

Background: ATP-binding cassette exporters utilize ATP binding and hydrolysis to switch between the inward-facing and outward-facing state during transport.
Results: Mutations in bacterial MsbA, in a conserved structural element termed the tetrahelix bundle, do not affect nucleotide binding but impair formation of the outward-facing conformation.
Conclusion: The ATP-dependent switch requires robust tetrahelix bundle interactions.
Significance: Agents that target the tetrahelix bundle might modulate clinically important ABC exporters.
Keywords: ABC Transporter, ATP, Energetics, Membrane Transport, Multidrug Transporters, “Power Stroke”, Tetrahelix Bundle
Abstract
ATP-binding cassette transporters affect drug pharmacokinetics and are associated with inherited human diseases and impaired chemotherapeutic treatment of cancers and microbial infections. Current alternating access models for ATP-binding cassette exporter activity suggest that ATP binding at the two cytosolic nucleotide-binding domains provides a power stroke for the conformational switch of the two membrane domains from the inward-facing conformation to the outward-facing conformation. In outward-facing crystal structures of the bacterial homodimeric ATP-binding cassette transporters MsbA from Gram-negative bacteria and Sav1866 from Staphylococcus aureus, two transmembrane helices (3 and 4) in the membrane domains have their cytoplasmic extensions in close proximity, forming a tetrahelix bundle interface. In biochemical experiments on MsbA from Escherichia coli, we show for the first time that a robust network of inter-monomer interactions in the tetrahelix bundle is crucial for the transmission of nucleotide-dependent conformational changes to the extracellular side of the membrane domains. Our observations are the first to suggest that modulation of tetrahelix bundle interactions in ATP-binding cassette exporters might offer a potent strategy to alter their transport activity.
Introduction
Multidrug-resistant tumors and infectious microbes render many chemotherapeutic interventions ineffective. One contributing cause of this phenomenon is the activity of multidrug ATP-binding cassette (ABC)5 membrane transport proteins that export multiple, structurally diverse drugs out of cells, away from intracellular targets. The design of inhibitors of multidrug exporters is of prime importance to overcome efflux-based multidrug resistance (1–3). As multidrug ABC exporters affect the pharmacokinetics of many drugs in mammals, their modulation might also be useful in regulating the bioavailability of drugs (4, 5). Furthermore, the dysfunction of ABC exporters has been linked to common genetic disease conditions, including cystic fibrosis, Stargardt disease, age-related macular degeneration, adrenoleukodystrophy, Tangier disease, Dubin-Johnson syndrome, and progressive familial intrahepatic cholestasis (6). Here, a detailed understanding of the mechanisms and functions of ABC transporters will greatly aid the development of by-pass and replacement strategies and will facilitate the generation of activators of these transport proteins. In vivo activation of ABC exporter function was recently obtained for the first time using an in vitro-selected DARPin modulator (7), demonstrating the importance of biochemical and functional studies on membrane transporters for drug development.
Many ABC exporters share a common homo- or heterodimer architecture with two nucleotide-binding domains (NBDs) and two membrane domains (MDs). The transmembrane helices (TMs) in the MDs are extended into the cytosol, where they form an interface with the NBDs (3, 8, 9). Structural and biochemical studies on bacterial and mammalian ABC exporters have suggested a conserved alternating access mechanism of substrate transport (8, 10–14). Briefly, substrate-binding surfaces formed by the membrane-spanning regions of the TMs are alternately exposed to the internal and external sides of the plasma membrane. This movement is guided by the ATP-binding driven dimerization of the NBDs and their dissociation following ATP hydrolysis. The two alternating conformations, termed “inward-facing” (Fig. 1A) and “outward-facing,” (Fig. 1B) mediate substrate binding and release, respectively, on opposite sides of the membrane (8, 12). Because of observations that the conformational “switch” from the inward- to outward-facing state is dependent on ATP binding at the NBDs, this step is often referred to as the “power stroke” of substrate export (15–19). This step might additionally require the formation of a “tight” ATP-occluded state following “loose” ATP binding (20).
FIGURE 1.

MsbA structures and the tetrahelix bundle. A, nucleotide-free inward-facing structure of dimeric MsbA (Protein Data Bank code 3B5W, full model). B, AMP-PNP-bound outward-facing structure (Protein Data Bank code 3B60) (12). Residues Thr-121–Glu-133 in TM 3 and TM 3′ are in blue, and Asn-195–His-214 in TM 4 and TM 4′ are in red. C, zoomed in snapshot of the tetrahelix bundle viewed from the cytoplasmic side, after rotation of the MsbA dimer in B by 90°. Residues Ser-324 to Gln-582 in each monomer (containing the NBD) are not shown for clarity of presentation. Residues Glu-208 and Lys-212 (in the cytoplasmic extensions of TM 4 and TM 4′) are shown as purple and yellow sticks, respectively. D, residues Ala-281–Ala-281′ (red spheres) in dimeric MsbA are in close proximity in the nucleotide-free inward-facing conformation and distant in the AMP-PNP-bound outward-facing state, whereas Glu-208 to Glu-208′ residues (purple spheres) follow the opposite pattern. For further explanation, see under “A281C-A281C′ Cross-linking in Dimeric MsbA-cl Responds to Nucleotides.”
Despite major advances in our understanding of the molecular mechanism of ABC exporters, the process of conformational coupling between the NBDs and MDs during the power stroke is surrounded by many mysteries. How are the conformational changes, resulting from dimerization or dissociation of the cytosolic NBDs, transmitted over a long distance to the membrane-spanning regions in the MDs? Previous studies on LmrA from Lactococcus lactis (21), mammalian ABCB1 (P-gp, MDR1) (22), and TAP1/2 (transporter associated with antigen processing) (23) point to a role of the cytosolic regions of the MDs in this process. Published crystal structures of mammalian ABCB1 homologs, the homodimeric lipid A/multidrug ABC exporter MsbA in Gram-negative bacteria (24–26) and multidrug transporter Sav1866 from Staphylococcus aureus (27), in their nucleotide-bound and outward-facing conformations reveal that, in addition to the NBDs, a dimer interface is also formed by the cytosolic extensions of TMs and their connecting loops at the NBD-MD interface. In particular, the extensions of TMs 3, 3′, 4, and 4′ are in close proximity and form what was recently denoted as a “tetrahelix bundle” that might mediate communication between the NBDs on one side and the TMs on the other side (Fig. 1C) (12, 16, 28). On closer inspection of the tetrahelix bundle, a unique pair of charged residues (Glu-208 and Lys-212) at the end of the extension of TM 4 and 4′ in MsbA, and equipositional polar residues (His-204 and Gln-208) in Sav1866, were observed to form part of a network of mostly polar interhelix interactions between side chains within and around the tetrahelix bundle (28, 29). In agreement with this, cysteine substitutions at Glu-208 and Glu-208′ in the MsbA-cl (Cys-less) dimer were shown to undergo nucleotide-responsive cysteine cross-linking in our previous study (10), revealing enhanced proximity of these residues at certain stages of the catalytic cycle.
Based on the above-described structural information and molecular dynamics simulations, we tested the hypotheses that the tetrahelix bundle network is important for stabilizing the outward-facing conformation and that disruption of the inter-monomer interactions that underlie the tetrahelix bundle network will block the transporter's power stroke. To biochemically test these hypotheses, we replaced critical residues in the tetrahelix bundle in MsbA by site-directed mutagenesis, and we investigated the effects of these replacements on ATP binding and hydrolysis and on substrate binding and transport. We also used inter-molecular cysteine cross-linking to test whether a weakened tetrahelix bundle affects the conformational cycle of the MsbA dimer.
EXPERIMENTAL PROCEDURES
Structural Analysis
The atomic coordinates deposited for the x-ray structures of MsbA (12) were analyzed using (Mac) PyMOL Molecular Viewer, version 1.3 (DeLano Scientific LLC).
Materials
M17 medium was obtained from Oxoid and LB medium from Formedium. Chemicals (reagent grade) were obtained from Sigma, and fluorescent dyes were purchased from Molecular Probes (Invitrogen), unless stated otherwise.
Bacterial Strains and Plasmids
L. lactis strain NZ9000 ΔlmrA ΔlmrCD (30), which is devoid of the endogenous MDR transporters LmrA and LmrCD, was used as a host for pNZ8048-derived plasmids harboring a chloramphenicol resistance marker, and it was grown in M17 medium supplemented with 5 μg/ml chloramphenicol where required. The pNZ8048 plasmids also contain a multiple cloning site downstream of a nisinA-inducible promoter (31). The nisinA producer strain L. lactis NZ9700 was grown in M17 medium without antibiotics. Its culture supernatant was used as a source of nisin for induction of protein expression (31). Escherichia coli strain XL1 Blue was used to transform and host the cloning vector pGEM®−5Zf(+) (Promega) harboring a carbenicillin resistance marker, and it was grown in LB medium containing 50 μg/ml carbenicillin where required (32).
Generation of Site-directed Mutants
pGEM plasmids harboring a wild type (WT) E. coli msbA or msbA-cl (Cys-less) genes coding for N-terminal hexa-His-tagged proteins were used as templates (10, 26, 32, 33). Site-directed mutants were constructed through QuikChange® (Stratagene, La Jolla, CA), using the following primers: MsbA E208A Fw, 5′-CAG CGC AGC ACA AAT GCT GAA G-3′, and Rv, 5′-CTT CAG CAT TTG TGC TGC GCT G-3′; E208Q Fw, 5′-CCA CCA GCG CAC AAC AAA TGC TG-3′, and Rv, 5′-CAG CAT TTG TTG TGC GCT GGT GG-3′; K212A Fw, 5′-CAA ATG CTG GCG GGC CAC AAA G-3′, and Rv, 5′-CTT TGT GGC CCG CCA GCA TTT G-3′; and A281C Fw, 5′-GAT AGC CTG ACT TGC GGT ACG ATT AC-3′, and Rv, 5′-GTA ATC GTA CCG CAA GTC AGG CTA TC-3′. pGEM MsbA K212A, pGEM MsbA-cl, and pGEM MsbA-cl A281C were used as templates to generate the double mutant E208A/K212A MsbA, A281C MsbA-cl, and the double mutant E208A/A281C MsbA-cl, respectively. Mutant msbA genes in pGEM were then subcloned as NcoI-SacI fragments into pNZ8048 downstream from the nisinA-inducible promoter nisA, to yield pNH MsbA E208A, pNH MsbA K212A, pNH MsbA E208A/K212A, pNH MsbA E208Q, pNH MsbA-cl A281C, and pNH MsbA-cl E208A/A281C. All inserted DNA fragments were sequenced to confirm that only the intended mutation was introduced.
Transport Assays
Preparation of inside-out membrane vesicles (ISOVs), Hoechst 33342 transport in ISOVs, and ethidium transport in intact cells were performed as described previously (27, 32).
Protein Purifications
His-tagged MsbA was purified by affinity chromatography. Once the protein expression was measured in ISOVs, total membrane proteins in the ISOVs (5 mg/ml, typically 30 mg) were solubilized in 50 mm KPi, pH 8.0, 10% glycerol, 0.2 m NaCl, and 1% n-dodecyl-β-d-maltoside (DDM) (Anatrace) for 3–4 h at 4 °C with gentle mixing on a rotating wheel. Unsolubilized protein was removed by ultracentrifugation at 164,000 × g for 40 min at 4 °C. Meanwhile, Ni2+-nitrilotriacetic acid resin (1 ml of resin per 10 mg of His-tagged protein) (Sigma) was equilibrated by washing three times with 5 resin volumes of MilliQ water and twice with 5 resin volumes of wash buffer A (50 mm KPi, pH 8.0, 10% glycerol, 0.1 m NaCl, 0.05% DDM, and 20 mm imidazole, pH 8.0) with the resin being spun down every time at 175 × g for 30 s at 4 °C. The solubilized membrane proteins in the supernatant were allowed to bind with the equilibrated resin by mixing on a rotating wheel overnight at 4 °C. Unbound proteins were removed by centrifugation, and the resin was then washed with 20 volumes of wash buffer A, followed by 30 volumes of wash buffer B (50 mm KPi, pH 7.0, 10% glycerol, 0.1 m NaCl, 0.05% DDM, and 20 mm imidazole, pH 8.0). Purified protein was eluted with 3–4 volumes of elution buffer (50 mm KPi, pH 7.0, 5% glycerol, 0.1 m NaCl, 0.05% DDM, and 150 mm imidazole, pH 8.0) using a Bio-Spin column (Bio-Rad). After discarding the first 0.5 resin volumes, the subsequent eluent was collected, maintained on ice, and used for experiments as immediately as possible. To determine the protein concentration in the presence of detergent, the MicroBCA (Pierce) assay was used. Purity of samples was assessed on Coomassie Brilliant Blue-stained SDS-polyacrylamide gels. If purified proteins were prepared for the determination of the ATPase activity in the malachite green method (see below), KPi in all the buffers was replaced by K-HEPES at the same pH.
Binding Assays
Hoechst 33342 and ethidium binding were measured in the absence of nucleotides by fluorescence spectroscopy as reported previously (32). For measurements of TNP-ATP (Molecular Probes) binding, 50 μg of purified protein was diluted in a 2-ml reaction buffer (50 mm KPi, pH 7.0, 10% glycerol, 0.01% DDM). Two aliquots, each of 1 μl of a 50 μm TNP-ATP solution (referred to as 2× 1 μl of 50 μm TNP-ATP), 2× 1 μl of 100 μm TNP-ATP, 5× 1 μl of 500 μm TNP-ATP, 5× 2 μl of 500 μm TNP-ATP, and 7× 5 μl of 500 μm TNP-ATP were subsequently added, giving a final concentration of 11.4 μm TNP-ATP. All additions were separated by 30 s. Equal volumes of elution buffer were used as a negative control. Data were fitted to a hyperbola using Equation 1,
in which y is the normalized TNP-ATP fluorescence (relative to the maximum achievable fluorescence, which corresponds to 159 ± 9 a.u. and is set at 100%); x is the TNP-ATP concentration; a is the maximum TNP-ATP binding (Bmax), and b is the TNP-ATP concentration giving ½Bmax (equal to the apparent dissociation constant, KdTNP-ATP). For the TNP-ATP binding data in Fig. 5, the mean predicted value for a was 82 ± 2%. For ADP-dependent TNP-ATP displacement, the binding assay was set up as above with 11.4 μm TNP-ATP, after which 1 μl of 4 m ADP, 2× 1 μl of 6 m ADP, 1 μl of 10 m ADP, 2 μl of 5 m ADP, 2 μl of 9 m ADP, and 5× 2 μl of 7 m ADP were subsequently added, giving a final concentration of 64 mm ADP. In the ATP-dependent TNP-ATP displacement experiments, the addition of TNP-ATP was followed by 2× 10 μl of 20 μm ATP, 2× 20 μl of 20 μm ATP, 5× 20 μl of 200 μm ATP, 5× 40 μl of 500 μm ATP, and 23× 25 μl of 4 mm ATP, giving a final concentration of 1.161 mm ATP. Equal volumes of elution buffer were used as a negative control. All additions were separated by 30 s. For analysis of the displacement data, the normalized TNP-ATP fluorescence (relative to the initial fluorescence in the absence of the displacing nucleotide, which is set at 100%) was fitted to Equation 2,
Equation 2 describes the displacement of TNP-ATP binding by ADP or ATP in the first term and the hyperbolic rise in the free TNP-ATP concentration in the second term, in which y is the normalized TNP-ATP fluorescence; x is the ADP or ATP concentration; a is the maximum TNP-ATP displacement; b is the ADP or ATP concentration giving ½ maximal displacement (IC50), and c is the fluorescence of displaced TNP-ATP. For the TNP-ATP displacement data in Fig. 5, the mean predicted value of a and c was 103 ± 1 and 38 ± 7%, respectively, in the displacement by ADP, and 93 ± 1 and 36 ± 3%, respectively, in the displacement by ATP. The mean predicted values for c correspond to an absolute fluorescence of about 57 a.u., which is consistent with the measured fluorescence of 58 ± 3 a.u. of free 11.4 μm TNP-ATP (in the absence of MsbA protein) under the experimental conditions. From the experimentally determined IC50, the apparent Kd for ADP binding (KdADP) or ATP binding (KdATP) by MsbA proteins was then calculated using Equation 3,
in which y is KdADP or KdATP; a is IC50; b is KdTNP-ATP (Table 1), and c is the fixed concentration of TNP-ATP in the displacement experiment (11.4 μm). Fluorescence was monitored in an LS 55B luminescence spectrometer (PerkinElmer Life Sciences) using excitation and emission wavelengths of 407 and 535 nm and slit widths of 10 and 15 nm, respectively.
FIGURE 5.

MsbA mutants used in this study exhibit WT-like binding affinities for nucleotides. TNP-ATP binding and ADP-dependent and ATP-dependent TNP-ATP displacement (inset) were determined in triplicate (n = 3) using purified MsbA proteins in detergent solution (large symbols for TNP-ATP binding and its displacement by ADP; small open and closed circles for displacement of TNP-ATP binding by ATP). ATP concentration scale is ×50 the actual ATP concentration. The IC50 values obtained in the TNP-ATP displacement experiments were used in Equation 3 under “Binding Assays” to calculate the dissociation constants for binding of ADP (KdADP) and ATP (KdATP). Values for KdADP, KdATP, and the dissociation constant for binding of TNP-ATP (KdTNP-ATP) are listed in the text and in Table 1. Error bars represent mean ± S.E.
TABLE 1.
Nucleotide binding affinities of WT MsbA are maintained in the MsbA mutants
| MsbA protein | TNP-ATP binding, KdTNP-ATP (μm)a | TNP-ATP displacement |
|||
|---|---|---|---|---|---|
| by ADP |
by ATP |
||||
| IC50 (mm)b | KdADP (mm)b | IC50 (μm)b | KdATP (μm)b | ||
| WT | 0.6 ± 0.1 | 43.9 ± 11.5 | 2.2 ± 0.7 | 261 ± 40 | 13.1 ± 3.0 |
| E208A | 0.8 ± 0.1 | 43.7 ± 12.9 | 2.9 ± 0.9 | 197 ± 40 | 12.9 ± 3.2 |
| K212A | 1.2 ± 0.1 | 66.3 ± 26.1 | 6.3 ± 3.6 | –c | – |
| A281C | 0.6 ± 0.0 | 29.9 ± 9.4 | 1.5 ± 0.5 | – | – |
| E208A/A281C | 0.6 ± 0.1 | 48.3 ± 7.7 | 2.4 ± 0.6 | – | – |
a KdTNP-ATP values were obtained from the main data in Fig. 5 by nonlinear regression analysis with Equation 1 under “Binding Assays.”
b IC50 values were obtained from the data in Fig. 5, inset, by nonlinear regression analysis with Equation 2 under “Binding Assays” and were used for calculation of KdADP and KdATP in Equation 3.
c – means not determined.
Intrinsic Tryptophan Fluorescence Measurements
Detergent-purified protein (45–65 μg) was used in 2 ml of 100 mm KPi, pH 7.0, containing 5 mm MgSO4 in a quartz cuvette. Equal volume of elution buffer was used as negative control. After 1-min incubation with stirring at RT, the sample was excited at a wavelength of 295 nm in an LS 55B luminescence spectrometer (PerkinElmer Life Sciences). An emission wavelength scan was performed from 300 to 420 nm. The excitation and emission slit widths were set at 5 nm each. Each scan was repeated three times, and the average fluorescence intensity at 328 nm was the peak intrinsic tryptophan fluorescence of the sample.
Trypsin Digestions
Purified protein (WT MsbA, E208A, K212A, or E208A/K212A) (9 μg in 20 μl) was mixed with trypsin at a trypsin/protein ratio (w/w) of 1:800 and incubated for 75 min at 37 °C. The reaction was stopped by the addition of soybean trypsin inhibitor at a concentration of 0.0045 mg/ml and incubated for 5 min at 37 °C. Subsequently, 10 μl of 3× sample loading buffer supplemented with 10 mm DTT was added. The samples were incubated at 37 °C for 15 min and then loaded on a 12% SDS-polyacrylamide gel supplemented with 2.5 mm DTT.
ATPase Activity Measurements
ATPase assays were performed on purified protein (1–5 μg per reaction) diluted in 100 mm K-HEPES buffer, containing 5 mm MgSO4 using the malachite green-ammonium molybdate method described previously (10, 30). Briefly, for each data point, the ATPase reaction was initiated by the addition of 1 μg of protein to 60 μl of 100 mm K-HEPES buffer, pH 7.0, supplemented with 5 mm MgSO4 and ATP (high grade ATP from Sigma) at concentrations as indicated in the legend to Fig. 6. The reactions were incubated at 30 °C after which the reactions were stopped by addition of malachite green-ammonium molybdate for 5 min. Absorbance at A600 was determined 25 min after the addition of 34% citric acid. Absorbance readings associated with Pi release between t = 1 and 2 min after the start of the ATPase reaction were converted into the amount of Pi released using a Pi calibration curve. To measure the Taxol-stimulated ATPase activity, Taxol was added from a 1 mm stock solution in absolute 98% ethanol to the reaction buffer before the addition of protein.
FIGURE 6.

E208Q mutation partially restores reduced catalytic activity of the E208A mutant. A, although the basal ATP hydrolysis activity of E208A MsbA at 1 mm ATP was reduced to 16.2 ± 3.9% of WT MsbA, E208Q MsbA was able to improve this performance to 44.4 ± 10.0% (100% activity of WT in these experiments equaled 158.5 ± 37.3 nmol ATP/min/mg) (n = 3; *, p < 0.05, Student's t test E208A mutant versus E208Q mutant). B, measurement of the basal ATP hydrolysis activity (n = 3) of detergent-purified WT MsbA (●) and E208A MsbA (○) over a range of ATP concentrations between 0.1 and 4 mm, and display of the data in a Lineweaver-Burk plot, revealed changes in the maximum rate of hydrolysis (Vmax) and Michaelis constant (Km) for E208A MsbA compared with WT MsbA, whereas the Vmax/Km ratio (= 1/slope) was relatively similar. See text for further details. C, inclusion of the MsbA substrate Taxol (32) stimulated the ATPase activity for WT MsbA but not for E208A MsbA. Experimental conditions and 100% value are as given in A, and symbols are as given in B. D, under conditions identical to those described for Fig. 3B, the E208Q mutant mediated the export of ethidium from intact cells. E, initial rates of ethidium efflux were measured over the first 50 s after the addition of glucose. These analyses showed that although E208Q MsbA could not efflux as well as the WT protein, its ethidium extrusion rate was significantly higher than that for E208A MsbA (WT versus E208A MsbA, *, p < 0.05; E208A MsbA versus E208Q MsbA, p < 0.09, Student's t tests). F, E208Q mutation improved Hoechst 33342 transport by MsbA in ISOVs compared with the E208A replacement. Experimental details are similar to those described in the legend to Fig. 3A. Consistent with ethidium transport measurements and ATPase assays, E208Q MsbA was never as efficient as WT MsbA. Traces in D and F and rates in E are based on observations in at least three independent experiments. Error bars represent mean ± S.E.
Cysteine Cross-linking, Atto590 Labeling, and Densitometric Analysis
MsbA-cl, E208C MsbA-cl, or A281C MsbA-cl-containing ISOVs against control (ISOVs from nonexpressing cells) were used for cysteine cross-linking and analyzed by Western blotting as described previously (10). For cysteine cross-linking combined with Atto590 labeling, ISOVs were diluted to 7 μg of total membrane protein/μl in 100 mm K-HEPES buffer, pH 7.0, containing 5 mm MgSO4 in a reaction volume of 90 μl in microcentrifuge tubes. Nucleotides (2 mm AMP-PNP or ADP, or 2 mm ATP plus 2 mm Vi) were then added, followed by incubation at room temperature for 3 min (and 6 and 12 min for the AMP-PNP-based experiments with E208A/A281C MsbA-cl). 20 μm of the rhodamine derivative Atto590-maleimide (Atto-tec) was then added (from a 2 mm stock in DMSO), and after incubation at room temperature for 5 min, cross-linking reactions were initiated by the addition of 0.5 mm copper phenanthroline solution, which was added from a 50 mm stock made by mixing CuSO4 and 1,10-phenanthroline in a ratio of 1:4 (w/w) in ultrapure H2O, as published (10). After piercing their lids, the tubes were kept in a 30 °C shaker (200 rpm) incubator for 15 min. The reactions were stopped by the addition of excess (10 mm, i.e. 9 μl from a freshly prepared stock of 100 mm in ultrapure H2O) of N-ethylmaleimide followed by incubation at RT for 1–2 min. 3–5 μl from each reaction was separated on SDS-polyacrylamide gels, which were first viewed under UV light to detect the Atto590-labeled protein bands and subsequently Coomassie Brilliant Blue-stained to visualize all protein bands. Monomer intensities from UV pictures and dimer intensities from Coomassie Brilliant Blue-stained gels were subjected to densitometric analyses using the ImageJ software, version 1.43 (National Institutes of Health).
Statistical Analyses
Equilibrium Hoechst 33342 binding and ATPase data were fitted to a hyperbola, B = Bmax·S/(Kd + S) or V = Vmax·S/(Km + S) in which B and V represent the binding of Hoechst 33342 and rate of the ATPase reaction, respectively; Bmax and Vmax are the maximum binding of Hoechst 33342 and maximum rate of ATP hydrolysis, respectively; Kd and Km represent the dissociation constant and Michaelis-Menten constant, respectively; and S is the substrate concentration. Error bars reported in this work represent ± S.E. For all densitometry/ATPase/binding data that are presented as values relative to a specific/standard condition taken as 1 or 100%, the standard condition was included (and set to 1 or 100%) in each experimental replicate. One-tailed, two-sample Student's t tests were performed on the datasets to test the significance of observed increases or decreases in relative band intensities or ATPase activities or ethidium efflux rates.
RESULTS
Transport-defective Tetrahelix Bundle Mutants of MsbA
When total membrane proteins in ISOVs were separated on an SDS-polyacrylamide gel and analyzed by Western blotting, it was observed that single and double alanine substitutions at the ionizable residues Glu-208 and Lys-212 in the tetrahelix bundle of E. coli MsbA dimer did not affect the expression level of the protein (Fig. 2A). WT MsbA and the K212A and E208A/K212A mutants had an apparent size of ∼56 kDa on SDS-polyacrylamide gels (Fig. 2B), as was observed previously for MsbA (10, 33). However, all three alanine mutants displayed greatly reduced ATP-dependent Hoechst 33342 transport activities in ISOVs compared with the WT protein, in an assay in which the fluorescence quenching of the dye was detected following its transport from the membrane into the acidic, aqueous lumen of the inside-out membrane vesicles (Fig. 3A) (32, 34). Consistent with this observation, we also found that each alanine mutant was defective in mediating metabolic energy-dependent export of the cationic substrate ethidium from cells, in an assay in which the fluorescence quenching of the dye was detected following its dissociation from genomic DNA during transport (Fig. 3B). It is interesting to note that for both Hoechst 33342 and ethidium, the transport phenotypes of the single mutants E208A and K212A were identical and reproducibly less affected than that of the double mutant E208A/K212A (Fig. 3, A and B). Thus, using two different substrates and experimental systems (Hoechst 33342 and ethidium, and cells and membrane vesicles) we confirmed that the alanine mutations, designed to weaken the tetrahelix bundle interaction network in MsbA, rendered the exporter defective in ATP-dependent extrusion of substrates.
FIGURE 2.
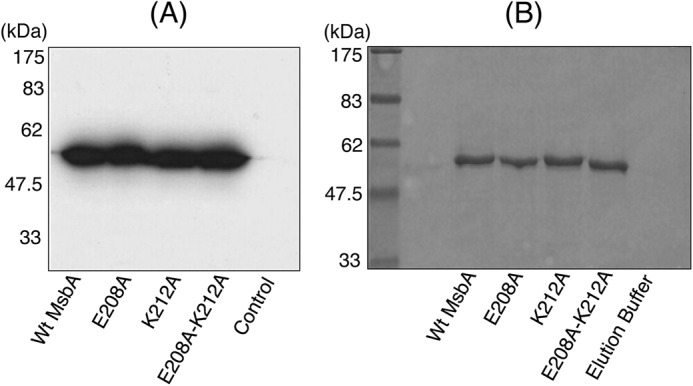
Expression and purification of MsbA mutants. A, Western blot analysis shows that E208A MsbA, K212A MsbA, and E208A/K212A MsbA mutants were equally well expressed as WT MsbA in the plasma membrane. B, Coomassie-stained SDS-polyacrylamide gel showing the apparent homogeneity of our preparations of purified protein. Approximate positions of the molecular mass marker are shown alongside in A and B.
FIGURE 3.

Effects of alanine mutations in the tetrahelix bundle on active substrate transport and substrate binding. A, when 0.25 μm Hoechst 33342 was added to WT MsbA, E208A MsbA, K212A MsbA, or E208A/K212A MsbA-containing ISOVs, followed by 2 mm ATP, all mutants displayed strongly reduced abilities to transport Hoechst 33342 compared with WT (n = 3). The double mutant E208A/K212A was more severely affected than the single mutants E208A and K212A. B, E208A MsbA, K212A MsbA, and E208A/K212A MsbA were also defective in mediating active export of ethidium from intact cells. Energy-depleted cells were pre-loaded with 2 μm ethidium, and at the arrow, 28 mm glucose was added to provide metabolic energy. Again, the double mutant E208A/K212A showed a larger defect than the single mutants E208A and K212A. Traces in A and B represent observations in at least three independent experiments. C, purified mutant proteins had WT-like Hoechst 33342 binding affinities (n = 3, see text).
Binding Properties and Protein Folding Are Unaffected by Alanine Mutations
The three alanine mutants along with WT MsbA were purified in detergent solution to homogeneity (Fig. 2B). The purified alanine mutant proteins could bind Hoechst 33342 as well as WT (Fig. 3C) with an apparent dissociation constant (Kd) for WT MsbA, E208A MsbA, K212A MsbA, and E208A/K212A MsbA of 0.8 ± 0.2, 0.6 ± 0.1, 1.1 ± 0.0, and 0.5 ± 0.2 μm, respectively, and maximal binding value (Bmax) of 48.2 ± 9.9, 41.9 ± 6.7, 50.9 ± 15.4, and 37.2 ± 9.0 a.u., respectively. Furthermore, the ethidium binding to E208A MsbA was similar to WT MsbA when measured in direct binding assays on purified protein based on fluorescence anisotropy and during MsbA-mediated facilitated ethidium uptake in intact cells (data not shown) (32). Intrinsic tryptophan fluorescence measurements and trypsin digestions were performed to compare the overall protein folding of WT MsbA and mutants. In contrast to heat-denatured MsbA (35), the alanine substitution mutants did not show any significant shifts in the emission maxima of their intrinsic tryptophan fluorescence spectra or decrease in the peak intrinsic tryptophan fluorescence intensities at 328 nm compared with WT (Fig. 4, A–C). Consistent with these observations, trypsin digestions of purified WT MsbA and the E208A, K212A, and E208A/K212A mutants gave identical digestion patterns on Coomassie-stained SDS-PAGE, with major signals at 31 and 7.5 kDa that were absent for heat-denatured MsbA (Fig. 4D), indicating that none of the mutations caused major protein misfolding. The mutants E208A and K212A were equally capable in binding the fluorescent ATP analog TNP-ATP as WT MsbA (apparent dissociation constant KdTNP-ATP: WT MsbA, 0.6 ± 0.1 μm; E208A MsbA, 0.8 ± 0.1 μm; K212A MsbA, 1.2 ± 0.1 μm) (Fig. 5 and Table 1). Furthermore, the apparent dissociation constant for binding of ADP by WT MsbA and mutants was determined in TNP-ATP displacement experiments and was found to be in a similar range (KdADP WT MsbA, 2.2 ± 0.7 mm; E208A MsbA, 2.9 ± 0.9 mm; K212A MsbA, 6.3 ± 3.6 mm) (Fig. 5, inset, and Table 1). In control experiments, the displacement of TNP-ATP from WT MsbA and E208A MsbA was also measured in the presence of ATP itself (Fig. 5, inset), yielding apparent dissociation constants for binding of ATP (KdATP) of 13.1 ± 3.0 μm for WT MsbA and 12.9 ± 3.2 μm for E208A MsbA (Table 1). Taken together, these results suggest that neither protein denaturation nor reduced substrate binding nor reduced nucleotide binding were responsible for reduced substrate transport by the mutant MsbA proteins with weakened interactions at the tetrahelix bundle.
FIGURE 4.

Alanine mutations in the tetrahelix bundle do not denature MsbA. WT MsbA and the mutant proteins showed similar overall protein folding that is different from heat-denatured MsbA, as deduced from intrinsic tryptophan fluorescence and trypsin digestion patterns. A, normalized spectral profiles of intrinsic tryptophan fluorescence measured for MsbA WT, without (black trace, labeled WT MsbA) or with (gray trace) denaturation for 1 min at 50 °C, revealed a denaturation-dependent right-shift in the emission maximum. B and C, alanine mutants E208A MsbA and K212A MsbA and the E208A/K212A double mutant gave overlapping tryptophan fluorescence traces (B) with similar peak fluorescence intensities at 328 nm as WT MsbA (n = 4) (C), and it did not show any signs of protein denaturation. Data are elution buffer-subtracted. Error bars represent mean ± S.E. D, trypsin digestions of purified MsbA proteins were examined on 12% SDS-PAGE (9 μg of protein/lane). Lane 1, molecular mass marker; lane 2, WT MsbA; lane 3, WT MsbA + trypsin; lane 4, E208A MsbA + trypsin; lane 5, K212A MsbA+ trypsin; lane 6, E208A/K212A MsbA + trypsin; lane 7, WT MsbA (denatured for 1 min at 50 °C) + trypsin. White triangles refer to major signals for WT MsbA (31 and 7.5 kDa) that were absent for heat-denatured MsbA. Band patterns are typical for observations in four independent experiments.
Carboxyl-to-Amide Mutation Partially Restores Transport and Catalytic Activity
Consistent with the inhibition of ATP-dependent substrate transport by the E208A mutation (Fig. 3, A and B), the basal ATP hydrolysis (ATPase) activity of purified E208A mutant at 1 mm ATP was reduced to 16.2 ± 3.9% of WT (Fig. 6A). We also measured the ATPase activity of purified proteins over a range of ATP concentrations between 0.1 and 4 mm, which revealed that the reduction was primarily in the maximum rate of hydrolysis (Vmax, E208A MsbA, 74.3 ± 1.0 nmol/min/mg protein versus 329.1 ± 10.8 nmol/min/mg protein for WT MsbA, corresponding to an apparent turnover number (kcat) of 9.6/min versus 42.7/min, respectively, whereas the Michaelis constant (Km) was 0.25 ± 0.01 mm ATP for E208A MsbA and 0.68 ± 0.07 mm ATP for WT MsbA) (Fig. 6B). As is evident from the slopes in the Lineweaver-Burk plots (Fig. 6B), these kinetic parameters give rise to similar Vmax/Km ratios (297 ± 16 μl/min/mg protein for E208A MsbA versus 484 ± 66 μl/min/mg protein for WT MsbA) and kcat/Km ratios (about 38/min/mm ATP for E208A MsbA versus 63/min/mm ATP for WT MsbA), with a relatively small difference of less than 2-fold. At concentrations of ATP close to the KdADP of WT MsbA and E208A MsbA, where these concentrations are almost 20-fold below Km, the kcat/Km ratio can be used as an estimate of the rate of ATP binding (v ≈(kcat/Km) [S]). Hence, these findings are in agreement with the similarity in KdATPfor both proteins (Fig. 5 and Table 1) and support our conclusion that the E208A mutation does not have major effects on ATP binding. Inhibition of the drug-stimulated ATPase by the E208A mutation was also observed (Fig. 6C). The maximum stimulation of the ATPase activity of WT MsbA at a concentration of 5 μm Taxol was absent for E208A MsbA.
To characterize tetrahelix bundle interactions in more detail, an E208Q mutant was tested along with the E208A mutant. As the hydrogen bond donor and acceptor functions of an ionizable Glu side chain can be retained upon substitution by the nonionizable Gln side chain, the E208Q substitution is potentially more conservative than the Glu-208 to nonpolar Ala replacement. Indeed, compared with the E208A replacement, the E208Q mutation improved the basal ATPase activity at 1 mm ATP to 44.4 ± 10.0% of the WT MsbA (p < 0.05, Student's t test E208A versus E208Q) (Fig. 6A). E208Q MsbA also displayed enhanced energy-dependent ethidium export activity in intact cells compared with E208A MsbA (Fig. 6, D and E) and ATP-dependent Hoechst 33342 transport in ISOVs (Fig. 6F). It is noteworthy that E208Q, although more efficient than E208A, could not fully restore WT-like ATPase or multidrug export activity (Fig. 6). It is also interesting to note here that cysteine substitutions in this region of ABCB1 and MsbA were previously reported to result in a higher basal ATPase activity compared with the respective Cys-less proteins due to covalent cysteine cross-linking (10, 36). Together, these data suggest that the strength of interactions at the tetrahelix bundle of MsbA determines the ability of the transporter to catalyze ATP-dependent substrate export and ATP hydrolysis but not the ability to bind substrate or ATP.
A281C-A281C′ Cross-linking in Dimeric MsbA-cl Responds to Nucleotides
We previously reported the conformational cycle of membrane-embedded dimeric MsbA during the ATPase reaction using cysteine cross-linking between the reporters E208C and E208C′, which are located near to the NBDs (10) and which are predicted to be directly accessible to each other in the AMP-PNP-bound MsbA structure (Fig. 1C) without obstruction by helices. To extend this study and introduce conformation reporters in the MDs, we engineered a cysteine mutation at Ala-281 located on the extracellular end of TM 6 of MsbA. The rationale for selecting this residue was to generate a complementary reporter near the external side of the MDs. Ala-281–Ala-281′ are in close proximity in the inward-facing conformation but separated by more than 20 Å due to the formation of the “wings” in the MDs in the outward-facing state. In contrast, Glu-208–Glu-208′ are distant in the inward-facing state and close in the outward-facing state (Fig. 1D).
A281C MsbA-cl was expressed equally well as MsbA-cl (Cys-less) (Fig. 7A) and was catalytically active in transporting Hoechst 33342 in ISOVs (see under “Mutation E208A Inhibits Formation of Outward-facing Conformation”). Consistent with the nucleotide-free inward-facing MsbA structure (Fig. 1D), membrane-embedded A281C MsbA-cl readily underwent A281C-A281C′ cross-linking under nonreducing nucleotide-free conditions (Fig. 7A). To test the predicted complementarity between the E208C-E208C′ and A281C-A281C′ cross-linking reactions, ISOVs harboring E208C MsbA-cl or A281C MsbA-cl were treated with an oxidizing agent (copper phenanthroline) in the absence of any added nucleotides and separated on an SDS-polyacrylamide gel without any reducing agents in the gel-loading buffer. Western blot analysis revealed that the proportion of cross-linked dimers (high molecular weight bands) was much higher for A281C MsbA-cl (∼75%) as compared with E208C MsbA-cl (∼40%) under nucleotide-free conditions (Fig. 7B). This observation is consistent with the nucleotide-free inward-facing state of MsbA, in which the NBDs are distant and MDs are proximal (Fig. 1D) (12). We also observed a small, but reproducible, difference between the migration of the cross-linked species of E208C MsbA-cl and A281C MsbA-cl (Fig. 7B), which is most likely due to differences in SDS binding to the cross-linked proteins.
FIGURE 7.

Cross-linking Atto590 labeling on E208C and A281C in dimeric MsbA-cl. A, when ISOVs containing A281C MsbA-cl were loaded on an SDS-polyacrylamide gel without the reducing agent DTT in the SDS sample-loading buffer and analyzed by Western blot, high molecular weight dimers were observed due to spontaneous cysteine cross-linking. These dimers were absent for MsbA-cl (Cys-less). Cysteine cross-linking was enhanced by incubation with the oxidizing agent copper phenanthroline (CuPhen), although cross-linking disappeared on the inclusion of DTT in the SDS sample-loading buffer. B, under no nucleotide conditions, E208C MsbA-cl (10) or A281C MsbA-cl in ISOVs was subjected to inter-molecular cysteine cross-linking, and MsbA-specific bands (monomers (M)/dimers (D)) were detected on Western blot probed with an anti-penta-His antibody (10). The approximate molecular mass marker positions are shown alongside. C, high saturating levels of Cys-Cys cross-linking were observed for A281C MsbA-cl in the no nucleotide condition. As the reduction in this high intensity after the transition from inward-facing to outward-facing MsbA is associated with a poor signal-to-noise ratio, we combined the reduction in A281C/A281C′ cross-linking with measurements on the increased accessibility of free A281C to the thiol-reactive fluorescent probe Atto590. To validate this method, we re-assayed A281C MsbA-cl and E208C MsbA-cl in the no nucleotide condition. On Coomassie Brilliant Blue staining of the gel, high molecular weight bands corresponding to E208C/E208C′ and A281C/A281C′ Cys-Cys cross-linked dimers (D) were observed, whereas such dimers were not obtained for MsbA-cl. A small but reproducible difference in migration was observed between dimers for the two mutants, which is attributed to anomalous SDS binding. The intensity of dimers for A281C MsbA-cl was found to be higher than for E208C MsbA-cl, consistent with the notion that the MsbA dimer will be in the inward-facing state in the absence of added nucleotides. D, when the same gel was visualized under UV light (prior to Coomassie-staining) only ISOVs containing E208C MsbA-cl or A281C MsbA-cl had the monomers (M) labeled with Atto590; labeled monomers were not observed for control and MsbA-cl ISOVs. Consistent with the enhanced dimer intensity for A281C MsbA-cl, the monomer Atto590-labeling intensity was reduced for this mutant compared with E208C MsbA-cl. The images in C and D pertain to one and the same gel/experiment, repeated at least three times, and were processed in parallel and cropped to increase clarity of presentation.
Next, we aimed to test the nucleotide dependence of cross-linking of A281C MsbA-cl in experiments that were complementary to those reported for E208C MsbA-cl (10). However, the high level of A281C-A281C′ cross-linking under no nucleotide conditions caused poor signal-to-background ratios (Fig. 7B) when analyzing the reduction in the high intensity of dimer signals associated with transitions from inward-facing to outward-facing states. We therefore combined cysteine cross-linking with the detection of the increased accessibility of the uncross-linked A281C side chain to the thiol-reactive dye Atto590-maleimide. Because of the fluorescence properties of Atto590, dye-labeled monomers on an SDS-polyacrylamide gel could easily be detected through in-gel UV fluorescence, after which the same gel was stained with Coomassie Brilliant Blue to detect the higher molecular weight dimeric MsbA species.
Being devoid of cysteine residues, MsbA-cl did not form a cross-linked dimeric species nor could monomers of MsbA-cl that were visible on the Coomassie-stained gel (Fig. 7C) be labeled with Atto590 (Fig. 7D). We reproduced the observation that under no nucleotide conditions the proportion of A281C-A281C′ cross-linked dimers was higher than that of E208C-E208C′ (Fig. 7C), indicative of an inward-facing conformation. Notably, the enhanced dimer intensity for A281C MsbA-cl was associated with a decrease in the Atto590-labeled monomer intensity and vice versa for E208C MsbA-cl (Fig. 7, C and D). Thus, having obtained complementary patterns of A281C-A281C′ and E208C-E208C′ cross-linking with (Fig. 7, C and D) or without (Fig. 7B) the use of the Atto590-labeling step, we conclude that the inclusion of Atto590 itself in the reaction does not change the overall protein conformation, i.e. MsbA is in the inward-facing conformation under nucleotide-free conditions.
We then tested the cross-linking properties of A281C MsbA-cl using the cysteine cross-linking Atto590-labeling method in the presence of various nucleotide conditions that represent intermediate steps of the ATP hydrolysis cycle, namely AMP-PNP (for the ATP-bound prehydrolysis state), ATP + sodium orthovanadate (ADP·Vi-trapping to mimic the ADP·Pi transition intermediate), and ADP (for post-hydrolysis and Pi release) (Fig. 8A). Densitometric analyses on the monomer and dimer signals revealed that AMP-PNP or ADP·Vi binding led to a decrease in the proportion of A281C-A281C′ cross-linked dimers and a concomitant increase in the Atto590-labeled monomers (Fig. 8B), compared with the no nucleotide condition. On the contrary, the dimer/monomer intensities for the ADP samples were more similar to those obtained for the no nucleotide condition (Fig. 8, A and B) (p < 0.03, Student's t test ADP·Vi or AMP-PNP versus ADP). As an additional control, we also performed the cross-linking Atto590-labeling experiment with E208C MsbA-cl in the presence of increasing concentrations of ADP up to 10 mm (Fig. 9A), which exceeds the KdADP of MsbA (Fig. 5 and Table 1) by a factor of at least 3. As we obtained an ADP dose-dependent decrease in the dimers and concomitant increase in monomer intensities (Fig. 9B), these data lend further support for the ADP binding-induced shift toward the inward-facing state of MsbA. They are also complementary to results previously reported for cysteine cross-linking of E208C-E208C′ in MsbA-cl (10) and are consistent with published crystallographic, cryo-EM, EPR, and mass spectrometry observations for MsbA (11–14, 37).
FIGURE 8.

Nucleotide-responsive cross-linking of A281C MsbA-cl and E208A/A281C MsbA-cl. A, intermolecular A281C-A281C′ cross-linking for A281C MsbA-cl was found to be nucleotide-responsive and thus could be used to monitor conformational changes associated with the ATPase cycle as a complement to previously published work with E208C MsbA-cl (10). All images here pertain to one and the same gel/experiment, processed in parallel to enhance readability (M, monomer; D, dimer). B, Atto590-labeled monomer and Coomassie Brilliant Blue-stained dimer intensities were analyzed by densitometry and are presented as -fold change in intensity relative to the no nucleotide condition (for which the -fold change was set at 1) (n = 3; **, p < 0.03, Student's t test). C, E208A mutation in the A281C background (E208A/A281C MsbA-cl) led to a reduction in Hoechst 33342 transport activity compared with the activity of MsbA-cl and A281C MsbA. A similar reduction was obtained for the E208A mutation in the WT MsbA background (see Fig. 3, A and B). D, compared with A281C MsbA-cl, the E208A/A281C MsbA-cl double mutant displayed a lack of changes in cross-linking Atto590 labeling in response to incubation with the nonhydrolysable ATP analog AMP-PNP (for 6 or 12 min). All panels pertain to one and the same gel/experiment, processed in parallel, and are cropped to enhance readability (M, monomer; D, dimer). E, E208A mutation interrupts the inward- to outward-facing conformational switch in MsbA. Densitometric analysis on the monomer and dimer signals for samples after 12 min of incubation without or with AMP-PNP was as indicated. Data are presented as -fold change in intensity, relative to the no nucleotide condition (for which the -fold change equals 1) (n = 3; *, p < 0.06; **, p < 0.04, Student's t test). Open bars refer A281C MsbA-cl, and gray bars refer to E208A/A281C MsbA-cl. Error bars represent mean ± S.E.
FIGURE 9.

ADP-responsive cross-linking Atto590 labeling of E208C MsbA-cl. A, to study the conformational changes affected by ADP binding, E208C MsbA-cl harboring ISOVs were assayed using cysteine cross-linking Atto590 labeling in the presence of increasing concentrations of ADP. Briefly, ISOVs at 5–7 mg/ml total membrane protein, in 100 mm K-HEPES buffer, pH 7.0, containing 5 mm MgSO4 were first mixed with 0.5 mm DTT for 3 min at room temperature to reduce preformed cross-links. ADP was added at the indicated concentrations. Following 3 min of incubation at RT, cross-linking was initiated by the addition of 0.5 mm copper phenanthroline. After 5 min of incubation in a 30 °C shaker incubator, 20 μm Atto590 was added, and the samples were further incubated for 10 min in the 30 °C shaker incubator. Reactions were stopped by the addition of 10 mm N-ethylmaleimide. 10–15 μg of protein from each sample was mixed with 6× SDS sample loading buffer devoid of reducing agents and separated on a 10% SDS-polyacrylamide gel. The gel was first viewed under UV light to detect Atto-labeled monomers (M; bottom panel), followed by Coomassie Brilliant Blue staining to detect dimers (D; top panel). B, monomer/dimer intensities are presented as percentage of total intensity of MsbA-related bands (monomers + dimers), relative to the percentage monomer/dimer intensity observed under the no nucleotide condition (which was set at as 1) (n = 3, data are mean ± S.E.). The results suggest that ADP binding to MsbA induces a conformational shift toward the inward-facing state.
Taken together, the evidence from reporter position Ala-281 in the MDs presented herein lends strong support for our earlier conclusion using the reporter position Glu-208 near the NBDs (10) that membrane-embedded MsbA goes from an inward-facing conformation to an outward-facing conformation upon ATP binding, that MsbA remains in the overall outward-facing state on hydrolyzing ATP to ADP·Pi, and that the transporter returns to an inward-facing conformation in the presence of ADP alone (38). Based on these nucleotide-dependent cross-linking/labeling data obtained under near-physiological conditions, we conclude that cysteines at position Ala-281 in MsbA can be used to detect the conformational switch associated with nucleotide-dependent wing formation in the MDs.
Mutation E208A Inhibits Formation of Outward-facing Conformation
Having characterized the importance of the tetrahelix bundle interactions for substrate export activity and ATP hydrolysis and having constructed a reporter mutant that could report the conformation of the MD wings, we asked the following question. Does a weak tetrahelix bundle affect the formation of the outward-facing conformation in MsbA? For this purpose, we generated the mutation E208A in the background of A281C in MsbA-cl (termed E208A/A281C MsbA-cl). Consistent with the phenotype observed for the mutation E208A in the WT background (Fig. 3A), the E208A/A281C double mutant displayed a strong reduction in ATP-dependent Hoechst 33342 export activity in ISOVs compared with MsbA-cl and A281C MsbA (Fig. 8C).
Following our confirmation of the reduced transport properties of E208A/A281C MsbA-cl in ISOVs, the above-described cross-linking Atto590-labeling assays were performed with E208A/A281C MsbA-cl-harboring ISOVs in the presence of 2 mm AMP-PNP, in parallel with A281C MsbA-cl ISOVs (Fig. 8D). Densitometry analyses confirmed that, although A281C MsbA-cl showed a marked decrease in the proportion of cross-linked dimers and an increase in Atto590-labeled monomers on AMP-PNP binding compared with the no nucleotide condition, these changes were nearly nonexistent for E208A/A281C MsbA-cl (Fig. 8E). An extension of the incubation period in the presence of AMP-PNP from 3 min up to 12 min did not alter the overall data (Fig. 8E). The absence of changes in the monomer/dimer signals was unlikely to be due to reduced binding of AMP-PNP, as E208A/A281C MsbA-cl was found to have a similar KdTNP-ATP (0.6 ± 0.1 μm) as WT MsbA, E208A MsbA, and A281C MsbA-cl (Fig. 5 and Table 1). Instead, the data suggest that the inward- to outward-facing conformational switch in MsbA is interrupted by the presence of the mutation E208A, or in other words, E208A MsbA represents a mutant protein that can bind ATP as well as the WT protein but is deficient in proceeding further in the conformational cycle. These findings underline the importance of robust interactions within the tetrahelix bundle for the transmission of ATP-dependent conformational changes from the NBDs to the extracellular end of TM 6 in the MDs. Thus, we biochemically demonstrate ATP-dependent conformational deficits in the E208A mutant and hence show that MsbA with a disrupted tetrahelix bundle has a reduced capacity to form a stable ATP-bound outward-facing state.
DISCUSSION
Many details are unknown regarding the mechanism by which multidrug ABC exporters mediate the ATP-dependent extrusion of amphiphilic compounds against the lipid-water partition coefficient. For example, the role of the intracellular regions of the MDs in the communication between the NBDs and membrane-spanning regions of the MDs is still poorly characterized. In these intracellular regions in the crystallized bacterial ABCB1 homologs MsbA and Sav1866, the cytoplasmic extensions of TMs 3 and 4 are in close proximity in the outward-facing state and form a central tetrahelix bundle at the dimer interface (28). In MsbA, this tetrahelix bundle contains a pair of charged residues (Glu-208 and Lys-212) in the extension of TM 4 that are proposed to be involved in a network of electrostatic and hydrogen-bonding interactions between the two half-transporters (28).
Structural data indicate that ATP binding causes NBD dimerization (18, 19) and formation of the wings in the outward-facing MDs (8, 12, 15, 16). Indeed, our cysteine cross-linking/labeling experiments using reporters E208C (near the NBD) (10) and A281C (in the wing) (this study) in MsbA-cl are consistent with these structural data. However, despite its capability to bind nucleotides with a similar affinity as WT, the E208A mutant of MsbA with disrupted tetrahelix bundle interactions exhibits a reduced ability to form the outward-facing conformation. Our biochemical data therefore imply that ATP binding-dependent conformational coupling between NBDs and membrane-spanning regions in the MDs occurs via robust tetrahelix bundle interactions. In addition to its reduced conformational repertoire and substrate transport capacities, E208A MsbA also displays a greatly diminished ATP hydrolysis activity compared with WT. The tetrahelix bundle is contacted by the X-loop glutamate residue (Glu-476) in the NBD (28, 29) and is therefore appropriately positioned to modulate ATPase activity at the NBDs. Our data imply that the basal ATPase activity of the full-length MsbA protein is aided by robust tetrahelix bundle interactions and the formation of a stable outward-facing conformation. These findings are in line with the recent molecular dynamics simulations on MsbA and Sav1866 (28, 29) predicting that side-chain interactions within the tetrahelix bundle are a critical determinant of the transport cycle. Indeed, our biochemical analyses of His-204 and Gln-208 substitution mutants in Sav1866, at positions equivalent to Glu-208 and Lys-212 in MsbA, revealed a similar functional importance of the tetrahelix bundle in Sav1866.
In conclusion, this paper presents the first biochemical investigation into the functional role of the tetrahelix bundle in an ABC exporter. We have found that disruption of molecular contacts within the tetrahelix bundle inhibits the formation of a stable, ATP-bound, outward-facing conformation. Tetrahelix assemblies are present in many, if not all, homo- and heterodimeric ABC exporters in eubacteria and eukaryotic organisms (23, 39) and show diversity in primary amino acid sequences. Our observations are therefore the first to suggest that agents that act on tetrahelix bundle formation and/or disassembly might be useful as selective modulators of clinically important ABC transporters.
Acknowledgments
We are grateful to the organizers and participants of the Gordon Research Conference on “Multidrug Efflux Systems” (2011) for awarding a best poster prize for part of this work. We thank Yvonne Khoo and Kelvin Agboh for critical reading of the manuscript.
This work was supported by the Biotechnology and Biological Sciences Research Council.
- ABC
- ATP-binding cassette
- MD
- membrane domain
- NBD
- nucleotide-binding domain
- TM
- transmembrane helix
- DDM
- n-dodecyl-β-d-maltoside
- TNP-ATP
- 2′,3′-O-(2,4,6-trinitrophenyl)adenosine-5′-triphosphate tetra(triethylammonium)
- AMP-PNP
- adenosine 5′-(β,γ-imido)triphosphate)
- Vi
- sodium orthovanadate
- Fw
- forward
- Rv
- reverse
- ISOV
- inside-out membrane vesicle
- a.u.
- arbitrary unit.
REFERENCES
- 1. Gottesman M. M., Fojo T., Bates S. E. (2002) Multidrug resistance in cancer. Role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58 [DOI] [PubMed] [Google Scholar]
- 2. Szakács G., Paterson J. K., Ludwig J. A., Booth-Genthe C., Gottesman M. M. (2006) Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 5, 219–234 [DOI] [PubMed] [Google Scholar]
- 3. Higgins C. F. (1992) ABC transporters. From microorganisms to man. Annu. Rev. Cell Biol. 8, 67–113 [DOI] [PubMed] [Google Scholar]
- 4. Borst P., Elferink R. O. (2002) Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 71, 537–592 [DOI] [PubMed] [Google Scholar]
- 5. Marquez B., Van Bambeke F. (2011) ABC multidrug transporters. Target for modulation of drug pharmacokinetics and drug-drug interactions. Curr. Drug Targets 12, 600–620 [DOI] [PubMed] [Google Scholar]
- 6. Dean M., Allikmets R. (2001) Complete characterization of the human ABC gene family. J. Bioenerg. Biomembr. 33, 475–479 [DOI] [PubMed] [Google Scholar]
- 7. Seeger M. A., Mittal A., Velamakanni S., Hohl M., Schauer S., Salaa I., Grütter M. G., van Veen H. W. (2012) Tuning the drug efflux activity of an ABC transporter in vivo by in vitro selected DARPin binders. PLoS One 7, e37845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gutmann D. A., Ward A., Urbatsch I. L., Chang G., van Veen H. W. (2010) Understanding polyspecificity of multidrug ABC transporters. Closing in on the gaps in ABCB1. Trends Biochem. Sci. 35, 36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seeger M. A., van Veen H. W. (2009) Molecular basis of multidrug transport by ABC transporters. Biochim. Biophys. Acta 1794, 725–737 [DOI] [PubMed] [Google Scholar]
- 10. Doshi R., Woebking B., van Veen H. W. (2010) Dissection of the conformational cycle of the multidrug/lipidA ABC exporter MsbA. Proteins 78, 2867–2872 [DOI] [PubMed] [Google Scholar]
- 11. Zou P., McHaourab H. S. (2009) Alternating access of the putative substrate-binding chamber in the ABC transporter MsbA. J. Mol. Biol. 393, 574–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ward A., Reyes C. L., Yu J., Roth C. B., Chang G. (2007) Flexibility in the ABC transporter MsbA. Alternating access with a twist. Proc. Natl. Acad. Sci. U.S.A. 104, 19005–19010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Borbat P. P., Surendhran K., Bortolus M., Zou P., Freed J. H., Mchaourab H. S. (2007) Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS Biol. 5, e271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mehmood S., Domene C., Forest E., Jault J. M. (2012) Dynamics of a bacterial multidrug ABC transporter in the inward- and outward-facing conformations. Proc. Natl. Acad. Sci. U.S.A. 109, 10832–10836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McDevitt C. A., Crowley E., Hobbs G., Starr K. J., Kerr I. D., Callaghan R. (2008) Is ATP binding responsible for initiating drug translocation by the multidrug transporter ABCG2? FEBS J. 275, 4354–4362 [DOI] [PubMed] [Google Scholar]
- 16. Dawson R. J., Locher K. P. (2007) Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581, 935–938 [DOI] [PubMed] [Google Scholar]
- 17. Higgins C. F., Linton K. J. (2004) The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol. 11, 918–926 [DOI] [PubMed] [Google Scholar]
- 18. Smith P. C., Karpowich N., Millen L., Moody J. E., Rosen J., Thomas P. J., Hunt J. F. (2002) ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell 10, 139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moody J. E., Millen L., Binns D., Hunt J. F., Thomas P. J. (2002) Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters. J. Biol. Chem. 277, 21111–21114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sauna Z. E., Ambudkar S. V. (2007) About a switch. How P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol. Cancer Ther. 6, 13–23 [DOI] [PubMed] [Google Scholar]
- 21. Vigano C., Grimard V., Margolles A., Goormaghtigh E., van Veen H. W., Konings W. N., Ruysschaert J.-M. (2002) A new experimental approach to detect long-range conformational changes transmitted between the membrane and cytosolic domains of LmrA, a bacterial multidrug transporter. FEBS Lett. 530, 197–203 [DOI] [PubMed] [Google Scholar]
- 22. Rosenberg M. F., Velarde G., Ford R. C., Martin C., Berridge G., Kerr I. D., Callaghan R., Schmidlin A., Wooding C., Linton K. J., Higgins C. F. (2001) Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J. 20, 5615–5625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oancea G., O'Mara M. L., Bennett W. F., Tieleman D. P., Abele R., Tampé R. (2009) Structural arrangement of the transmission interface in the antigen ABC transport complex TAP. Proc. Natl. Acad. Sci. U.S.A. 106, 5551–5556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou Z., White K. A., Polissi A., Georgopoulos C., Raetz C. R. (1998) Function of Escherichia coli MsbA, an essential ABC family transporter, in lipid A and phospholipid biosynthesis. J. Biol. Chem. 273, 12466–12475 [DOI] [PubMed] [Google Scholar]
- 25. Doerrler W. T., Reedy M. C., Raetz C. R. (2001) An Escherichia coli mutant defective in lipid export. J. Biol. Chem. 276, 11461–11464 [DOI] [PubMed] [Google Scholar]
- 26. Reuter G., Janvilisri T., Venter H., Shahi S., Balakrishnan L., van Veen H. W. (2003) The ATP binding cassette multidrug transporter LmrA and lipid transporter MsbA have overlapping substrate specificities. J. Biol. Chem. 278, 35193–35198 [DOI] [PubMed] [Google Scholar]
- 27. Velamakanni S., Yao Y., Gutmann D. A., van Veen H. W. (2008) Multidrug transport by the ABC transporter Sav1866 from Staphylococcus aureus. Biochemistry 47, 9300–9308 [DOI] [PubMed] [Google Scholar]
- 28. Weng J.-W., Fan K.-N., Wang W.-N. (2010) The conformational transition pathway of ATP binding cassette transporter MsbA revealed by atomistic simulations. J. Biol. Chem. 285, 3053–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aittoniemi J., de Wet H., Ashcroft F. M., Sansom M. S. (2010) Asymmetric switching in a homodimeric ABC transporter. A simulation study. PLoS Comput. Biol. 6, e1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Venter H., Velamakanni S., Balakrishnan L., van Veen H. W. (2008) On the energy dependence of Hoechst 33342 transport by the ABC transporter LmrA. Biochem. Pharmacol. 75, 866–874 [DOI] [PubMed] [Google Scholar]
- 31. de Ruyter P. G., Kuipers O. P., de Vos W. M. (1996) Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Appl. Environ. Microbiol. 62, 3662–3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Woebking B., Velamakanni S., Federici L., Seeger M. A., Murakami S., van Veen H. W. (2008) Functional role of transmembrane helix 6 in drug binding and transport by the ABC transporter MsbA. Biochemistry 47, 10904–10914 [DOI] [PubMed] [Google Scholar]
- 33. Woebking B., Reuter G., Shilling R. A., Velamakanni S., Shahi S., Venter H., Balakrishnan L., van Veen H. W. (2005) Drug-lipid A interactions on the Escherichia coli ABC transporter MsbA. J. Bacteriol. 187, 6363–6369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schaedler T. A., Tong Z., van Veen H. W. (2012) The multidrug transporter LmrP protein mediates selective calcium efflux. J. Biol. Chem. 287, 27682–27690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eckford P. D., Sharom F. J. (2008) Functional characterization of Escherichia coli MsbA. Interaction with nucleotides and substrates. J. Biol. Chem. 283, 12840–12850 [DOI] [PubMed] [Google Scholar]
- 36. Loo T. W., Bartlett M. C., Clarke D. M. (2010) Human P-glycoprotein is active when the two halves are clamped together in the closed conformation. Biochem. Biophys. Res. Commun. 395, 436–440 [DOI] [PubMed] [Google Scholar]
- 37. Ward A., Mulligan S., Carragher B., Chang G., Milligan R. A. (2009) Nucleotide-dependent packing differences in helical crystals of the ABC transporter MsbA. J. Struct. Biol. 165, 169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Doshi R., Gutmann D. A., Khoo Y. S., Fagg L. A., van Veen H. W. (2011) The choreography of multidrug export. Biochem. Soc. Trans. 39, 807–811 [DOI] [PubMed] [Google Scholar]
- 39. Zolnerciks J. K., Wooding C., Linton K. J. (2007) Evidence for a Sav1866-like architecture for the human multidrug transporter P-glycoprotein. FASEB J. 21, 3937–3948 [DOI] [PubMed] [Google Scholar]