

Abstract
Nearly 70 inherited human glycosylation disorders span a breathtaking clinical spectrum, impacting nearly every organ system and launching a family-driven diagnostic odyssey. Advances in genetics, especially next generation sequencing, propelled discovery of many glycosylation disorders in single and multiple pathways. Interpretation of whole exome sequencing results, insights into pathological mechanisms, and possible therapies will hinge on biochemical analysis of patient-derived materials and animal models. Biochemical diagnostic markers and readouts offer a physiological context to confirm candidate genes. Recent discoveries suggest novel perspectives for textbook biochemistry and novel research opportunities. Basic science and patients are the immediate beneficiaries of this bidirectional collaboration.
Keywords: Glycobiology, Glycosylation, Glycosylation Inhibitors, Glycosyltransferases, Golgi, Congenital Disorders of Glycosylation, GPI Anchor, N-Glycosylation, Dystroglycanopathy, Whole Exome Sequencing
Orientation
Recent progress in exome sequencing (1, 2) places biochemistry in the enviable position of having to explain how patients with inherited glycosylation disorders develop their symptoms and to suggest therapies to treat them. The technology to identify defective genes is no longer rate-limiting; it is available, inexpensive, and in demand. Informatics effectively sifts through predictions of damaging mutations in hundreds of cases of patients with unknown genetic disorders (3, 4). Now, biochemistry must step in to provide context and functional information for genes, some known only by letters and numbers.
Each of these developments increases biochemical momentum. The recent report from the National Academy of Sciences National Research Council on the future of glycosciences (5) alerts the scientific community, funding agencies, and politicians to this often overlooked field. The creation of the National Center for Advancing Translational Sciences marks a fuller commitment of the National Institutes of Health to translational medicine. The goal of the newly formed Centers for Mendelian Genomics is to solve the genetic basis of >3500 rare disorders. About 70 known genetic disorders affect glycan synthesis, and it is estimated that 2% of the genome encodes currently known glycosylation reactions (7). The bottom line is that many glycosylation disorders are known, many more will be found, and gene sequencing technology will deliver diverse medical specialties to glycobiology in the search for biochemical validation and a deeper understanding of therapeutic options. This minireview focuses on how clinical medicine and basic science, now more than ever, generate an immediate, symbiotic, cross-fertilizing partnership. Several recently discovered glycosylation disorders will also raise important questions for further biochemical investigation.
The collection of glycosylation disorders causes abnormalities in nearly every organ system (7, 8). This means that physicians from every specialty will likely encounter patients who carry glycosylation defects. Although near-term exome (or genome) sequencing will undoubtedly indicate either known or predictable glycosylation genes within their clinical arena, other genes may appear on the “fringes” of the current understanding of what constitutes a “glycosylation gene.” Examples follow. Essentially all of the known glycosylation biosynthetic pathways are included in these disorders. Rather than cover all of them, this minireview focuses on recent discoveries that generate novel perspectives and orient future areas for research. A general point to remember is that although many of the disorders are restricted to a specific biosynthetic pathway, such as the assembly of the precursor glycan for N-glycosylation, others, such as those that generate metabolic precursors or Golgi trafficking complexes, can impact multiple pathways.
Glycosylation Pathways
Mammals have eight major glycosylation pathways in the endoplasmic reticulum (ER)2-Golgi (7–9). Three of these will be highlighted here because they house most of the newly discovered glycosylation disorders. N-Glycosylation (Fig. 1) occurs in the ER during or soon after the synthesis of nascent proteins. UDP-GlcNAc, GDP-Man, dolichol-phosphate (Dol-P)-Man, and Dol-P-Glc provide the activated precursors to construct a glycan composed of Glc3Man9GlcNAc2, which is built stepwise onto a dolichol acceptor embedded in the membrane. The glycan from this lipid-linked oligosaccharide (LLO) precursor is transferred en bloc to asparagine within an NX(T/S) context of the protein acceptors. Remodeling (processing) of the protein-bound chain excises glucose in the ER and a variable number of mannose units (ER and Golgi), and this can be followed by the addition of variable amounts of GlcNAc, Gal, Fuc, and sialic acid (Golgi). Some chains are decorated with sulfate or phosphate, and the glycoproteins are sent to destinations within the cell, on its surface, or beyond (10).
FIGURE 1.

Schematic of the N-glycosylation pathway. The upper oval represents the ER. The early steps involve GlcNAc (blue squares), Glc (blue circles), and Man (green circles) addition to an LLO assembly on Dol-P (red circles with wavy green lines) using UDP-GlcNAc (orange star with blue square) and GDP-Man (orange star with green circle) donors on the cytoplasmic face. The partially completed glycan flips to the luminal side, where it is completed using the donors Dol-P-Man (large green circle with small red circle and wavy green line) and Dol-P-Glc (large blue circle with small red circle and wavy green line) to form Glc3Man9GlcNAc2-P-P-Dol, which is then transferred to proteins in the ER. The lower oval denotes processing of the N-glycans using a series of glycosidases and glycosyltransferases that require UDP-GlcNAc (orange star with blue square), UDP-Gal (orange star with yellow circle), and CMP-Sia (orange star with purple diamond) transported into the Golgi. Purple diamonds, sialic acid; yellow circles, galactose.
Glycosylphosphatidylinositol (GPI) anchors are assembled stepwise on phosphatidylinositol in the ER membrane (11), starting with transfer of GlcNAc by a protein complex on the cytoplasmic face (PIG-A), followed by de-N-acetylation (PIG-L). Flip to the luminal side for addition of an extra acyl chain to inositol (PIG-W). This is followed by the addition of 2 mannose units (PIG-M and PIG-V), ethanolamine phosphate (PIG-N), another mannose (PIG-B), and two ethanolamine phosphates (PIG-O and PIG-F). The entire sugar-lipid unit is transferred to proteins with the appropriate C-terminal amino acid sequence using a multisubunit transamidase complex. A deacylase removes the acyl chain generated by PIG-W (Fig. 2).
FIGURE 2.

GPI anchor synthesis. This schematic shows the stepwise pathway for GPI anchor assembly, including the reaction and the names of the enzymes and genes involved. Known glycosylation disorders are highlighted in red. White and blue squares, glucosamine; EtNP (N connected to P with bent lines), ethanolamine phosphate; GlcNAc-PI, N-acetylglucosaminylphosphatidylinositol. This figure was adapted from Ref. 11.
O-Mannose-based glycosylation structures and biosynthetic pathways are incomplete, and studies are ongoing. Selected Ser/Thr residues on target proteins (primarily α-dystroglycan (α-DG)) use Dol-P-Man and a POMT1-POMT2 complex to begin the assembly with mannose (12). The addition of GlcNAc (POMGnT1) and Gal (β1,4 Galactosyltransferase) and sialic acid makes simple structures. More complex branched glycans add GlcNAc and sulfated glucuronic acid (GlcUA) (13). Beyond this point, the biosynthetic pathways are unclear. Some functionally important chains are phosphorylated to generate Man-6-P and receive a glycosaminoglycan-like polymer containing alternating α1,3-Xyl and β1,3-GlcUA residues (14). Man-6-P is converted to a diester of unknown composition (15). The prerequisite glycan structures, order of addition, and donor substrates are undefined. Human glycosylation disorders, the α-dystroglycanopathies, have been key to solving this pathway. Mutations in the fukutin (FKTN) and FKRP genes that define glycosylation-related muscular dystrophies encode putative glycosyltransferases, but both are enzymes in search of donor and acceptor substrates (13).
Clinical and Genetic Nomenclature
Many human glycosylation disorders were first described by physicians and based on their patients' clinical presentations because the genetic basis was unknown. A good example is the severity-based categories of α-dystroglycanopathies that affect the addition of O-mannose-based glycans on the α-DG complex in muscle cells (12). A revised nomenclature was proposed (16). However, many cases still lack specific diagnoses as the search for new gene defects continues.
One group of glycosylation disorders was called carbohydrate-deficient glycoprotein syndromes and then congenital disorders of glycosylation (CDG) (17), subdivided into two groups based on whether the mutated genes affected the addition of N-glycans (type I) or their processing (type II). The latest simplified version (18) employs a non-italicized gene name followed by CDG, e.g. CDG-Ia is now PMM2-CDG. Both systems will likely co-exist for some time.
Biochemical Markers for Glycosylation Disorders
Potential glycosylation disorders can be assessed with biochemical biomarkers (19–21). However, markers do not identify the genetic defect. Serum transferrin (Tf) is the best marker for detecting most disorders affecting the N-glycosylation pathway (8, 22). Tf has two N-linked glycans, each containing two sialic acids. Mass spectrometry, HPLC, or isoelectric focusing easily identifies disorders in which one or both of the sites are unoccupied. The same methods yield different abnormal patterns when the defects alter N-glycan processing. Defects in GPI anchor synthesis can often be identified using antibodies (20, 23) against the GPI anchor itself or more commonly GPI-anchored proteins, such as CD59 on leukocytes. The α-dystroglycanopathies caused by defects in O-mannose-based glycans can be recognized in muscle biopsies using monoclonal antibodies (IIH6 and VIA4) directed against the glycan itself (24). Obviously, obtaining muscle is more invasive than a simple blood test and would not be routinely done as an inexpensive first step. There are no simple markers for defects in glycosaminoglycan chain synthesis.
Revolution in Gene Discovery
Previously, the genes responsible for most glycosylation disorders were identified by biochemical analysis of fibroblasts and serum glycans (8, 25). Now, genetic mapping techniques have largely replaced this approach (1, 7, 8). This is especially important for finding defects in consanguineous families. Plummeting sequencing costs, lightning speed, improved informatic analysis, and widespread availability of technology put these approaches in hand for solving glycosylation and other rare disorders. However, employing biomarkers as a first step quickly focuses the search on glycosylation, and more importantly, it provides a metabolic context for candidate genes. In the past 2 years, three defects were solved with traditional biochemical analysis and five by autozygosity mapping, linkage analysis, or targeted genomic arrays. Ten defects were solved with state-of-the-art whole exome analysis (Table 1).
TABLE 1.
Glycosylation disorders identified in 2011–2012

The mutated gene must be shown to impair the function of the protein or cause pathology. In many cases, the effects of the mutation on RNA splicing or protein structure are predicted based on multiple programs, such as SIFT (26), PolyPhen (27), and Condel (28), along with evolutionary conservation (phyloP) (29) and frequency of the occurrence of the non-synonymous SNP. A physiologically relevant approach is to complement the patient's fibroblasts with the normal allele of the mutated gene and show it corrects abnormal insufficient glycosylation (30–33). Another is to complement yeast or mammalian cells with defects in the candidate gene with the normal human allele but not the mutated version. Restoration of cell growth, lectin binding, or normal glycosylation of target proteins is typically done. In more complex systems (flies, fish, worms, and mice), functional knockdown of the gene that generates a similar gross phenotype is taken as evidence that the mutations in the gene are damaging (34–36).
Rediscovering Sugar Metabolism
The well established metabolic pathways connecting monosaccharide metabolism to protein glycosylation are incomplete (Fig. 3). They show potential reactions but do not reflect contributions of different sources to convergent pathways, cell and organ preferences, or allowance for metabolic states. Glycosylation disorders and models of these diseases are adding unexpected dimensions to the two-dimensional pathways (10, 37–39).
FIGURE 3.
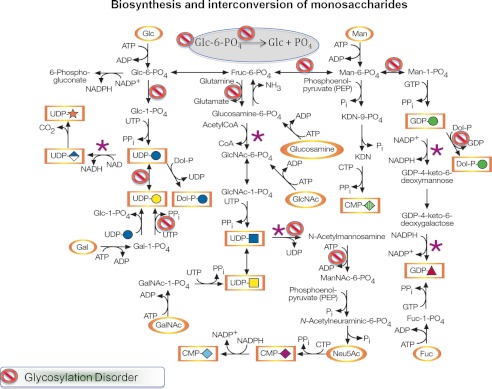
Monosaccharide metabolism in mammals and human glycosylation disorders. The gray oval indicates the ER, and all other reactions are thought to occur in the cytoplasm. The purple asterisks indicate known points of metabolic regulations. This figure was adapted from Ref. 10.
Patients deficient in mannose-6-phosphate isomerase (Fru-6-P ↔ Man-6-P) lack sufficient Man-6-P for full N-glycosylation. Clearly, glucose is a vital source of this precursor. However, modest daily supplements of mannose correct most of the patients' glycosylation deficiencies (40, 41). On the other hand, hypomorphic PMM2-deficient patients (Man-6-P → Man-1-P) do not benefit from mannose treatment. In mice, null alleles of either gene are embryonic lethal (embryonic days 2–10.5) (42, 43). Compound heterozygous mice carrying patient-equivalent mutations in Pmm2 with ∼10% residual enzymatic activity also die in midgestation. Surprisingly, providing mannose to the dams in their drinking water (intake of ∼25 mg/day) bypasses a critical block and produces viable full-term embryos that continue to thrive beyond weaning without mannose (38). This result suggests that the critical period is a gestational glycosylation insufficiency. It is premature to advise at-risk parents to consider mannose supplements in an attempt to ameliorate potential defects in their glycosylation-deficient pathways (7).
Zebrafish morphants deficient in pmm2 or mpi have significant morphological abnormalities in many ways comparable to the patients (35, 44). The mpi morphants could be substantially rescued with 50 mm mannose in the water, but supplementation was only required in the first 24 h. Removal of mannose or continued treatment beyond that time did not alter the outcome. The pmm2 morphant fish were not given mannose, but reducing the metabolic flux through the glycosylation pathway by generating double morphants deficient in both pmm2 and mpi actually improved the pmm2 phenotype. A curious aspect is that pmm2 morphants have increased amounts of Man-6-P. The accumulation correlates with the loss of LLO and appearance of free oligosaccharides, presumably released from the LLO (45, 46). These results recall important in vitro studies showing that increased Man-6-P levels lead to degradation of mature LLO precursor and release of the intact glycan. This suggests that Man-6-P may function as an intracellular sensor or signaling molecule. The mechanism is unknown.
Congenital myasthenic syndromes result from impaired signal transmission at the neuromuscular synapse (47). Using genetic linkage, one study (48) identified 13 unrelated families with mutations in GFPT1 (glutamine:fructose-6-phosphate transaminase 1), used for UDP-GlcNAc synthesis supplying most glycosylation pathways. No specific pathway was shown to cause pathology, but knockdown of the zebrafish ortholog gfpt1 altered muscle fiber morphology and impaired neuromuscular junction development in embryos. A surprising feature of this study was that some GFPT1 mutations had no effect on enzymatic activity, suggesting that the organization or localization of the enzyme in the cytoplasm is important for normal function.
Another whole exome study (49) describes five patients with a limb-girdle myasthenia syndrome and mutations in DPAGT1, the first enzyme in LLO synthesis and well known cause of CDG (7, 8). The deficiency may be due to a failure to export acetylcholine receptors to the end plate. No enzymatic assay was performed, but patients had abnormal Tf and a much milder phenotype than seen in previous CDG patients (50).
Whole exome and other genetic mapping studies showed glycosylation abnormalities due to mutations in G6PT1, G6PG3, and PGM1. All of these involve Glc-6-P metabolism (Fig. 3). G6PT1 encodes the Glc-6-P (51) translocator, which causes glycogen storage disease Ib; and G6PC3 encodes glucose-6-phosphatase catabolic-3. These disorders have profound effects on neutrophils: neutropenia, ER stress, and abnormal N- and O-linked glycans. Abnormal glycosylation in particular affects gp91phox, the electron-transporting component of the NADPH oxidase that is critical for the oxidative burst. This leads to a poor oxidative response, and increased ER stress causes excessive apoptosis. Both N- and O-glycan chains were truncated, with many lacking galactose and sialic acid. However, it was unclear whether N-glycosylation sites were fully occupied. This is important because tunicamycin causes similar ER stress by generating unoccupied sites.
How these two genes exert their effects on glycosylation is unknown, but deficiency of galactose and sialic acid on both N- and O-linked glycans suggests that it involves insufficient supply of UDP-Gal. Exome sequencing showed that mutations in PGM1 (Glc-1-P ↔ Glc-6-P) in two patients cause hypoglycemia and liver abnormalities and result in both absence of N-glycan chains and insufficient galactosylation/sialylation (52, 53). Incomplete glycan chains could be due to PGM1 effects on the UDP-Gal pool through the well known metabolic steps (Fig. 3). There is no explanation for how PGM1 deficiency causes the absence of entire glycan chains from proteins. However, uncontrolled galactosemia that leads to Gal-1-P accumulation (54) and hereditary fructose intolerance and Fru-1-P accumulation produce similar glycosylation abnormalities (55). Intracellular accumulation of various sugar phosphates or other metabolic byproducts appears to prevent full N-glycosylation in selected cells and tissues. Only Man-6-P has been shown to have an effect on LLO levels (45). It is clear that mutations in fundamental glucose utilization pathways must now consider additional cell-specific effects on glycosylation.
Although Tf analysis led to the identification of 35 glycosylation disorders spanning a large clinical spectrum, it focused most of the attention on the lack of N-glycans. Accumulation of toxic incomplete or unnatural products (as seen in uncontrolled galactosemia) is seldom considered. Disorders involving dolichol biosynthesis or activation of mannose and its downstream donors could also impair synthesis of GPI anchors, O-mannose glycans, and C-mannosylation (56), but few studies have probed other types of glycans (57). Synthesis of Dol-P-Man requires a complex containing DPM1–3, each encoded by a different gene. DPM1 is the catalytic subunit, and DPM3 tethers the complex to the ER membrane (58), whereas DPM2 stabilizes DPM1 and enhances binding of Dol-P-Man (59). A single patient with mutations in DPM3 showed a mild and late onset muscular dystrophy but did not have any of the typical symptoms seen in Dol-P-Man-deficient patients. The mutations disrupt the DPM1/DPM3 binding interface (57). Two families with much more severe pathology typical of CDG patients had mutations in DPM2 with muscular dystrophy, with reduced O-mannose staining in muscle (60).
α-Dystroglycanopathies
An entire set of congenital muscular dystrophies (CMDs) with variable severity results from defects in the biosynthetic pathway that adds O-mannose-linked glycans to α-DG. This peripheral membrane component of the dystrophin-glycoprotein complex is located in muscle, nerve, heart, and brain. α-DG is one of the two subunits of the dystrophin-glycoprotein complex, bridging the extracellular matrix to the cytoskeleton. α-DG and β-DG are derived from a single gene, DAG1. In muscle, cytoskeletal actin is linked to β-DG, which spans the cell membrane. The extracellular domain of β-DG binds to α-DG, which in turn binds to laminin in the extracellular matrix via its glycan-containing domain. The degree and types of α-DG glycosylation vary in different tissues. Monoclonal antibodies against the glycans have been key to identifying glycosylation-related defects that affect α-DG.
Collectively, these disorders, called α-dystroglycanopathies, result from mutations in seven genes, and more will likely be found. Other proteins probably contain these glycans and might contribute to CMD pathology in the brain because brain-specific deletion of α-DG does not reduce the amount of those glycans. Recently, Dwyer et al. (61) reported that these glycans occur on a receptor protein-tyrosine phosphatase and the secreted form, phosphacan, and that mutations in POMGnT1, the second enzyme in the pathway, in a mutant mouse strain result in a lower molecular weight and loss of the glycan antigen.
Defining the structure of the critical glycan(s) and their location on the protein has been challenging (34, 62). The key laminin-binding glycans contain Man-6-P residues in an acid-resistant diester linkage. Details of biosynthesis are nebulous, but Man-6-P addition is unrelated to the lysosomal enzyme-targeting pathway. LARGE is a protein with two putative glycosyltransferase domains and functions as a co-polymerase that adds a variable number of alternating units of α1,3-Xyl and β1,3-GlcUA to the protein (14). The acceptor sugar and structure of the glycan are unknown. Solving this long-standing enigma relied on compositional analysis of expressed α-DG and testing a series of glycosides as acceptors for expressed LARGE.
Walker-Warburg syndrome (WWS) is a clinically defined, severe CMD. Only about 50% of these cases result from mutations in POMT1 or POMT2. One study showed that a putative glycosyltransferase, GTDC2, is mutated in some WWS patients based on whole exome analysis and homozygosity mapping of consanguineous families (63). Morpholino knockdown of gtdc2 in zebrafish duplicated the WWS phenotype. Two additional studies using a combination of linkage analysis and exome sequencing identified a large number of patients with recessive mutations in ISPD (isoprenoid synthase domain-containing). One study complemented patients' fibroblasts with wild-type ISPD (62), and the other used zebrafish morphants to recapitulate the human phenotype, including hydrocephalus, smaller eye size, muscle degeneration, and reduced α-DG glycosylation (34). The gene ISPD is the human homolog of a series of genes found in plants and prokaryotes in the non-mevalonate (2-C-methyl-d-erythritol 4-phosphate) pathway of isoprenoid synthesis (64). However, this pathway does not exist in chordates, so its biochemical function, presumably in the biosynthesis of the O-mannosyl glycan, is unknown. Two homologs in bacteria have cytidylyltransferase activity and are used in the synthesis of CDP-methylerythritol and CDP-ribitol. One suggestion (34) is that the gene is involved in CTP-driven substrate activation to a precursor in the pathway, but this idea awaits more structural information on the glycan.
Human glycosylation defects in the isoprenoid-derived dolichol pathway are well established. Mutations in SRD5A3, the long-sought polyprenol reductase, show the existence of another unknown route of dolichol biosynthesis (65, 66). Mutations in DHDDS, a cis-isoprenyltransferase, cause retinitis pigmentosa but no other clinical deficits (67, 68). Knockdown of NUS1, the Nogo-B receptor that contains a cis-isoprenyltransferase domain, reduces the LLO level and decreases N-glycosylation (69). To date, no defects are known in this likely CDG target gene.
Defects in GPI Anchor Synthesis
Whole sequencing and autozygosity mapping identified six disorders in GPI anchor biosynthesis: PIG-A, PIG-L, PIG-M, PIG-N, PIG-O, and PIG-V. PIG-A catalyzes the first step in GPI anchor synthesis, and somatic mutations in the X-linked gene cause the well known hematological disorder paroxysmal nocturnal hemoglobinuria, which results in erythrocyte lysis (70). A lethal germ-line mutation in PIGA was found in one patient who appeared to retain residual activity (71).
PIG-L carries out the second step of the pathway, de-N-acetylation of N-acetylglucosaminylphosphatidylinositol, and mutations in it cause CHIME syndrome, with ocular coloboma, heart defects, ichthyosis, mental retardation, and ear anomalies. PIG-M is a mannosyltransferase that adds the first mannose to the core GPI (72). Its decrease causes venous thrombosis and seizures (73). However, the mutation occurs at an SP1 transcription factor-binding site. Butyrate restores normal PIGM transcription and cell surface GPI expression in patients' lymphoblasts. Two-week treatment with phenylbutyrate eliminates seizures and improves motor skills (74). PIGN encodes the ethanolamine phosphate transferase, which adds the ethanolamine phosphate to the first mannose on the GPI anchor. Mutations in PIGN cause multiple congenital anomalies, including hypotonia and seizures (23).
Mutations in the second mannosyltransferase, PIGV, were first identified as the cause of hyperphosphatasia and mental retardation syndrome (75). Patients with a similar phenotype who lacked mutations in PIGV had mutations in PIGO, an ethanolamine phosphate transferase (76). Both had reduced level of GPI-anchored substrates at the cell surface. Alkaline phosphatase (ALP) is normally GPI-anchored, but it is secreted into the blood, accounting for the characteristic of these two disorders. The other GPI disorders do not result in ALP secretion. It appears that secretion of ALP depends on GPI transamidase removal of the C-terminal GPI attachment signal peptide and GPI addition. Defects in which shorter, non-mannosylated GPI chains accumulate result in ALP degradation, whereas it is secreted from cells with incomplete mannose-containing chains. Transamidase appears to recognize the presence of incomplete mannose-containing chains and cleaves a hydrophobic signal peptide, resulting in secretion (77).
Fringes of Glycosylation Disorders
Most glycosylation disorders are caused by defects in genes that conform to our current concepts. Others suggest that an expanded view might be better. One case in point is a glycosylation disorder caused by mutations in the gene TMEM165 (TPARL) (78). Tf and total serum N- and O-glycans from patients are deficient in sialic acid and galactose, suggesting a defect in the Golgi. Staining with Golgi markers TGN46 and GM130 showed a dilated morphology and fragmented trans-Golgi network. TMEM165 encodes a 324-amino acid membrane protein with six predicted transmembrane regions. It is ubiquitously expressed, and ∼230 residues are highly conserved in eukaryotes and many bacteria. Several motifs emerge, but none indicates function. Patients' fibroblasts are deficient in the late Golgi-localized protein. This deficiency leads to modest decreased sialylation in patients' cells and siRNA knockdown in HEK cells. The function eluded detection, and the best guess was a cation (proton, calcium?) pump. Conservation from bacteria to human suggests a very ancient function, and previous glycosylation disorders in the Golgi are known to affect pH homoeostasis.
Poor N-glycosylation causes ER stress (79), and cellular responses are aimed at reducing the stress. Glycosylation inhibitors, such as tunicamycin, or genetic defects up-regulate the ER stress response (80), which includes reducing protein synthesis or up-regulating various molecular chaperones to restore homeostasis. Hopelessly misfolded ER-located glycoproteins are dispatched to the cytoplasm for proteasomal degradation, but first, the N-glycan chains must be stripped. The gene NGLY1 encodes a stripper enzyme that clears the glycan, and exome mapping identified a patient with mutations that eliminated the protein (81). This may be considered the first “congenital disorder of deglycosylation” and is predicted to cause accumulation of N-glycosylated proteins in the cytoplasm and possibly ER stress. Accumulation of the undegraded material in the cytoplasm could have separate toxic effects.
Conclusions and Perspectives
A thorough whole exome sequencing study (3) focused on 100 patients with intellectual disability and solved about half of the cases by employing sophisticated informatics to identify the gene and Sanger sequencing for confirmation. Causality was based on programs used to predict the effects of mutations on the protein structure or analogy to known genes in the same or related pathway. Surprisingly, the study identified 22 patients with potentially causative de novo mutations in novel candidate genes. Sixteen patients had mutations in known intellectual disability genes.
Whole exome/genome sequencing will continue to identify new glycosylation disorders. Biochemical markers will help focus the search, but these approaches will also identify glycosylation-related genes that the serum biomarkers missed or patients who were never tested for the markers. Both approaches will continue to focus on new genes that impact glycosylation, and biochemical analysis will continue its prominent role to provide a physiological context and basis for therapies.
Acknowledgments
I thank Bobby Ng for help in preparing the figures and Amy Zimmon for administrative assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DK55615. This work was also supported by The Rocket Fund and The Bertrand Might Research Fund. This is the fifth article in the Thematic Minireview Series on Glycobiology and Extracellular Matrices: Glycan Functions Pervade Biology at All Levels.
- ER
- endoplasmic reticulum
- Dol-P
- dolichol-phosphate
- LLO
- lipid-linked oligosaccharide
- GPI
- glycosylphosphatidylinositol
- α-DG
- α-dystroglycan
- GlcUA
- glucuronic acid
- CDG
- congenital disorders of glycosylation
- Tf
- transferrin
- CMD
- congenital muscular dystrophy
- WWS
- Walker-Warburg syndrome
- ALP
- alkaline phosphatase.
REFERENCES
- 1. Matthijs G., Rymen D., Millón M. B., Souche E., Race V. (2013) Approaches to homozygosity mapping and exome sequencing for the identification of novel types of CDG. Glycoconj. J. 30, 67–76 [DOI] [PubMed] [Google Scholar]
- 2. Saunders C. J., Miller N. A., Soden S. E., Dinwiddie D. L., Noll A., Alnadi N. A., Andraws N., Patterson M. L., Krivohlavek L. A., Fellis J., Humphray S., Saffrey P., Kingsbury Z., Weir J. C., Betley J., Grocock R. J., Margulies E. H., Farrow E. G., Artman M., Safina N. P., Petrikin J. E., Hall K. P., Kingsmore S. F. (2012) Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci. Transl. Med. 4, 154ra135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de Ligt J., Willemsen M. H., van Bon B. W., Kleefstra T., Yntema H. G., Kroes T., Vulto-van Silfhout A. T., Koolen D. A., de Vries P., Gilissen C., del Rosario M., Hoischen A., Scheffer H., de Vries B. B., Brunner H. G., Veltman J. A., Vissers L. E. (2012) Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929 [DOI] [PubMed] [Google Scholar]
- 4. Mefford H. C. (2012) Diagnostic exome sequencing–are we there yet? N. Engl. J. Med. 367, 1951–1953 [DOI] [PubMed] [Google Scholar]
- 5. Committee on Assessing the Importance and Impact of Glycomics and Glycosciences, Board on Chemical Sciences and Technology, Board on Life Sciences, Division on Earth and Life Studies, and National Research Council (2012) Transforming Glycoscience: A Roadmap for the Future, The National Academies Press, Washington, D.C. [PubMed] [Google Scholar]
- 6. Hu H., Eggers K., Chen W., Garshasbi M., Motazacker M. M., Wrogemann K., Kahrizi K., Tzschach A., Hosseini M., Bahman I., Hucho T., Mühlenhoff M., Gerardy-Schahn R., Najmabadi H., Ropers H. H., Kuss A. W. (2011) ST3GAL3 mutations impair the development of higher cognitive functions. Am. J. Hum. Genet. 89, 407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Freeze H. H., Eklund E. A., Ng B. G., Patterson M. C. (2012) Neurology of inherited glycosylation disorders. Lancet Neurol. 11, 453–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hennet T. (2012) Diseases of glycosylation beyond classical congenital disorders of glycosylation. Biochim. Biophys. Acta 1820, 1306–1317 [DOI] [PubMed] [Google Scholar]
- 9. Freeze H. H., Haltiwanger R. S. (2009) in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., eds) 2nd Ed., pp. 162–170, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 10. Freeze H. H., Elbein A. D. (2009) in Essentials of Glycobiology Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., eds) 2nd Ed., pp. 47–61, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 11. Ferguson M. A. J., Kinoshita T., Hart G. W. (2009) in Essentials of Glycobiology Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., eds) 2nd Ed., pp. 143–161, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 12. Muntoni F., Torelli S., Brockington M. (2008) Muscular dystrophies due to glycosylation defects. Neurotherapeutics 5, 627–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stalnaker S. H., Stuart R., Wells L. (2011) Mammalian O-mannosylation: unsolved questions of structure/function. Curr. Opin. Struct. Biol. 21, 603–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Inamori K., Yoshida-Moriguchi T., Hara Y., Anderson M. E., Yu L., Campbell K. P. (2012) Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 335, 93–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoshida-Moriguchi T., Yu L., Stalnaker S. H., Davis S., Kunz S., Madson M., Oldstone M. B., Schachter H., Wells L., Campbell K. P. (2010) O-Mannosyl phosphorylation of α-dystroglycan is required for laminin binding. Science 327, 88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Amberger J., Bocchini C., Hamosh A. (2011) A new face and new challenges for Online Mendelian Inheritance in Man (OMIM(R)). Hum. Mutat. 32, 564–567 [DOI] [PubMed] [Google Scholar]
- 17. Aebi M., Helenius A., Schenk B., Barone R., Fiumara A., Berger E. G., Hennet T., Imbach T., Stutz A., Bjursell C., Uller A., Wahlström J. G., Briones P., Cardo E., Clayton P., Winchester B., Cormier-Dalre V., de Lonlay P., Cuer M., Dupré T., Seta N., de Koning T., Dorland L., de Loos F., Kupers L., et al. , (1999) Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: an updated nomenclature for CDG. Glycoconj. J. 16, 669–671 [DOI] [PubMed] [Google Scholar]
- 18. Jaeken J., Hennet T., Matthijs G., Freeze H. H. (2009) CDG nomenclature: time for a change! Biochim. Biophys. Acta 1792, 825–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brockington M., Torelli S., Sharp P. S., Liu K., Cirak S., Brown S. C., Wells D. J., Muntoni F. (2010) Transgenic overexpression of LARGE induces α-dystroglycan hyperglycosylation in skeletal and cardiac muscle. PLoS ONE 5, e14434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ng B. G., Hackmann K., Jones M. A., Eroshkin A. M., He P., Wiliams R., Bhide S., Cantagrel V., Gleeson J. G., Paller A. S., Schnur R. E., Tinschert S., Zunich J., Hegde M. R., Freeze H. H. (2012) Mutations in the glycosylphosphatidylinositol gene PIGL cause CHIME syndrome. Am. J. Hum. Genet. 90, 685–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O'Brien J. F., Lacey J. M., Bergen H. R., 3rd (2007) Detection of hypo-N-glycosylation using mass spectrometry of transferrin. Curr. Protoc. Hum. Genet. Chapter 17, Unit 17.14 [DOI] [PubMed] [Google Scholar]
- 22. Sturiale L., Barone R., Garozzo D. (2011) The impact of mass spectrometry in the diagnosis of congenital disorders of glycosylation. J. Inherit. Metab. Dis. 34, 891–899 [DOI] [PubMed] [Google Scholar]
- 23. Maydan G., Noyman I., Har-Zahav A., Neriah Z. B., Pasmanik-Chor M., Yeheskel A., Albin-Kaplanski A., Maya I., Magal N., Birk E., Simon A. J., Halevy A., Rechavi G., Shohat M., Straussberg R., Basel-Vanagaite L. (2011) Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J. Med. Genet. 48, 383–389 [DOI] [PubMed] [Google Scholar]
- 24. Muntoni F., Torelli S., Wells D. J., Brown S. C. (2011) Muscular dystrophies due to glycosylation defects: diagnosis and therapeutic strategies. Curr. Opin. Neurol. 24, 437–442 [DOI] [PubMed] [Google Scholar]
- 25. Eklund E. A., Freeze H. H. (2006) The congenital disorders of glycosylation: a multifaceted group of syndromes. NeuroRx 3, 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kumar P., Henikoff S., Ng P. C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 [DOI] [PubMed] [Google Scholar]
- 27. Adzhubei I. A., Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., Kondrashov A. S., Sunyaev S. R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. González-Pérez A., López-Bigas N. (2011) Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 88, 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pollard K. S., Hubisz M. J., Rosenbloom K. R., Siepel A. (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lübke T., Marquardt T., Etzioni A., Hartmann E., von Figura K., Körner C. (2001) Complementation cloning identifies CDG-IIc, a new type of congenital disorders of glycosylation, as a GDP-fucose transporter deficiency. Nat. Genet. 28, 73–76 [DOI] [PubMed] [Google Scholar]
- 31. Jones M. A., Ng B. G., Bhide S., Chin E., Rhodenizer D., He P., Losfeld M. E., He M., Raymond K., Berry G., Freeze H. H., Hegde M. R. (2012) DDOST mutations identified by whole-exome sequencing are implicated in congenital disorders of glycosylation. Am. J. Hum. Genet. 90, 363–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kranz C., Ng B. G., Sun L., Sharma V., Eklund E. A., Miura Y., Ungar D., Lupashin V., Winkel R. D., Cipollo J. F., Costello C. E., Loh E., Hong W., Freeze H. H. (2007) COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum. Mol. Genet. 16, 731–741 [DOI] [PubMed] [Google Scholar]
- 33. Wu X., Steet R. A., Bohorov O., Bakker J., Newell J., Krieger M., Spaapen L., Kornfeld S., Freeze H. H. (2004) Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 10, 518–523 [DOI] [PubMed] [Google Scholar]
- 34. Roscioli T., Kamsteeg E.-J., Buysse K., Maystadt I., van Reeuwijk J., van den Elzen C., van Beusekom E., Riemersma M., Pfundt R., Vissers L. E. L. M., Schraders M., Altunoglu U., Buckley M. F., Brunner H. G., Grisart B., Zhou H., Veltman J. A., Gilissen C., Mancini G. M. S., Delrée P., Willemsen M. A., Ramadža D. P., Chitayat D., Bennett C., Sheridan E., Peeters E. A. J., Tan-Sindhunata G. M. B., de Die-Smulders C. E., Devriendt K., Kayserili H., El-Hashash O. A. E.-F., Stemple D. L., Lefeber D. J., Lin Y.-Y., van Bokhoven H. (2012) Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat. Genet. 44, 581–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cline A., Gao N., Flanagan-Steet H., Sharma V., Rosa S., Sonon R., Azadi P., Sadler K. C., Freeze H. H., Lehrman M. A., Steet R. (2012) A zebrafish model of PMM2-CDG reveals altered neurogenesis and a substrate-accumulation mechanism for N-linked glycosylation deficiency. Mol. Biol. Cell 23, 4175–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Geisler C., Kotu V., Sharrow M., Rendić D., Pöltl G., Tiemeyer M., Wilson I. B., Jarvis D. L. (2012) The Drosophila neurally altered carbohydrate mutant has a defective Golgi GDP-fucose transporter. J. Biol. Chem. 287, 29599–29609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Freeze H. H., Sharma V. (2010) Metabolic manipulation of glycosylation disorders in humans and animal models. Semin. Cell Dev. Biol. 21, 655–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schneider A., Thiel C., Rindermann J., DeRossi C., Popovici D., Hoffmann G. F., Gröne H. J., Körner C. (2012) Successful prenatal mannose treatment for congenital disorder of glycosylation-Ia in mice. Nat. Med. 18, 71–73 [DOI] [PubMed] [Google Scholar]
- 39. Grigorian A., Araujo L., Naidu N. N., Place D. J., Choudhury B., Demetriou M. (2011) N-Acetylglucosamine inhibits T-helper 1 (Th1)/T-helper 17 (Th17) cell responses and treats experimental autoimmune encephalomyelitis. J. Biol. Chem. 286, 40133–40141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Niehues R., Hasilik M., Alton G., Körner C., Schiebe-Sukumar M., Koch H. G., Zimmer K. P., Wu R., Harms E., Reiter K., von Figura K., Freeze H. H., Harms H. K., Marquardt T. (1998) Carbohydrate-deficient glycoprotein syndrome type Ib. Phosphomannose isomerase deficiency and mannose therapy. J. Clin. Invest. 101, 1414–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harms H. K., Zimmer K. P., Kurnik K., Bertele-Harms R. M., Weidinger S., Reiter K. (2002) Oral mannose therapy persistently corrects the severe clinical symptoms and biochemical abnormalities of phosphomannose isomerase deficiency. Acta Paediatr. 91, 1065–1072 [DOI] [PubMed] [Google Scholar]
- 42. DeRossi C., Bode L., Eklund E. A., Zhang F., Davis J. A., Westphal V., Wang L., Borowsky A. D., Freeze H. H. (2006) Ablation of mouse phosphomannose isomerase (Mpi) causes mannose 6-phosphate accumulation, toxicity, and embryonic lethality. J. Biol. Chem. 281, 5916–5927 [DOI] [PubMed] [Google Scholar]
- 43. Thiel C., Lübke T., Matthijs G., von Figura K., Körner C. (2006) Targeted disruption of the mouse phosphomannomutase 2 gene causes early embryonic lethality. Mol. Cell. Biol. 26, 5615–5620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chu J., Mir A., Gao N., Rosa S., Monson C., Sharma V., Steet R., Freeze H. H., Lehrman M. A., Sadler K. C. (2013) A zebrafish model of congenital disorders of glycosylation with phosphomannose isomerase deficiency reveals an early opportunity for corrective mannose supplementation. Dis. Model. Mech. 6, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gao N., Shang J., Huynh D., Manthati V. L., Arias C., Harding H. P., Kaufman R. J., Mohr I., Ron D., Falck J. R., Lehrman M. A. (2011) Mannose 6-phosphate regulates destruction of lipid-linked oligosaccharides. Mol. Biol. Cell 22, 2994–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gao N., Shang J., Lehrman M. A. (2005) Analysis of glycosylation in CDG-Ia fibroblasts by fluorophore-assisted carbohydrate electrophoresis. Implications for extracellular glucose and intracellular mannose 6-phosphate. J. Biol. Chem. 280, 17901–17909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Farrugia M. E. (2011) Myasthenic syndromes. J. R. Coll. Physicians Edinb. 41, 43–47 [DOI] [PubMed] [Google Scholar]
- 48. Senderek J., Müller J. S., Dusl M., Strom T. M., Guergueltcheva V., Diepolder I., Laval S. H., Maxwell S., Cossins J., Krause S., Muelas N., Vilchez J. J., Colomer J., Mallebrera C. J., Nascimento A., Nafissi S., Kariminejad A., Nilipour Y., Bozorgmehr B., Najmabadi H., Rodolico C., Sieb J. P., Steinlein O. K., Schlotter B., Schoser B., Kirschner J., Herrmann R., Voit T., Oldfors A., Lindbergh C., Urtizberea A., von der Hagen M., Hübner A., Palace J., Bushby K., Straub V., Beeson D., Abicht A., Lochmüller H. (2011) Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am. J. Hum. Genet. 88, 162–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Belaya K., Finlayson S., Slater C. R., Cossins J., Liu W. W., Maxwell S., McGowan S. J., Maslau S., Twigg S. R., Walls T. J., Pascual Pascual S. I., Palace J., Beeson D. (2012) Mutations in DPAGT1 cause a limb-girdle congenital myasthenic syndrome with tubular aggregates. Am. J. Hum. Genet. 91, 193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carrera I. A., Matthijs G., Perez B., Cerdá C. P. (2012) DPAGT1-CDG: report of a patient with fetal hypokinesia phenotype. Am. J. Med. Genet. A 158A, 2027–2030 [DOI] [PubMed] [Google Scholar]
- 51. Hayee B., Antonopoulos A., Murphy E. J., Rahman F. Z., Sewell G., Smith B. N., McCartney S., Furman M., Hall G., Bloom S. L., Haslam S. M., Morris H. R., Boztug K., Klein C., Winchester B., Pick E., Linch D. C., Gale R. E., Smith A. M., Dell A., Segal A. W. (2011) G6PC3 mutations are associated with a major defect of glycosylation: a novel mechanism for neutrophil dysfunction. Glycobiology 21, 914–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Timal S., Hoischen A., Lehle L., Adamowicz M., Huijben K., Sykut-Cegielska J., Paprocka J., Jamroz E., van Spronsen F. J., Körner C., Gilissen C., Rodenburg R. J., Eidhof I., Van den Heuvel L., Thiel C., Wevers R. A., Morava E., Veltman J., Lefeber D. J. (2012) Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum. Mol. Genet. 21, 4151–4161 [DOI] [PubMed] [Google Scholar]
- 53. Pérez B., Medrano C., Ecay M. J., Ruiz-Sala P., Martínez-Pardo M., Ugarte M., Pérez-Cerdá C. (2013) A novel congenital disorder of glycosylation type without central nervous system involvement caused by mutations in the phosphoglucomutase 1 gene. J. Inherit. Metab. Dis., doi 10.1007/s10545-012-9525-7 [DOI] [PubMed] [Google Scholar]
- 54. Tang M., Odejinmi S. I., Vankayalapati H., Wierenga K. J., Lai K. (2012) Innovative therapy for classic galactosemia–tale of two HTS. Mol. Genet. Metab. 105, 44–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pronicka E., Adamowicz M., Kowalik A., Płoski R., Radomyska B., Rogaszewska M., Rokicki D., Sykut-Cegielska J. (2007) Elevated carbohydrate-deficient transferrin (CDT) and its normalization on dietary treatment as a useful biochemical test for hereditary fructose intolerance and galactosemia. Pediatr. Res. 62, 101–105 [DOI] [PubMed] [Google Scholar]
- 56. Cantagrel V., Lefeber D. J. (2011) From glycosylation disorders to dolichol biosynthesis defects: a new class of metabolic diseases. J. Inherit. Metab. Dis. 34, 859–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lefeber D. J., Schönberger J., Morava E., Guillard M., Huyben K. M., Verrijp K., Grafakou O., Evangeliou A., Preijers F. W., Manta P., Yildiz J., Grünewald S., Spilioti M., van den Elzen C., Klein D., Hess D., Ashida H., Hofsteenge J., Maeda Y., van den Heuvel L., Lammens M., Lehle L., Wevers R. A. (2009) Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am. J. Hum. Genet. 85, 76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ashida H., Maeda Y., Kinoshita T. (2006) DPM1, the catalytic subunit of dolichol-phosphate mannose synthase, is tethered to and stabilized on the endoplasmic reticulum membrane by DPM3. J. Biol. Chem. 281, 896–904 [DOI] [PubMed] [Google Scholar]
- 59. Maeda Y., Tanaka S., Hino J., Kangawa K., Kinoshita T. (2000) Human dolichol-phosphate-mannose synthase consists of three subunits, DPM1, DPM2 and DPM3. EMBO J. 19, 2475–2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Barone R., Aiello C., Race V., Morava E., Foulquier F., Riemersma M., Passarelli C., Concolino D., Carella M., Santorelli F., Vleugels W., Mercuri E., Garozzo D., Sturiale L., Messina S., Jaeken J., Fiumara A., Wevers R. A., Bertini E., Matthijs G., Lefeber D. (2012) DPM2-CDG: a muscular dystrophy-dystroglycanopathy syndrome with severe epilepsy. Ann. Neurol. 72, 550–558 [DOI] [PubMed] [Google Scholar]
- 61. Dwyer C. A., Baker E., Hu H., Matthews R. T. (2012) RPTPζ/phosphacan is abnormally glycosylated in a model of muscle-eye-brain disease lacking functional POMGnT1. Neuroscience 220, 47–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Willer T., Lee H., Lommel M., Yoshida-Moriguchi T., de Bernabe D. B., Venzke D., Cirak S., Schachter H., Vajsar J., Voit T., Muntoni F., Loder A. S., Dobyns W. B., Winder T. L., Strahl S., Mathews K. D., Nelson S. F., Moore S. A., Campbell K. P. (2012) ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat. Genet. 44, 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Manzini M. C., Tambunan D. E., Hill R. S., Yu T. W., Maynard T. M., Heinzen E. L., Shianna K. V., Stevens C. R., Partlow J. N., Barry B. J., Rodriguez J., Gupta V. A., Al-Qudah A. K., Eyaid W. M., Friedman J. M., Salih M. A., Clark R., Moroni I., Mora M., Beggs A. H., Gabriel S. B., Walsh C. A. (2012) Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am. J. Hum. Genet. 91, 541–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Heuston S., Begley M., Gahan C. G., Hill C. (2012) Isoprenoid biosynthesis in bacterial pathogens. Microbiology 158, 1389–1401 [DOI] [PubMed] [Google Scholar]
- 65. Cantagrel V., Lefeber D. J., Ng B. G., Guan Z., Silhavy J. L., Bielas S. L., Lehle L., Hombauer H., Adamowicz M., Swiezewska E., De Brouwer A. P., Blümel P., Sykut-Cegielska J., Houliston S., Swistun D., Ali B. R., Dobyns W. B., Babovic-Vuksanovic D., van Bokhoven H., Wevers R. A., Raetz C. R., Freeze H. H., Morava E., Al-Gazali L., Gleeson J. G. (2010) SRD5A3 is required for converting polyprenol to dolichol and is mutated in a congenital glycosylation disorder. Cell 142, 203–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stiles A. R., Russell D. W. (2010) SRD5A3: A surprising role in glycosylation. Cell 142, 196–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Züchner S., Dallman J., Wen R., Beecham G., Naj A., Farooq A., Kohli M. A., Whitehead P. L., Hulme W., Konidari I., Edwards Y. J., Cai G., Peter I., Seo D., Buxbaum J. D., Haines J. L., Blanton S., Young J., Alfonso E., Vance J. M., Lam B. L., Peričak-Vance M. A. (2011) Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am. J. Hum. Genet. 88, 201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zelinger L., Banin E., Obolensky A., Mizrahi-Meissonnier L., Beryozkin A., Bandah-Rozenfeld D., Frenkel S., Ben-Yosef T., Merin S., Schwartz S. B., Cideciyan A. V., Jacobson S. G., Sharon D. (2011) A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am. J. Hum. Genet. 88, 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Harrison K. D., Park E. J., Gao N., Kuo A., Rush J. S., Waechter C. J., Lehrman M. A., Sessa W. C. (2011) Nogo-B receptor is necessary for cellular dolichol biosynthesis and protein N-glycosylation. EMBO J. 30, 2490–2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pu J. J., Brodsky R. A. (2011) Paroxysmal nocturnal hemoglobinuria from bench to bedside. Clin. Transl. Sci. 4, 219–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Johnston J. J., Gropman A. L., Sapp J. C., Teer J. K., Martin J. M., Liu C. F., Yuan X., Ye Z., Cheng L., Brodsky R. A., Biesecker L. G. (2012) The phenotype of a germline mutation in PIGA: the gene somatically mutated in paroxysmal nocturnal hemoglobinuria. Am. J. Hum. Genet. 90, 295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Maeda Y., Watanabe R., Harris C. L., Hong Y., Ohishi K., Kinoshita K., Kinoshita T. (2001) PIG-M transfers the first mannose to glycosylphosphatidylinositol on the lumenal side of the ER. EMBO J. 20, 250–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Almeida A. M., Murakami Y., Layton D. M., Hillmen P., Sellick G. S., Maeda Y., Richards S., Patterson S., Kotsianidis I., Mollica L., Crawford D. H., Baker A., Ferguson M., Roberts I., Houlston R., Kinoshita T., Karadimitris A. (2006) Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat. Med. 12, 846–851 [DOI] [PubMed] [Google Scholar]
- 74. Almeida A. M., Murakami Y., Baker A., Maeda Y., Roberts I. A., Kinoshita T., Layton D. M., Karadimitris A. (2007) Targeted therapy for inherited GPI deficiency. N. Engl. J. Med. 356, 1641–1647 [DOI] [PubMed] [Google Scholar]
- 75. Krawitz P. M., Schweiger M. R., Rödelsperger C., Marcelis C., Kölsch U., Meisel C., Stephani F., Kinoshita T., Murakami Y., Bauer S., Isau M., Fischer A., Dahl A., Kerick M., Hecht J., Köhler S., Jäger M., Grünhagen J., de Condor B. J., Doelken S., Brunner H. G., Meinecke P., Passarge E., Thompson M. D., Cole D. E., Horn D., Roscioli T., Mundlos S., Robinson P. N. (2010) Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat. Genet. 42, 827–829 [DOI] [PubMed] [Google Scholar]
- 76. Krawitz P. M., Murakami Y., Hecht J., Krüger U., Holder S. E., Mortier G. R., Delle Chiaie B., De Baere E., Thompson M. D., Roscioli T., Kielbasa S., Kinoshita T., Mundlos S., Robinson P. N., Horn D. (2012) Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am. J. Hum. Genet. 91, 146–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Murakami Y., Kanzawa N., Saito K., Krawitz P. M., Mundlos S., Robinson P. N., Karadimitris A., Maeda Y., Kinoshita T. (2012) Mechanism for release of alkaline phosphatase caused by glycosylphosphatidylinositol deficiency in patients with hyperphosphatasia mental retardation syndrome. J. Biol. Chem. 287, 6318–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Foulquier F., Amyere M., Jaeken J., Zeevaert R., Schollen E., Race V., Bammens R., Morelle W., Rosnoblet C., Legrand D., Demaegd D., Buist N., Cheillan D., Guffon N., Morsomme P., Annaert W., Freeze H. H., Van Schaftingen E., Vikkula M., Matthijs G. (2012) TMEM165 deficiency causes a congenital disorder of glycosylation. Am. J. Hum. Genet. 91, 15–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cao S. S., Kaufman R. J. (2012) Unfolded protein response. Curr. Biol. 22, R622–R626 [DOI] [PubMed] [Google Scholar]
- 80. Shang J., Körner C., Freeze H., Lehrman M. A. (2002) Extension of lipid-linked oligosaccharides is a high-priority aspect of the unfolded protein response: endoplasmic reticulum stress in type I congenital disorder of glycosylation fibroblasts. Glycobiology 12, 307–317 [DOI] [PubMed] [Google Scholar]
- 81. Need A. C., Shashi V., Hitomi Y., Schoch K., Shianna K. V., McDonald M. T., Meisler M. H., Goldstein D. B. (2012) Clinical application of exome sequencing in undiagnosed genetic conditions. J. Med. Genet. 49, 353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rafiq M. A., Kuss A. W., Puettmann L., Noor A., Ramiah A., Ali G., Hu H., Kerio N. A., Xiang Y., Garshasbi M., Khan M. A., Ishak G. E., Weksberg R., Ullmann R., Tzschach A., Kahrizi K., Mahmood K., Naeem F., Ayub M., Moremen K. W., Vincent J. B., Ropers H. H., Ansar M., Najmabadi H. (2011) Mutations in the α1,2-mannosidase gene, MAN1B1, cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 89, 176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]