

Background: Neutrophil extracellular traps (NETs) composed of DNA and proteins form a scaffold in thrombi, supplementing the fibrin matrix.
Results: DNA and histones modify the structure of fibrin and render it resistant to mechanical and enzymatic destruction.
Conclusion: NET components are essential factors in thrombus stability.
Significance: Therapeutic strategies could be optimized to enhance fibrinolysis in clots containing DNA and histones.
Keywords: DNA, Fibrinolysis, Histones, Neutrophil, Tissue Plasminogen Activator (tPA), Neutrophil Extracellular Trap
Abstract
Neutrophil extracellular traps are networks of DNA and associated proteins produced by nucleosome release from activated neutrophils in response to infection stimuli and have recently been identified as key mediators between innate immunity, inflammation, and hemostasis. The interaction of DNA and histones with a number of hemostatic factors has been shown to promote clotting and is associated with increased thrombosis, but little is known about the effects of DNA and histones on the regulation of fibrin stability and fibrinolysis. Here we demonstrate that the addition of histone-DNA complexes to fibrin results in thicker fibers (increase in median diameter from 84 to 123 nm according to scanning electron microscopy data) accompanied by improved stability and rigidity (the critical shear stress causing loss of fibrin viscosity increases from 150 to 376 Pa whereas the storage modulus of the gel increases from 62 to 82 pascals according to oscillation rheometric data). The effects of DNA and histones alone are subtle and suggest that histones affect clot structure whereas DNA changes the way clots are lysed. The combination of histones + DNA significantly prolongs clot lysis. Isothermal titration and confocal microscopy studies suggest that histones and DNA bind large fibrin degradation products with 191 and 136 nm dissociation constants, respectively, interactions that inhibit clot lysis. Heparin, which is known to interfere with the formation of neutrophil extracellular traps, appears to prolong lysis time at a concentration favoring ternary histone-DNA-heparin complex formation, and DNase effectively promotes clot lysis in combination with tissue plasminogen activator.
Introduction
Neutrophil extracellular traps (NETs)2 are networks of DNA decorated with histones and other proteins, proteases, and other antimicrobial factors (1). They are produced when neutrophils, basophils, or mast cells release nucleosomes after stimulation by inflammatory cytokines or LPS for example (1) or by interaction with platelets after stimulation of platelet toll-like receptors 4 and 2 by microbial structures (2, 3). It is proposed that NET formation is the first line of defense of the innate immune system, providing an effective way of trapping pathogenic microbes and removing them from the circulation (4).
The DNA and histones of NETs provide a scaffold for cell localization (including neutrophils and red blood cells), platelet aggregation, and activation that serves to promote coagulation and thrombosis (5). Thus, NETs are a focus of significant cross-talk between innate immunity, inflammation, and hemostasis, and there are many points of contact including bidirectional interactions with platelets, inflammatory cytokines (IL-8), fibrinogen, fibronectin, von Willebrand factor, tissue factor pathway inhibitor, protein C, thrombomodulin, factor XII, and neutrophil-derived serine proteases (3–6). Histones H3 and H4 have been identified as critical players and mediators of damage able to stimulate phosphatidylserine exposure and factor Va expression on platelets and interact with polyphosphates to promote clotting even in the absence of factor XII (3). Histones may come from NETs or dying cells and are implicated in the progression of sepsis. Stimulation of coagulation by NETs can result in unwanted thrombosis (7), and infection is a common event in the development of deep vein thrombosis (8, 9). Targeting the release of nucleosomes, development of NETs, and availability of circulating histones could be a strategy for prevention or therapeutic intervention for venous thromboembolism, sepsis, and other diseases involving cell death and lysis.
However, although there are extensive studies on the interaction between NET components and coagulation, little is known about the effects of DNA and histones on fibrinolysis. Bacterial infection by Streptococcus and Staphylococcus spp. is accompanied by secretion of plasminogen-binding and activating proteins as the bacteria hijack the host fibrinolytic system (10, 11). Streptokinase is a classic example of a plasminogen activator from streptococci, and staphylokinase is produced by staphylococci. Interestingly, streptococci also release a DNase, streptodornase, which is linked to bacterial virulence (12), suggesting that streptokinase and streptodornase can work together to break down fibrin clots and NETs and promote bacterial dissemination. DNases have been shown to degrade NETs and prevent platelet adhesion and activation (5) and to prevent thrombosis in in vivo models (2, 13).
Systemic infection triggers coagulation and an immune response involving neutrophils and NETs that work together to restrict the movement of microorganisms within the vasculature and at the same time promote thrombin generation and thrombosis. As suggested previously (5), it is important to understand the effects that DNA and histones might have on fibrin structure and mechanical properties and the implications for fibrinolysis, thrombus breakdown, and embolism. These topics are the subject of the current study with the main focus on the endogenous fibrinolysis system regulated by tissue plasminogen activator (tPA).
EXPERIMENTAL PROCEDURES
Generation and Purification of tPA Variants and Other Activators
tPA was modified to express a C-terminal fusion of enhanced green fluorescent protein using pFastBac-tPA (and derivatives) as described previously (14).
Fibrinolysis Kinetics Methods
Plasminogen activation and fibrin lysis assays were performed as described previously (15) to measure the effect of DNA and histones on clot formation and lysis in microtiter plates by mixing equal volumes of solutions of (a) thrombin, tPA, and DNA (where present) with (b) fibrinogen, plasminogen, and histones (where present) to give a 100-μl reaction mixture of 25 nm thrombin, 70 pm tPA, 8 μm fibrinogen, and ranges of DNA and histone concentrations. Where heparin was present in reaction mixtures (over a range of concentrations shown under “Results”), it was added first to reaction wells followed by solution a and then solution b. Fibrinogen and DNA (calf thymus) were from Calbiochem; plasminogen was from Hyphen-Biomed, Neuville-sur-Oise, France; histones (calf thymus, type IIIS) were from Sigma-Aldrich; and α-thrombin (01/580), tPA (98/714), and heparin (07/328) were from the National Institute for Biological Standards and Control, South Mimms, UK. Selectable endpoints for analysis (such as clotting maximum absorbance, time to clotting maximum absorbance, time to 50% lysis, time to 100% lysis, and time to 50% lysis from clotting maximum) were extracted from absorbance versus time data and analyzed using bespoke software written using the free statistical software package R (16) (script available on request). tPA activity was also determined in tubes by mixing 200 μl of a solution of histones (50 μg/ml), thrombin (30 nm), and tPA with 1.0 ml of fibrinogen (1.3 mg/ml), plasminogen (200 nm), and DNA (70 μg/ml) (final concentrations given) and monitoring the collapse of clots by release of bubbles (17). tPA was present at 0.4, 0.2, and 0.1 nm so potency relative to conditions without DNA and histones and 95% confidence limits could be calculated using parallel line bioassay methods (17). Time lapse videos of lysis were prepared under the same conditions with 0.2 nm tPA by following the reactions for 2–3 h. DNase I was from Thermo Scientific, Rockford, IL.
Scanning Electron Microscope (SEM) Imaging of Thrombi and Fibrin
Immediately (within 5 min) after surgery or the preparation of fibrin, 5 × 5 × 10-mm pieces of thrombi or fibrin clots of 100-μl volume were placed into 10 ml of 100 mm sodium cacodylate, pH 7.2 buffer for 24 h at 4 °C. Following repeated washes with the same buffer, samples were fixed in 1% (v/v) glutaraldehyde for 16 h. The fixed samples were dehydrated in a series of ethanol dilutions (20–96% (v/v)), a 1:1 mixture of 96% (v/v) ethanol/acetone, and pure acetone followed by critical point drying with CO2 in an E3000 Critical Point Drying Apparatus (Quorum Technologies, Newhaven, UK). The specimens were mounted on adhesive carbon discs and sputter-coated with gold in an SC7620 Sputter Coater (Quorum Technologies), and images were taken with scanning electron microscope EVO40 (Carl Zeiss GmbH, Oberkochen, Germany).
Immunohistochemistry
After surgery, the removed thrombus samples were frozen immediately at −70 °C and stored until examination. Cryosections (6-μm thickness) of thrombi were attached to lysine-coated slides. Sections were fixed in acetone at 4 °C for 10 min and air-dried for 5 min at room temperature followed by incubation in 100 mm sodium phosphate, 100 mm NaCl, pH 7.5 buffer (PBS) containing 5% (w/v) bovine serum albumin to eliminate nonspecific binding of antibodies. Subsequently, slides were washed in PBS three times, and DNA was stained with the dimeric cyanine nucleic acid dye TOTO-3® (T-3604, Invitrogen; excitation, 640 nm, emission, 660 nm) at 1:5000 dilution with PBS containing 10% glycerol and 0.02% Tween 20 for 15 min followed by three washes in 50 mm Tris-HCl, 100 mm NaCl, 0.02% (w/v) NaN3, pH 7.4 buffer (TBS). For double immunostaining, the sections were incubated with 2 μg/ml mouse monoclonal anti-human fibrin antibody (ADI313, American Diagnostica, Pfungstadt, Germany) and 2 μg/ml rabbit anti-human histone H1 antibody (Sigma-Aldrich) in TBS. Following washing with TBS, sections were treated with Alexa Fluor® 488 (excitation, 495 nm; emission, 519 nm) goat anti-mouse immunoglobulin antibody (Invitrogen) at 1:100 dilution and Alexa Fluor 546 (excitation, 556 nm; emission, 573 nm) goat anti-rabbit immunoglobulin antibody (Invitrogen) at 1:100 dilution. Following three washes, glass coverslips were affixed over a drop of 50% (v/v) glycerol in TBS. Confocal fluorescence images were taken using a Zeiss LSM510 confocal laser scanning microscope equipped with a 20× 1.4 objective (Carl Zeiss GmbH, Jena, Germany) at 488-nm excitation laser line (20% intensity) and emission in the 500–530-nm wavelength range, 543-nm excitation laser line (100% intensity) and emission in the 565–615-nm wavelength range, 633-nm excitation laser line (100% intensity) and emission in the range over 650-nm wavelength.
Isothermal Titration Calorimetry (ITC)
Enthalpy changes accompanying the interaction of DNA and proteins (fibrin degradation products (FDPs), fibrinogen, histones, and plasminogen) were measured using an isothermal titration method on a VP-ITC microcalorimeter (MicroCal Inc., Northampton, MA). The proteins were injected in a series of 25 aliquots (10 μl each) into the cell of the calorimeter containing DNA or protein, and the heat increment of each addition was recorded by the instrument. Dilutions of protein into buffer were carried out in a separate series of injections, and these heat increments were subtracted from the raw data. Heat data for the interactions were evaluated according to the single site algorithm with ITC Data Analysis version 7.0 software (MicroCal Inc.). For the calculation of equilibrium parameters, the mass concentration of DNA was converted to molar concentration of nucleotides using an average molecular mass of 500 Da. The molar concentration of the FDPs of 150 kDa or larger was estimated from the mass concentration and densitometric data of the polyacrylamide gel electrophoretic (PAGE) pattern of the FDPs used for the binding experiments.
Preparation of FDPs
Clotting of fibrinogen (human, plasminogen-depleted; Calbiochem) and fibrinolysis were initiated simultaneously with thrombin (90 nm) and plasmin (5 nm). The final concentration of fibrinogen was 6 μm for preparation of extensively degraded products of fibrin digestion and 12 μm for partial digestion and generation of large FDPs. Plasmin action was stopped by the addition of 4-(2-aminoethyl)benzenesulfonyl fluoride (Pefabloc® from Roche Applied Science) at a final concentration of 0.05 mm immediately after a steel ball placed on the surface of the clot reached the bottom of the tube (∼16–18 h after the start of lysis). For more extensive degradation Pefabloc was added 2–4 h later when the visible fibrin gel had totally disappeared. The fluid phases were withdrawn from each of the tubes after centrifugation at 6000 × g for 5 min, and the total protein contents were determined from the values of absorbance of the supernatants at 280 nm (A280 of 1.6 corresponds to 1 g/liter non-clottable fibrin degradation products measured under identical conditions (18)). The supernatant was subjected to SDS electrophoresis on a 4–15% polyacrylamide gel under non-reducing and reducing conditions and silver-stained. Concentrations of large degradation fragments (over 150 kDa) were calculated as a fraction of total protein based on quantitative gel analysis using SigmaGel software (Jandel Scientific, Erkrath, Germany).
Confocal Microscopy Imaging
Fibrin clots were prepared from 6 μm fibrinogen, 2% of which was Alexa Fluor 546-conjugated fibrinogen (Invitrogen), and 1.5 μm plasminogen clotted with 16 nm thrombin for 30 min at room temperature in sterile, uncoated Ibidi VI 0.4 μ-Slides (Ibidi GmbH, Martinsried, Germany). In certain cases, 50 or 100 μg/ml DNA (human; isolated from granulocytes) and/or 3.45 μm histone IIIS (from calf thymus; Sigma-Aldrich) was also added to the mixture. Thereafter, 4 μg/ml tPA-GFP was added to the edge of the clot, and the fluorescence (excitation wavelength, 488 nm; emission wavelength, 525 nm for tPA-GFP detection; excitation wavelength, 543 nm; emission wavelength, 575 nm for Alexa Fluor 546-fibrinogen detection) was monitored with Confocal Laser Scanning System LSM510 (Carl Zeiss GmbH, Jena, Germany) taking sequential images of the fluid-fibrin interface at a distance of ∼50 μm from the glass surface with identical exposures and laser intensities using a Plan-Neofluar×20/0.5 objective.
Preparation of Neutrophil DNA
Neutrophil granulocytes were isolated from the buffy coat fraction of human blood (Hungarian Blood Supply Service, Budapest, Hungary) (19) according to a procedure described previously (20). Following cell lysis, DNA was extracted using 25:24:1 phorbol/chloroform/isoamyl alcohol reagent (Sigma-Aldrich); precipitated out of the water phase in 0.3 m sodium acetate, pH 5.2 buffer and 96% (v/v) ethanol; and resuspended in 25 mm NaH2PO4/Na2HPO4, pH 7.4 buffer containing 75 mm NaCl. The ratio of absorbance at 260 and 280 nm was 1.88–1.95 in the final preparation. The concentration of DNA was determined from absorbance at 260 nm using calf thymus DNA (Calbiochem) as a reference.
Evaluation of Fibrin Rigidity
140 μl of 10 mg/ml fibrinogen was premixed with 60 or 120 μl of 0.5 mg/ml DNA and supplemented with 10 mm HEPES, pH 7.4 buffer containing 150 mm NaCl to a 500-μl final volume. Clotting was initiated with 50 μl of 100 nm thrombin added to 410 μl of fibrinogen solution, and 410 μl of the clotting mixture was transferred to the plate of the HAAKE RheoStress 1 oscillation rheometer (Thermo Scientific, Karlsruhe, Germany) thermostatted at 37 °C. The cone (titanium, 2° angle, 35-mm diameter) of the rheometer was brought to the gap position, and a shear strain (γ) of 0.015 was imposed exactly at 2 min after the addition of thrombin. Measurements of storage modulus (G′) and loss modulus (G″) were taken at 1 Hz in the course of 15 min with HAAKE RheoWin data manager software v. 3.50.0012 (Thermo Scientific, Karlsruhe, Germany) (21). Following this 15-min clotting phase, determination of the flow limit of fibrin gels was performed in the same samples by increasing the applied shear stress (τ) from 0.01 to 500.0 pascals stepwise in 150 s, and the resulting strain measured was used for calculation of the viscosity modulus (the critical shear stress τ0 determined by extrapolation of the decline in viscosity to 0 that was used as an indicator of the gel/fluid transition in the fibrin structure).
Structural Characterization of Fibrin by Small Angle X-ray Scattering (SAXS)
Fibrin samples with the same composition as those used for the evaluation of rigidity and additional samples containing unfractionated heparin were also examined by SAXS measurements performed on the four-crystal monochromator beamline of Physikalisch-Technische Bundesanstalt (PTB) (Berlin, Germany) supplemented by the SAXS setup of Helmholtz-Zentrum Berlin (HZB German Patent DE 2006 029 449) at the synchrotron radiation facility BESSY II (HZB, Berlin, Germany) (22). The samples were filled into glass capillaries with 1.0-mm diameter. The energy of the incoming x-ray beam was 7 keV, and the two-dimensional scattering patterns were collected with a gas-filled area detector. Measurements were performed at sample-to-detector distances of 1.4 and 4 m to cover the range of momentum transfer of q = 0.7–3 nm−1 (q = 4π/λ sinθ where θ is half the scattering angle and λ is the wavelength of the incident x-ray beam). All measurements were carried out at room temperature. The scattering curves were obtained by radial averaging of the two-dimensional patterns using SASREDTOOL program v. 1.2 (Sylvio Haas, Helmholtz-Zentrum, Berlin, Germany). For quantitative analysis of the scattering pattern, non-linear least square fitting of an empirical model function to the scattering curves was performed using SASfit program v. 0.93.3 (Joachim Kohlbrecher and Ingo Bressler, Paul Scherrer Institute, Villigen, Switzerland) in which the peaks were approximated by Lorentzian functions and the decay trend was taken into account by different power law functions.
Statistical Procedures in Morphometric Analysis
The SEM images of fibrin were analyzed to determine the diameter of the fibrin fibers using self-designed scripts running under Image Processing Toolbox v. 7.0 of Matlab 7.10.0.499 (R2010a) (The Mathworks, Natick, MA) (23). For the diameter measurements, a grid was drawn over the image with 10–15 equally spaced horizontal lines, and all fibers crossed by them were included in the analysis. The diameters were measured by manually placing the pointer of the Distance tool over the end points of transverse cross-sections of 300 fibers from each image (always perpendicularly to the longitudinal axis of the fibers). Two images of two independent samples were analyzed in a single global procedure. The distribution of the data on fiber diameter was analyzed using an algorithm described in detail previously (23) where theoretical distributions were fitted to the empirical data sets and compared using the Kuiper test and Monte Carlo simulation procedures.
RESULTS
Thrombi from Patients
Little is known about the distribution of DNA and histones in thrombi found in patients. Fig. 1 shows staining for DNA and histones found in three representative thrombi recovered from patients. There was variable but widespread staining for DNA. Histones were also present although not so widely dispersed and in some cases were coincident with fibrin aggregates. The thrombi rich in red blood cells (Fig. 1, TO) or in fibrin (GI) according to the SEM images showed limited DNA- and histone-positive regions in contrast to the extensively stained areas in the leukocyte-rich (TJ) thrombus.
FIGURE 1.

Fibrin, histone, and DNA content of arterial thrombi. Following thrombectomy, thrombus samples were either frozen for immunostaining or washed, fixed, and dehydrated for SEM processing as detailed under “Experimental Procedures.” Sections of frozen samples were doubly immunostained for fibrin (green) and histone 1 (red) as well as with a DNA dye, TOTO-3 (blue). Images were taken at an original magnification of ×20 with a confocal laser microscope. SEM images were taken from the fixed samples of the same thrombi. TO, a thrombus from popliteal artery; GI, a thrombus from infrarenal aorta aneurysm; TJ, a thrombus from femoropopliteal graft. Scale bars, 2 μm in SEM panels and 50 μm in all other panels.
Fibrin Structure in Purified Systems
Further detailed analysis of fibrin structure and the kinetics of clotting and lysis was performed using a simple model system of purified proteins. When fibrinogen was clotted in the presence of DNA and/or histones, morphometric analysis of SEM images showed significant changes in fibrin fiber diameter as summarized in Table 1. Statistical analysis of fibrin fiber diameter was performed, and probability density distributions were calculated, for fibrin clots with no additions or with DNA and/or histones as indicated in Table 1. The general trend apparent from these studies was that DNA alone produced small effects on fibrin structure, whereas in the presence of histones or DNA + histones, the fibrin fiber thickness increased.
TABLE 1.
Effect of DNA and histones on fiber diameter in composite fibrin networks
SEM images of fibrin clots prepared from 6 μm fibrinogen and the indicated additives and clotted with 30 nm thrombin were used for the measurement of fiber diameter as described previously (14). The fiber size is reported in nm as median and bottom-top quartile values (in parentheses) of the theoretical distributions fitted to the measured diameter values (data from four SEM images with 300 measured diameters in each). H1, histone 1.
| No DNA | 50 μg/ml DNA | 100 μg/ml DNA | |
|---|---|---|---|
| No H1 | 84 (64–110) | 94 (74–120) | 92 (76–111) |
| 50 μg/ml H1 | 119 (91–154) | 122 (97–153) | 114 (92–140) |
| 100 μg/ml H1 | 108 (88–132) | 122 (93–157) | 123 (98–149) |
The SEM data characterize the protein content of individual fibrin fibers, but this technique cannot resolve nanometer-scale structure of fibrin in its natural hydrated state. Small angle x-ray and neutron scattering proved to be a powerful tool in the characterization of the longitudinal arrangement of the monomers in the protofibrils and the lateral alignment of protofibrils in fibers (24). The general decay trend of the scattering curves (Fig. 2) reflects the fractal structure of the fibrin clot, and its effect can be modeled as a background signal with empirical power law functions in the form of C0 + C4 × q−α for clots containing fibrin, DNA, and heparin or with an additional function with a fixed exponent of −1 for samples with histones (supplemental table). The peaks arising above this background reflect the longitudinal and cross-sectional alignment of fibrin monomers. A small but sharp peak in pure fibrin at a q value of ∼0.285 nm−1 (Fig. 2 and supplemental table, Peak 2) corresponds to a longitudinal periodicity of d = 2π/q′ = 22 nm, which is in agreement with earlier SAXS studies (24) and a little bit lower than the values reported for dried samples in transmission electron microscopy investigations (25). This peak cannot be resolved in fibrin containing DNA or heparin, indicating that these additives disrupt the regular longitudinal alignment of the monomeric building blocks. In contrast, the addition of histone does not interfere with the longitudinal periodicity, and the related scattering peak is even more pronounced (Fig. 2 and supplemental table). In pure fibrin, two additional broad scattering peaks can be resolved, spanning over the q ranges of ∼0.2–0.5 and ∼0.6–1.5 nm−1 (supplemental table, Peak 1 and Peak 3, respectively). The first peak can be attributed to a periodicity of ∼12.5–31 nm in cluster units of the fibers, whereas the second peak corresponds to a periodicity of ∼4–10 nm characteristic for the average protofibril-to-protofibril distance based on the structural models of Yang et al. (26) and Weisel (25). Both of these broad peaks are most profoundly affected by the presence of histone (a 10-fold decrease in the area of Peak 1 and complete loss of Peak 3; supplemental table), suggesting that this additive interferes with the lateral organization of protofibrils, resulting in lower protofibril density. Earlier studies (27) have shown that lower protofibril density can correspond to thicker fiber diameter, which is in qualitative agreement with our SEM results (Table 1). The structure-modifying effects of histone are preserved in the presence of DNA, but these effects are completely reversed in the quaternary system of fibrin/DNA/histone/heparin (Fig. 2).
FIGURE 2.

Small angle x-ray scattering of fibrin clots containing 100 μg/ml DNA, 300 μg/ml histone, 10 IU/ml heparin, or their combinations at the same concentrations. Curves are shifted vertically by the factors indicated at their origin for better visualization. Symbols represent the measured intensity values, and solid lines show the fitted empirical functions as described in the supplemental table. The dashed vertical line indicates the longitudinal periodicity of about 22 nm, and the solid vertical lines show the boundaries of the broad peaks that characterize the lateral structure of the fibrin fibers.
Further evidence that DNA and histones can affect the behavior of fibrin clots was obtained from rheology studies. Fibrin clots were formed to contain pure fibrin or 50 or 100 μg/ml DNA, and the effect of added histones (300 μg/ml) was also investigated. The most striking differences seen in rheology parameters were in the shear stress necessary to disassemble the fibrin as presented in Fig. 3 where two opposing effects are clearly demonstrated. In the presence of DNA alone, the curves can be interpreted as increased sensitivity of fibrin to mechanical shear so that the shear force needed to disassemble fibrin (where viscosity approaches 0) is reduced in comparison with the situation without DNA. However, when histones are added to fibrin and to a greater extent when histones are added to fibrin + DNA, the clots become more stable and resistant to shear forces. Similarly, the softening of the fibrin caused by DNA (decrease in the storage modulus G′; see Table 2) was reversed by the addition of histones. The changes in the loss modulus G″ followed the trend of the storage modulus G′, but DNA on its own did not alter their ratio, whereas the presence of histones resulted in a relatively greater increase in the loss modulus, indicating a fibrin structure that dissipates more energy during deformation (presumably due to more significant rearrangements in fibrin protofibrils or fibers (see Ref. 21).
FIGURE 3.

Rheology studies showing the effect of DNA, histones, and DNA-histone on the critical shear stress needed to disassemble fibrin. Curves are shown for pure fibrin (red), fibrin containing 50 μg/ml DNA (green), and 100 μg/ml DNA (magenta). The addition of histones leads to increased clot stability under sheer stress shown in the presence of 300 μg/ml histone (blue) and 300 μg/ml histone + 100 μg/ml DNA (black). Pa, pascals.
TABLE 2.
Rigidity and viscoelastic parameters of composite fibrin clots
Clots containing 2.5 mg/ml fibrin and the indicated additives were prepared, and their rigidity was evaluated in an oscillation rheometer as described under “Experimental Procedures.” The values of the storage modulus (G′), the loss modulus (G″), and the loss tangent (tanδ = G″/G′) were determined after 15 min of clotting when they reached a plateau, whereas the critical shear stress τ0 was determined by extrapolation of the fall in viscosity to 0 as illustrated in Fig. 3. Mean and S.D. of three measurements are shown. Pa, pascals.
| Additive |
|||||
|---|---|---|---|---|---|
| None | 50 μg/ml DNA | 100 μg/ml DNA | 300 μg/ml histone | 300 μg/ml histone + 100 μg/ml DNA | |
| G′ (Pa) | 61.79 (13.57) | 41.07 (7.29)a | 32.42 (2.53)a | 67.13 (5.42) | 82.06 (5.81)a |
| G″ (Pa) | 4.03 (1.12) | 2.75 (0.42)a | 2.06 (0.35)a | 7.70 (0.21)a | 9.46 (0.38)a |
| G″/G′ | 0.063 (0.019) | 0.065 (0.014) | 0.063 (0.010) | 0.115 (0.008)a | 0.115 (0.008)a |
| τ0 (Pa) | 147.9 (15.8) | 79.6 (16.2)a | 51.9 (5.9)a | 270.4 (15.5)a | 370.8 (9.3)a |
a p < 0.05 according to Kolmogorov-Smirnov test in comparison with pure fibrin.
Fibrinolysis
tPA-catalyzed fibrinolysis was monitored in several systems to investigate the effects of DNA and histones. Fibrin turbidity was monitored in a microtiter plate format, providing information on clotting and lysis. In agreement with the morphometric analysis, adding a range of DNA concentrations alone to a clot resulted in small changes in clot turbidity with minor increases in lysis times (assessed as times to 50 and 100% lysis) as shown in Fig. 4A. The apparent potency of tPA when assessed over a range of tPA concentrations (5–40 ng/ml) is reduced by 15–20% at an optimal DNA concentration of 0.25 mg/ml. When histones were incorporated into clotting fibrin, there appeared to be more significant changes in fibrin structure as shown by maximum turbidity and fibrinolysis as shown in Fig. 4B. Time to maximum absorbance changed from 1400 to 1000 s at 0.04–0.1 mg/ml histones, and time to 50% lysis was apparently extended from around 2000 to 4000 s as histone concentration increased up to 1.0 mg/ml. The combination of both DNA and histones had a marked effect on clot lysis profiles as can be seen in Fig. 4C where curves for 0, 0.04, 0.33, and 1 mg/ml histones all in the presence of 0.03 mg/ml DNA are shown. The typically smooth, reproducible lysis curves seen without DNA or histones or with DNA or histones individually became highly variable when DNA and histones were added together. Clots could take many hours to be lysed completely. A different method that does not rely on clot turbidity was used to assess the apparent potency of tPA under similar conditions (17). Setting tPA activity in the absence of DNA or histones to 100%, the relative activity (and 95% confidence intervals) with additions was 85.4% (78.8–92.6%) with 70 μg/ml DNA and 146.8% (138.6–155.5%) with 50 μg/ml histones. The potency of tPA in the presence of both DNA and histones could not be determined as this method relies on identifying the collapse of the fibrin clot and release of trapped bubbles, but this end point was highly variable and subjective in the presence of both DNA and histones, mirroring the pattern seen in Fig. 4C.
FIGURE 4.

Effect of DNA and histones included within fibrin clots on clotting and lysis profiles. A shows the effects of 0 (circles), 0.03 (squares), 0.13 (triangles), and 0.5 mg/ml (diamonds) DNA on clotting and lysis in the absence of histones. B shows the effects of 0 (circles), 0.04 (squares), 0.33 (triangles), and 1 mg/ml histones (diamonds) in the absence of DNA. C shows the same range of histones as in B but in the presence of 0.03 mg/ml DNA. In all cases, only every tenth point is shown for clarity
The profound effect of DNA and histones on clot lysis in this system is illustrated in Fig. 5 and the accompanying time lapse videos (supplemental Videos 1 and 2). The consistent pattern observed was that clots were initially more opaque but were lysed more quickly in the presence of 50 μg/ml histones (tube 2); complete lysis was slightly delayed in the presence of 70 μg/ml DNA (tube 3). However, the onset of observable changes within the clot was not delayed by the presence of DNA, but the fibrin structure held together longer. Tube 5 shows a much delayed lysis in the presence of DNA + histones accompanied by heterogeneous clot formation and lysis. Tube 6 demonstrates how 5 units/ml DNase I could reverse some of the effects of DNA + histones when mixed with the forming clot (Fig. 5A) or when DNA was pretreated with DNase I for 30 min before clotting (Fig. 5B). Control clots without tPA were stable for days, indicating that lysis was not affected by contaminating proteases in the DNA or histone preparations.
FIGURE 5.
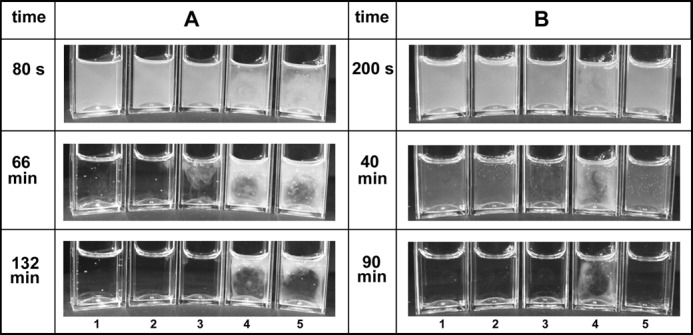
Effects of DNA and histones on appearance of fibrin clots and rates of lysis. Clot lysis was monitored in the presence of (final concentrations) 1.4 mg/ml fibrinogen and 200 nm plasminogen clotted with 30 nm thrombin in the presence of 0.2 nm tPA. Tubes in positions 1–5 have the following additions: none, 50 μg/ml histones, 70 μg/ml DNA, DNA + histones, DNA + histones + 5 units/ml DNase I, respectively. In A, the DNase was separated from the DNA until clotting was initiated, and in B, the DNA solution was pretreated with DNase I for 30 min before clotting. The reaction was monitored for 3 h during which 2000 pictures were taken to form a time lapse video (supplemental Videos 1 and 2). The images shown here were selected at the times given to illustrate significant stages of the reaction.
To focus on the microscopic behavior of tPA during clot lysis, we used confocal microscopy in conjunction with green fluorescent protein fusions of tPA (tPA-GFP) with fluorescently modified fibrinogen (14). Results are summarized in Fig. 6. The top row shows the typical pattern (14) in which fibrinolysis is accompanied by the generation of fibrin aggregates near the clot surface around the lysis front that strongly bind tPA-GFP. The second row with the addition of DNA shows some fibrin aggregate formation but strikingly a diffuse clot structure that remains behind the advancing tPA-GFP front. The lower two rows both containing histones show further changes in binding and fibrinolysis, primarily suggesting much poorer binding of tPA-GFP even though there is fibrin aggregate formation. Nevertheless, measurements of the rates of movement of the tPA-GFP front through fibrin show that although DNA may impede clot lysis, histones with or without DNA speed up the advance of the lysis front.
FIGURE 6.

Confocal microscopy studies using tPA-GFP and red fluorescent fibrin after 25 min of fibrinolysis. Each column of micrographs from left to right shows green tPA-GFP fluorescence, red Alexa Fluor 546-conjugated fibrin fluorescence, and the merged image. The first row shows the accumulation of fibrin aggregates that co-localize with tPA-GFP. The second row with the addition of 70 μg/ml DNA shows less fibrin aggregate formation but a diffuse fibrin clot that remains behind the advancing tPA-GFP front. The lower two rows where clots contain 50 μg/ml histones and 50 μg/ml histones + 70 μg/ml DNA, respectively, demonstrate reduced formation of fibrin aggregates within fibrin and less binding of tPA-GFP. The numbers in the last column indicate the relative distance for penetration of tPA-GFP in the clot at 25 min (the mean value for pure fibrin is 1, mean and S.E. from at least six samples, p < 0.05 for all additives according to the Kolmogorov-Smirnov test in comparison with pure fibrin). Scale bars, 20 μm.
Binding Studies on Fibrin Degradation Products and DNA
Given the apparent widespread distribution of DNA in thrombi noted in Fig. 1 and the suggestion from Fig. 6 that fibrin clot structure and the behavior of aggregates formed from FDPs are affected by the presence of DNA and/or histones, further studies were performed to investigate the interactions between DNA and histones with fibrin degradation products using ITC. These studies, summarized in Fig. 7, clearly showed that FDPs bind to both DNA (Kd = 136.1 nm) and histones (Kd = 190.7 nm) with higher affinity than with fibrinogen and plasminogen (Table 3). The size of the binding site in the DNA correlated with the size of the interacting ligand; fibrinogen required the largest binding site (2146 nucleotides), and plasminogen required the smallest binding site (682 nucleotides). Further studies (not shown) indicated that only larger FDPs with molecular masses >150 kDa demonstrated this high affinity binding, whereas smaller FDPs or fibrinogen did not.
FIGURE 7.
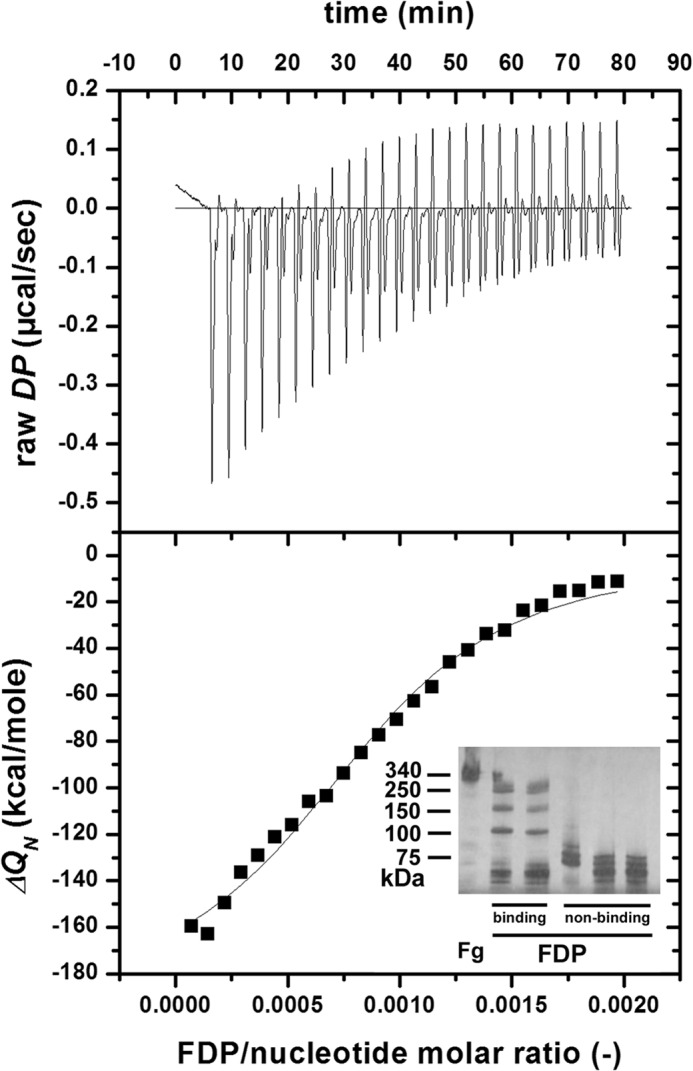
Binding of FDPs and DNA studied using ITC. The cell (1.43 ml) of the titration calorimeter was filled with 0.5 mg/ml DNA, 25 successive aliquots (10 μl each) of 6 μm FDPs were injected into the cell at 25 °C, and the heat increments of each addition (raw differential power (DP)) were measured (top panel). The base line-corrected, peak-integrated, and concentration-normalized enthalpy changes (ΔQN; bottom panel, squares) were evaluated according to the single site algorithm, and the best fitting binding isotherm is shown. The inset shows a non-reducing SDS-PAGE gel of typical FDP preparations consisting of high molecular weight fibrin fragments (binding) and low molecular weight fibrin fragments (non-binding).
TABLE 3.
Binding data from isothermal titration calorimetry
The intermolecular interactions of the indicated ligands were measured as described under “Experimental Procedures” and illustrated in Fig. 7. Fg, fibrinogen; Plg, plasminogen; N, size of binding site (in number of nucleotides when DNA is the binding partner or number of molecules when histone is used); Kd, dissociation equilibrium constant; ΔH, enthalpy change. Mean and S.D. of at least four measurements are shown.
| FDP/DNA |
FDP/histone |
Fg/DNA |
Plg/DNA |
|||||
|---|---|---|---|---|---|---|---|---|
| Mean | S.D. | Mean | S.D. | Mean | S.D. | Mean | S.D. | |
| N | 1315.8 | 136.3 | 1.6 | 0.5 | 2145.9 | 1903.0 | 682.0 | 110.9 |
| Kd (nm) | 136.1 | 111.7 | 190.7 | 91.7 | 534.3 | 52.8 | 524.1 | 72.7 |
| ΔH (kcal/mol) | −239.5 | 28.8 | −12.8 | 2.9 | −1116.3 | 1874.4 | −153.0 | 135.6 |
Heparin is another highly negative charged molecule like DNA and may be able to disrupt DNA-histone complexes. The effects of adding heparin in the microtiter plate system used in Fig. 4 are shown in Fig. 8. Fig. 8A confirms that there is an effect of DNA and histones on fibrin structure where clots are formed with a range of histones with constant DNA because maximum absorbance (related to fibrin fiber thickness) increases with increasing histones in the presence of DNA. If heparin is present, maximum clot absorbance is reduced, and the effect of histones and DNA is nullified. The effect of heparin on fibrin lysis is more complicated in the presence of DNA and histones. Although increasing heparin appears to have only a small effect on time to 50% lysis in the absence of histones, there is a major effect of heparin when DNA and histones are both present. The plots in Fig. 8, B–F, like those in A, are consistent with a pattern where heparin can displace DNA from the complex with histones and higher concentrations of heparin are required as the histone concentration is increased. However, the obvious peak in lysis times seen at an optimum heparin concentration in Fig. 8, B–F, suggests that there is a heparin-histone-DNA ternary complex that has a major effect on clot structure and lysis.
FIGURE 8.

Heparin displaces DNA in the histone-DNA complex. A shows the effect of unfractionated heparin concentration on maximum clot absorbance for clots containing 0.015 mg/ml DNA and 0 (crosses), 0.02 (circles), 0.05 (diamonds), 0.1 (triangles), 0.25 (squares), and 0.3 mg/ml (inverted triangles) histone. B–F show the effects of increasing unfractionated heparin concentration on the times to 50% lysis (not with same ranges but all in seconds) in clots containing 0.015 mg/ml DNA and the same range of histone concentrations as in A. In all cases, the crosses show the effect of heparin alone (lysis times with DNA but no histones), and the x axis is heparin concentration (log scale IU/ml in reaction mixture).
DISCUSSION
Based on the demonstration of neutrophil elastase-specific fibrin degradation products, we have recently provided ex vivo evidence for the proteolytic contribution of neutrophils to fibrinolysis in arterial thrombi (28). A different aspect of leukocyte functionality in thrombolysis is suggested by the presence of DNA and histones in clots from arteries revealed by the present study (Fig. 1). These observations on arterial clots add to previous work on the contribution of DNA and histones to the pathogenesis of deep vein thrombosis in animal models, for example baboons (5) and mice (13), as well as other conditions such as sepsis (29) and inflammatory and autoimmune diseases (30). The influence of DNA and histones on arterial thrombi warranted further investigation, and our results presented above suggest that DNA, histones, DNA + histones, and heparin can have different, sometimes opposing, effects, which are now considered below in turn.
Histones
Histones appear to have a significant effect on clot structure as shown in Table 1 and suggested in Figs. 2, 4, and 8. The trend is for more opaque fibrin, correlating with thicker fibrin fibers. As illustrated in Fig. 3, the impact is to provide a more robust clot structure in line with earlier data from pure fibrin clots (21) in which changes in fiber diameter had a significant impact on the G″/G′ ratio (dissipated/stored energy in deformation; Table 2). The fibrinolysis data for time to clot collapse (results summarized in Fig. 5) and confocal microscopy studies in Fig. 6 suggest that the activity of tPA may be up to 50% higher in the presence of 50 μg/ml histones. The microtiter plate system (Fig. 4) provided data that histones appear to delay lysis at the higher concentrations approaching 1 mg/ml. However, at lower concentrations, the relative rate of fibrinolysis is higher in the presence of histones if we take into account the higher initial peak absorbance and time to lysis. These results agree with previous observations that thicker fibers promote more rapid clot lysis (31). There is evidence from the ITC studies (Fig. 7) that histones can bind FDPs, and this may account for inhibition of lysis at higher histone concentrations (>110 μg/ml).
DNA
In contrast to histones, the effects of DNA on clot structure are minor (Table 1 and Figs. 2 and 4). However, rheology data suggest that clots containing DNA were less stable in response to mechanical shear forces, had reduced stiffness (G′), and were more able to dissipate energy in response to deformation (G″), suggesting “weak, floppy” clots (Table 2). The SAXS data (Fig. 2) reveal the structural background of these effects: disrupted longitudinal alignment of the monomers (loss of Peak 2; supplemental table) and preserved protofibril-to-protofibril distance (no change in Peak 3; supplemental table). Fibrinolysis may also be inhibited in these clots according to data presented in Fig. 4 and from the clot collapse assay, but here it may be important to differentiate between fibrin lysis and plasminogen activation in clots that incorporate DNA. Previous work involving binding studies and enzyme assays has shown that DNA and oligonucleotides can promote plasminogen activation by tPA and urokinase-type plasminogen activator with both Glu- and Lys-plasminogen (32). The mechanism of stimulation is proposed to be via template formation between plasminogen, activator, and DNA such that the apparent Km for plasminogen reaction with tPA or to a lesser extent urokinase-type plasminogen activator is reduced. Potentially significant increases up to 480- or 17-fold in stimulation of kcat/Km for plasminogen activation by tPA and urokinase-type plasminogen activator, respectively, were found, but the interaction was also found to be dependent on ionic strength. It was proposed that DNA and fibrin compete to bind plasminogen and activator (32). We have made similar observations using an assay system in which plasminogen activation is measured with a plasmin-specific chromogenic substrate in the presence of fibrin (33). However, fibrinolysis assays do not show any increased lysis due to DNA but rather delayed clot dissolution. Significantly, it appears from the confocal microscopy studies in Fig. 6 (second row) coupled with the ITC data shown in Fig. 7 and observations in Fig. 5 that DNA may bind to large FDPs and effectively hold the network together. Further digestion of large FDPs to lower molecular weight forms is then required to achieve complete clot dissolution.
DNA + Histones
The combination of DNA with histones provides effects that are greater than either component alone. Structural changes seen with histones are retained with the addition of DNA as shown in Table 1 and SAXS studies (Fig. 2) for example. Clot rigidity is enhanced in rheology studies (Fig. 3) by the addition of DNA to histones as evidenced by improved stability, rigidity, an increase in loss modulus G″, and a significant impact on the G″/G′ ratio (dissipated/stored energy in deformation; Table 2). Increased fiber diameter has been identified as a significant factor in increasing clot stability and network stiffness (21), and it has also been proposed that clot rigidity might be a predisposing factor for increased myocardial infarction (34). The striking changes seen in Fig. 4C are immediately visually apparent in Fig. 5, tubes 4 and 5. Thus, the incorporation of DNA and histones into fibrin clots extended the time to lysis by tPA by many hours. Nevertheless, close inspection of these tubes suggests that the onset of fibrinolysis was rapid, and the trapped bubbles monitored in Fig. 5 began to move early in the lysis process before those in the tubes containing DNA or histones alone, suggesting efficient plasminogen activation and fibrin breakdown. Furthermore, confocal studies (Fig. 6) also suggest that histones and DNA together do not hinder the rate of fibrin lysis, the background of which can be identified in the decreased fiber density (loss of Peak 3; Fig. 2 and supplemental table). Taken together with the ITC data that indicate that both DNA and histones may bind FDPs, it may be proposed that the slow breakdown seen in Fig. 4C and Fig. 5, tube 4, is due to a long lived network of DNA, FDPs, and histones. That DNA is critical in this proposed network is demonstrated by tube 5, which contained DNase I either added during clot formation (Fig. 5A) or mixed with DNA 30 min prior to clotting (Fig. 5B). Close inspection of lysis in supplemental Videos 1 and 2 shows that the DNase causes a different pattern of breakdown of the network in Fig. 5A whereby large fragments of fibrin can be seen raining down (without DNase, the fibrin network simply fades). In Fig. 5B, tube 5, the DNase pretreatment prevents formation of the heterogeneous clot structure seen in tube 4, and lysis is efficient and only slightly prolonged relative to control clots with no DNA or histones. Thus, DNase is an effective fibrinolytic agent in this system in combination with tPA.
Heparin
Heparin has been suggested as a potential treatment for sepsis as it can potentially bind to and neutralize histones in circulation, and heparin is known to interfere with formation of NETs (5, 35). Non-anticoagulant heparins have been explored, and evidence has been presented that they act in more complex ways than simply binding positively charged histones (3, 36). In the current study, unfractionated heparin was included in clot lysis experiments to investigate its effects on clot structure and lysis. A summary of results is presented in Fig. 8. Both clotting and lysis were affected, indicating that heparin, histones, and DNA can modify fibrin formation and structure. Over a range of heparin concentrations in a system containing DNA and histones, there was a pronounced peak in clot lysis time at a heparin concentration that was dependent on the histone concentration. This pattern of response where there is an optimum heparin concentration and a bell-shaped profile on a log heparin concentration scale suggests a template mechanism and formation of a heparin-histone-DNA trimeric complex, which has a profound impact on clot behavior. As the heparin concentration is increased, the data suggest that DNA is displaced, and the histone-heparin dimer complex predominates. The displacement of DNA is supported by the SAXS data (Fig. 2). The nanostructural pattern of the quaternary fibrin-DNA-histone-heparin complex resembles that of the fibrin-DNA clots but not of the pure fibrin clots (supplemental table). The proposition that the heparin-histone-DNA trimer may have distinct and significant effects on clot structure and lysis requires further investigation if heparin and heparin-like molecules are to be used in the clinical setting of sepsis and other situations where DNA and histones are present in the circulation.
In Vivo Implications
The diverse methods utilized in the current work mostly require purified components in relatively simple systems, and obviously caution is required when extrapolating to the in vivo situation. Nevertheless, these data add to previous work implicating DNA and histones in disturbances in coagulation and promotion of deep vein thrombosis (3, 5, 36). We have extended these studies to include arterial clots and now focus on fibrinolysis. These earlier studies involved histones within the same concentration range used in the present study (around 40 μg/ml for example (3, 36)), and concentrations up to 70 μg/ml have been measured in baboon models of sepsis (29). It is difficult to estimate the amounts of DNA that might be found in venous or arterial blood clots, but very high local concentrations around dead cells are likely as observed previously (5). The heterogeneous distribution of DNA and histones observed in arterial clots shown in Fig. 1 suggests that it will be difficult to predict how they affect clot stability and lysis in vivo. In addition to situations where NET formation is likely, DNA and histones may accumulate in the vicinity of atherosclerotic plaques, which contain dead cells. Thrombosis is believed to occur here after necrotic core expansion causes weakening of the atheroma cap to generate thrombogenic debris (37). Inflammatory signals may also recruit additional leukocytes to blood clots, providing an increased pool of DNA and histones (38). We propose that DNA release may result in weakened clots more prone to embolize, whereas histones might strengthen clot structure. DNA and histones may enhance plasminogen activation, but the most important observation in the present study is the slow breakdown of fibrin clots containing DNA + histones, which appear to stabilize the network by binding large FDPs. We find that heparin can counter the effects of DNA in clot lysis, but further work is required to investigate the activity of DNA-histone-heparin complexes. Prolonged clot lysis in the presence of histones + DNA may be reversed by DNase, providing a rationale for the secretion of streptodornase along with streptokinase by streptococci as mentioned above. If arterial clots do contain significant levels of histones and DNA derived from atheroma or released by apoptotic cells within the clots it raises the prospect that adding DNase to plasminogen activators used for thrombolytic treatment may lead to improved therapeutic outcomes.
Acknowledgments
We are grateful to Györgyi Oravecz for technical assistance and to Dr. András Kovács for immunostaining of thrombus sections. We are also grateful to Rizwan Yusuf and Paul Metcalfe for help with photography.
This work was supported by Wellcome Trust Grant 083174, Hungarian Scientific Research Fund Grants OTKA 75430 and 83023, and German Academic Exchange Service Grant DAAD A/12/01760.

This article contains a supplemental table and Videos 1 and 2.
- NET
- neutrophil extracellular trap
- tPA
- tissue plasminogen activator
- SEM
- scanning electron microscope
- ITC
- isothermal titration calorimetry
- FDP
- fibrin degradation product
- SAXS
- small angle x-ray scattering.
REFERENCES
- 1. Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D. S., Weinrauch Y., Zychlinsky A. (2004) Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 [DOI] [PubMed] [Google Scholar]
- 2. Clark S. R., Ma A. C., Tavener S. A., McDonald B., Goodarzi Z., Kelly M. M., Patel K. D., Chakrabarti S., McAvoy E., Sinclair G. D., Keys E. M., Allen-Vercoe E., Devinney R., Doig C. J., Green F. H., Kubes P. (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 13, 463–469 [DOI] [PubMed] [Google Scholar]
- 3. Semeraro F., Ammollo C. T., Morrissey J. H., Dale G. L., Friese P., Esmon N. L., Esmon C. T. (2011) Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 118, 1952–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Massberg S., Grahl L., von Bruehl M. L., Manukyan D., Pfeiler S., Goosmann C., Brinkmann V., Lorenz M., Bidzhekov K., Khandagale A. B., Konrad I., Kennerknecht E., Reges K., Holdenrieder S., Braun S., Reinhardt C., Spannagl M., Preissner K. T., Engelmann B. (2010) Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 16, 887–896 [DOI] [PubMed] [Google Scholar]
- 5. Fuchs T. A., Brill A., Duerschmied D., Schatzberg D., Monestier M., Myers D. D., Jr., Wrobleski S. K., Wakefield T. W., Hartwig J. H., Wagner D. D. (2010) Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. U.S.A. 107, 15880–15885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ruf W., Ruggeri Z. M. (2010) Neutrophils release brakes of coagulation. Nat. Med. 16, 851–852 [DOI] [PubMed] [Google Scholar]
- 7. Borissoff J. I., ten Cate H. (2011) From neutrophil extracellular traps release to thrombosis: an overshooting host-defense mechanism? J. Thromb. Haemost. 9, 1791–1794 [DOI] [PubMed] [Google Scholar]
- 8. Esmon C. T. (2009) Basic mechanisms and pathogenesis of venous thrombosis. Blood Rev. 23, 225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smeeth L., Cook C., Thomas S., Hall A. J., Hubbard R., Vallance P. (2006) Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet 367, 1075–1079 [DOI] [PubMed] [Google Scholar]
- 10. Gladysheva I. P., Sazonova I. Y., Chowdhry S. A., Liu L., Turner R. B., Reed G. L. (2002) Chimerism reveals a role for the streptokinase β-domain in nonproteolytic active site formation, substrate, and inhibitor interactions. J. Biol. Chem. 277, 26846–26851 [DOI] [PubMed] [Google Scholar]
- 11. Walker M. J., McArthur J. D., McKay F., Ranson M. (2005) Is plasminogen deployed as a Streptococcus pyogenes virulence factor? Trends Microbiol. 13, 308–313 [DOI] [PubMed] [Google Scholar]
- 12. Aziz R. K., Ismail S. A., Park H. W., Kotb M. (2004) Post-proteomic identification of a novel phage-encoded streptodornase, Sda1, in invasive M1T1 Streptococcus pyogenes. Mol. Microbiol. 54, 184–197 [DOI] [PubMed] [Google Scholar]
- 13. Brill A., Fuchs T. A., Savchenko A. S., Thomas G. M., Martinod K., De Meyer S. F., Bhandari A. A., Wagner D. D. (2012) Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 10, 136–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Longstaff C., Thelwell C., Williams S. C., Silva M. M., Szabó L., Kolev K. (2011) The interplay between tissue plasminogen activator domains and fibrin structures in the regulation of fibrinolysis: kinetic and microscopic studies. Blood 117, 661–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Silva M. M., Thelwell C., Williams S. C., Longstaff C. (2012) Regulation of fibrinolysis by C-terminal lysines operates through plasminogen and plasmin but not tissue plasminogen activator (tPA). J. Thromb. Haemost. 10, 2354–2360 [DOI] [PubMed] [Google Scholar]
- 16. R Development Core Team (2011) R, a Program for Statistical Computing, The R Foundation for Statistical Computing, Wien, Austria [Google Scholar]
- 17. Sands D., Whitton C. M., Merton R. E., Longstaff C. (2002) A collaborative study to establish the 3rd International Standard for tissue plasminogen activator. Thromb. Haemost. 88, 294–297 [PubMed] [Google Scholar]
- 18. Komorowicz E., Kolev K., Machovich R. (1998) Fibrinolysis with des-kringle derivatives of plasmin and its modulation by plasma protease inhibitors. Biochemistry 37, 9112–9118 [DOI] [PubMed] [Google Scholar]
- 19. Rácz Z., Baróti C. (1993) Technical aspects of buffy coat removal from whole blood and those of platelet production from single buffy coat units. Biomed. Tech. 38, 266–269 [DOI] [PubMed] [Google Scholar]
- 20. Wohner N., Keresztes Z., Sótonyi P., Szabó L., Komorowicz E., Machovich R., Kolev K. (2010) Neutrophil granulocyte-dependent proteolysis enhances platelet adhesion to the arterial wall under high-shear flow. J. Thromb. Haemost. 8, 1624–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ryan E. A., Mockros L. F., Weisel J. W., Lorand L. (1999) Structural origins of fibrin clot rheology. Biophys. J. 77, 2813–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gleber G., Cibik S., Haas S., Hoell A., Muller P., Krumrey M. (2010) Traceable size determination of PMMA nanoparticles based on Small Angle X-ray Scattering (SAXS). J. Phys. Conf. Ser. 247, doi: 10.1088/1742-6596/247/1/012027 [DOI] [Google Scholar]
- 23. Nikolova N. D., Toneva-Zheynova D., Kolev K., Tenekedjiev K. (2012) in Theory and Applications of Monte Carlo Simulations (Chan W. K., ed) InTech, Rijeka, Croatia [Google Scholar]
- 24. Yeromonahos C., Polack B., Caton F. (2010) Nanostructure of the fibrin clot. Biophys. J. 99, 2018–2027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weisel J. W. (1986) The electron microscope band pattern of human fibrin: various stains, lateral order, and carbohydrate localization. J. Ultrastruct. Mol. Struct. Res. 96, 176–188 [DOI] [PubMed] [Google Scholar]
- 26. Yang Z., Mochalkin I., Doolittle R. F. (2000) A model of fibrin formation based on crystal structures of fibrinogen and fibrin fragments complexed with synthetic peptides. Proc. Natl. Acad. Sci. U.S.A. 97, 14156–14161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guthold M., Liu W., Stephens B., Lord S. T., Hantgan R. R., Erie D. A., Taylor R. M., Jr., Superfine R. (2004) Visualization and mechanical manipulations of individual fibrin fibers suggest that fiber cross section has fractal dimension 1.3. Biophys. J. 87, 4226–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rábai G., Szilágyi N., Sótonyi P., Kovalszky I., Szabó L., Machovich R., Kolev K. (2010) Contribution of neutrophil elastase to the lysis of obliterative thrombi in the context of their platelet and fibrin content. Thromb. Res. 126, e94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu J., Zhang X., Pelayo R., Monestier M., Ammollo C. T., Semeraro F., Taylor F. B., Esmon N. L., Lupu F., Esmon C. T. (2009) Extracellular histones are major mediators of death in sepsis. Nat. Med. 15, 1318–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Esmon C. T., Xu J., Lupu F. (2011) Innate immunity and coagulation. J. Thromb. Haemost. 9, Suppl. 1, 182–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weisel J. W., Litvinov R. I. (2008) The biochemical and physical process of fibrinolysis and effects of clot structure and stability on the lysis rate. Cardiovasc. Hematol. Agents Med. Chem. 6, 161–180 [DOI] [PubMed] [Google Scholar]
- 32. Komissarov A. A., Florova G., Idell S. (2011) Effects of extracellular DNA on plasminogen activation and fibrinolysis. J. Biol. Chem. 286, 41949–41962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Longstaff C., Whitton C. M. (2004) A proposed reference method for plasminogen activators that enables calculation of enzyme activities in SI units. J. Thromb. Haemost. 2, 1416–1421 [DOI] [PubMed] [Google Scholar]
- 34. Scrutton M. C., Ross-Murphy S. B., Bennett G. M., Stirling Y., Meade T. W. (1994) Changes in clot deformability—a possible explanation for the epidemiological association between plasma fibrinogen concentration and myocardial infarction. Blood Coagul. Fibrinolysis 5, 719–723 [DOI] [PubMed] [Google Scholar]
- 35. Fuchs T. A., Bhandari A. A., Wagner D. D. (2011) Histones induce rapid and profound thrombocytopenia in mice. Blood 118, 3708–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ammollo C. T., Semeraro F., Xu J., Esmon N. L., Esmon C. T. (2011) Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 9, 1795–1803 [DOI] [PubMed] [Google Scholar]
- 37. Rocha V. Z., Libby P. (2009) Obesity, inflammation, and atherosclerosis. Nat. Rev. Cardiol. 6, 399–409 [DOI] [PubMed] [Google Scholar]
- 38. Saha P., Humphries J., Modarai B., Mattock K., Waltham M., Evans C. E., Ahmad A., Patel A. S., Premaratne S., Lyons O. T., Smith A. (2011) Leukocytes and the natural history of deep vein thrombosis: current concepts and future directions. Arterioscler. Thromb. Vasc. Biol. 31, 506–512 [DOI] [PMC free article] [PubMed] [Google Scholar]