

Background: Mcl-1 inhibits apoptosis and promotes survival of cancer cells.
Results: The Hsp70 co-chaperone, BAG3, prevents Mcl-1 degradation by the proteasome.
Conclusion: BAG3 sustains Mcl-1 expression in cancer cells, promoting its antiapoptotic activity.
Significance: Mcl-1 expression is a major determinant of resistance in human cancer, so BAG3 is a potential target for anticancer drug discovery.
Keywords: Apoptosis, Bcl-2 Family Proteins, Cancer, Proteasome, siRNA, Cochaperone
Abstract
Members of the Bcl-2 family of proteins are important inhibitors of apoptosis in human cancer and are targets for novel anticancer agents such as the Bcl-2 antagonists, ABT-263 (Navitoclax), and its analog ABT-737. Unlike Bcl-2, Mcl-1 is not antagonized by ABT-263 or ABT-737 and is considered to be a major factor in resistance. Also, Mcl-1 exhibits differential regulation when compared with other Bcl-2 family members and is a target for anticancer drug discovery. Here, we demonstrate that BAG3, an Hsp70 co-chaperone, protects Mcl-1 from proteasomal degradation, thereby promoting its antiapoptotic activity. Using neuroblastoma cell lines, with a defined Bcl-2 family dependence, we found that BAG3 expression correlated with Mcl-1 dependence and ABT-737 resistance. RNA silencing of BAG3 led to a marked reduction in Mcl-1 protein levels and overcame ABT-737 resistance in Mcl-1-dependent cells. In ABT-737-resistant cells, Mcl-1 co-immunoprecipitated with BAG3, and loss of Mcl-1 after BAG3 silencing was prevented by proteasome inhibition. BAG3 and Mcl-1 were co-expressed in a panel of diverse cancer cell lines resistant to ABT-737. Silencing BAG3 reduced Mcl-1 protein levels and overcame ABT-737 resistance in several of the cell lines, including triple-negative breast cancer (MDA-MB231) and androgen receptor-negative prostate cancer (PC3) cells. These studies identify BAG3-mediated Mcl-1 stabilization as a potential target for cancer drug discovery.
Introduction
Apoptotic cell death is critical for the maintenance of tissue homeostasis in a healthy organism, providing an efficient and safe mechanism to remove unwanted cells. Altered control of apoptosis contributes to many diseases including neoplasia (1). Dysregulated and/or defective cell death is one of the hallmarks of cancer (2). Bcl-2 family proteins are key regulators of apoptosis, and alterations in their expression and function are associated with cancer development (3, 4). The family includes both proapoptotic and antiapoptotic members that contain one to four Bcl-2 homology (BH)2 domains, which are involved in the interaction between family members. Oligomerization of the multidomain apoptotic members, Bax and Bak, results in mitochondrial outer membrane permeabilization, allowing the release of proapoptogenic proteins, such as cytochrome c, and subsequent activation of caspases and induction of cell death. The antiapoptotic family members, Bcl-2, Bcl-xL, Mcl-1, and Bcl-w, bind to proapoptotic members of the family, preventing the oligomerization (5) of Bax/Bak and thereby inhibiting programmed cell death.
Other proapoptotic members that contain only one Bcl-2 homology domain, known as BH3-only proteins, include Bad, Bid, Bik, Noxa, Puma, Hrk, Bim, and more (6). In response to a wide variety of damage signals, such as DNA damage, growth factor withdrawal, or oncogene activation, BH3-only proteins are released and promote apoptosis by either freeing Bax/Bak from antiapoptotic Bcl-2 proteins (7) or direct activation of Bax/Bak (8).
Bcl-2 and related antiapoptotic proteins have emerged as key therapeutic targets in cancer due to their role in its genesis and their contribution to drug resistance. In fact, Bcl-2 down-regulation using antisense oligonucleotides has been shown to sensitize cancer cells to chemotherapeutic agents (9). This formed the basis for the design of small molecule Bcl-2 antagonists, such as ABT-737 and ABT-263 (Navitoclax), which mimic BH3-only proteins. ABT-737 and ABT-263 are Bad mimetics that bind to and neutralize Bcl-2, Bcl-xL, and Bcl-w. In some cancer cell lines, ABT-737 shows single-agent activity, whereas in others, it increases sensitivity to chemotherapeutics (10). ABT-737 does not bind to Mcl-1, so Mcl-1 expression is believed to be responsible for ABT-737 resistance in certain settings (11, 12). Similar to Bcl-2, Mcl-1 overexpression has been observed in a variety of human hematopoietic (13–15) and solid tumors (16).
Although both Mcl-1 and Bcl-2 promote cell survival, there is increasing evidence that suggests Mcl-1 may play a distinct role in the control of apoptosis (17). Differences in tissue distribution as well as timing of expression have been observed, implying that Mcl-1 and Bcl-2 can be independently regulated (18). Mcl-1 is structurally different from the other Bcl-2 antiapoptotic members at the amino terminus, which includes PEST sequences characteristic of short-lived proteins (19). Expression of Mcl-1 is highly regulated post-translationally, and many phosphorylation sites are described at the amino terminus along with one caspase cleavage site (20–24). Mcl-1 can be ubiquitylated and degraded by the proteasome (25) although mutation of all lysine residues does not prevent degradation (26). Unlike Bcl-2, many apoptotic stimuli promote Mcl-1 degradation by the proteasome or caspases (24, 27, 28). For example, Mcl-1 loss is required for the initiation of apoptosis after ultraviolet irradiation (29). Recently, it has been suggested that proteins that stabilize this otherwise labile protein could play a role in Mcl-1-driven cancers (30). Although Mcl-1 expression is regulated at many levels, clearly, proteasomal degradation plays an important role, unique among antiapoptotic family members.
Our laboratory has shown that in a colon cancer cell line subjected to electrophile stress, BAG3 plays an important role in the adaptive response regulated by heat shock factor 1 (HSF1) (31). Although the mechanism of BAG3 action is still unknown, preliminary results correlated decreases of BAG3 levels with decreases in the levels of Bcl-2, Bcl-xL, and Mcl-1. BAG3 belongs to the BAG family of co-chaperones that interact with proteins of the heat shock family (e.g. Hsp70). BAG1 (Bcl-2-associated athanogene 1) was initially discovered as a Bcl-2-interacting protein (32) and later described as an Hsp70-binding protein (33). The BAG family includes six members that share an evolutionarily conserved BAG domain, which binds to the ATPase domain of Hsp70, and acts as a nucleotide exchange factor for the chaperone (34). Although all BAG proteins bind to Hsp70, their modulation of its activity differs and depends upon multiple factors involving co-chaperones and subcellular localization. Although BAG1 interacts with the proteasome and increases Hsp70 client protein degradation (35), BAG3 inhibits proteasomal degradation of Hsp70 clients (36) and participates in the recruitment for autophagy (37, 38). High expression of BAG3 has been observed in chronic lymphocytic leukemia, (39), thyroid carcinoma (40) and pancreatic cancer (41), where it is associated with cancer resistance. BAG3 is described in the literature as an antiapoptotic protein (42), although the mechanism of apoptosis inhibition remains unclear.
To study the role of BAG3 in the regulation of Mcl-1, we used neuroblastoma cell lines. Neuroblastoma is a pediatric solid tumor derived from developing sympathetic neuroblasts (43), and high risk neuroblastoma is associated with high morbidity and mortality (44). Suppression of apoptosis is a common theme in high risk neuroblastoma, and gain of prosurvival Bcl-2 function is an important mechanism (45, 46). Neural tissues, including many neuroblastomas, express high levels of Mcl-1, and Mcl-1 maps to a region in chromosome 1q that shows frequent copy number gain in high risk neuroblastoma (47–49). In fact, Mcl-1 has been suggested to function as an oncogene in this subset (46). A study by the pediatric preclinical testing program demonstrated that ABT-263 has limited single-agent activity in neuroblastoma, which may be related to the high frequency of Mcl-1 dependence in this tumor type. In the present study, we used neuroblastoma cell lines with a well characterized dependence on prosurvival Bcl-2 proteins (46, 50, 51) to study the role of BAG3 in Mcl-1-driven cancer cells. Using this cellular model along with a panel of different types of cancer cell lines, we demonstrate that BAG3 sustains Mcl-1 expression by inhibiting its proteasomal degradation, thereby promoting survival of cancer cells and resistance to ABT-737.
EXPERIMENTAL PROCEDURES
Reagents
ABT-737 was purchased from ChemiTek (Indianapolis, IN). Doxorubicin was purchased from Sigma. Geldanamycin, cycloheximide, and MG-132 were purchased from Enzo Life Sciences (Plymouth Meeting, PA). Z-VAD-FMK was purchased from BD Biosciences. Cell culture medium was purchased from Invitrogen. Fetal bovine serum was purchased from Atlas Biologicals (Fort Collins, CO).
Cell Culture and Treatment
All cancer cells, except PC3, were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and 5 mm glutamine. PC3 cells were cultured in DMEM F12 supplemented with 10% fetal bovine serum. HEK293T cells were cultured in DMEM-GlutaMAX supplemented with 10% fetal bovine serum. Cells were grown in humidified cell culture incubators under 5% CO2, 95% air. Drug or DMSO (vehicle control) was added, and cells were incubated for the indicated lengths of time. DMSO concentration never exceeded 0.1%.
Viability Assays
Cells were seeded in 96-well plates at different cell densities (8 × 103 IMR5, 10 × 103 NLF, 20 × 103 SK-N-AS, 40 × 103 SMS-SAN, 10 × 103 MB231, 5 × 103 H292, 10 × 103 PC3, 10 × 103 LNCap), allowed to adhere overnight, and then treated with DMSO or drugs. After 48 h, relative viability was determined using WST-1 (Roche Applied Science) according to the manufacturer's protocol. For viability assays including transfection, cells were transfected in 96-well plates 24 h before the start of drug treatment.
siRNA Transfections
Transfections of neuroblastoma cells were performed using 10 nm Stealth RNAi siRNA (Invitrogen) and 0.2 μl of DharmaFECT 1 (Thermo-Scientific, Lafayette, CO) per well in 96-well plates using a reverse transfection. Briefly, lipid-siRNA complexes were prepared in Opti-MEM (Invitrogen), and a cell suspension in culture medium (10% fetal bovine serum) was added later. After 24 h, medium was replenished. For protein collection, cells were transfected in a 100-mm dish using 50 pmol of siRNA and 8 μl of DharmaFECT 1 in a final volume of 5 ml. Fresh medium was added after 24 h, and samples were collected at different time points. The siRNA sequences used are shown in supplemental Table S1. Transfection of MDA-MB231, H292, PC3, and LNCaP cell lines was done as described above with the following modifications: MB231 (0.1 μl of DharmaFECT 4), H292 (0.2 μl of Lipofectamine 2000), and PC3 and LNCaP (0.2 μl of DharmaFECT 2).
Plasmid Constructs and Co-transfection
A cDNA clone of BAG3 (Mammalian Gene Collection accession number BC006418) was obtained from the Vanderbilt Microarray Shared Resource cDNA clone collection. BAG3 was PCR-amplified from pOTB7 using the primers 5′-GGGATCCTCCACCATGGATTACAAGGATGACGACGATAAGAGCGCCCGCCACCCACTCG-3′ (includes an N-terminal FLAG tag) and 5′-CCTCGAGCTACGGTGCTGCTGGGTTA-3′. The PCR product was digested with BamHI and XhoI and ligated into pcDNA3.1 Hygro(+) (Invitrogen). Site-directed mutagenesis was used to mutate Arg-480 into Ala (BAG3R480A) and Pro-209 into Leu (BAG3P209L). Primers used to create nucleotide substitution are listed below, and the substituted codon is underlined: BAG3R480A, 5′-GTGCGTCAGGCCGCGAGAGACGGTGTC-3′, and BAG3P209L, 5′-TACATCTCCATTCTTGTGATACACGAG-3′. For co-transfection, HEK293T cells (2.5 × 106) were transfected using 25 nm Stealth RNAi siRNA (Invitrogen) and 10 μl of Lipofectamine 2000 (Invitrogen). After 24 h, cells were transfected using 0.5 μg of expression construct using 15 μl of Lipofectamine 2000 (Invitrogen).
Total Protein Extraction and Western Blotting
Total proteins were collected using M-PER lysis buffer (Thermo Scientific) containing mammalian protease inhibitor mixture (Sigma). Lysates were centrifuged at 16,000 × g for 10 min and stored at −80 °C. Protein concentration was determined by the BCA assay (Thermo Scientific). For Western blotting, equal amounts of protein were resolved by SDS-PAGE and transferred onto a 0.2-mm nitrocellulose membrane. Membranes were blocked (20 mm Tris, pH 7.6, 140 mm NaCl, 0.05% Tween 20, 5% nonfat dry milk or BSA) prior to incubation with antibodies. Luminol-based detection was performed using SuperSignal West Pico reagents (Thermo Scientific). Primary antibodies were obtained from the following sources: actin, Bcl-2 (sc-509), Mcl-1 (sc-20679), Hsp70 (sc-24), and ubiquitin (sc-166553) were from Santa Cruz Biotechnology (Santa Cruz, CA); Akt, BAG1, caspase 3, and lamin A/C were from Cell Signaling (Danvers, MA); BAG3, BAG4, BAG5, and BAG6 were from Abcam (Cambridge, MA); BAG2 was from Novus (Littleton, CO); Hsp90 and poly(ADP-ribose) polymerase (PARP) were from BD Biosciences; and Noxa was from Enzo Life Sciences. The following secondary HRP-conjugated antibodies were used: anti-rabbit and anti-mouse from Promega (Madison, WI) and anti-goat from Santa Cruz Biotechnology.
Cell Fractionation
Cells were fractionated with the Qproteome cell compartment kit (Qiagen, Valencia, CA) according to manufacturer's protocol. The fractionated lysates were quantified using the BCA assay (Thermo Scientific), and equal amounts of fractionated lysates were analyzed by immunoblot.
Immunoprecipitation
Cell lysates were prepared in immunoprecipitation lysis buffer (0.025 m Tris, 0.15 m NaCl, 1 mm EDTA, 1% Nonidet P-40, 5% glycerol, mammalian protease inhibitor mixture (Sigma), pH 7.4) and clarified by centrifugation (16,000 × g, 10 min). The following antibodies were used for immunoprecipitation: Mcl-1 (BD Biosciences), BAG3 (goat 112016, Abcam, Cambridge, MA), and Bcl-2 (sc-509, Santa Cruz Biotechnology). Antibodies and protein A/G-agarose beads (Pierce) were incubated with cell lysates (300–600 μg of protein) in immunoprecipitation coupling buffer (10 mm sodium phosphate, 0.155 m NaCl, mammalian protease inhibitor mixture (Sigma), pH 7.4) overnight at 4 °C. Beds were washed three times with immunoprecipitation lysis buffer and one time with conditioning buffer (classic immunoprecipitation kit, Pierce). Proteins were eluted using acidic conditions and eluates analyzed by immunoblot.
Ubiquitination Studies
IMR5 cells were transfected with BAG3 siRNA or negative control siRNA (NEG) as described above. Cells were treated at 24 h after transfection with 10 μm MG132 during 4 h and used to assess Mcl-1 ubiquitination as described elsewhere (26).
Real Time PCR Analysis
Cell lysates were homogenized using QIAshredder (Qiagen). Total RNA was then isolated using the RNeasy collection kit (Qiagen). RNA samples were quantified by absorbance at λ260 and λ280, and 1 μg of RNA was reverse-transcribed in a 20-μl reaction using iScript (Bio-Rad). Ten percent of each reaction (2 μl) was used per well in subsequent real time PCR analysis using iQ SYBR Green Supermix (Bio-Rad). Real time reactions were performed on a Bio-Rad iCycler. Real time PCR primers were as follows: BAG3, forward, 5′-CAACAGCCGCACCACTAC-3′; reverse, 5′-CATTGGCAGAGGATGGAGTC-3′; MCL-1, forward, 5′-AGCGACGGCGTAACAAAC-3′; reverse, 5′-AAGAACTCCACAAACCCATCC-3′.
RESULTS
BAG3 Expression in ABT-737-sensitive and -resistant Cell Lines
BH3-only proteins have different binding affinities for antiapoptotic Bcl-2 proteins. Although some BH3-only proteins (e.g. Bim) bind to all antiapoptotic family members, others (e.g. Noxa and Bik) bind only to a subset. For example, Bad binds to Bcl-2, Bcl-xL, and Bcl-w, but it does not bind to Mcl-1, whereas Noxa binds preferentially to Mcl-1. Peptides derived from BH3-only proteins can be used to determine the BH3 profile of a cell line by treating isolated mitochondria and quantifying the amount of cytochrome c released. This profile identifies the dependence of the tumor cell on specific Bcl-2 antiapoptotic proteins (52). The neuroblastoma cell lines used in the present study belong to three different BH3 profile groups (Fig. 1A) (51). At diagnosis, neuroblastoma cells demonstrate competent Bak/Bax activation but require the activities of Bcl-2 or Mcl-1 for survival. In the former, tonic neutralization of Bim by Bcl-2 provides the survival bias, and these cells (e.g. SMS-KCN, SMS-SAN, and NB-1643) are exquisitely sensitive to the Bcl-2 antagonist ABT-737. In the latter, Mcl-1 provides the survival bias by neutralizing Bim, and these cells (e.g. IMR5 and NLF) are ABT-737-resistant unless Mcl-1 activity is antagonized (such as by Mcl-1 knockdown). Finally, tumor cells derived at the time of acquired therapy resistance (after relapse) have repressed Bak/Bax activation. They maintain their Bcl-2 or Mcl-1 activities (e.g. SK-NAS) but are resistant to ABT-737 and other cytotoxic stressors.
FIGURE 1.
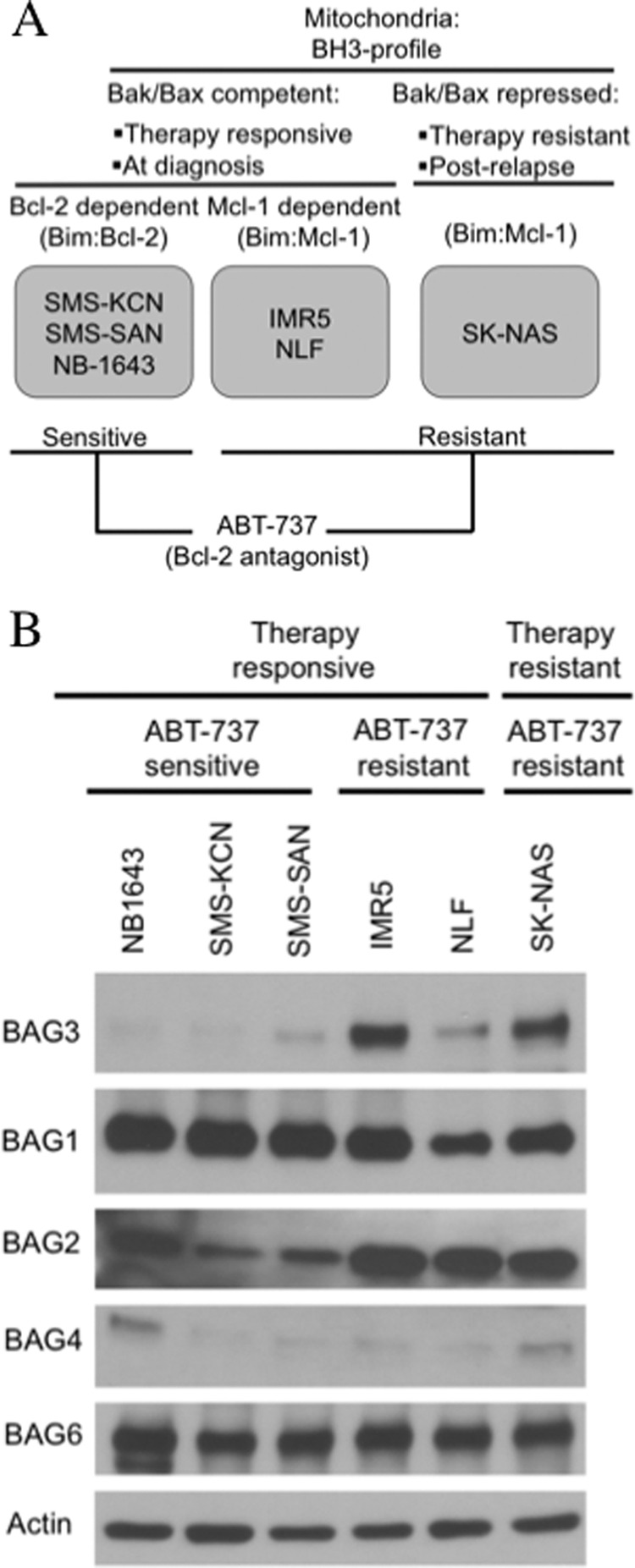
BAG3 expression is related to ABT-737 resistance in neuroblastoma. A, classification of neuroblastoma cell lines according to BH3 profile and sensitivity to the Bcl-2 antagonist, ABT-737. B, protein immunoblot showing the levels of BAG proteins in neuroblastoma cell lines; BAG5 was not detected. Actin was included as a loading control.
We assessed the expression of BAG proteins in the neuroblastoma cell lines and found a relationship between BAG3 expression and ABT-737 resistance (Fig. 1B). BAG3 was almost exclusively expressed in ABT-737-resistant cell lines, including Mcl-1-dependent ones. BAG5 was not detected in any of the cell lines, and BAG4 was weakly expressed in all of them. Unlike BAG3, BAG1 and BAG6 were uniformly expressed at high levels in both ABT-737-resistant and ABT-737-sensitive groups. BAG2 was the only other co-chaperone with a higher expression in ABT-737-resistant cell lines, but it was also highly expressed in the ABT-737-sensitive cell line NB1643.
Subcellular localization of BAG3 and Mcl-1 in ABT-737-resistant cell lines was studied by cell fractionation (supplemental Fig. 1). In agreement with previous findings in other cell lines (18, 53, 54), BAG3 was predominantly cytosolic, whereas Mcl-1 was mainly observed in the membrane fraction. The same pattern was observed for Mcl-1-dependent and BH3-resistant cell lines.
BAG3 Binds to Mcl-1 in ABT-737-resistant Cells
To study the binding of Mcl-1 to BAG3, we immunoprecipitated endogenous proteins using an antibody directed to BAG3. In ABT-737-resistant cell lines, Mcl-1 co-immunoprecipitated with BAG3 (Fig. 2A). Although these cell lines express high levels of Bcl-2, the interaction of Bcl-2 with BAG3 was only observed in IMR5 cells (Fig. 2A and supplemental Fig. 2A). The BAG3-Mcl-1 interaction was also studied using an antibody to precipitate Mcl-1 (Fig. 2B). BAG3 co-immunoprecipitated with Mcl-1 in the ABT-737-resistant cells. In NLF cells, where BAG3 appeared as two bands, only one band co-immunoprecipitated with Mcl-1. BAG3 has been observed as two bands in different cancer lines or when overexpressed, and the origin of these bands is uncertain (55). The BH3-only protein, Noxa, also co-immunoprecipitated with Mcl-1 in the three cell lines.
FIGURE 2.
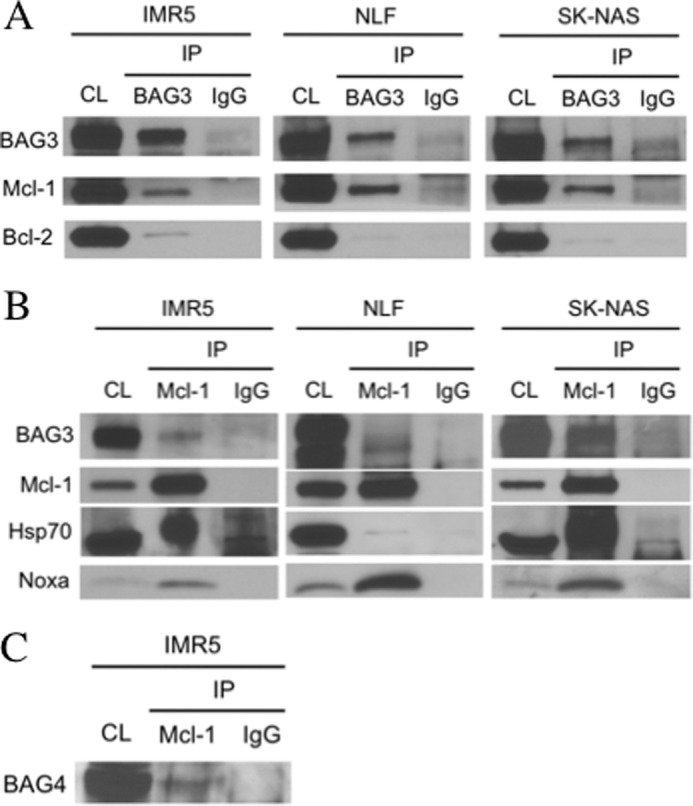
BAG3 binds to Mcl-1 in ABT-737-resistant cell lines. Endogenous proteins were immunoprecipitated using specific antibodies for BAG3 (A) or Mcl-1 (B and C). Co-immunoprecipitated proteins were detected by immunoblot. Normal sera IgG were used as control. CL, cell lysate; IP, immunoprecipitate.
Previous work showed that Bcl-xL interacts with BAG3 and Hsp70 in colon cancer cells (31). A similar result was obtained for Mcl-1 with the ABT-737-resistant neuroblastoma cell lines, IMR5 and SK-NAS. The significant amount of Hsp70 co-immunoprecipitated, when compared with BAG3, suggests that Mcl-1 interaction with Hsp70 is not occurring through BAG3. In fact, in NLF cells, where BAG3 co-immunoprecipitated with Mcl-1, the level of Hsp70 co-immunoprecipitated was close to background, which suggests that it is not complexed with Mcl-1 and BAG3 (Fig. 2B).
BAG4 was the only other BAG family member that co-immunoprecipitated with Mcl-1 in IMR5 cells (Fig. 2C). BAG1 and BAG2, although highly expressed in this cell line, showed no association with Mcl-1 (supplemental Fig. 2B). The BAG domains of BAG4 and BAG3 share a 62% identity, the highest among family members (56).
BAG3 Prevents Mcl-1 Proteasomal Degradation
We used small interfering RNA to study the effect of BAG3 on Mcl-1 expression. The down-regulation of BAG3 resulted in a rapid loss of Mcl-1, which was more pronounced in the Mcl-1-dependent cell lines, IMR5 and NLF, 48 h after transfection (Fig. 3A). Although BAG3 also interacted with Bcl-2 in IMR5 cells (Fig. 2A and supplemental Fig. 2A), silencing BAG3 had no effect on Bcl-2 levels. This difference may be related to the longer half-life of Bcl-2 when compared with Mcl-1 (54). To confirm that this effect was specific to BAG3 silencing, we tested six different siRNA oligonucleotides that target different regions of BAG3 mRNA. Although we did not see a linear correlation between BAG3 and Mcl-1 levels, five out of six siRNA sequences directed to BAG3 were able to down-regulate Mcl-1, supporting that the effect is specific to targeted BAG3 reduction. (supplemental Fig. 3).
FIGURE 3.

BAG3 silencing reduces Mcl-1 protein levels. A, neuroblastoma cells were transfected with nontargeting siRNA (NEG) or BAG3 siRNA (BAG3), and after 48 h, the levels of Mcl-1 and Bcl-2 were determined by immunoblot. Actin was included as a loading control. B, IMR5 cells were transfected with nontargeting siRNA (NEG) or BAG3 siRNA (BAG3), and the expression of BAG3 and Mcl-1 was determined by real time PCR (RT-PCR) 24 h after transfection. Data are expressed relative to nontargeting siRNA as mean ± S.E. (n = 3). C, IMR5 cells were treated with 10 μg/ml cycloheximide (CHX) 24 h after transfection with nontargeting siRNA (NEG) or BAG3 siRNA (BAG3), and the level of Mcl-1 was determined by immunoblot. Actin was included as a loading control. D, the Mcl-1 level in the cycloheximide chase experiment was quantified by densitometry, and data are expressed as mean ± S.D. (n = 3). Mcl-1 half-life was calculated in each experiment, and comparison between groups was done using an unpaired Student's t test (nontargeting siRNA versus BAG3, p value < 0.05).
BAG3 silencing had little effect on Mcl-1 mRNA levels (Fig. 3B), indicating that the mechanism of BAG3 action on Mcl-1 is not transcriptional. This was further studied using cycloheximide to inhibit protein synthesis. IMR5 cells were treated with BAG3 siRNA or negative control siRNA (NEG) for 24 h and then incubated with cycloheximide. Over a period of 60 min, samples were taken, and the level of Mcl-1 was analyzed by immunoblot to determine a relative half-life (Fig. 3, C and D). BAG3 silencing increased reduced Mcl-1 half-life from 49 to 19 min.
BAG3 has been shown to inhibit the proteasomal degradation of Hsp70 client proteins, so next we assessed the role of the proteasome in Mcl-1 loss. Again, IMR5 cells were treated with BAG3 siRNA or negative control siRNA (NEG) for 48 h and then incubated with the proteasome inhibitor MG132 for 4 h. Inhibition of the proteasome for this short period of time increased Mcl-1 protein levels, implying that the proteasome is involved in Mcl-1 loss after BAG3 knockdown (Fig. 4A). Although Mcl-1 level returned to control level (Fig. 4A, NEG) after MG132 treatment in BAG3 siRNA-treated cells, it was lower than the level observed when MG132 was used in combination with negative control siRNA (NEG). This difference could be explained by the accumulation of new Mcl-1 on top of extant Mcl-1 in the case of cells treated with the negative control siRNA (NEG). However, this result could also suggest the existence of some Mcl-1 degradation independent of the proteasome. To confirm the specificity of this result, we used a second approach to inhibit the proteasome. We knocked down PSMD14, a part of the regulatory subunit of the 26 S proteasome that is required for deubiquitination of proteins, in order for them to be degraded. In IMR5 cells, PSMD14 knockdown increased the level of Mcl-1, suggesting that it is involved in Mcl-1 degradation by the proteasome (Fig. 4B). When PSMD14 siRNA was used in combination with BAG3 siRNA, it completely prevented the loss of Mcl-1 (Fig. 4B).
FIGURE 4.
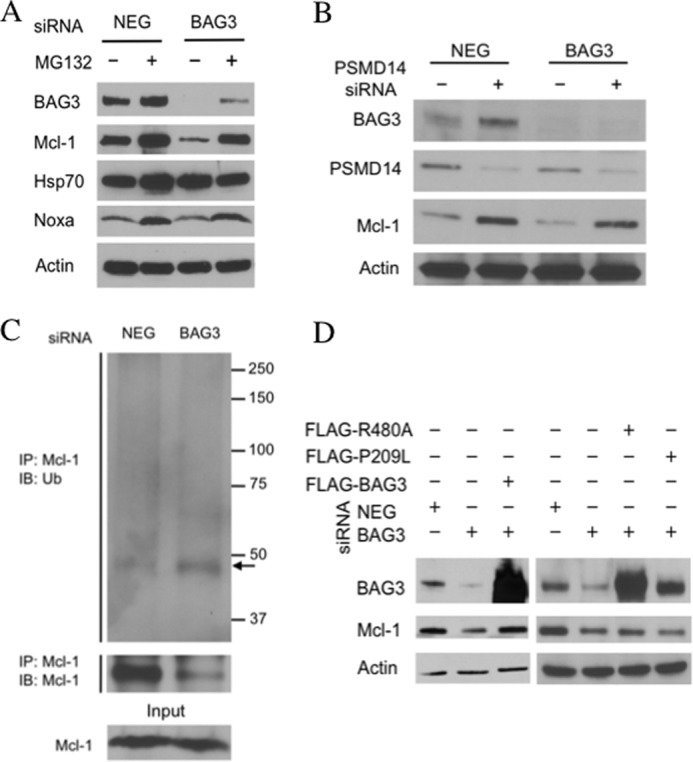
BAG3 prevents Mcl-1 degradation by the proteasome. A, IMR5 cells were transfected with nontargeting siRNA (NEG) or BAG3 siRNA (BAG3). After 48 h, cells were treated for 4 h with MG-132 (10 μm) or DMSO, and protein levels were determined by immunoblot. B, IMR5 cells were transfected with nontargeting siRNA (NEG) or BAG3 siRNA (BAG3) with or without PSMD14 siRNA. After 48 h, proteins levels were assessed by immunoblot. C, IMR5 cells were transfected with nontargeting siRNA (NEG) or BAG3 siRNA (BAG3). After 24 h, cells were treated with MG-132 (10 μm, 4 h) and then collected, and Mcl-1 was immunoprecipitated (IP). Ubiquitin conjugates were detected by immunoblot (IB). A band possibly corresponding to mono-ubiquitinated Mcl-1 is indicated with an arrow. D, HEK293T cells were transfected with nontargeting siRNA (NEG) or BAG3 siRNA (BAG3) alone or along with the corresponding FLAG-tagged BAG3 constructs: FLAG-BAG3 (WT), FLAG-R480A (BAG3R480A), and FLAG-P209L (BAG3P209L). Proteins levels were determined by immunoblot. Actin was included as a loading control.
To further address the role of BAG3 in Mcl-1 degradation by the proteasome, we assessed Mcl-1 ubiquitination. Mcl-1 was immunoprecipitated in IMR5 cells previously treated with BAG3 siRNA or negative control siRNA (NEG) for 24 h, and the presence of ubiquitin conjugates was analyzed by immunoblot. Under these conditions, no Mcl-1 was immunoprecipitated in the cells treated with BAG3 siRNA (data not shown). Because the lower levels of Mcl-1 after BAG3 silencing could be affecting the immunoprecipitation, we added MG132 4 h before collecting the cells. Although MG132 treatment prevented Mcl-1 loss, the amount of Mcl-1 detected in the immunoprecipitates from BAG3-silenced cells was less than in the nontargeting siRNA control (Fig. 4C, NEG), suggesting that modification of Mcl-1 after BAG3 silencing, such as ubiquitination, may interfere with the binding of the antibody. Indeed, an increase in Mcl-1 ubiquitination was observed after BAG3 knockdown, with the appearance of a band at ∼50 kDa, which may be mono-ubiquitinated Mcl-1 (Fig. 4C). The same band is observed when total ubiquitinated proteins were immunoprecipitated and when Mcl-1 was detected by immunoblot (supplemental Fig. 4).
To probe the role of Hsp70 in BAG3 action, we transfected recipient cells with BAG3 mutants. HEK293T cells were used because neuroblastoma cells did not transfect well. HEK293T cells express both BAG3 and Mcl-1, and siRNA silencing of BAG3 led to down-regulation of Mcl-1 (Fig. 4D). Reduction in Mcl-1 levels after transfection with siRNA directed to the BAG3 3′-UTR was reversed by ectopic expression of a BAG3 construct lacking the 3′-UTR (Fig. 4D). However, ectopic expression of a BAG3 R480A mutant, which is unable to bind to Hsp70 (57), did not rescue Mcl-1 levels, suggesting that BAG3 binding to Hsp70 is required to stabilize Mcl-1. The only clinically described BAG3 mutation is P209L, which is associated with a severe form of myofibrillar myopathy (58). Although the mechanism of the disease is unknown, it was associated with an increase in apoptosis (58). We found that ectopic expression of the BAG3 P209L mutant was unable to rescue Mcl-1 levels (Fig. 4D).
BAG3 Silencing Induces Apoptosis in Mcl-1-dependent Cells, but Mcl-1 Degradation Is Caspase-independent
To compare the effect of BAG3 and Mcl-1 in apoptosis, we used siRNA to silence protein expression and then evaluated the number of viable cells and PARP cleavage (Fig. 5, A and B) as an indicator of caspase activation. As expected, both Mcl-1-dependent cell lines showed a decrease in the relative viability after Mcl-1 knockdown, although the extent of cytotoxicity differed. IMR5 cells showed more than 80% reduction in the relative viability, whereas only 20% reduction was observed with NLF cells. Silencing BAG3 in IMR5 cells caused a 50% reduction in the relative viability 72 h after transfection, and PARP cleavage was observed as early as 24 h after transfection (Fig. 5B). In the BH3-resistant cell line SK-NAS, neither Mcl-1 silencing nor BAG3 silencing had any effect on the relative viability (Fig. 5A).
FIGURE 5.
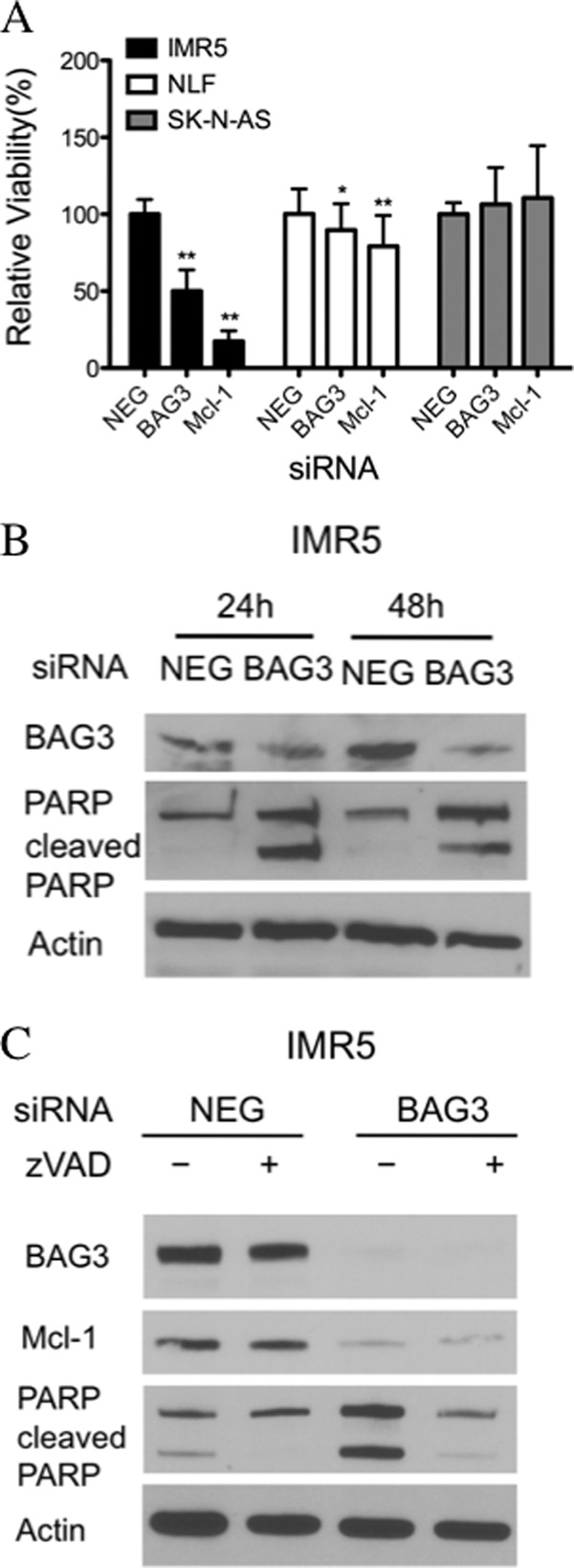
Silencing BAG3 increases apoptosis in Mcl-1-dependent cells. A, neuroblastoma cells were transfected with BAG3 siRNA (BAG3), Mcl-1 siRNA (Mcl-1), or nontargeting siRNA (NEG). Relative viability was determined using WST-1 72 h after transfection. Data are expressed relative to the corresponding negative control as mean ± S.D. (n ≥ 8). Statistically significant values relative to the negative control are marked with asterisks (**, p value < 0.01, *, p value < 0.05). B, immunoblot of IMR5 cells showing the levels of PARP and cleaved PARP 24–48 h after transfection with BAG3 siRNA (BAG3) or nontargeting siRNA (NEG). C, IMR5 cells were pretreated with 10 μm Z-VAD-FMK (zVAD) or DMSO 1 h before transfection with nontargeting siRNA (NEG) or BAG3 siRNA (BAG3), and the pan-caspase inhibitor was maintained in the medium until cell collection (48 h). Protein levels were determined by immunoblot. Actin was included as loading control.
As mentioned in the Introduction, Mcl-1 can be cleaved by caspases after induction of apoptosis. Considering that BAG3 silencing led to caspase activation in IMR5 cells, as verified by PARP cleavage, we evaluated the role of caspases in Mcl-1 degradation. To do this, IMR5 cells were preincubated with the pan-caspase inhibitor Z-VAD-FMK and treated with BAG3 siRNA or negative control siRNA during 48 h in the presence of the caspase inhibitor. Although Z-VAD-FMK prevented PARP cleavage after BAG3 silencing, the reduction in Mcl-1 level remained unchanged (Fig. 5C).
BAG3 Silencing Overcomes ABT-737 Resistance in Mcl-1-dependent Cell Lines
Because Mcl-1 expression causes ABT-737 resistance in cancer cells, we assessed the effect of BAG3 in the cell response to this Bcl-2 antagonist. As expected, the Bcl-2-dependent cell line SMS-SAN was sensitive to ABT-737, which exhibited an IC50 of 16 nm. However, both Mcl-1-dependent cell lines (IMR5, NLF) and the BH3-resistant cell line (SK-NAS) were refractory to ABT-737 at a concentration 3 orders of magnitude higher (Fig. 6A). Others have shown that Mcl-1 neutralization, either by Noxa overexpression or by siRNA down-regulation, sensitizes cancer cells to ABT-737 (59, 60). We found that BAG3 knockdown led to a 10-fold increase in ABT-737 sensitivity in Mcl-1-dependent cells, very close to the increase observed with direct Mcl-1 knockdown (Fig. 6, B and C). On the other hand, SK-NAS cells, which are resistant to ABT-737 and other stressors by a mechanism downstream of Mcl-1 activity, were not sensitized to ABT-737 by BAG3 silencing (Fig. 6D). Although an increase in ABT-737 toxicity was observed at 10 μm for both knockdowns, this is not a clinically meaningful activity, and no further increase was observed at higher concentration (up to 40 μm, data not shown). ABT-737 is an effective Bcl-2 antagonist at nanomolar concentrations, but other mechanisms may be responsible for its cytotoxicity at micromolar concentrations. Altogether, these results suggest that BAG3 can play a role in ABT-737 resistance through modulation of Mcl-1.
FIGURE 6.

Silencing BAG3 increases ABT-737 sensitivity in Mcl-1-dependent cell lines. A, representative concentration-dependent response curves for ABT-737 in neuroblastoma cell lines. B–D, cells were transfected with nontargeting siRNA (NEG), BAG3 siRNA (BAG3), or Mcl-1 siRNA (Mcl-1). After 24 h, ABT-737 was added, and cells were treated for an additional 48 h. Relative viability was determined by WST-1. Data are expressed relative to the corresponding DMSO-treated cells as mean ± S.E. (n = 8).
BAG3 Regulates Mcl-1 Expression and ABT-737 Response in Diverse Cancer Cell Types
The correlation between BAG3 expression and ABT-737 resistance was not restricted to neuroblastoma cell lines. We observed BAG3 and Mcl-1 co-expression in a panel of diverse cancer cell lines resistant to ABT-737 (Fig. 7A). This panel includes breast cancer (MB231), ovarian cancer (OVCAR3), lung cancer (H292), prostate cancer (PC3, LNCap), colon cancer (HCT116), head and neck carcinoma (1483), and renal cancer (CAKI-1). Although BAG3 and Mcl-1 were both expressed in these cell lines, their relative levels varied, underscoring the existence of multiple mechanisms controlling Mcl-1 levels. siRNA silencing of BAG3 led to Mcl-1 down-regulation (Fig. 7B) and an increased sensitivity to ABT-737 (Fig. 7C and supplemental Fig. 5) in all tested cell lines. The most dramatic effect was observed for the androgen receptor-positive prostate cancer cell line, LNCap, which exhibited an increase in ABT-737 sensitivity of nearly 3 orders of magnitude (Fig. 7C).
FIGURE 7.

BAG3 regulates Mcl-1 levels and is related to ABT-737 resistance in cancer cells. A, protein immunoblot showing the levels of BAG3 and Mcl-1 in cancer cell lines that are resistant to ABT-737. Actin was included as a loading control. B, selected cancer cell lines were transfected with nontargeting siRNA (NEG) or BAG3 siRNA (BAG3), and 48 h later, the level of Mcl-1 was determined by immunoblot. Actin was included as a loading control. C, cells were transfected with nontargeting siRNA (NEG) or BAG3 siRNA (BAG3). After 24 h, ABT-737 was added, and cells were treated for an additional 48 h. Relative viability was determined by WST-1. Data are expressed relative to the corresponding DMSO-treated cells as mean ± S.E. (n = 8).
DISCUSSION
Previous work from our group showed that BAG3 knockdown reduced the level of the antiapoptotic Bcl-2 proteins, Bcl-xL and Mcl-1, in a colon cancer cell line by an unknown mechanism (31). In the present work, we found that BAG3 modulates Mcl-1 expression by preventing its proteasomal degradation. In neuroblastoma cells, BAG3 interacted with Mcl-1 (Fig. 2), and silencing BAG3 increased Mcl-1 ubiquitination (Fig. 4C) and reduced Mcl-1 protein levels (Fig. 3A). A possible mechanism to explain BAG3 activity is shown in Fig. 8. BAG3 interaction with Mcl-1 and Hsp70 may prevent or destabilize the binding of Mcl-1 to Hsp70, reducing the delivery of Mcl-1 to the proteasome. Previous work showed that BAG3 may function as a negative regulator of Hsp70-driven proteasomal degradation by binding to the chaperone and leading to the release of the client protein (36). The results obtained in HEK293T with the BAG3 R480A mutant suggested that BAG3 interaction with Hsp70 is required for Mcl-1 stabilization (Fig. 4D). Although it is possible that BAG3 is acting as a general inhibitor of proteasomal degradation, some evidence suggests otherwise. For example, the BH3-only protein Noxa is a labile protein with a half-life of 1–2 h (61). Short term treatment of IMR5 cells with the proteasome inhibitor, MG132, increased Noxa levels (Fig. 4A), confirming that is being actively degraded by the proteasome. However, siRNA silencing of BAG3 had no effect on Noxa protein levels (Fig. 4A), implying that BAG3 inhibition of proteasome degradation may be selective for some Hsp70 clients, such as Mcl-1.
FIGURE 8.

BAG3 supports Mcl-1 antiapoptotic function in cancer cells. Mcl-1 is a short-lived protein that is actively degraded by the proteasome after binding to Hsp70. In Mcl-1-dependent cells, BAG3 expression prevents Mcl-1 degradation in the proteasome and supports Mcl-1 antiapoptotic activity. Neutralization of BAG3 (dashed arrows) increases Mcl-1 degradation by the proteasome, rendering the cell sensitive to the Bcl-2 antagonist ABT-737.
Several lines of evidence suggest that BAG3 has a role in the regulation of apoptosis. In fact, BAG3 was initially described as a Bcl-2-interacting protein and was named Bis (Bcl-2-interacting death suppressor) due to its synergism with Bcl-2 in preventing cell death (53). Until now, however, a direct connection between the BAG3 antiapoptotic activity and Bcl-2 family proteins has not been established. Using neuroblastoma cells with a defined BH3 profile, we were able to unambiguously link Mcl-1 stabilization with the BAG3 antiapoptotic activity in Mcl-1-dependent cells. This finding has important implications in cancer, where Mcl-1 overexpression is commonly observed in association with drug resistance (62, 63). Further, BAG3 regulation of apoptosis through Mcl-1 can be relevant in other settings than cancer. BAG3 is abundantly expressed in normal cardiac and skeletal muscle tissue, where Mcl-1 is expressed, and BAG3 knock-out mice have been shown to develop a fulminant myopathy with apoptotic features (64, 65). Further, a severe form of myofibrillar myopathy is observed in patients with a BAG3 P209L mutation (58), and we showed that this mutation impaired the ability of BAG3 to stabilize Mcl-1 in HEK293T cells (Fig. 4D). These findings suggest that BAG3 may play a role in regulation of Mcl-1 levels and apoptosis in cardiac tissue.
Unlike others members of the BAG family, BAG3 is induced by stress and growth factors and is part of the stress response regulated by the transcription factor HSF1 (42). HSF1 orchestrates a protective response that increases cell survival under many adverse conditions. Cells in a tumor are exposed to a rapidly changing and harsh microenvironment, where adaptive responses are required for survival. In addition, their cellular metabolism is aberrant because of the high degree of genomic instability, which places significant stress on transformed cells (66). In fact, HSF1 has been shown to play a role as a facilitator of transformation, and silencing HSF1 in diverse cancer cell lines (including MDA-MB231 and PC3) reduces cell growth and survival (67). Furthermore, genetic deletion of HSF1 in mice drastically reduces their sensitivity to skin tumorigenesis in the two-stage, initiation-promotion model (67). In our panel of cancer cells, BAG3 siRNA increased ABT-737-induced apoptosis in MDA-MB231 and PC3 cell lines (Fig. 7C). Furthermore, BAG3 silencing induced apoptosis in the prostate cancer cell line LNCap (supplemental Fig. 6). Prostate cancer cells are known to overexpress HSF1, where it promotes drug resistance and metastasis (68). We have shown previously that BAG3 mediates the antiapoptotic effect of HSF1 in 4-hydroxynonenal-induced apoptosis of colon cancer cells (31), and recent studies showed that BAG3 is part of the genetic program regulated by HSF1 in cancer cells (69). The results presented here suggest that BAG3 plays an important role in the protective response regulated by HSF1 in several different cancer cell lines, providing a link between two hallmarks of cancer, the ability to deal with proteotoxic stress and resistance to apoptosis, through its interaction with antiapoptotic Bcl-2 family proteins, especially Mcl-1.
The stresses that induce adaptive responses in cancer cells can also lead to the release of death signaling molecules such as BH3-only proteins (Fig. 8). Thus, a common trait observed in cancer cells is the increase of antiapoptotic Bcl-2 proteins to counteract BH3-only proteins and avoid apoptosis. This is the rationale for the development of Bcl-2 antagonists, such as ABT-737, that neutralize antiapoptotic Bcl-2 proteins, allowing apoptosis to follow. However, the inability of ABT-737 to bind Mcl-1 leads to resistance in Mcl-1-driven cancers. Here, we report a clear association between BAG3 expression and ABT-737 resistance in certain cancer cell lines demonstrating an Mcl-1 survival dependence, related to the ability of BAG3 to maintain Mcl-1 levels. Using Mcl-1-dependent cancer cells, we found that BAG3 neutralization (siRNA) overcomes ABT-737 resistance (Fig. 8). Furthermore, the same effect was observed in a panel of diverse cancer cell lines resistant to ABT-737, although cells selected for profound therapy resistance in situ (that is, those progressing after failed therapy) have additional apoptotic defects downstream of Mcl-1 activity. Taken together, our results reveal a new mechanism to sustain Mcl-1 expression and afford ABT-737 resistance in cancer. The results obtained with BAG3 down-modulation identify BAG3-mediated Mcl-1 stabilization as a potential target for cancer drug discovery.
Acknowledgment
We thank Stephen Fesik for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants ES013125 (to L. J. M.) and CA97323 (to M. D. H.) from the NIEHS and an Innovator Award from Alex's Lemonade Stand Foundation (to M. D. H.).

This article contains supplemental Table S1 and Figs. 1–6.
- BH
- Bcl-2 homology
- BAG
- Bcl-2-associated athanogene
- HSF1
- heat shock factor 1
- DMSO
- dimethyl sulfoxide
- PARP
- poly(ADP-ribose) polymerase
- Z
- benzyloxycarbonyl
- FMK
- fluoromethyl ketone.
REFERENCES
- 1. Green D. R., Evan G. I. (2002) A matter of life and death. Cancer Cell 1, 19–30 [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D., Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 [DOI] [PubMed] [Google Scholar]
- 3. Reed J. C., Cuddy M., Slabiak T., Croce C. M., Nowell P. C. (1988) Oncogenic potential of bcl-2 demonstrated by gene transfer. Nature 336, 259–261 [DOI] [PubMed] [Google Scholar]
- 4. Yip K. W., Reed J. C. (2008) Bcl-2 family proteins and cancer. Oncogene 27, 6398–6406 [DOI] [PubMed] [Google Scholar]
- 5. Yang J., Liu X., Bhalla K., Kim C. N., Ibrado A. M., Cai J., Peng T. I., Jones D. P., Wang X. (1997) Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129–1132 [DOI] [PubMed] [Google Scholar]
- 6. Lomonosova E., Chinnadurai G. (2008) BH3-only proteins in apoptosis and beyond: an overview. Oncogene 27, Suppl. 1, S2–S19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Willis S. N., Fletcher J. I., Kaufmann T., van Delft M. F., Chen L., Czabotar P. E., Ierino H., Lee E. F., Fairlie W. D., Bouillet P., Strasser A., Kluck R. M., Adams J. M., Huang D. C. (2007) Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315, 856–859 [DOI] [PubMed] [Google Scholar]
- 8. Letai A., Bassik M. C., Walensky L. D., Sorcinelli M. D., Weiler S., Korsmeyer S. J. (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192 [DOI] [PubMed] [Google Scholar]
- 9. Reed J. C., Stein C., Subasinghe C., Haldar S., Croce C. M., Yum S., Cohen J. (1990) Antisense-mediated inhibition of BCL2 protooncogene expression and leukemic cell growth and survival: comparisons of phosphodiester and phosphorothioate oligodeoxynucleotides. Cancer Res. 50, 6565–6570 [PubMed] [Google Scholar]
- 10. Oltersdorf T., Elmore S. W., Shoemaker A. R., Armstrong R. C., Augeri D. J., Belli B. A., Bruncko M., Deckwerth T. L., Dinges J., Hajduk P. J., Joseph M. K., Kitada S., Korsmeyer S. J., Kunzer A. R., Letai A., Li C., Mitten M. J., Nettesheim D. G., Ng S., Nimmer P. M., O'Connor J. M., Oleksijew A., Petros A. M., Reed J. C., Shen W., Tahir S. K., Thompson C. B., Tomaselli K. J., Wang B., Wendt M. D., Zhang H., Fesik S. W., Rosenberg S. H. (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 [DOI] [PubMed] [Google Scholar]
- 11. Konopleva M., Contractor R., Tsao T., Samudio I., Ruvolo P. P., Kitada S., Deng X., Zhai D., Shi Y. X., Sneed T., Verhaegen M., Soengas M., Ruvolo V. R., McQueen T., Schober W. D., Watt J. C., Jiffar T., Ling X., Marini F. C., Harris D., Dietrich M., Estrov Z., McCubrey J., May W. S., Reed J. C., Andreeff M. (2006) Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 10, 375–388 [DOI] [PubMed] [Google Scholar]
- 12. Tahir S. K., Yang X., Anderson M. G., Morgan-Lappe S. E., Sarthy A. V., Chen J., Warner R. B., Ng S. C., Fesik S. W., Elmore S. W., Rosenberg S. H., Tse C. (2007) Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 67, 1176–1183 [DOI] [PubMed] [Google Scholar]
- 13. Aichberger K. J., Mayerhofer M., Krauth M. T., Skvara H., Florian S., Sonneck K., Akgul C., Derdak S., Pickl W. F., Wacheck V., Selzer E., Monia B. P., Moriggl R., Valent P., Sillaber C. (2005) Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood 105, 3303–3311 [DOI] [PubMed] [Google Scholar]
- 14. Derenne S., Monia B., Dean N. M., Taylor J. K., Rapp M. J., Harousseau J. L., Bataille R., Amiot M. (2002) Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-xL is an essential survival protein of human myeloma cells. Blood 100, 194–199 [DOI] [PubMed] [Google Scholar]
- 15. Cho-Vega J. H., Rassidakis G. Z., Admirand J. H., Oyarzo M., Ramalingam P., Paraguya A., McDonnell T. J., Amin H. M., Medeiros L. J. (2004) MCL-1 expression in B-cell non-Hodgkin's lymphomas. Hum. Pathol. 35, 1095–1100 [DOI] [PubMed] [Google Scholar]
- 16. Sieghart W., Losert D., Strommer S., Cejka D., Schmid K., Rasoul-Rockenschaub S., Bodingbauer M., Crevenna R., Monia B. P., Peck-Radosavljevic M., Wacheck V. (2006) Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J. Hepatol. 44, 151–157 [DOI] [PubMed] [Google Scholar]
- 17. Akgul C. (2009) Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol. Life Sci. 66, 1326–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang T., Kozopas K. M., Craig R. W. (1995) The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J. Cell Biol. 128, 1173–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 20. Ding Q., Huo L., Yang J. Y., Xia W., Wei Y., Liao Y., Chang C. J., Yang Y., Lai C. C., Lee D. F., Yen C. J., Chen Y. J., Hsu J. M., Kuo H. P., Lin C. Y., Tsai F. J., Li L. Y., Tsai C. H., Hung M. C. (2008) Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res. 68, 6109–6117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Domina A. M., Vrana J. A., Gregory M. A., Hann S. R., Craig R. W. (2004) MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 23, 5301–5315 [DOI] [PubMed] [Google Scholar]
- 22. Maurer U., Charvet C., Wagman A. S., Dejardin E., Green D. R. (2006) Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 21, 749–760 [DOI] [PubMed] [Google Scholar]
- 23. Inoshita S., Takeda K., Hatai T., Terada Y., Sano M., Hata J., Umezawa A., Ichijo H. (2002) Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J. Biol. Chem. 277, 43730–43734 [DOI] [PubMed] [Google Scholar]
- 24. Weng C., Li Y., Xu D., Shi Y., Tang H. (2005) Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J. Biol. Chem. 280, 10491–10500 [DOI] [PubMed] [Google Scholar]
- 25. Zhong Q., Gao W., Du F., Wang X. (2005) Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121, 1085–1095 [DOI] [PubMed] [Google Scholar]
- 26. Stewart D. P., Koss B., Bathina M., Perciavalle R. M., Bisanz K., Opferman J. T. (2010) Ubiquitin-independent degradation of antiapoptotic MCL-1. Mol. Cell Biol. 30, 3099–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Craig R. W. (2002) MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia 16, 444–454 [DOI] [PubMed] [Google Scholar]
- 28. Clohessy J. G., Zhuang J., Brady H. J. (2004) Characterisation of Mcl-1 cleavage during apoptosis of haematopoietic cells. Br. J. Haematol. 125, 655–665 [DOI] [PubMed] [Google Scholar]
- 29. Nijhawan D., Fang M., Traer E., Zhong Q., Gao W., Du F., Wang X. (2003) Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 17, 1475–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schwickart M., Huang X., Lill J. R., Liu J., Ferrando R., French D. M., Maecker H., O'Rourke K., Bazan F., Eastham-Anderson J., Yue P., Dornan D., Huang D. C., Dixit V. M. (2010) Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463, 103–107 [DOI] [PubMed] [Google Scholar]
- 31. Jacobs A. T., Marnett L. J. (2009) HSF1-mediated BAG3 expression attenuates apoptosis in 4-hydroxynonenal-treated colon cancer cells via stabilization of anti-apoptotic Bcl-2 proteins. J. Biol. Chem. 284, 9176–9183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takayama S., Sato T., Krajewski S., Kochel K., Irie S., Millan J. A., Reed J. C. (1995) Cloning and functional analysis of BAG-1: a novel Bcl-2-binding protein with anti-cell death activity. Cell 80, 279–284 [DOI] [PubMed] [Google Scholar]
- 33. Takayama S., Bimston D. N., Matsuzawa S., Freeman B. C., Aime-Sempe C., Xie Z., Morimoto R. I., Reed J. C. (1997) BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J. 16, 4887–4896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Doong H., Vrailas A., Kohn E. C. (2002) What's in the 'BAG'?–A functional domain analysis of the BAG-family proteins. Cancer Lett. 188, 25–32 [DOI] [PubMed] [Google Scholar]
- 35. Lüders J., Demand J., Höhfeld J. (2000) The ubiquitin-related BAG-1 provides a link between the molecular chaperones Hsc70/Hsp70 and the proteasome. J. Biol. Chem. 275, 4613–4617 [DOI] [PubMed] [Google Scholar]
- 36. Doong H., Rizzo K., Fang S., Kulpa V., Weissman A. M., Kohn E. C. (2003) CAIR-1/BAG-3 abrogates heat shock protein-70 chaperone complex-mediated protein degradation: accumulation of poly-ubiquitinated Hsp90 client proteins. J. Biol. Chem. 278, 28490–28500 [DOI] [PubMed] [Google Scholar]
- 37. Gamerdinger M., Hajieva P., Kaya A. M., Wolfrum U., Hartl F. U., Behl C. (2009) Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 28, 889–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gamerdinger M., Kaya A. M., Wolfrum U., Clement A. M., Behl C. (2011) BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 12, 149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen H. Y., Liu P., Sun M., Wu L. Y., Zhu H. Y., Qiao C., Dong H. J., Zhu D. X., Xu W., Li J. Y. (2010) [Bag3 gene expression in chronic lymphocytic leukemia and its association with patients' prognosis]. Zhongguo Shi Yan Xue Ye Xue Za Zhi 18, 838–842 [PubMed] [Google Scholar]
- 40. Chiappetta G., Ammirante M., Basile A., Rosati A., Festa M., Monaco M., Vuttariello E., Pasquinelli R., Arra C., Zerilli M., Todaro M., Stassi G., Pezzullo L., Gentilella A., Tosco A., Pascale M., Marzullo L., Belisario M. A., Turco M. C., Leone A. (2007) The antiapoptotic protein BAG3 is expressed in thyroid carcinomas and modulates apoptosis mediated by tumor necrosis factor-related apoptosis-inducing ligand. J. Clin. Endocrinol. Metab. 92, 1159–1163 [DOI] [PubMed] [Google Scholar]
- 41. Liao Q., Ozawa F., Friess H., Zimmermann A., Takayama S., Reed J. C., Kleeff J., Büchler M. W. (2001) The anti-apoptotic protein BAG-3 is overexpressed in pancreatic cancer and induced by heat stress in pancreatic cancer cell lines. FEBS Lett. 503, 151–157 [DOI] [PubMed] [Google Scholar]
- 42. Rosati A., Ammirante M., Gentilella A., Basile A., Festa M., Pascale M., Marzullo L., Belisario M. A., Tosco A., Franceschelli S., Moltedo O., Pagliuca G., Lerose R., Turco M. C. (2007) Apoptosis inhibition in cancer cells: a novel molecular pathway that involves BAG3 protein. Int. J. Biochem. Cell Biol. 39, 1337–1342 [DOI] [PubMed] [Google Scholar]
- 43. Schwab M., Westermann F., Hero B., Berthold F. (2003) Neuroblastoma: biology and molecular and chromosomal pathology. Lancet Oncol. 4, 472–480 [DOI] [PubMed] [Google Scholar]
- 44. Landis S. H., Murray T., Bolden S., Wingo P. A. (1999) Cancer statistics, 1999. CA Cancer J. Clin. 49, 8–31 [DOI] [PubMed] [Google Scholar]
- 45. Goldsmith K. C., Hogarty M. D. (2005) Targeting programmed cell death pathways with experimental therapeutics: opportunities in high-risk neuroblastoma. Cancer Lett. 228, 133–141 [DOI] [PubMed] [Google Scholar]
- 46. Lestini B. J., Goldsmith K. C., Fluchel M. N., Liu X., Chen N. L., Goyal B., Pawel B. R., Hogarty M. D. (2009) Mcl1 downregulation sensitizes neuroblastoma to cytotoxic chemotherapy and small molecule Bcl2-family antagonists. Cancer Biol. Ther. 8, 1587–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Janoueix-Lerosey I., Schleiermacher G., Michels E., Mosseri V., Ribeiro A., Lequin D., Vermeulen J., Couturier J., Peuchmaur M., Valent A., Plantaz D., Rubie H., Valteau-Couanet D., Thomas C., Combaret V., Rousseau R., Eggert A., Michon J., Speleman F., Delattre O. (2009) Overall genomic pattern is a predictor of outcome in neuroblastoma. J. Clin. Oncol. 27, 1026–1033 [DOI] [PubMed] [Google Scholar]
- 48. Mosse Y. P., Greshock J., Margolin A., Naylor T., Cole K., Khazi D., Hii G., Winter C., Shahzad S., Asziz M. U., Biegel J. A., Weber B. L., Maris J. M. (2005) High-resolution detection and mapping of genomic DNA alterations in neuroblastoma. Genes Chromosomes Cancer 43, 390–403 [DOI] [PubMed] [Google Scholar]
- 49. Hackett C. S., Hodgson J. G., Law M. E., Fridlyand J., Osoegawa K., de Jong P. J., Nowak N. J., Pinkel D., Albertson D. G., Jain A., Jenkins R., Gray J. W., Weiss W. A. (2003) Genome-wide array CGH analysis of murine neuroblastoma reveals distinct genomic aberrations which parallel those in human tumors. Cancer Res. 63, 5266–5273 [PubMed] [Google Scholar]
- 50. Goldsmith K. C., Lestini B. J., Gross M., Ip L., Bhumbla A., Zhang X., Zhao H., Liu X., Hogarty M. D. (2010) BH3 response profiles from neuroblastoma mitochondria predict activity of small molecule Bcl-2 family antagonists. Cell Death Differ. 17, 872–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goldsmith K. C., Gross M., Peirce S., Luyindula D., Liu X., Vu A., Sliozberg M., Guo R., Zhao H., Reynolds C. P., Hogarty M. D. (2012) Mitochondrial Bcl-2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res. 72, 2565–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Certo M., Del Gaizo Moore V., Nishino M., Wei G., Korsmeyer S., Armstrong S. A., Letai A. (2006) Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365 [DOI] [PubMed] [Google Scholar]
- 53. Lee J. H., Takahashi T., Yasuhara N., Inazawa J., Kamada S., Tsujimoto Y. (1999) Bis, a Bcl-2-binding protein that synergizes with Bcl-2 in preventing cell death. Oncogene 18, 6183–6190 [DOI] [PubMed] [Google Scholar]
- 54. Akgul C., Moulding D. A., White M. R., Edwards S. W. (2000) In vivo localisation and stability of human Mcl-1 using green fluorescent protein (GFP) fusion proteins. FEBS Lett. 478, 72–76 [DOI] [PubMed] [Google Scholar]
- 55. Iwasaki M., Homma S., Hishiya A., Dolezal S. J., Reed J. C., Takayama S. (2007) BAG3 regulates motility and adhesion of epithelial cancer cells. Cancer Res. 67, 10252–10259 [DOI] [PubMed] [Google Scholar]
- 56. Takayama S., Xie Z., Reed J. C. (1999) An evolutionarily conserved family of Hsp70/Hsc70 molecular chaperone regulators. J. Biol. Chem. 274, 781–786 [DOI] [PubMed] [Google Scholar]
- 57. Gentilella A., Khalili K. (2011) BAG3 expression in glioblastoma cells promotes accumulation of ubiquitinated clients in an Hsp70-dependent manner. J. Biol. Chem. 286, 9205–9215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Selcen D., Muntoni F., Burton B. K., Pegoraro E., Sewry C., Bite A. V., Engel A. G. (2009) Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann. Neurol. 65, 83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen S., Dai Y., Harada H., Dent P., Grant S. (2007) Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 67, 782–791 [DOI] [PubMed] [Google Scholar]
- 60. van Delft M. F., Wei A. H., Mason K. D., Vandenberg C. J., Chen L., Czabotar P. E., Willis S. N., Scott C. L., Day C. L., Cory S., Adams J. M., Roberts A. W., Huang D. C. (2006) The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10, 389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Baou M., Kohlhaas S. L., Butterworth M., Vogler M., Dinsdale D., Walewska R., Majid A., Eldering E., Dyer M. J., Cohen G. M. (2010) Role of NOXA and its ubiquitination in proteasome inhibitor-induced apoptosis in chronic lymphocytic leukemia cells. Haematologica 95, 1510–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Quinn B. A., Dash R., Azab B., Sarkar S., Das S. K., Kumar S., Oyesanya R. A., Dasgupta S., Dent P., Grant S., Rahmani M., Curiel D. T., Dmitriev I., Hedvat M., Wei J., Wu B., Stebbins J. L., Reed J. C., Pellecchia M., Sarkar D., Fisher P. B. (2011) Targeting Mcl-1 for the therapy of cancer. Expert Opin. Investig. Drugs 20, 1397–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Beroukhim R., Mermel C. H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J. S., Dobson J., Urashima M., Mc Henry K. T., Pinchback R. M., Ligon A. H., Cho Y. J., Haery L., Greulich H., Reich M., Winckler W., Lawrence M. S., Weir B. A., Tanaka K. E., Chiang D. Y., Bass A. J., Loo A., Hoffman C., Prensner J., Liefeld T., Gao Q., Yecies D., Signoretti S., Maher E., Kaye F. J., Sasaki H., Tepper J. E., Fletcher J. A., Tabernero J., Baselga J., Tsao M. S., Demichelis F., Rubin M. A., Janne P. A., Daly M. J., Nucera C., Levine R. L., Ebert B. L., Gabriel S., Rustgi A. K., Antonescu C. R., Ladanyi M., Letai A., Garraway L. A., Loda M., Beer D. G., True L. D., Okamoto A., Pomeroy S. L., Singer S., Golub T. R., Lander E. S., Getz G., Sellers W. R., Meyerson M. (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Homma S., Iwasaki M., Shelton G. D., Engvall E., Reed J. C., Takayama S. (2006) BAG3 deficiency results in fulminant myopathy and early lethality. Am. J. Pathol. 169, 761–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Krajewski S., Bodrug S., Krajewska M., Shabaik A., Gascoyne R., Berean K., Reed J. C. (1995) Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. Am. J. Pathol. 146, 1309–1319 [PMC free article] [PubMed] [Google Scholar]
- 66. Luo J., Solimini N. L., Elledge S. J. (2009) Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dai C., Whitesell L., Rogers A. B., Lindquist S. (2007) Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130, 1005–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hoang A. T., Huang J., Rudra-Ganguly N., Zheng J., Powell W. C., Rabindran S. K., Wu C., Roy-Burman P. (2000) A novel association between the human heat shock transcription factor 1 (HSF1) and prostate adenocarcinoma. Am. J. Pathol. 156, 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mendillo M. L., Santagata S., Koeva M., Bell G. W., Hu R., Tamimi R. M., Fraenkel E., Ince T. A., Whitesell L., Lindquist S. (2012) HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150, 549–562 [DOI] [PMC free article] [PubMed] [Google Scholar]