proteins. Biochemistry and Molecular Biology International 35: 307–315. [PubMed] [Google Scholar]
- 73.Geoghegan W, Jordon R (1992) Anti-mitochondrial IgG1 subclass antibodies in pemphigus foliaceus. Journal of Investigative Dermatology 98: 589, Abstr. #225.
- 74. Marchenko S, Chernyavsky AI, Arredondo J, Gindi V, Grando SA (2010) Antimitochondrial autoantibodies in pemphigus vulgaris: a missing link in disease pathophysiology. J Biol Chem 285: 3695–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yesilova Y, Ucmak D, Selek S, Dertlioglu SB, Sula B, et al.. (2012) Oxidative stress index may play a key role in patients with pemphigus vulgaris. J Eur Acad Dermatol Venereol. [DOI] [PubMed]
- 76. Naziroglu M, Kokcam I, Simsek H, Karakilcik AZ (2003) Lipid peroxidation and antioxidants in plasma and red blood cells from patients with pemphigus vulgaris. J Basic Clin Physiol Pharmacol 14: 31–42. [DOI] [PubMed] [Google Scholar]
- 77. Yazdanpanah MJ, Ghayour-Mobarhan M, Taji A, Javidi Z, Pezeshkpoor F, et al. (2011) Serum zinc and copper status in Iranian patients with pemphigus vulgaris. International Journal of Dermatology 50: 1343–1346. [DOI] [PubMed] [Google Scholar]
- 78. Arredondo J, Chernyavsky AI, Karaouni A, Grando SA (2005) Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in pemphigus. Am J Pathol 167: 1531–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gil MP, Modol T, Espana A, Lopez-Zabalza MJ (2012) Inhibition of FAK prevents blister formation in the neonatal mouse model of pemphigus vulgaris. Experimental Dermatology 21: 254–259. [DOI] [PubMed] [Google Scholar]
- 80. Rainteau D, Mansuelle P, Rochat H, Weinman S (1995) Characterization and ultrastructural localization of annexin VI from mitochondria. FEBS Lett 360: 80–84. [DOI] [PubMed] [Google Scholar]
- 81. Tigalonowa M, Bjerke JR, Livden JK, Matre R (1990) The distribution of Fc gamma RI, Fc gamma RII and Fc gamma R III on Langerhans’ cells and keratinocytes in normal skin. Acta Derm Venereol 70: 385–390. [PubMed] [Google Scholar]
- 82. Lim PL, Zouali M (2006) Pathogenic autoantibodies: emerging insights into tissue injury. Immunol Lett 103: 17–26. [DOI] [PubMed] [Google Scholar]
- 83. Ihrie RA, Marques MR, Nguyen BT, Horner JS, Papazoglu C, et al. (2005) Perp is a p63-regulated gene essential for epithelial integrity. Cell 120: 843–856. [DOI] [PubMed] [Google Scholar]
- 84. Davies L, Gray D, Spiller D, White MR, Damato B, et al. (2009) P53 apoptosis mediator PERP: localization, function and caspase activation in uveal melanoma. J Cell Mol Med 13: 1995–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hu Z, Bonifas JM, Beech J, Bench G, Shigihara T, et al. (2000) Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet 24: 61–65. [DOI] [PubMed] [Google Scholar]
- 86. Sudbrak R, Brown J, Dobson-Stone C, Carter S, Ramser J, et al. (2000) Hailey-Hailey disease is caused by mutations in ATP2C1 encoding a novel Ca(2+) pump. Hum Mol Genet 9: 1131–1140. [DOI] [PubMed] [Google Scholar]
- 87. Behne MJ, Tu CL, Aronchik I, Epstein E, Bench G, et al. (2003) Human keratinocyte ATP2C1 localizes to the Golgi and controls Golgi Ca2+ stores. J Invest Dermatol 121: 688–694. [DOI] [PubMed] [Google Scholar]
- 88. Seishima M, Esaki C, Osada K, Mori S, Hashimoto T, et al. (1995) Pemphigus IgG, but not bullous pemphigoid IgG, causes a transient increase in intracellular calcium and inositol 1,4,5-triphosphate in DJM-1 cells, a squamous cell carcinoma line. Journal of Investigative Dermatology 104: 33–37. [DOI] [PubMed] [Google Scholar]
- 89. Kitajima Y (2002) Mechanisms of desmosome assembly and disassembly. Clin Exp Dermatol 27: 684–690. [DOI] [PubMed] [Google Scholar]
- 90. Ware J, Russell S, Ruggeri ZM (1997) Cloning of the murine platelet glycoprotein Ibalpha gene highlighting species-specific platelet adhesion. Blood Cells Mol Dis 23: 292–301. [DOI] [PubMed] [Google Scholar]
- 91. Taylor VC, Buckley CD, Douglas M, Cody AJ, Simmons DL, et al. (1999) The myeloid-specific sialic acid-binding receptor, CD33, associates with the protein-tyrosine phosphatases, SHP-1 and SHP-2. Journal of Biological Chemistry 274: 11505–11512. [DOI] [PubMed] [Google Scholar]
- 92. Gerard C, Gerard NP (1994) C5A anaphylatoxin and its seven transmembrane-segment receptor. Annu Rev Immunol 12: 775–808. [DOI] [PubMed] [Google Scholar]
- 93. Jordon RE, Kawana S, Fritz KA (1985) Immunopathologic mechanisms in pemphigus and bullous pemphigoid. J Invest Dermatol 85: 72s–78s. [DOI] [PubMed] [Google Scholar]
- 94. Lessey E, Li N, Diaz L, Liu Z (2008) Complement and cutaneous autoimmune blistering diseases. Immunol Res 41: 223–232. [DOI] [PubMed] [Google Scholar]
- 95. Kawana S, Geoghegan WD, Jordon RE, Nishiyama S (1989) Deposition of the membrane attack complex of complement in pemphigus vulgaris and pemphigus foliaceus skin. J Invest Dermatol 92: 588–592. [DOI] [PubMed] [Google Scholar]
- 96. Vermeer BJ, Kardaun SH, Koerten HK, Claas FH, Ploem JS (1985) Model for detection of membrane-associated antigens on epithelial cells (HLA-A, -B, -C alloantigens and pemphigus antigens). Br J Dermatol 113 Suppl 28 118–123. [DOI] [PubMed] [Google Scholar]
- 97. Thwaites DT, Anderson CM (2011) The SLC36 family of proton-coupled amino acid transporters and their potential role in drug transport. British Journal of Pharmacology 164: 1802–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Grando SA (2006) Pemphigus in the XXI Century: New life to an old story. Autoimmunity 39: 521–530. [DOI] [PubMed] [Google Scholar]
- 99. Desai BV, Harmon RM, Green KJ (2009) Desmosomes at a glance. J Cell Sci 122: 4401–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Reactivities of patient and control sera on protein micoarrays.
(PDF)
Reactivities of Dsg3-positive patients’ sera on protein micoarrays.
(PDF)