

Abstract
Standard microbial evolutionary ontology is organized according to a nested hierarchy of entities at various levels of biological organization. It typically detects and defines these entities in relation to the most stable aspects of evolutionary processes, by identifying lineages evolving by a process of vertical inheritance from an ancestral entity. However, recent advances in microbiology indicate that such an ontology has important limitations. The various dynamics detected within microbiological systems reveal that a focus on the most stable entities (or features of entities) over time inevitably underestimates the extent and nature of microbial diversity. These dynamics are not the outcome of the process of vertical descent alone. Other processes, often involving causal interactions between entities from distinct levels of biological organisation, or operating at different time scales, are responsible not only for the destabilisation of pre-existing entities, but also for the emergence and stabilisation of novel entities in the microbial world. In this article we consider microbial entities as more or less stabilised functional wholes, and sketch a network-based ontology that can represent a diverse set of processes including, for example, as well as phylogenetic relations, interactions that stabilise or destabilise the interacting entities, spatial relations, ecological connections, and genetic exchanges. We use this pluralistic framework for evaluating (i) the existing ontological assumptions in evolution (e.g. whether currently recognized entities are adequate for understanding the causes of change and stabilisation in the microbial world), and (ii) for identifying hidden ontological kinds, essentially invisible from within a more limited perspective. We propose to recognize additional classes of entities that provide new insights into the structure of the microbial world, namely “processually equivalent” entities, “processually versatile” entities, and “stabilized” entities.
Keywords: Ontology, Microbial evolution, Process philosophy, Tree of life, Network
Introduction: biological ontology
Fundamental to any scientific view of the natural world is an ontology: a view of the kinds of things, their most important properties and capacities, and their typical interactions, that constitute the domain of nature under consideration. Ultimately we assume that such an ontology must be empirically grounded. However, the central role that ontological assumptions play in the articulation of the scientific investigation of a domain is such that they are not easily questioned. They are deep within the Quinean web of belief, or, to adapt a phrase from (Wimsatt 2007) to a different purpose, generatively entrenched. Empirical results that appear to threaten this basic ontology are liable to be reinterpreted or treated with suspicion. It can easily seem that the central ontological assumptions are a priori foundations for the scientific domain. Most fundamental to the ontological framework of a scientific field will be categories of entities and the kinds of relations that hold between these entities. So, for example, the basic ontology of chemistry might be atoms and the bonds that occur between atoms. Needless to say, things will be more complicated and diverse for the life sciences.
As a quick preliminary, we should mention our understanding of what makes an entity ‘real’ as opposed to a mere artefact of our representation. We assume that real entities are those that have causal powers; complex entities are real if they have causal powers that are not merely aggregates of the causal powers of their parts. Organisms, for example, can do things that none of their parts can manage on their own. Similarly functional proteins have capacities—catalytic, structural, etc.—that are not exhibited by any of the amino acids of which they are composed. We don’t want to commit to any particular account of causation; perhaps ‘having a causal power’ means just ‘making a difference to something else’.1 Making a difference to something is a minimal necessary condition for being the kind of entity we have any interest in recognising in formulating a biological ontology.
A more abstract metaphysical distinction is also central to our thinking about biological ontology (and perhaps ontology generally). We understand living things to be most fundamentally the consequences of numerous interweaving (occasionally nested) processes. Although it is common to describe the domain of biology as consisting of things, for example organisms, cells, genes, and so on, we understand even these as ultimately processual. As recent thinking in evolutionary biology, notably the rapidly growing field of evolutionary developmental biology (evo-devo) has emphasised, an organism is a developmental process. When we use a set of properties to describe the adult state of an organism, perhaps for taxonomic purposes, we are abstracting a particular time slice from this developmental process. For these reasons, the ontology we aim to describe is an ontology of processes. A processual ontology should characterize entities in terms of how they emerge, are maintained and are stabilized. As evolution is uncontroversially a process, an evolutionary ontology will quite naturally be processual. An evolutionary ontology of the living world should distinguish the real evolutionary players, the units with causal powers resulting from or contributing to evolutionary processes.
A further premise of our argument, which leads directly from the preceding point, is that the naturalness with which we see the biological world as composed of relatively stable things needs to be explained in terms of a variety of processes that stabilise these entities. Such processes range from the multiple homeostatic mechanisms that maintain metabolisms within viable parameters, to the stabilising natural selection that maintains the viability of a population across generations. This will be discussed in more detail below. Here we just reiterate that what we are inclined to think of as biological things are, on more careful inspection, specific temporal stages of stabilised biological processes. Furthermore, since these stabilising processes take place at very different time scales—from many thousands of years for the stabilising selection of a metazoan lineage, to as little as milliseconds for the stabilisation of a functional macromolecule—whether an entity appears as thing-like will depend on the time scale with which we are concerned. Again, these ideas will be taken up in much more detail below.
While we believe that the argument of this paper would apply equally to the ontogeny and phylogeny of multicellular organisms, in this paper we will focus on the evolutionary ontology of the microbial world, where its application is most clear-cut. By ‘microbes’ we mean what are generally referred to as unicellular organisms: the prokaryotes, Bacteria and Archaea; a wide variety of protists and fungi; and subcellular entities such as viruses and plasmids. This restriction does still leave us with about 80 % of the history of life and the vast majority of entities that have existed in the more recent 20 %. The complexity of microbes, we would add, is often underappreciated. Microbes commonly engage in multicellular and multilineage organisations, such as multispecies biofilms, in which microbial cells undergo cellular differentiation, and exhibit some form of division of labor (Ghigo 2001; Hall-Stoodley et al. 2004; Reisner et al. 2006; Ereshefsky and Pedroso 2012). So the limitation of our analysis to the microbial realm is a minor one.2
Evolutionary ontology: the standard model
The standard evolutionary ontology of biology is hierarchical. Starting with the intuitively central category of organisms, one can work downwards through a hierarchy of organs, cells, organelles, and molecules. This is a hierarchy in which entities at each level are constituents of entities at the next higher level. Moreover it is possible to move upwards from the organism to the level of species, which are themselves widely considered to be concrete individual entities of which organisms are the constituent parts (Hull 1989). It has also been thought that this hierarchy is the key to a proper scientific epistemology, since the scientific understanding of entities at one level should be a consequence of discovering the properties of its constituents at the next lower level. This reductionist view, however, has become highly controversial and is anyhow not the topic of this essay. Here we are concerned merely with how this standard evolutionary ontology, based on a genealogy emphasizing vertical inheritance, has had a profound influence on the structure of evolutionary thinking. The kind of thinking we have in mind is most distinctively represented by the construction of genealogical trees, whether of species, organisms, cells, genomes, genes, or whatever else.
Central to the tree model are the assumptions that through evolutionary time entities replicate themselves (or are replicated), and that this process of replication produces the reproductively linked sequences of similar entities that constitute lineages. These lineages, then, are everywhere constrained within the limits of the branch of the tree in which they are located. With what is generally considered the minor exception of hybrids (Heliconius Genome Consortium 2012), lineages of genes move between different organisms through reproduction and sexual recombination, but only so long as the destination organism is located within the same branch. The whole branch can be seen as an entity held together (at least for organisms to which this applies) by the sexually mediated flow of genes between organisms. Intermediate entities such as cells or genomes are simply carried along by their organismic hosts, and for cladists, at least, the tree of species is just a higher level representation of this same tree of organisms.
We certainly do not mean to deny that this evolutionary ontology has been extremely productive for many parts of biology. It has, for example, justified and guided the search for more representatives of the classes it recognises, notably genes, organisms and species. Just one example would be the discrimination of morphologically very similar sibling species on the basis of genetic separation (Mayr 1963). More recently it has been used to justify the use of lower level entities as proxies for investigating the structure and history of the biological world as constituted by higher level entities. For example, the use of genetic analysis to map the phylogenetic history of metazoa has provided many insights. But it must be noted that only in so far as lineages of genes and lineages of species really are constrained within the same branch of the tree are inferences from the history of genes to the history of species legitimate. This condition implies that there are severe limits to the model, and areas, for example phylogenetic analysis of prokaryotes, where is has proved less effective (Bapteste et al. 2009).
Consequently, we will argue that in spite of its major contribution to evolutionary studies, the tree picture has also provided us with some problematic ontological entities, e.g. classes that are not causally real processes. In particular, it has often encouraged the identification of evolutionary players—in our understanding the units with causal powers either resulting from or contributing to evolutionary processes—with phylogenetic species, and clades. But while clades have a historical coherence, it is not clear that they constitute entities with causal powers of their own. Hence, a processual evolutionary perspective should provide something quite distinct from the standard evolutionary ontology, by distinguishing the emergence and stabilization of objects irrespective of any a priori partitions between distinct levels of biological organisation (as when interactions between levels contribute to the emergence/stabilization of new entities during evolution), and irrespective of possibly artificial genealogical partitions (as when attention solely to genealogical lineages ignores the origin and sustainability of associations between multiple lineages into a functional unit).
Central to the standard model, and also central to the argument of this paper, is the concept of a lineage. We will not attempt to offer any detailed analysis of this important concept, but one uncontentious aspect is that it refers to sequences of more or less similar entities over generally long periods of time. Sequences often thought of as lineages could be of organisms, cells, genes, or genomes. But temporal successions of organs, proteins, or indeed anything that occurs regularly in an organism, could also be identified and might be considered as lineages. We shall make just two points about such potential lineages. First, the processes that maintain and stabilise these sequences are quite diverse. Certainly there must be such processes to sustain a lineage, processes that explain the constant reproduction of very similar entities, but we should not assume there is anything common to these processes beyond their capacity to produce this outcome. Second, some lineages are physically embedded within lineages of more complex entities. Thus lineages of livers are entirely embedded in lineages of metazoan organisms. This is, no doubt, the reason why lineages of livers do not attract much theoretical attention. If livers could occasionally evolve in a manner spatially uncoupled from the whole that they partially compose, they would be of great interest, no matter whether they reproduce by themselves or are reproduced as a byproduct of the reproduction of some containing entity (i.e. whether they are simple reproducers or scaffolded reproducers (Godfrey-Smith 2009)). Gene lineages are precisely such embedded, yet evolutionarily potentially independent, lineages in microbial organisms. When laterally transferred genes encode for molecular “organs” (Forterre 2010), organs, too, can move from one bacterium to another (e.g. a flagellum can be introduced into a nonmotile bacterium (Diene et al. 2012)).
Like an organism, an organ such as the bacterial flagellum requires particular genes to be reproduced, and mutations in these genes are physical marks of possible transformations of this organ. Such mutations will track the lineage of flagella. When such genes transfer laterally from one cell to another, the evolution of the lineage of flagella as tracked by these mutations is uncoupled from that of the lineage of its previous carrier, and thus becomes a potential object of study in its own right: its evolution is now partly autonomous. If a particular flagellar organisation lasted longer than the bacterial species in which it was first evolved it, or any other part with a lifespan uncoupled from that of its embedding host, would have an evolutionary history distinct not only from that of its original host, but also from any subsequent host.
In the microbial world, lineages of genes, or genomes, are not fully embedded in lineages of organisms, or even of species; lineages of genes can be independent from lineages of genomes. The interweaving of independent though sometimes coincident lineages will be central to the general picture we hope to present. These will include, in addition to those mentioned, lineages of mobile acellular genetic elements such as viruses and plasmids, and lineages of symbiotic communities of interacting organisms, though the complexity of the processes that sustain the latter may make their lineage formation less central to the analysis. This interacting multiplicity of kinds of lineage, with distinct stabilisation time scales and different degrees of obligate physical connection, introduces important limitations to the standard model that our alternative presentation of an extended evolutionary ontology aims to address.
Problems with the standard model
The starting point for our understanding of microbial ontology is that the tree of life as conventionally understood, far from being a universal framework, is a model of limited usefulness for comprehending the microbial world. This is primarily because of the phenomenon of lateral gene transfer (LGT): transfer of genes between often very different kinds of organisms or, in the present context, cells (Bapteste et al. 2009). What this phenomenon implies is that, contrary to the simple ontological vision embodied in the tree of life, the origins of the genetic components that are found in a biological entity may be quite disparate (Bapteste and Burian 2010; Baquero 2011; Dagan et al. 2008; Fondi and Fani 2010; Lima-Mendez et al. 2008; Moustafa et al. 2009; Puigbo et al. 2010; Skippington and Ragan 2011a, b; Smillie et al. 2011).
This, we think, is exemplary of a quite general characteristic of biological entities. Rather than coming into being in a unitary way through a unique path (e.g., a series of ever smaller branches in the tree of life) biological entities typically involve the coming together of a range of constituents often from diverse sources (Bapteste et al. 2012; Bouchard 2010; Hatfull et al. 2008; Lane and Archibald 2008; Lima-Mendez et al. 2008; Martin and Embley 2006; Moustafa et al. 2009; Zhaxybayeva et al. 2009). Another, perhaps more controversial, example is of the organism. If one thinks of the organism not within the ontological framework provided by the tree of life, but rather in terms of the functional wholes that interact with their wider biological and abiotic contexts, then it is rapidly apparent that these wholes typically involve a variety of entities with quite disparate origins. Typical metazoans require diverse and numerous microbial symbionts to function normally (Greenblum et al. 2012; Lozupone et al. 2008; Qu et al. 2008). Microbes themselves are most commonly found in complex collaborations such as biofilms (Ghigo 2001; Hall-Stoodley et al. 2004; Reisner et al. 2006). There is an increasingly compelling case for taking the whole symbiotic system as the most basic referent for the term ‘organism’ (Dupré and O’Malley 2009).
So the problem with the tree of life grounding our ideas about evolutionary ontology is that it privileges one particular biological relation, that of vertical inheritance from parent to offspring, and one type of entity, namely those with genealogical coherence. Hence this model, which is focused exclusively on genealogical relations, will give a very partial evolutionary ontology, a deficient inventory of the causal players in the evolutionary process. Of course, all scientific representations in biology are to some degree abstractions from the full complexity of living systems, so it is no sufficient criticism of the tree of life that it emphasises one set of relations over others (Wimsatt 2007; Levins 1984). And undoubtedly its genealogical focus has provided important insights in some areas of biology, notably eukaryote systematics. Abstraction also raises an ever-present danger, however, which is that part of the truth will be taken as the whole truth (Cartwright 1983; Dupré 1993). A particularly interesting instance of this danger is the possibility of substantial distortion of the basic ontology of a field, in this case the reduction of an evolutionary ontology to a genealogical ontology. The tree of life, we think, by insisting on the predominant importance of vertically transmitted origin as the defining feature of biological entities, has tended to promote just such an error, marginalising evolutionarily significant entities that did not evolve along the single, privileged tree.
The prevalence of lateral gene transfer among microbes gives the tree of life an additional disadvantage of being almost impossible to identify (Bapteste et al. 2008; Dagan and Martin 2006). It is true that there must, in principle, be an actual historical tree of cells, since cells do, barring some very rare if evolutionarily decisive events such as the origin of the eukaryote cell, always bifurcate in reproduction. But extensive lateral gene transfer makes this tree of cells impossible to discern since any genetic marker we use to trace the relevant reproductive relations will give us only a gene tree, a history of that gene. And lateral gene transfer implies that different gene trees cannot be relied upon to coincide on any unique tree of cells. Moreover, even if it were accurately reconstructible, such a tree of cells would be of dubious utility, since lateral gene transfer also implies that the genetically derived capacities of a cell could not be inferred from its position in the tree of cells.
The problem can best be understood in terms of different time scales, a perspective we shall emphasise throughout this paper. Even in the absence of lateral gene transfer, the branching pattern of the gene lineage (produced by mutations) cannot be directly translated into the branching pattern of the cell lineage (produced by cellular divisions). Mutation events occur at a different time scale than do events of cellular division. Sometimes, mutations accumulate faster in the cell than the cell divides; sometimes (most of the time) the cell divides faster than mutations accumulate in the gene. Therefore, the gene lineage generally evolves more slowly than the cell lineage. But whatever the specific rates of evolution, there is no reason to assume a direct correspondence between the gene lineage and the cellular lineage in which genes from that family were embedded. A gene tree, in sum, is a tree of genes not a tree of cells. Consequently, it seems that we need a more complex representation of the interactions and processes within the microbiological world than can be provided by giving ontological priority to either the tree of life (if such there be) or the tree of cells. We intend to sketch such a model in this paper.
In moving to an ontological framework that goes beyond the limitations of the standard model, and that recognises a variety of distinct and non-coincident genealogies, we aim to avoid the monism that frequently infects the standard ontological framework. We recognise, for example, that entities may have quite different roles in different systems and that the ontology may seem quite different when we adopt perspectives that emphasise different features. As an extreme illustration of the first point we might think of DNA sequences that mimic proteins (Zack et al. 1995; Dryden and Tock 2006), often to counteract various kinds of immune response. Such sequences thus belong in some contexts in the same functional categories as the proteins they mimic. Of course in other contexts, such as DNA sequence replication, DNA protein mimics function as normal DNA. Less exotic are so-called moonlighting proteins, proteins that function in quite different ways in different cellular contexts. The use of the term “moonlighting” nicely displays the deep assumption that normally a molecule has one proper function, and something is out of the ordinary when it is discovered doing something different (Henderson and Martin 2011; Huberts and van der Klei 2010; Collingridge et al. 2010). More familiar, perhaps, is the realisation that genes, far from having one specific function may, by virtue of such mechanisms as alternative splicing, end up with many different protein products serving a range of functions (Bondos and Hsiao 2012; Toor et al. 2006). We take this to be a self-evidently sensible strategy for biological systems to evolve: surely it is efficient to use entities that an organism has the resources to produce for as many functions as they can be made to serve. Monism is not a necessary concomitant of the standard ontology, but we think that it fits easily with the linear focus on vertical inheritance: the function of an entity is its role in driving the evolving lineage along its branch of the tree. Though it is well known that homology alone may not provide a reliable guideline for functional classification, it is nevertheless often the case that homology is used to infer functional information, for example in the. functional annotation of genes using COG, or KEGG databases3 (Tanabe and Kanehisa 2012; Tatusov et al. 2001).
The issue of entities with multiple functions—a possibility that may easily be obscured by the attribution of a single place in a hierarchical ontology—is one aspect of a much broader pluralism that we think an adequate evolutionary ontology must encompass, and one central reason for rejecting the standard ontology that has attributed overwhelming importance to just one process, phylogeny. We need an ontology that can represent a diverse set of processes including, for example, as well as phylogenetic relations, interactions that stabilise or destabilise the interacting entities, spatial relations, ecological connections, genetic exchanges, etc. In the next section we will offer a sketch of a network that aims to represent a variety of processes connecting different entities. The network model we have in mind should be able to detect, for example, cases in which the functional signature is stronger than or even contradicts the phylogenetic signal of an entity (Dinsdale et al. 2008; Kav et al. 2012; Lozupone et al. 2008). Indeed, what we are aiming to describe, at least as a theoretical ideal, is a synoptic picture including both microbial entities at multiple levels and multiple connecting processes. A network seems a natural way to represent diverse kinds of entities (as nodes) and diverse kinds of relations (by edges), even though this representation will only be able to provide a static representation of the processes affecting biological entities. In principle, the dynamics of these processes (and how they unfold over time) can be investigated by the reconstruction of series of temporally delimited networks, each of them corresponding to a particular time slice. However, in practice this is still too complex an enterprise, but the introduction of a static apparatus will already improve how microbiologists capture the variety of processes that stabilise entities within any of a wide range of time scales. Such networks are already being used, and have met with increasing interest in the microbiological community (Skippington and Ragan 2011b; Skippington and Ragan 2012).
We do not suggest that any imaginable model could capture every possible perspective or set of processes. But representing several that we take to be especially important will at least give us a reasonable sense of the partiality of particular perspectives, and the kinds of ways in which different perspectives overlap and interact.4 Finally, though, we also want to set some limits on the lush ontology we are proposing. We hope that it maintains a proper naturalness, and avoids the completely artificial or artefactual. The key idea here is that we are looking for entities that have, as briefly mentioned above, some distinctive causal power. A pattern in the model we describe provides a candidate for a significant entity. Confirmation of that status requires that its causal efficacy in vivo, or at least in vitro, be demonstrated.
A crucial motivation for adopting the framework we propose is that the standard phylogenetic model obscures the implications of the different rates of biological processes, and we conclude this section with some further remarks on that issue. We have noted that the stabilisations of process that result in what we standardly treat as biological objects occur in specific and diverse time frames. This creates a problem for the inferences generally licensed by the standard model, a problem that is independent of, and perhaps even deeper than, the problem of intersecting lineages. This problem arises from the fact that these inferences, for example from patterns of genes to patterns of species, implicitly make assumptions about the relevant time scales of the processes that stabilise the entities in these processes, and these assumptions may often be incorrect.
It will be helpful to approach the problem by thinking of a much simpler example, and one that starts with something far from obviously conceived as the temporally stabilized result of processes, a mountain. While a mountain may be unequivocally part of the stable background of largely unchanging things from the perspective of a hiker ascending its slopes, from the point of view of geology it is a very slow process. Consider the small mountains or hills on Dartmoor in South-West England. These are typically topped by tors, impressive piles of huge granite rocks, left behind by the erosion of the softer material that originally covered them. Eventually, perhaps, these granite extrusions may be all that is left of these features. Though these were parts of the relevant, or ancestral, features for as long as they existed (since they were extruded), it would be wrong to infer from attention to these maximally stable constituents over time that the entity to which they belong had at all times been a pile of granite. Indeed it is currently a more complex mixture of different kinds of parts. And even more importantly, it would clearly be wrong to assume that the bits of granite had always been part of a tor or mountain at all. For example, during the late Cretaceous Dartmoor was submerged by the rising sea level, and the granite was covered with a limestone deposit; subsequently sea levels fell, the land re-emerged, and the limestone covering was eroded, re-exposing the granite. In the Cretaceous, then, the lumps of granite were parts of the sea bed. If lumps of granite are more stable entities than mountains or sea beds, the history of a lump of granite does not correspond to the history of mountains or sea beds. We suggest that such an inference from stablest constituent to a stable kind of entity of which it is part is a central, very common, and problematic style of inference within the standard model.
Consider the use of ribosomal genes in phylogenetic inference. These genes were selected precisely because of their assumed high level of stability—or more precisely the stability of their lineages—over long periods of time. The stabilisation process is assumed to be selection for a vital feature of all cells. The problem is with the attempt to infer general characteristics of entities, evolving at a different time scale and often at other levels (organisms, species), from these maximally stabilised features. We know that lineages of organisms and species are stabilised over much shorter time scales; so any such inference runs the risk of a mistake parallel to the conclusion that Dartmoor tors were always piles of bare granite or that pieces of this granite were always parts of tors. We offer two brief examples of such errors.
The first relates to the question of microbial species. Increasingly many microbiologists will now deny that there is any interesting analogue of the eukaryote species in the prokaryote world, but this remains a hotly contested issue (Doolittle and Zhaxybayeva 2009; Gevers et al. 2005; Mora et al. 2011). Resistance to abandoning the notion of species for prokaryotes rests in part, we suggest, on the implicit idea that if, using highly stable markers such as ribosomal genes, we are able to trace evolutionary history into the prokaryote past, we must be tracing essentially the same homogeneous process involving throughout the same kinds of entities. But in fact we know that the history of the entities containing these relatively most stable genes is composed of many different time scales of stabilisation, so a gene tree provides no reason for assuming a similar general structure of species throughout the evolutionary time span of its existence. The problem here is not merely the familiar one that genes (or even non-coding DNA segments) must be selected with an appropriate rate of evolution for tackling a particular phylogenetic issue (Yang 1998). It is rather that the standard evolutionary ontology is used in ways that ignore the fundamental difficulty in inferring the ontological status of rapidly changing wholes from the study of slowly evolving parts. The use of existing ontological categories to interpret the branching of a gene tree as if these categories were fixed throughout the entire history of the gene can lead to error. For instance, sexual and asexual eukaryotes may have histones with the exact same sequence. However, sexual and asexual species should be considered as having distinct ontologies: they are not generated, affected or stabilized by the same processes. There need not be any branchings in the histone (or equally ribosomal) tree that correspond to a divergence between sexual and asexual species. Because the standard evolutionary ontology encourages the unwarranted assumption of an evolutionary coupling between parts and wholes, it is particularly ill-suited to making inferences about the general structure of living processes across major evolutionary transitions (Fig. 1).
Fig. 1.

Decoupling of changes in sequence and carrier-ontology. The evolution of the relatively most stable entities used to track evolutionary changes in the microbial world (e.g. sequences from a well conserved gene family) can be represented by a tree. The branching and ancestral nodes in such a gene tree are commonly used to infer the presence of a particular type of evolutionary event (e.g. speciation, transfer, etc.), affecting a particular class of less stable entities (e.g. ancestral organism, species, etc.). However, the difference of time scales for the stabilisation of the most stable entities and that for the other entities that these slowly evolving sequences are supposed to be tracking makes it impossible to infer a direct correspondence between the changes recorded in the gene tree and those that have occurred in other entities. Dichotomies and divergences in the gene tree cannot be directly coupled to changes in the ontology of the gene carriers, and the evolutionary events affecting less stable entities of which this gene is a component. There is not necessarily information on the gene tree alone about what type of entities carried a particular ancestral gene form, and about whether and when these types of entities may have dramatically changed, i.e. when the evolution of novel classes of sequence-based entities resulted in major (hypothetical) evolutionary transitions. (Interested readers can see Gross and Bhattacharya (2010), Koonin and Martin (2005), and Woese (1990) for more details on some of these transitions.) Such decoupling means that gene trees are informative about gene history, but may not be informative about the evolution of other entities, contrary to what is assumed in a standard ontology, which couples the evolutionary history of parts and wholes. Fig. 1 represents important ontological transitions by changes in branch format. Because of these changes, relatively similar sequences in a branching lineage may reside in different types of (sometimes novel) entities with different time scales for their stabilisation. Interpreting a sequence tree with a single ontology from present to past will then provide a severely distorted view of evolution
Our second example shows that the problem is not limited to inferences about the past. Consider the human gut microbiome. At a certain point in the investigation of this entity it appeared that there were considerable differences in the populations of microbes found in humans in different parts of the world as measured by standard 16S rRNA methods. One might easily have imagined that by comparing these different microbiota we could discover, for instance, which were the essential symbionts and which were more opportunistic visitors. However, as metagenomic methods were developed and applied to these populations it emerged that the total genetic constitution of different gut microbiomes was far more similar than indicated by 16S rRNA taxonomy (Kav et al. 2012; Lozupone et al. 2008). The explanation is fairly clear. The object that is stabilised by the constraints of the animal gut is the metagenome of the whole community: this provides the genetic resources required for normal animal functions. Lineages of microbes are much less stable and more diverse, with many genes moving between them. So the relatively stable genetic resources required for the animal gut community can be provided by a wide variety of relatively less stable collections of lineages. Inferences from the discovery that a bacterial lineage serves a necessary function in a particular gut, to the conclusion that this is an essential animal symbiont, would be quite unjustified. Proper inference in this area requires identifying the relevant stabilised entities serving the various functions of the gut microbiome, and this is a higher level entity than the lineages of bacteria that seem the natural objects of interest within the standard model. We suspect that failure to pay attention to the different time scales at which entities are stabilised can lead to serious distortions in our inferences about the existence of biological entities at particular times. Understanding the linkages and uncouplings between nested hierarchies of entities absolutely requires such attention: time scales for the stabilisation of entities should feature as an integral aspect of the evolutionary ontology.
A multidimensional framework for microbial ontology
Microbiologists have so far grounded their ontologies largely on a single process. Vertical descent with modification can be taken to underlie various taxonomic projects: the classification of organisms into a particular species; the classification of genes into functional categories (Tatusov et al. 2001); stabilization of sets of metabolic capacities to classify entities with specific impacts on geochemical processes, for example multiple species as denitrifiers; and so on. Occasionally, investigations go beyond the vertical inheritance model to enhance the understanding of complex microbial systems, for example to determine which functional categories of genes were vertically inherited or laterally transferred in denitrifiers yet not in other species (Falkowski et al. 2008). However, we argue that such cross-fertilisation of ontological information should be made more systematic in order to expand the explanatory power of studies of the microbial world. We propose a model that starts with as wide a range of currently recognised entities as possible, at multiple levels of organisations, and makes room for multiple processes affecting these entities. Such a model could provide a powerful framework for evaluating (i) our existing ontological assumptions (e.g. whether the currently distinguished entities are adequate for understanding the causes of change and stabilisation in the microbial world), as well as (ii) for identifying potentially hidden ontological classes, thereby recognising additional entities that provide new insights into the structure of the microbial world (Skippington and Ragan 2012, 2011b).
Consideration of well-known features of microbial organisation makes the recourse to such a multidimensional framework compelling. The evolution of the majority of known microbial communities and coalitions involves entities from multiple levels of biological organisation, participating in a wide range of biological processes (Ghigo 2001; Hall-Stoodley et al. 2004; Overmann 2010; Reisner et al. 2006; Smillie et al. 2011). Take for instance the well-studied coalition between oceanic photosynthetic bacteria and their phages carrying photosynthetic genes. In this coalition, infection results in an increase of photosynthetic productivity in infected bacteria, thereby preventing the extinction of the bacterial population (Alperovitch-Lavy et al. 2011; Sullivan et al. 2006; Hellweger 2009). An accurate (and dynamic) model of the evolution of lineages of such cyanobacteria requires (at least) the consideration of three biological levels: mobile genetic elements, bacterial cells, and the integrated community formed by the coalition of mobile elements and bacterial cells. It also requires the comprehension of several processes: the transfer of genetic material (e.g. the psbA photosynthetic gene) between cyanophages, between cyanophages and bacterial cells, and between bacterial cells; the stabilising selection against changes in that photosynthetic gene in all the entities carrying the genes; the replication of cells and of phages; and arguably group selection favouring these coalitions (Villarreal 2009) over populations of cyanobacteria infected by phages that do not carry photosynthetic genes. Not all of these processes can be directly mapped onto the hierarchy based on vertical descent among entities within a single branch of the tree of life.
We propose to replace the hierarchical one-dimensional framework with a multidimensional framework, in which processual interactions between these microbial entities, crossing over what are generally seen as distinct levels of biological organisations, can be explicitly modelled (Fig. 2).
Fig. 2.

Schematic representation of a multidimensional network. Nodes are previously recognized entities, from different levels of biological organisation (indicated by different symbols). The value ‘1’ means that two entities were inferred to result from, or be involved in, a particular process (represented by an edge with a particular line type), ‘0’ means that no such relationship could be inferred. The matrix to the left would result in the network to the right. This picture could provide a less biased and partial representation that will enable us both to analyse, and to suggest methods for the detection of, the ontology for this kind of common example from microbiology
As a first step, we propose the construction of networks with nodes of distinct types, representing different ontological categories of entities, and edges of distinct types, representing different types of causal interactions or other biologically significant relations (e.g., homology, collocation–the tendency to appear in the same place) occurring between these entities. This graph contrasts with the graph used in the standard ontology: the evolutionary tree is an acyclic graph on which all nodes are necessarily connected, while our network can be cyclic and disconnected.
To make this approach analytically tractable, the number of types of nodes and of edges should be limited. We propose that nodes should represent gene sequences, proteins, cells, mobile genetic elements, and communities. Three main types of edges should be sufficient to provide useful information about the relations within systems that include representatives of these types of entities. First, edges reflecting processes responsible for particular similarities between these nodes could offer an initial structuring of the network. These similarity-generating processes could be further distinguished into (i) processes of vertical inheritance, resulting in a global similarity between genealogically related entities (i.e. two gene sequences that would align along all their sequence) (Adai et al. 2004); (ii) processes of recombination introducing partial, local similarities between entities resulting from these combinations and the lineages that recombined (i.e. two gene sequences that would only align over part of their sequences); (iii) selective pressures leading to convergent phenotypes (e.g. enrichment in hydrophobic amino acid in transmembrane proteins (Koehler et al. 2009), or GC-biased gene conversion (Hildebrand et al. 2010)). Second, edges reflecting causal interactions between nodes, such as the transfer of biological material (e.g. DNA, protein, cells) between two entities, or processes resulting in conflicts between these entities (e.g. predation, arms races, etc.) could be searched for. These interactions could be further characterized as stabilizing or destabilizing (Bapteste et al. 2012). Third, and finally, edges should represent collocation, the fact that two entities are typically found close together or in the same place.
As one example, a simple approach to the detection of stabilizing selection at a molecular level in a network of microbial entities could be to explore whether the genes shared by these entities (each represented by a node) are under purifying selection. The result of classical KA/KS analysis between groups of connected sequences (i.e. the ratio of the number of non-synonymous substitutions per non-synonymous site (KA) to the number of synonymous substitutions per synonymous site (KS), which provides an indicator of selective pressure acting on a protein-coding gene), could thus be incorporated in the network by reporting edges of different colours for different values of KA/KS. The detection of groups of entities sharing sequences undergoing similar selection could be linked by an edge, which would reveal groups of entities affected by the same stabilizing selection on particular genes. It would also be possible with such a network to represent communities of genetic exchanges (compare Skippington and Ragan 2011a) based on edges of stabilizing selection at the gene level. Finally, edges reflecting processes responsible for the collocation of entities in the microbial world would introduce relevant information on the coming together of these entities at a given spatiotemporal scale, or on ecological selective processes causing these entities to inhabit a given type of environment. Each of these types of edges introduces a dimension along which the behaviour of entities from the microbial world (e.g. their patterns of connection to other entities in the network) can be studied.
Such a rich network would offer a much closer approximation to a synoptic picture of the causal interactions between, and longer causal chains connecting, microbial entities, and their dynamics. Some causal chains are made of a single type of interaction; other causal chains result from a mixture of several different types. Our network model therefore offers a natural framework to represent the cascading effects or positive feedback loops that sustain the existence of biological entities.
Such a multidimensional network can be directly applied to tackle several of the ontological issues described above. First, it makes explicit both the causal relations that initiate change and those that foster stability; it therefore highlights the ultimately processual character of the entities in our basic ontological framework. Moreover, representing processes at different time-scales brings out the fact that what are treated in the standard evolutionary model as stable, maximally coherent, entities are in reality only units sufficiently stabilised to be treatable as fixed relative to a particular time-scale for the purposes of a particular problem. (Recalling our illustrative example, while a walker can very properly treat a mountain as a fixed feature of the environment, from the perspective of tectonics it is a particular phase in a process.)
Furthermore, our networks can represent both the causal interactions of parts of an entity, when these parts are represented by nodes, and the causal interactions of the whole with different entities (and different parts of different entities). In that framework it is natural to expect that the relationships between higher level entities change when the relationships between their component parts change. Such a network thus provides a more comprehensively dynamic vision of microbial entities, allowing for modifications to an entity introduced by changes in the relationships between its parts, either by changes in the relationships of the whole to other entities, or by relations of its parts to other entities. This is a strength of the general framework, because it does not privilege the most stable categories in understanding a fundamentally dynamic biological world. This framework also offers a basis on which to model the stabilisation and destabilisation of an entity over time. These changes are caused by distinct influences (both internal and external) affecting a given entity, and are represented by different types of edges. We thus explicitly acknowledge the way in which features of the context in which it is placed contribute to determining the identity (or indeed change of identity) of a higher level entity. It should then become possible to determine when changes in the external or internal relationships of an entity, visualized as features in the multidimensional topology of some part of the causal network, may justify distinguishing new entities perhaps not recognised in pre-existing ontological classes.
One example, in which moving to a more complex multidimensional perspective might motivate a major change in ontology, is the distinction microbiologists make between core and shell (peripheral) genes (Charlebois and Doolittle 2004; Medini et al. 2005; Lukjancenko et al. 2010) to provide a criterion for ontological change: provided an entity includes the core genes of E. coli, then an E. coli is what it is. A more useful approach might be to recognise that changes in peripheral genes, connected also to changes in the environment of a particular E. coli strain, produce such radical modifications in its causal relations that we should recognise it as a quite distinct kind of entity. We hope that the model we are sketching, by giving a wider picture of these causal relations, would provide a better motivated basis for making such decisions. While the connections established between entities by core genes are unlikely to change (unless all the core genes are lost), connections established by peripheral genes may vary significantly between individuals, when for instance a gene acquisition opens a new ecological niche to its carrier or exposes it to novel selective processes (Lopez and Bapteste 2009; Fondi and Fani 2010; Kloesges et al. 2011; Popa et al. 2011).
Expanding microbial evolutionary ontology
The framework offered by our network model makes possible the observation of patterns of connections orthogonal to the expected ones for entities of a particular pre-existing class. This could lead to the recognition of additional ontological classes, in terms of which aspects of biological diversity could also be measured. More specifically, we anticipate three major types of significant pattern that might be observed in the network, which might lead us to postulate the existence of specific types of entities: (i) “processually equivalent” entities (PE-entities), (ii) “processually versatile” entities (PV-entities), and (iii) “stabilized” entities (S-entities) (Fig. 3).
Fig. 3.
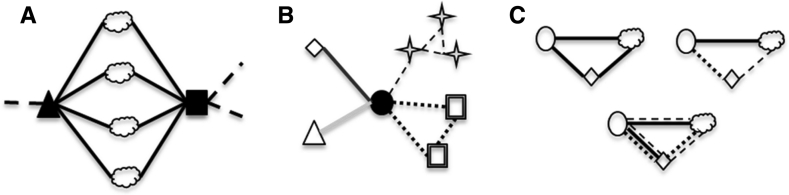
Examples of some patterns that can be found in a multidimensional network. Nodes are previously recognised entities. Edges indicates similarity, collocation or causal relationships between two entities. Entities from different levels of biological organisation are represented by different shapes. When edges are observed in different conditions, or result from, or are involved in, different causal processes, they are represented by a different line type. a In black, two PE-entities, sharing a significant fraction of direct neighbours. b In black, a PV-entity, involved in different causal chains, in different conditions. When this node is removed, the graph disconnects in multiple subgraphs, therefore the PV-entity is a local clique minimal separator of the graph. c Three sets of S-entities. On the left, S-entities are stabilized by the same process. On the right, S-entities are stabilized by the interplay of different processes. At the bottom, S-entities are stabilized by multiple processes
PE-entities are entities that are seen to play a similar role in the network model, because they display significant common topological local properties (i.e. they share the same direct neighbours) (Fig. 3a). More precisely, entities could be called equivalent if they share at least one causal chain (e.g. one path in common in the network). It might be useful to treat processual equivalence as a matter of degree: the higher the proportion of shared paths between two entities among all the paths in the network model, the higher their degree of processual equivalence.
Defining classes of PE-entities is, of course, one thing the standard ontology aims to achieve, and sometimes succeeds in achieving. For example, to the extent that belonging to a species (i.e. being part of the same genealogical lineage) determines organisms as having the same causal powers and relations, then this is a useful way of generating processually equivalent classes. The same possibility arises for lineages of genes and gene families. When the assumption of processual equivalence within a genealogical class fails, however, reasons for their processual heterogeneity can be searched for in (at least) two directions. First, they may just be classes with minimal explanatory interest or significance. The proposal of such classes is not uncommon in systematics. For instance, the category of Chromalveolates (a large hypothetical clade of eukaryotes, allegedly deriving from an event of secondary endosymbiosis of a red alga within a eukaryotic cell with two flagella) has recently been shown to correspond to an artefactual grouping of unicellular eukaryotes, about which it would be difficult to justify any systematic generalizations (Hackett et al. 2007). More interestingly, this heterogeneity may point toward a genuine processual versatility within that class of entities (see below).
Another very significant possibility is that we may unexpectedly distinguish classes of PE-entities that are highly diverse either structurally or genealogically or both. Such classes might include entities from distinct levels of organisation, or distantly related entities, that nevertheless play the same causal roles. Paradigmatic for this case is the evolution of mimics, for example a protein that mimics DNA to overcome the defences of restriction-modification systems (McMahon et al. 2009). Such cases would be displayed in the multidimensional network when a bit of DNA and a protein not coded by that DNA, or several genealogically unrelated proteins, exhibit largely overlapping sets of direct neighbours. This pattern indicates that these entities have largely similar causal effects, suggesting that they play very similar or identical roles in the workings of the microbial world.
PV-entities are entities that are homogeneous in a traditional sense (structural or genealogical) but are involved in a number of different causal processes. In our network model, the set of direct neighbours of a PV-entity will change in different contexts (Fig. 3b). As with processual equivalence, processual versatility could be quantified based on the topological properties of these entities in the network. For instance, the number of disconnected causal chains to which that entity belongs can be computed by removing the node corresponding to that entity, followed by the count of the n locally disconnected causal chains that result from this procedure. The larger the value of n, the more versatile the entity is.
The observation that accepted categories such as species, or lineages, are PV-entities is important for two reasons. First, if these categories are genuinely explanatory units, this versatility may very well help to explain their evolutionary success. Second, if a class of entities is processually versatile, we need to be cautious about its suitability for broad generalization. For instance, if being part of a species (e.g. E. coli) entailed that E. coli individuals would all behave similarly, then individual E. coli cells in the network would be expected to have a rather similar set of topological properties (e.g. similar patterns of gene transfer). If, on the other hand, contextual differences could affect the causal powers exhibited by distinct E. coli cells, then the class defined by this criterion would be shown to be of seriously limited significance. If the goal of classification is to provide classes with members that possess similar properties or, in terms of our network model, that display the same pattern of connection, then processual versatility is a defect in a putative class. More constructively, the detection of PV-entities emphasizes the fact that no quasi-essentialist assumption should be made about prokaryotic species. Not all E. coli are pathogens, because pathogenicity can come from externally acquired (or internal) changes in the individual cells, for instance as the result of the gain or loss of mobile elements such as pathogenicity islands (Bezuidt et al. 2011; Beauregard-Racine et al. 2011; Lukjancenko et al. 2010).
Entities at a lower level than the organism or species may also prove processually versatile. It is sometimes supposed that a given gene sequence, for instance, should be expected always to code for the same (set of) function(s), and a protein always to perform the same function(s). Many genes indeed may very well carry invariant genetic instructions, so that if these genes are laterally transferred from one organism to another, they will, if expressed, code for proteins achieving the same effect in the two organisms (Smillie et al. 2011; Alperovitch-Lavy et al. 2011). The nature of the gene (or the protein), in short, is often seen as largely independent of the context, robust to external variations. Changes in the context may make the gene/protein useless or toxic (Sorek et al. 2007), but will not change the fundamental functional nature of that molecule.
But by contrast to this highly stable—almost essentialist—view of the gene or protein, it is also possible that a given genetic sequence does not encode a single (set of) instruction(s), but that the context largely determines what the gene or protein does, and therefore even what it is. And in fact it is well-known that genes are capable of doing different things in different contexts, as is shown, for instance, by the phenomenon of alternative splicing. For the case of proteins we have the phenomenon of moonlighting, described above. Our network model would be expected to distinguish such versatile entities (or classes) as belonging to two or more distinct and disconnected causal clusters. Dynamic analysis of the network might enable us to distinguish features that function as differentiating context, and features that emerge as consequences of the particular context.
S-entities, finally, are another very important ontological category. They correspond to sets of entities that are detectable because their connections are stabilized, either by a single type of process, or by multiple types of processes (e.g. cliques5 or quasi-cliques of causally related nodes, Fig. 3c). In the latter case, either the interplay of multiple processes is responsible for the stabilized unit, or the superposition of several independent but distinct processes. (An example here would be the various distinct mechanisms that promote the formation of microtubules in the elaboration of the mitotic spindle (Duncan and Wakefield 2011)). The coexistence of several distinct stabilising mechanisms is an especially strong indication of a robust and biologically significant stabilised entity. This is potentially important in determining the boundaries of complex assemblies such as, for example, biofilms (Hall-Stoodley et al. 2004). The extent to which stabilising links connect a particular kind of entity to the assembly should be a guide to whether that entity should be considered a part of the larger whole, or a distinct entity interacting with it.
In general, to highlight novel instances of ontological classes that are not the usual families of entities sharing a common ancestry, we propose the detection of “clubs” (Bapteste et al. 2012): stabilized groups of entities with one or more detectable causal powers, resulting from the interplay of processes of evolution and development orthogonal to processes generating the tree of cells. In theory, if networks from successive temporal slices could be reconstructed, they would further provide clues about the dynamics of stabilisation of such clubs.
One interesting kind of club includes bacteria and their phages. Typically, when a free-living alphaproteobacterium acquires an endosymbiotic plasmid that confers the ability to grow in nodules of plants, roots will acquire peripheral genes that introduce this free-living alphaproteobacterium into the bacterial community that is selected within the plant root nodule (Sullivan et al. 2002). The local topological properties around that bacterium in the gene sharing network were changed by the acquisition of these mobile genes, since new partnerships with a densely connected set of endosymbiotic bacteria were thereby introduced.
Another example of traces left in temporal slices of the network as these entities become stabilized could be found by contrasting the relationships between entities in a network before and after the emergence of a novel chimerical organism, such as a lichen (Grube and Hawksworth 2007), or the first eukaryotes (Martin and Müller 1998; Moreira and Lopez-Garcia 1998). The local neighbourhood of such novel nodes, i.e. the set of other nodes to which the super-organismal node is directly connected, would be unprecedented in the graph. Most importantly, it would include nodes that were not direct neighbours before the emergence of the super-organism. It could capture such phenomena as the aggregation of micro-organisms and mobile elements such as plasmids into a growing biofilm, or their selection as members of a stable multilevel club, as for instance in the case of marine cyanobacteria and cyanophages (Alperovitch-Lavy et al. 2011; Sullivan et al. 2006). It could also detect the process of fragmentation in monophyletic groups (such as species) as their members undergo some kind of divergence. Causes of these divergences might even be revealed, if for instance members of a species show connections indicating that they are under the influence of different groups of phages. This situation is indeed known to lead to the evolution of populations of microbes with uneven fitnesses, and eventually to the extinction of some of these diversifying populations, when members of the species carrying a particular phage migrate or come in contact with other members of the same species that are not immune to that phage (Villarreal 2009). Importantly, robust communities in the network that do not match any simple category of the standard ontology (i.e. communities involving entities from multiple levels of biological organisation or distantly related lineages) will likely constitute S-entities that would be invisible from within more traditional perspectives (i.e. communities of genetic exchanges involving multiple lineages (Skippington and Ragan 2011a).
Explanatory use of non-standard ontological classes
The identification of the most significant set of entities is an essential precondition for the scientific analysis of the microbial world. It determines the types of statements that can be made about nature, because it provides the objects about which statements can be made. In the case of the standard ontology it not only provides the entities, but since it is grounded in a single process, it also provides a favoured set of relationships in terms of which these entities are likely to be better understood. Typically, evolutionary scenarios in microbial evolution are focused on the genealogical relationships between monophyletic groups. They firstly seek to explain the structures observed in microbial diversity in terms of sister-groups, divergence, and common ancestry. The recourse to different or additional ontological classes could (at least in principle) make possible new kinds of scientific hypotheses. These hypotheses should be better suited to the distinctive character of the microbial world, and thereby likely to enhance our knowledge of it, including our knowledge of its evolution.
We will briefly sketch some novel types of scientific claims (and even scientific research programmes) that could quite naturally follow from the recognition of the ontological classes described above: processually equivalent entities, processually versatile entities, and stabilized entities.
PE-entities are defined by the detection of high degrees of similarity in the causal chains in which a set of entities occurs. The cases of present interest are those in which a particular class of PE-entities includes members that are structurally or genealogically quite distinct. PE-entities may be found within one level of biological organisation (e.g. when genes from distinct gene families code for the same step of a metabolic pathway), but they can also belong to distinct levels of biological organisations (e.g. when phage or plasmid protein mimics DNA (McMahon et al. 2009)). Either kind of equivalence suggests that the functions shared by the various PE entities are significant for the sustainable functioning of entities within the microbial world, since the functions have evolved, and presumably have been selected for, on more than one occasion.
An interesting empirical question to explore is whether the number of PE-entities increases over evolutionary time. Are PE-entities favoured by selection as providing resources that can be used in diverse ways or should one, rather, expect natural selection to find, eventually, the unique entity that best serves each particular biological function? In the case of the flu virus mimicking histones with another protein (Marazzi et al. 2012), the evolution of histones preceded that of its mimic, suggesting that processual equivalence has evolved in relatively recent time. Hypotheses about the lineages, the environments and the functions in which processual equivalence is more common could also be used to delineate crucial features underlying the diversity of the microbial world. These features could then help to identify the entities that humans could most effectively target in their attempts to control, or counteract, pathogens in the microbial world.
Moonlighting, or PV-, entities raise different types of questions. Their flexibility, their ability to act in diverse and distinct causal chains, suggests the hypothesis that PV-entities are ancient generalists with high evolvability, which has contributed to the success of a wide range of lineages. Alternatively, however, such entities could be quite recent evolutionary innovations in the microbial world, an idea that seems at least plausible, since the evolution of single entities able to fulfil multiple distinct roles can be advantageous in terms of cellular economy. These competing possibilities suggest the desirability of scientific research programmes that try to quantify the prevalence of versatility among known entities, in order to test whether some higher level entities are particularly rich in “moonlighting” lower level entities (for example, whether there exist species particularly well equipped with multifunctional proteins).
The distribution of PV-entities might be relevant to investigation of the biogeographical distribution of microbial entities. It could encourage investigation of whether moonlighting entities are required to explain the wide distribution of microbial kinds: if ‘everything is everywhere, but the environment selects’ (Baas-Becking 1934, cited in O’Malley 2008), moonlighting entities might be selected more frequently than other kinds. A higher proportion of versatile components would indeed enable related organisms to be successful in a wide range of different environments and consortia. Further, when entities move between higher level entities (for example genes moving between genomes) the moonlighting potential of the lower level entity might prove crucial in explaining the success of the entity in being viable or advantageous in new contexts, and hence globally successful. Of course, these two classes of entities, PE and PV are not mutually exclusive. Two entities may be processually equivalent in one context, whereas each of them may be capable of performing quite different functions in other contexts, and hence may be processually versatile.
Stabilized entities could raise even more fundamental questions for evolutionary microbiology. In fact, stabilisation is not really a problem for a standard ontology, because this ontology assumes the stability of entities. By contrast, in our extended ontology the evolution of stabilisation (and of mechanisms of stabilisation) in the microbial world becomes a major issue. In the example of integrated cyanophage and cyanobacterial communities, in which entities from at least three levels of biological organisation are involved, stabilisation is not achieved just by one particular entity, or at a single level. While cyanobacteria certainly need to limit the infection rate by cyanophages, an absolute end to the genetic exchanges with these phages might be detrimental. Questions of stabilisation concern fine-tuning of the interactions between these entities through which their stability is optimized. Recognition of such stabilized entities might then inspire a more general search for mechanisms facilitating stabilisation (e.g. non homologous repair systems able to integrate foreign genes in genomes without killing the host (Weller et al. 2002; Shuman and Glickman 2007)), and lead to investigations of how these mechanisms are distributed, and how eventually they can be moderated or bypassed (to prevent integration, or slow down the rate of evolution of an entity).
More fundamentally, however, the consideration of stabilisation processes in the microbial world indicates that the entities scientists work with are only relatively stable, their stability being dependent on the processes that sustain the functional integration of their parts. This observation has an important practical consequence on questions of origins, such as for instance the origin of eukaryotes or the origin of microbial species. Answers to such questions should not be phrased in terms of stable entities: there is no such thing as the first eukaryote, or the first representative of a particular species. There is, however, a process through which a certain level of stabilised functional integration is eventually reached (Lawrence and Retchless 2010). In the process of stabilisation that leads to a new kind of entity (and which happens at variable rates for different entities), we could have no way to distinguish the putative first entity from the entity one generation before. It would also be crucial to study the time scales of the stabilisation of the (relatively stable) phenotypes that are analyzed by evolutionary biologists (e.g. the stabilisation of the eukaryotic organisation from the initial merger of distinct prokaryotic partners, the stabilisation of a particular metabolic pathway, etc.).
Conclusion
In this paper we have called for a more expansive evolutionary ontology for microbiology and a more egalitarian treatment of the diverse kinds of entities and processes familiar in the mainstream of microbiology, but sometimes downplayed in importance due to excessive concern with the specific perspective of vertical inheritance. We have also sketched a mode of representation that could facilitate the move away from a narrowly phylogenetic framework for biology generally. While the phylogenetic framework usually relies on a tree-based representation, we suggested that networks could provide a broader framework, in particular because these latter graphs can represent both cyclic and acyclic relationships, and do not assume that all entities under study are connected. Even if one does not want to endorse all aspects of the ontological expansion we propose, our network model might still offer a powerful alternative to the more traditional evolutionary framework. Underlying these proposals is an exploration of the sometimes fundamental implications of taking seriously the idea that biology is, or at least can be usefully conceived as, process all the way down. One motivation for advocating this perspective is that it allows us to raise a wide variety of important questions that do not readily arise within a more traditional ontological framework. Indeed, many of these questions would simply not make sense in the context of the traditional evolutionary ontology. They could, however, lead to the discovery of general features crucial for the maintenance of the microbial world, flexible features central to explaining the success of diverse lineages of entities, and mechanisms that facilitate the integration of lower level entities into a higher level entity. The ideas we propose are likely to be regarded as controversial; but we think that the potential payoffs that they might offer are sufficiently impressive to make the attempt to explore them in more detail worth the effort.
Acknowledgments
We thank M. O’Malley for inviting us to contribute to this special issue on microbes, and P. Lopez for critical discussions about microbial evolution. We are also most grateful to three anonymous referees who provided detailed comments that led to major improvements in the article. J.D. acknowledges the support of the ESRC (Economic and Social Research Council, UK). His contribution to this work was part of the programme of Egenis, the ESRC Centre for Genomics in Society.
Footnotes
The classic exposition of such a difference-making account of causation is Woodward (2003). More robust accounts of causal powers can be found in Cartwright (1989) and Mumford and Anjum (2011).
The opposite neglect of the microbial world is, we would argue, a much commoner and more serious fault. For a general argument for the importance of the microbial world, and the limitations imposed on philosophy of biology by its neglect, see O'Malley and Dupré (2007).
Clusters of Orthologous Groups (COGs) of proteins are generated by comparing the protein sequences of complete prokaryotic genomes; each of these clusters is classified into one or several functional categories, such as, for instance, RNA processing and modification, or cell-cycle control and mitosis. The KEGG database is an integrated database in which molecular-level information is classified in ways that facilitate the systemic study of the molecular interactions and chemical reactions in which genes are involved in an organism.
The pluralism defended earlier by one of us (Dupré (1993)) insufficiently emphasised the important problem of bringing multiple perspectives to bear on particular problems. This problem is addressed by Mitchell (2003).
A clique in an undirected graph is a subset of its vertices such that every two vertices in the subset are connected by an edge.
Contributor Information
Eric Bapteste, Phone: +33-1-44272164, FAX: +33-1-44275801, Email: eric.bapteste@snv.jussieu.fr.
John Dupré, Email: j.a.dupre@ex.ac.uk.
References
- Adai AT, Date SV, Wieland S, Marcotte EM. LGL: creating a map of protein function with an algorithm for visualizing very large biological networks. J Mol Biol. 2004;340(1):179–190. doi: 10.1016/j.jmb.2004.04.047. [DOI] [PubMed] [Google Scholar]
- Alperovitch-Lavy A, Sharon I, Rohwer F, Aro EM, Glaser F, Milo R, Nelson N, Beja O. Reconstructing a puzzle: existence of cyanophages containing both photosystem-I and photosystem-II gene suites inferred from oceanic metagenomic datasets. Environ Microbiol. 2011;13(1):24–32. doi: 10.1111/j.1462-2920.2010.02304.x. [DOI] [PubMed] [Google Scholar]
- Bapteste E, Burian RM. On the need for integrative Phylogenomics, and some steps toward its creation. Biol Philos. 2010;25:711–736. doi: 10.1007/s10539-010-9218-2. [DOI] [Google Scholar]
- Bapteste E, Susko E, Leigh J, Ruiz-Trillo I, Bucknam J, Doolittle WF. Alternative methods for concatenation of core genes indicate a lack of resolution in deep nodes of the prokaryotic phylogeny. Mol Biol Evol. 2008;25(1):83–91. doi: 10.1093/molbev/msm229. [DOI] [PubMed] [Google Scholar]
- Bapteste E, O’Malley MA, Beiko RG, Ereshefsky M, Gogarten JP, Franklin-Hall L, Lapointe FJ, Dupré J, Dagan T, Boucher Y, Martin W. Prokaryotic evolution and the tree of life are two different things. Biol Direct. 2009;4:34. doi: 10.1186/1745-6150-4-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste E, Burian RM, Bouchard F (2012) Philosophy and evolution: minding the gap between evolutionary patterns and tree-like patterns. In: Anisimova M (ed) Evolutionary genomics: statistical and computational methods, vol 2. Humana Press, New York, pp 81–110 [DOI] [PubMed]
- Bapteste E, Lopez P, Bouchard F, Baquero F, McInerney JO, Burian RM (2012) Evolutionary analyses of non-genealogical bonds produced by introgressive descent. Proc Natl Acad Sci USA. doi:10.1073/pnas.1206541109 [DOI] [PMC free article] [PubMed]
- Baquero F. The 2010 Garrod Lecture: the dimensions of evolution in antibiotic resistance: ex unibus plurum et ex pluribus unum. J Antimicrob Chemother. 2011;66(8):1659–1672. doi: 10.1093/jac/dkr214. [DOI] [PubMed] [Google Scholar]
- Beauregard-Racine J, Bicep C, Schliep K, Lopez P, Lapointe FJ, Bapteste E. Of woods and webs: possible alternatives to the tree of life for studying genomic fluidity in E. coli. Biol Direct. 2011;6(1):39. doi: 10.1186/1745-6150-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezuidt O, Pierneef R, Mncube K, Lima-Mendez G, Reva ON. Mainstreams of horizontal gene exchange in enterobacteria: consideration of the outbreak of enterohemorrhagic E. coli O104:H4 in Germany in 2011. PLoS ONE. 2011;6(10):e25702. doi: 10.1371/journal.pone.0025702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondos SE, Hsiao HC. Roles for intrinsic disorder and fuzziness in generating context-specific function in Ultrabithorax, a Hox transcription factor. Adv Exp Med Biol. 2012;725:86–105. doi: 10.1007/978-1-4614-0659-4_6. [DOI] [PubMed] [Google Scholar]
- Bouchard F. Symbiosis, lateral function transfer and the (many) saplings of life. Biol Philos. 2010;25(4):623–641. doi: 10.1007/s10539-010-9209-3. [DOI] [Google Scholar]
- Cartwright ND. How the Laws of Physics lie. Oxford: Oxford University Press; 1983. [Google Scholar]
- Cartwright ND. Nature’s Capacities and their Measurement. New York: Oxford University Press; 1989. [Google Scholar]
- Charlebois RL, Doolittle WF. Computing prokaryotic gene ubiquity: rescuing the core from extinction. Genome Res. 2004;14(12):2469–2477. doi: 10.1101/gr.3024704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge PW, Brown RW, Ginger ML. Moonlighting enzymes in parasitic protozoa. Parasitology. 2010;137(9):1467–1475. doi: 10.1017/S0031182010000259. [DOI] [PubMed] [Google Scholar]
- Dagan T, Martin W. The tree of one percent. Genome Biol. 2006;7(10):118. doi: 10.1186/gb-2006-7-10-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagan T, Artzy-Randrup Y, Martin W. Modular networks and cumulative impact of lateral transfer in prokaryote genome evolution. Proc Natl Acad Sci USA. 2008;105(29):10039–10044. doi: 10.1073/pnas.0800679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diene SM, Merhej V, Henry M, El Filali A, Roux V, Robert C, Azza S, Gavory F, Barbe V, La Scola B, Raoult D, Rolain JM (2012) The rhizome of the multidrug-resistant Enterobacter aerogenes genome reveals how new killer bugs are created because of a sympatric lifestyle. Mol Biol Evol. doi:10.1093/molbev/mss236 [DOI] [PubMed]
- Dinsdale EA, Edwards RA, Hall D, Angly F, Breitbart M, Brulc JM, Furlan M, Desnues C, Haynes M, Li L, McDaniel L, Moran MA, Nelson KE, Nilsson C, Olson R, Paul J, Brito BR, Ruan Y, Swan BK, Stevens R, Valentine DL, Thurber RV, Wegley L, White BA, Rohwer F. Functional metagenomic profiling of nine biomes. Nature. 2008;452(7187):629–632. doi: 10.1038/nature06810. [DOI] [PubMed] [Google Scholar]
- Doolittle WF, Zhaxybayeva O. On the origin of prokaryotic species. Genome Res. 2009;19(5):744–756. doi: 10.1101/gr.086645.108. [DOI] [PubMed] [Google Scholar]
- Dryden DT, Tock MR. DNA mimicry by proteins. Biochem Soc Trans. 2006;34(Pt 2):317–319. doi: 10.1042/BST20060317. [DOI] [PubMed] [Google Scholar]
- Duncan T, Wakefield JG. 50 ways to build a spindle: the complexity of microtubule generation during mitosis. Chromosome Res. 2011;19(3):321–333. doi: 10.1007/s10577-011-9205-8. [DOI] [PubMed] [Google Scholar]
- Dupré J. The disorder of things: metaphysical foundations of the disunity of science. Cambridge MA: Harvard University Press; 1993. [Google Scholar]
- Dupré J, O’Malley MA. Varieties of living things: life at the intersection of lineage and metabolism. Philosophy and Theory in Biology. 2009;1:e003. [Google Scholar]
- Dupré J, O’Malley MA. Metagenomics and biological ontology. Stud Hist Philos Biol Biomed Sci. 2007;38:834–846. doi: 10.1016/j.shpsc.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Ereshefsky M, Pedroso M (2012) Biological individuality: the case of biofilms. Biol Philos. doi:10.1007/s10539-012-9340-4
- Falkowski PG, Fenchel T, Delong EF. The microbial engines that drive earth’s biogeochemical cycles. Science. 2008;320(5879):1034–1039. doi: 10.1126/science.1153213. [DOI] [PubMed] [Google Scholar]
- Fondi M, Fani R. The horizontal flow of the plasmid resistome: clues from inter-generic similarity networks. Environ Microbiol. 2010;12(12):3228–3242. doi: 10.1111/j.1462-2920.2010.02295.x. [DOI] [PubMed] [Google Scholar]
- Forterre P. Defining life: the virus viewpoint. Orig Life Evol Biosph. 2010;40(2):151–160. doi: 10.1007/s11084-010-9194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Cohan FM, Lawrence JG, Spratt BG, Coenye T, Feil EJ, Stackebrandt E, Van de Peer Y, Vandamme P, Thompson FL, Swings J. Opinion: re-evaluating prokaryotic species. Nat Rev Microbiol. 2005;3(9):733–739. doi: 10.1038/nrmicro1236. [DOI] [PubMed] [Google Scholar]
- Ghigo JM. Natural conjugative plasmids induce bacterial biofilm development. Nature. 2001;412(6845):442–445. doi: 10.1038/35086581. [DOI] [PubMed] [Google Scholar]
- Godfrey-Smith PG. Darwinian populations and natural selection. Oxford: Oxford University Press; 2009. [Google Scholar]
- Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci USA. 2012;109(2):594–599. doi: 10.1073/pnas.1116053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross J, Bhattacharya D. Uniting sex and eukaryote origins in an emerging oxygenic world. Biol Direct. 2010;5:53. doi: 10.1186/1745-6150-5-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grube M, Hawksworth DL. Trouble with lichen: the re-evaluation and re-interpretation of thallus form and fruit body types in the molecular era. Mycol Res. 2007;111(Pt 9):1116–1132. doi: 10.1016/j.mycres.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Hackett JD, Yoon HS, Li S, Reyes-Prieto A, Rummele SE, Bhattacharya D. Phylogenomic analysis supports the monophyly of cryptophytes and haptophytes and the association of rhizaria with chromalveolates. Mol Biol Evol. 2007;24(8):1702–1713. doi: 10.1093/molbev/msm089. [DOI] [PubMed] [Google Scholar]
- Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2(2):95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- Hatfull GF, Cresawn SG, Hendrix RW. Comparative genomics of the mycobacteriophages: insights into bacteriophage evolution. Res Microbiol. 2008;159(5):332–339. doi: 10.1016/j.resmic.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellweger FL. Carrying photosynthesis genes increases ecological fitness of cyanophage in silico. Environ Microbiol. 2009;11(6):1386–1394. doi: 10.1111/j.1462-2920.2009.01866.x. [DOI] [PubMed] [Google Scholar]
- Henderson B, Martin A. Bacterial moonlighting proteins and bacterial virulence. Curr Top Microbiol Immunol. 2011 doi: 10.1007/82_2011_188. [DOI] [PubMed] [Google Scholar]
- Hildebrand F, Meyer A, Eyre-Walker A. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 2010;6(9):e1001107. doi: 10.1371/journal.pgen.1001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberts DH, van der Klei IJ. Moonlighting proteins: an intriguing mode of multitasking. Biochim Biophys Acta. 2010;1803(4):520–525. doi: 10.1016/j.bbamcr.2010.01.022. [DOI] [PubMed] [Google Scholar]
- Hull DL. The metaphysics of evolution. Albany: SUNY:Press; 1989. [Google Scholar]
- Kav AB, Sasson G, Jami E, Doron-Faigenboim A, Benhar I, Mizrahi I. Insights into the bovine rumen plasmidome. Proc Natl Acad Sci USA. 2012;109(14):5452–5457. doi: 10.1073/pnas.1116410109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloesges T, Popa O, Martin W, Dagan T. Networks of gene sharing among 329 proteobacterial genomes reveal differences in lateral gene transfer frequency at different phylogenetic depths. Mol Biol Evol. 2011;28(2):1057–1074. doi: 10.1093/molbev/msq297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler J, Woetzel N, Staritzbichler R, Sanders CR, Meiler J. A unified hydrophobicity scale for multispan membrane proteins. Proteins. 2009;76(1):13–29. doi: 10.1002/prot.22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Martin W. On the origin of genomes and cells within inorganic compartments. Trends Genet. 2005;21(12):647–654. doi: 10.1016/j.tig.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane CE, Archibald JM. The eukaryotic tree of life: endosymbiosis takes its TOL. Trends Ecol Evol. 2008;23(5):268–275. doi: 10.1016/j.tree.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Lawrence JG, Retchless AC. The myth of bacterial species and speciation. Biol Philos. 2010;25:569–588. doi: 10.1007/s10539-010-9215-5. [DOI] [Google Scholar]
- Levins R. The strategy of model-building in population biology. In: Sober E, editor. Conceptual issues in evolutionary biology. 1. Cambridge: MIT Press; 1984. pp. 18–27. [Google Scholar]
- Lima-Mendez G, Van Helden J, Toussaint A, Leplae R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol Biol Evol. 2008;25(4):762–777. doi: 10.1093/molbev/msn023. [DOI] [PubMed] [Google Scholar]
- Lopez P, Bapteste E. Molecular phylogeny: reconstructing the forest. C R Biol. 2009;332(2–3):171–182. doi: 10.1016/j.crvi.2008.07.003. [DOI] [PubMed] [Google Scholar]
- Lozupone CA, Hamady M, Cantarel BL, Coutinho PM, Henrissat B, Gordon JI, Knight R. The convergence of carbohydrate active gene repertoires in human gut microbes. Proc Natl Acad Sci USA. 2008;105(39):15076–15081. doi: 10.1073/pnas.0807339105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukjancenko O, Wassenaar TM, Ussery DW. Comparison of 61 sequenced Escherichia coli genomes. Microb Ecol. 2010;60(4):708–720. doi: 10.1007/s00248-010-9717-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J, The Heliconius Genome Consortium (2012). Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature (published online) doi: 10.1038/nature11041 [DOI] [PMC free article] [PubMed]
- Marazzi I, Ho JS, Kim J, Manicassamy B, Dewell S, Albrecht RA, Seibert CW, Schaefer U, Jeffrey KL, Prinjha RK, Lee K, Garcia-Sastre A, Roeder RG, Tarakhovsky A. Suppression of the antiviral response by an influenza histone mimic. Nature. 2012;483(7390):428–433. doi: 10.1038/nature10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin W, Embley TM. Eukaryotic evolution, changes and challenges. Nature. 2006;430:623–630. doi: 10.1038/nature04546. [DOI] [PubMed] [Google Scholar]
- Martin W, Müller M. The hydrogen hypothesis for the first eukaryote. Nature. 1998;392(6671):37–41. doi: 10.1038/32096. [DOI] [PubMed] [Google Scholar]
- Mayr E. Animal species and evolution. Cambridge: Harvard University Press; 1963. [Google Scholar]
- McMahon SA, Roberts GA, Johnson KA, Cooper LP, Liu H, White JH, Carter LG, Sanghvi B, Oke M, Walkinshaw MD, Blakely GW, Naismith JH, Dryden DT. Extensive DNA mimicry by the ArdA anti-restriction protein and its role in the spread of antibiotic resistance. Nucleic Acids Res. 2009;37(15):4887–4897. doi: 10.1093/nar/gkp478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev. 2005;15(6):589–594. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Mitchell SD. Biological complexity and integrative pluralism. Cambridge: Cambridge University Press; 2003. [Google Scholar]
- Mora C, Tittensor DP, Adl S, Simpson AG, Worm B. How many species are there on Earth and in the ocean? PLoS Biol. 2011;9(8):e1001127. doi: 10.1371/journal.pbio.1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira D, Lopez-Garcia P. Symbiosis between methanogenic archaea and delta-proteobacteria as the origin of eukaryotes: the syntrophic hypothesis. J Mol Evol. 1998;47(5):517–530. doi: 10.1007/PL00006408. [DOI] [PubMed] [Google Scholar]
- Moustafa A, Beszteri B, Maier UG, Bowler C, Valentin K, Bhattacharya D. Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science. 2009;324(5935):1724–1726. doi: 10.1126/science.1172983. [DOI] [PubMed] [Google Scholar]
- Mumford S, Anjum RL. Getting causes from powers. Oxford: Oxford University Press; 2011. [Google Scholar]
- O’Malley MA. ‘Everything is everywhere: but the environment selects’: ubiquitous distribution and ecological determinism in microbial biogeography. Stud Hist Philos Biol Biomed Sci. 2008;39(3):314–325. doi: 10.1016/j.shpsc.2008.06.005. [DOI] [PubMed] [Google Scholar]
- O’Malley MA, Dupré J. Size doesn’t matter: towards a more inclusive philosophy of biology. Biol Philos. 2007;22:155–191. doi: 10.1007/s10539-006-9031-0. [DOI] [Google Scholar]
- Overmann J. The phototrophic consortium “Chlorochromatium aggregatum”—a model for bacterial heterologous multicellularity. Adv Exp Med Biol. 2010;675:15–29. doi: 10.1007/978-1-4419-1528-3_2. [DOI] [PubMed] [Google Scholar]
- Popa O, Hazkani-Covo E, Landan G, Martin W, Dagan T. Directed networks reveal genomic barriers and DNA repair bypasses to lateral gene transfer among prokaryotes. Genome Res. 2011;21(4):599–609. doi: 10.1101/gr.115592.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigbo P, Wolf YI, Koonin EV. The tree and net components of prokaryote evolution. Genome Biol Evol. 2010;2:745–756. doi: 10.1093/gbe/evq062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu A, Brulc JM, Wilson MK, Law BF, Theoret JR, Joens LA, Konkel ME, Angly F, Dinsdale EA, Edwards RA, Nelson KE, White BA. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS ONE. 2008;3(8):e2945. doi: 10.1371/journal.pone.0002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisner A, Holler BM, Molin S, Zechner EL. Synergistic effects in mixed Escherichia coli biofilms: conjugative plasmid transfer drives biofilm expansion. J Bacteriol. 2006;188(10):3582–3588. doi: 10.1128/JB.188.10.3582-3588.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S, Glickman MS. Bacterial DNA repair by non-homologous end joining. Nat Rev Microbiol. 2007;5(11):852–861. doi: 10.1038/nrmicro1768. [DOI] [PubMed] [Google Scholar]
- Skippington E, Ragan MA. Lateral genetic transfer and the construction of genetic exchange communities. FEMS Microbiol Rev. 2011;35(5):707–735. doi: 10.1111/j.1574-6976.2010.00261.x. [DOI] [PubMed] [Google Scholar]
- Skippington E, Ragan MA. Within-species lateral genetic transfer and the evolution of transcriptional regulation in Escherichia coli and Shigella. BMC Genomics. 2011;29(12):532. doi: 10.1186/1471-2164-12-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skippington E, Ragan MA. Evolutionary dynamics of small RNAs in 27 Escherichia coli and Shigella a genomes. Genome Biol Evol. 2012;4(3):330–345. doi: 10.1093/gbe/evs001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- Sorek R, Zhu Y, Creevey CJ, Francino MP, Bork P, Rubin EM. Genome-wide experimental determination of barriers to horizontal gene transfer. Science. 2007;318(5855):1449–1452. doi: 10.1126/science.1147112. [DOI] [PubMed] [Google Scholar]
- Sullivan JT, Trzebiatowski JR, Cruickshank RW, Gouzy J, Brown SD, Elliot RM, Fleetwood DJ, McCallum NG, Rossbach U, Stuart GS, Weaver JE, Webby RJ, De Bruijn FJ, Ronson CW. Comparative sequence analysis of the symbiosis island of Mesorhizobium loti strain R7A. J Bacteriol. 2002;184(11):3086–3095. doi: 10.1128/JB.184.11.3086-3095.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MB, Lindell D, Lee JA, Thompson LR, Bielawski JP, Chisholm SW. Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol. 2006;4(8):e234. doi: 10.1371/journal.pbio.0040234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe M, Kanehisa M (2012) Using the KEGG database resource. Curr Protoc Bioinformatics. Chapter 1: Unit 1.12 [DOI] [PubMed]
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29(1):22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor N, Robart AR, Christianson J, Zimmerly S. Self-splicing of a group IIC intron: 5′ exon recognition and alternative 5′ splicing events implicate the stem-loop motif of a transcriptional terminator. Nucleic Acids Res. 2006;34(22):6461–6471. doi: 10.1093/nar/gkl820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal LP. Persistence pays: how viruses promote host group survival. Curr Opin Microbiol. 2009;12(4):467–472. doi: 10.1016/j.mib.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weller GR, Kysela B, Roy R, Tonkin LM, Scanlan E, Della M, Devine SK, Day JP, Wilkinson A, d’Adda di Fagagna F, Devine KM, Bowater RP, Jeggo PA, Jackson SP, Doherty AJ. Identification of a DNA nonhomologous end-joining complex in bacteria. Science. 2002;297(5587):1686–1689. doi: 10.1126/science.1074584. [DOI] [PubMed] [Google Scholar]
- Wimsatt WC. Re-engineering philosophy for limited beings: piecewise approximations to reality. Chicago: University of Chicago Press; 2007. [Google Scholar]
- Woese CR. Evolutionary questions: the “progenote”. Science. 1990;247(4944):789. doi: 10.1126/science.2305249. [DOI] [PubMed] [Google Scholar]
- Woodward J. Making Things Happen: A Theory of Causal Explanation. Oxford: Oxford University Press; 2003. [Google Scholar]
- Yang Z. On the best evolutionary rate for phylogenetic analysis. Syst Biol. 1998;47(1):125–133. doi: 10.1080/106351598261067. [DOI] [PubMed] [Google Scholar]
- Zack DJ, Yamamoto K, Wong AL, Stempniak M, French C, Weisbart RH. DNA mimics a self-protein that may be a target for some anti-DNA antibodies in systemic lupus erythematosus. J Immunol. 1995;154(4):1987–1994. [PubMed] [Google Scholar]
- Zhaxybayeva O, Swithers KS, Lapierre P, Fournier GP, Bickhart DM, DeBoy RT, Nelson KE, Nesbo CL, Doolittle WF, Gogarten JP, Noll KM. On the chimeric nature, thermophilic origin, and phylogenetic placement of the Thermotogales. Proc Natl Acad Sci USA. 2009;106(14):5865–5870. doi: 10.1073/pnas.0901260106. [DOI] [PMC free article] [PubMed] [Google Scholar]