

Background: The POK proteins play roles in the regulation of the cell cycle, oncogenesis, and tumor suppression. A novel POK protein, BOZF1, is overexpressed in cancer.
Results: BOZF1 represses transcription of CDKN1A by inhibition of p53 acetylation and Sp1 binding.
Conclusion: BOZF1 stimulates cell proliferation by repressing p21 expression.
Significance: BOZF1 is a negative regulator of tumor suppressors p53 and p21.
Keywords: Cell Cycle, Cell Proliferation, p300, p53, Sp1, BOZF1, CDKN1A, ZBTB8A, p21WAF, p53 Acetylation
Abstract
The human POZ domain and Krüppel-like zinc finger (POK) family proteins play important roles in the regulation of apoptosis, cell proliferation, differentiation, development, oncogenesis, and tumor suppression. A novel POK family transcription factor, BTB/POZ and zinc finger domains factor on chromosome 1 (BOZF-1; also called ZBTB8A), contains a POZ domain and two C2H2-type Krüppel-like zinc fingers and is localized at nuclear speckles. Compared with paired normal tissues, BOZF1 expression is increased in cancer tissues of the prostate, breast, and cervix. BOZF1 repressed the transcription of p21WAF/CDKN1A by acting on the proximal promoter concentrated with Sp1-binding GC boxes. BOZF1 competed with Sp1 in binding to GC boxes 1–5/6 of the CDKN1A proximal promoter. In addition, BOZF1 interacted with p53 and decreased the acetylation of p53 by p300, which reduced the DNA binding activity of p53 at the far distal p53-binding element. BOZF1 blocked the two major molecular events that are important in both constitutive and inducible transcription activation of CDKN1A. BOZF1 is unique in that it bound to all the proximal GC boxes to repress transcription, and it inhibited p53 acetylation without affecting p53 stability. BOZF1 might be a novel proto-oncoprotein that stimulates cell proliferation.
Introduction
The POZ2 domain and Krüppel-like zinc finger (POK) proteins play critical roles in apoptosis, cell differentiation, cell cycle regulation, development, tumor suppression, and oncogenesis (1, 2). The POK transcription factors that have been relatively well characterized include BCL6 (3, 4), promyelocytic leukemia zinc finger (5, 6), leukemia/lymphoma-related factor/FBI-1 (7–9), hypermethylated in cancer 1 (10, 11), and Myc-interacting zinc finger 1 (MIZ-1) (12–14). Some of the novel POK proteins have been reported as transcription regulators of the genes that control the cell cycle. BCL6 interacts with MIZ-1 to repress CDKN1A transcription and increases cell cycle progression in germinal center B cells (14). Promyelocytic leukemia zinc finger is a negative regulator of cell cycle progression and suppresses cell growth through the expression of cyclin A (15). The proto-oncogene FBI-1 (also called Pokemon; ZBTB7A) is a specific transcription repressor of human CDKN2A, which encodes tumor suppressor alternate reading frame (ARF) and CDKN1A (9, 16). ZBTB4 suppresses cell cycle arrest through the regulation of p21 expression (17).
Under DNA damaging conditions, progression of the cell cycle can be controlled by the genes of the p53 pathway (ARF-Mdm2-TP53-CDKN1A) (18, 19). DNA damaging signals induce p53 expression, which serves as the “guardian of the genome” through the induction of cell cycle arrest and/or apoptosis depending on the DNA damage level. As a tumor suppressor, p53 protects the cell from DNA damage, abnormal cell proliferation, angiogenesis, and the loss of survival factors. The inactivation or loss of function of p53 could lead to oncogenesis (20, 21).
p53 controls the expression of a number of downstream target genes to determine cell fate, and depending on the posttranslational modification of p53, a particular subset of genes is expressed to perform specific functions related to cell fate (22, 23). One of the important regulators of the cell cycle and a downstream target of p53 is CDKN1A, which encodes p21. The expression of p21 is induced by p53 (24–26). p21 is a potent cyclin-dependent kinase inhibitor that binds to and inhibits the activity of cyclin-CDK2 or -CDK4 complexes and thus functions as a negative regulator of cell cycle progression (26–29).
In addition to inducible p53, Sp1 transcription factor activates CDKN1A transcription through binding at the six Sp1-binding elements (GC boxes 1–6) and plays an important role in the basal expression of p21 (30, 31). The interaction between p53 bound at the distal p53-binding elements and Sp1 bound at GC box 3 synergistically activates CDKN1A transcription. Mutations in GC box 3 greatly affect not only the basal transcription of the CDKN1A promoter but also the synergistic transcription activation by p53 and Sp1, suggesting the importance of the element (32).
Stress signals activate various protein kinases (ATM and ATR) and acetyltransferases (p300, PCAF, and Tip60), which phosphorylate and acetylate p53, respectively (33–37). The posttranslational modification of p53 results in the stabilization and/or activation of p53 in the nucleus. Modified p53 binds to the DNA elements of target genes and regulates diverse cellular responses, such as apoptosis, cell cycle arrest, and DNA repair. For example, the acetylation of p53 at Lys-373 and Lys-382 increases the stability, site-specific DNA binding, and p53 target gene expression (e.g. CDKN1A) (38, 39). The PCAF-mediated acetylation of p53 at Lys-320 is induced through DNA damage and results in the inhibition of apoptosis (40). Deacetylases (SIRT1 and histone deacetylases) and ubiquitinases have also been shown to affect p53 stability (41, 42).
BOZF1 is an uncharacterized member of the POK family transcription factors. BOZF1 mRNA encodes polypeptides composed of 441 amino acids and containing a POZ domain at the N terminus and two Krüppel-like zinc fingers at the C terminus. The gene located at chromosome 1p35.1 is 67 kb in size and is composed of five exons and four introns. BOZF1 is highly expressed in cancer tissues of the prostate, breast, cervix, etc. We found that BOZF1 increased cell proliferation by repressing CDKN1A expression through a molecular mechanism that involved the inhibition of p53 and Sp1 activity.
EXPERIMENTAL PROCEDURES
Plasmids, Antibodies, and Reagents
To express BOZF1 protein with Myc and poly(His) tags, BOZF1 cDNA fragment was cloned into pcDNA3.1. The pGL2-CDKN1A-Luc plasmid series was reported elsewhere (43). pGL2-CDKN1A-Luc-mC/EBPα, -β, and -mRAR were prepared by site-directed mutagenesis. Oligonucleotide primers used in mutagenesis are described in the supplemental table. pGL2-CDKN1A-Luc-ΔGC-boxes was prepared by deleting GC boxes 1–4 of the proximal promoter. To prepare the recombinant GST-POZ-BOZF1 and GST-ZF-BOZF1 proteins, cDNA fragments encoding the POZ domain (amino acids 1–92) and zinc fingers (amino acids 282–333) were cloned into pGEX4T1. All plasmid constructs were verified by sequencing.
Antibodies against p21, p53, acetyl-p53 (Lys-320), acetyl-p53 (Lys-382), acetyl-lysine, Sp1, Myc tag, GAPDH, and His tag were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), Millipore (Billerica, MA), R&D Systems (Minneapolis, MN), Upstate Biotechnology (Lake Placid, NY), and Cell Signaling Technology (Beverly, MA). The rabbit polyclonal antibody against BOZF1 was prepared in our laboratory using recombinant POZ domain polypeptide (amino acids 1–92) of BOZF1 as antigen. Most of the chemical reagents were purchased from Sigma.
Cell Cultures and Transcription Analysis
Cells were cultured in the media recommended by ATCC (Manassas, VA). For the analysis of transcriptional regulation of various promoters by BOZF1, promoter-reporter fusion plasmids, pcDNA3.1-BOZF1, or the pcDNA3.1 vector in various combinations were transiently transfected into HEK293 and HCT116 p53+/+ cells using Lipofectamine Plus reagent (Invitrogen) and analyzed for luciferase activity as reported elsewhere (43).
Knockdown of BOZF1 mRNA by siRNA and Quantitative Real Time PCR Analysis of BOZF1 mRNA
Four siRNA against BOZF1 mRNA were synthesized in duplicate and purchased from Bioneer (Daejeon, Korea). Approximately 100 pmol of siRNA was transfected into HEK293 cells using Lipofectamine 2000 (Invitrogen). The siRNA sequences used were: siBOZF1-1, 5′-GCAAGGAACAGGUAUAAUA-3′; siBOZF1-2, 5′-CUUCAAAGCACAUCGAAAU-3′; siBOZF1-3, 5′-GAUCCUAUGUGGAGAUUGU-3′; siBOZF1-4, 5′-UCUAACAGGGCAAGUGGUA-3′.
Total RNA was isolated from the transfected cells using TRIzol reagent (Invitrogen). cDNAs were synthesized using 5 μg of total RNA, oligo(dT) primer, and Superscript reverse transcriptase II (200 units/μl) (Invitrogen). The RT-qPCR was performed using SYBR Green Master Mix (Applied Biosystems, Foster City, CA). The following oligonucleotide PCR primers were used for RT-qPCR: BOZF1: forward, 5′-GCGATTCAAGTGCCCGTACT-3′; reverse, 5′-CTTGACATGGATAGGGCCTTTC-3′; CDKN1A: forward, 5′-AGGGGACAGCAGAGGAAG-3′; reverse, 5′-GCGTTTGGAGTGGTAGAAATCTG-3′; GAPDH: forward, 5′-CCCCTTCATTGACCTCAACTAC-3′; reverse, 5′-TCTCGCTCCTGGAAGATGG-3′. To analyze BOZF1 mRNA expression in a C57BL/6N mouse, total RNA was isolated from various mouse tissues, and RT-PCR was carried out using the following oligonucleotide primer sets: murine BOZF1: forward, 5′-CCTCCTGCAGCAACTGAATGA-3′; reverse, 5′-CACGAGAATACTGCAGTCACAAAAC-3′; murine CDKN1A: forward, 5′-CATTCCCTGCCTGGTTCCTT-3′; reverse, 5′-CCTGTTCTAGGCTGTGACTGCTT-3′; murine GAPDH: forward, 5′-CCCCTTCATTGACCTCAACTAC-3′; reverse, 5′-TCTCGCTCCTGGAAGATGG-3′.
Western Blot Analysis and Co-immunoprecipitation Assays
Western blot analyses and co-immunoprecipitation were performed as reported elsewhere (16). The blotted PVDF membranes were incubated with antibodies against the His tag, GAPDH, p21, p53, Sp1, Myc tag, and BOZF1. The membranes were further incubated with anti-mouse or -rabbit secondary antibodies conjugated with HRP (Vector Laboratories, Burlingame, CA). The protein bands were visualized using ECL solution (PerkinElmer Life Sciences).
For co-immunoprecipitation assays, the HEK293 cell lysate was precleared, and the supernatant was incubated with antibodies at 4 °C overnight and further incubated with protein A/G-agarose beads for 4 h at room temperature. Beads were washed and resuspended in 5× SDS loading buffer. Immunoprecipitated proteins were separated by 10 or 12% SDS-PAGE and analyzed for interacting proteins by following a standard Western protocol.
Immunocytochemistry
HEK293 cells transfected with pcDNA3.1-BOZF1 were cultured in 6-well culture dishes overlaid with slide coverglasses. The cells were washed, fixed with cold methanol/formaldehyde (99:1), permeabilized with 0.2% Triton X-100, blocked with horse serum, and incubated with mouse anti-His primary antibody for 1 h at room temperature. After washing, the cells were further incubated with FITC-conjugated anti-mouse secondary antibody. Finally, the cells were soaked with solution containing 4,6-diamidino-2-phenylindole (1 mg/ml), washed, and mounted. The cells were examined with a Carl Zeiss LSM 510 confocal laser-scanning microscope (Carl Zeiss, Germany).
Quantitative Chromatin Immunoprecipitation (ChIP)-qPCR Assays
The molecular interaction between the transcription factors (BOZF1, p53, and Sp1) and the DNA elements of the CDKN1A promoter and histone modification around the proximal promoter in HEK293 cells were analyzed using a standard qChIP assay protocol as described elsewhere (44, 45).
The quantitative PCR analysis of chromatin immunoprecipitated DNA was performed using the following oligonucleotide primer sets designed to amplify the upstream regulatory regions flanking the p53 binding sites and the proximal promoter: p53RE-1 primers (bp 2307 to −1947): forward, 5′-CTGTGGCTCTGATTGGCTTT-3′; reverse, 5′-GGGTCTTTAGAGGTCTCCTGTCT-3′; p53RE-2 binding primers (bp −1462 to −1128): forward, 5′-CCACAGCAGAGGAGAAAGAAG-3′; reverse, 5′-GCTGCTCAGAGTCTGGAAATC-3′; proximal promoter (bp −133 to +100): forward, 5′-CGCTGGGCAGCCAGGAGCCT-3′; reverse, 5′-TCGTCACCCGCGCACTTAGA-3′.
FACS Analysis
HEK293 cells were transfected with the BOZF1 expression or control vector. Cells were fixed with 100% methanol and washed with PBS, and cellular DNA was stained with propidium iodide (50 μg/ml) in RNase A (100 μg/ml) solution (43). A cell cycle profile and forward scatter were determined using a BD FACSCalibur, and the data were analyzed using the CellQuest program (BD Biosciences).
Focus Formation Assay
NIH3T3 and HEK293 cells were transfected with pcDNA3.1 or pcDNA3.1-BOZF1 and cultured overnight. The cells were collected, plated, and cultured on 6-well plates. The cells stably overexpressing BOZF1 were selected by culturing the cells in a medium containing G418 (500 μg/ml) for 14 days. The cells were washed two times with ice-cold 1× PBS, fixed with ice-cold methanol, and stained with 0.5% crystal violet solution.
Cell Growth Curves and 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide Assays of Cell Proliferation
The growth of HEK293 cells was measured by directly counting the number of cells using a hemocytometer. Confluent HEK293 cells were transfected with the BOZF1 expression or control vector, cultured overnight, and plated at 2 × 103 cells/well in 24-well dishes. The cell numbers were counted by harvesting cells daily for 6 days.
Confluent HEK293 cells were grown on 10-cm culture dishes and transfected with the BOZF1 expression or control vector. The cells (5 × 103) were seeded in each well of a 96-well plate and cultured for 0–4 days. At 0, 2, and 4 days, the cells were incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (2 mg/ml) for 2–4 h at 37 °C. Cell proliferation was determined from the conversion of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide to formazan using a SpectraMAX 250 (Molecular Devices) at 570 nm.
BrdUrd Incorporation
Confluent HEK293 cells were transfected with pcDNA3.1 or pcDNA3.1-BOZF1, incubated for 24 h, and cultured in serum-free medium overnight. Cells were exposed to BrdUrd (20 μm) for 1 h, washed, and fixed. Cells were permeabilized, incubated for 2 h with an anti-BrdUrd monoclonal antibody, washed, and incubated for 1 h with an Alexa Fluor 488 goat anti-mouse IgG secondary antibody. Nuclei were stained with DAPI for 10 min. Three random images per sample were acquired using an Olympus BX51 fluorescence microscope (Olympus, Tokyo, Japan).
Oligonucleotide Pulldown Assays of p53, Sp1, and BOZF1 Binding
The transfected cells were lysed in HKMG buffer (10 mm HEPES, pH 7.9, 100 mm KCl, 5 mm MgCl2, 10% glycerol, 1 mm DTT, 0.5% Nonidet P-40, and protease inhibitors). The cellular extracts were incubated with 1 μg of biotinylated double-stranded oligonucleotides (p53RE-1, p53RE-2, and GC boxes 1, 2, 3, 4, and 5/6) for 12 h. The mixtures were incubated with streptavidin-agarose beads for 4 h, washed three times with HKMG buffer, and precipitated by centrifugation. The precipitated biotin-streptavidin oligonucleotide protein complexes were analyzed by Western blot assay using antibodies against p53, Sp1, and BOZF1. The sequences of the oligonucleotides were (only the top strands are shown): p53RE-1, 5′-GTCAGGAACATGTCCCAACATGTTGAGCTC-3′; p53RE-2, 5′-TAGAGGAAGAAGACTGGGCATGTCTGGGCA-3′; GC box 1, 5′-GATCGGG AGGGCGGTCCCG-3′; GC box 2, 5′-GATCTCCCGGGCGGCGCG-3′; GC box 3, 5′-GATCCGAGCGCGGGTCCCGCCTC-3′; GC box 4, 5′-GATCCTTGAGGCGGGCCCG-3′; GC box 5/6, 5′-GATCGGGCGGGGCGGTTGTATATCA-3′.
Immunohistochemistry of BOZF1 Expression in Cancer Tissues
A human prostate cancer tissue array (45 cases), uterine cervix cancer-metastasis-normal array (55 cases; CZA2) and breast cancer-metastasis-normal array (55 cases; CBA4) were purchased from Accumax (Array number A222III) (ISU AXIS, Seoul, Korea) and SUPER BIO CHIPS (Seoul, Korea). Cancer tissues with corresponding normal tissues were stained for the immunohistochemical detection of BOZF1 expression using the Vectastain Elite ABC (Vector Laboratories) and Peroxidase Substrate Kit (3,3′-diaminobenzidine) (Vector Laboratories) by standard protocol. The tissue slides were deparaffinized in xylene, rehydrated, and boiled for 20 min in sodium citrate solution (10 mm sodium citrate, pH 6.0) for antigen retrieval. The slides were washed, permeabilized with 0.2% Triton X-100 in PBS, and blocked using BSA (5%) and goat serum (1%) for 30 min at room temperature. The slides were incubated overnight at 4 °C with a primary antibody against BOZF1 (dilution, 1:10), washed, and subsequently incubated with a biotinylated universal secondary antibody for 1 h at room temperature. The cancer tissue slides were examined using an Olympus BX51 light microscope. The BOZF1-positive cells were counted and recorded by a pathologist. The immunohistochemical staining intensity was scored as follows: no staining, 0; weak, 1; moderate, 2; and strong, 3, respectively.
Electrophoretic Mobility Shift Assay (EMSA) of BOZF1
EMSAs were performed as described previously by incubating the probes with recombinant GST-ZF-BOZF1 protein and the probes (16). The sequences of the six 32P-labeled GC box probes on the CDKN1A proximal promoter and the two p53 response element probes on the distal promoter are reported elsewhere (16).
GST Fusion Protein Pulldown Assay of Protein Interaction between BOZF1 and p53 or p300
Recombinant GST and GST-POZ-BOZF1 and GST-ZF-BOZF1 fusion proteins were prepared from Escherichia coli BL21(DE3) by glutathione-agarose 4 bead affinity chromatography (Peptron, Taejeon, Korea). p53 polypeptides were prepared using an in vitro transcription and translation system (TnT system, Promega, Madison, WI) in the presence of [35S]methionine (PerkinElmer Life Sciences). GST fusion protein-agarose bead complexes were incubated with 35S-labeled p53 polypeptides or p300 polypeptides at 4 °C for 4 h in HEMG buffer (40 mm HEPES, 100 mm KCl, 0.2 mm EDTA, 5 mm MgCl2, 0.1% Nonidet P-40, 10% glycerol, 1.5 mm DTT, protease inhibitors). The reaction mixtures were centrifuged, pellets were rinsed, and the bound proteins were separated using 12% SDS-PAGE. The gels were exposed to x-ray film (Eastman Kodak Co.).
In Vitro Acetylation Assay of p53 by the p300 Protein
Recombinant p53 protein (6 μg) and acetyl coenzyme A (0.83 mm) were incubated with p300 (300 ng) (Active Motif, Carlsbad, CA) or p300 + BOZF1 (6 μg) in 1× HAT assay buffer (50 mm Tris-HCl, pH 8.0, 10% glycerol, 0.1 mm EDTA, and 1 mm dithiothreitol) (Upstate Biotechnology) at 37 °C for 1 h. Aliquots () of the reaction mixtures were resolved by SDS-PAGE and analyzed by Western blot using anti-lysine antibody to evaluate the acetylation of p53.
Ubiquitination Assay
H1299 cells were co-transfected with 3 μg of pcDNA3-p53 and 2 μg of pcDNA3-His-ubiquitin in the presence or absence of pcDNA3.1-BOZF1. The cells were cultured overnight and treated with 20 μm MG132 for 3 h before harvest. The cell lysates (500 μg) in radioimmune precipitation assay buffer was incubated with the MagneHisTM nickel particles (Promega), and the precipitated pellets were washed, resolved by 10% SDS-PAGE, and analyzed by Western blotting using anti-p53 antibody.
Statistical Analysis
Student's t test was used for the statistical analyses. p values of <0.05 were considered statistically significant.
RESULTS
BOZF1 Increases Proliferation of NIH3T3 and HEK293 Cells
We isolated and characterized a new member of the human POK protein family, BOZF1, which encodes a protein of 441 amino acid residues. BOZF1 has a POZ domain at its N terminus (amino acids 24–92) and two zinc finger domains at its C terminus (amino acids 282–333). Immunocytochemistry showed that BOZF1 is located in the nucleus of HEK293 cells, particularly in nuclear speckle-like structures (supplemental Fig. 1, A and B). Although Bozf1 mRNA is expressed in most mouse tissues, it is relatively abundantly expressed in the duodenum, pancreas, stomach, and testis and highly expressed in the white adipose tissue (supplemental Fig. 2A). BOZF1 mRNA was expressed in several human cell lines tested and was relatively high in HEK293 and HCT116 p53+/+ cells (supplemental Fig. 2B).
We examined whether BOZF1 could form larger colony foci by cellular transformation into rapidly growing cells. NIH3T3 and HEK293 cells transfected with the BOZF1 expression vector formed a substantial number of large foci, suggesting that BOZF1 caused cellular transformation into rapidly growing cells (Fig. 1A and supplemental Fig. 1D). Analysis of HEK293 cell growth showed that ectopic BOZF1 increased cell proliferation by 1.5-fold at day 6, and the knockdown of BOZF1 expression decreased cell proliferation by 1.6-fold (Fig. 1B). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays also showed that ectopic BOZF1 increased cell growth by 1.4-fold at day 4 compared with the control, and the siRNA-mediated knockdown of BOZF1 expression decreased cell growth by 1.8-fold (supplemental Fig. 1C). The FACS analyses of the HEK293 cells showed that BOZF1 increased the number of the cells in the S phase from 27.7 to 49.6%. The knockdown of endogenous BOZF1 expression decreased the cells in S phase (Fig. 1C). The percentage of cells incorporating BrdUrd (as an indicator of cell proliferation) also increased by more than 13-fold in HEK293 cells transfected with BOZF1 expression vector compared with the control cells (Fig. 1D). Overall, these results suggested that BOZF1 promotes cell proliferation.
FIGURE 1.

BOZF1 increases cell proliferation and increases number of cells in S phase. A, focus formation assays. NIH3T3 cells were transfected with BOZF1 expression or control vector and selected with medium containing G418 for 2 weeks. The foci were stained with 0.5% crystal violet. B, growth curve of HEK293 cells. The cells were transfected with either the BOZF1 expression vector (left) or siRNA against BOZF1 mRNA (right). Cell numbers were counted daily for 6 days. Mean values of three independent experiments are shown. *, p < 0.05. Error bars represent S.D. C, FACS analysis of cell cycle. HEK293 cells were transfected with the BOZF1 expression vector or BOZF1 siRNA and cultured for 2 days. The cells were fixed, stained with propidium iodide, and analyzed by FACS. Mean values of three independent experiments are shown. *, p < 0.05. Error bars represent S.D. D, BrdUrd incorporation assay. HEK293 cells were transfected with pcDNA3.1 or pcDNA3.1-BOZF1. One day after transfection, cells were cultured in serum-free medium and incubated for 1 h with BrdUrd in DMEM with 10% FBS. The cells actively incorporating BrdUrd were counted by immunocytochemistry using a fluorescence microscope. Mean values of three independent experiments are shown. *, p < 0.05. Error bars represent S.D. Primary anti-BrdUrd antibody and secondary anti-mouse Ig fluorescein were used. Magnification, 200×. N.C., negative control.
BOZF1 Represses Expression of a Negative Cell Cycle Regulator, p21, Encoded by CDKN1A
BOZF1 expression was increased in several cancer tissues tested, and BOZF1 increased cell proliferation. Accordingly, we examined whether BOZF1 could regulate the expression of p21, which is important in the regulation of the cell cycle. BOZF1 repressed the transcription of pGL2-CDKN1A-Luc (Fig. 2A), and this repression was strong. We also examined whether BOZF1 regulates endogenous p21 expression in HEK293 cells. Ectopic BOZF1 suppressed p21 expression by 55% (Fig. 2, B and C) in HEK293 cells. RT-qPCR and Western blot analyses revealed that the knockdown of endogenous BOZF1 mRNA by siRNAs 1, 2, 3, and 4 resulted in the derepression of p21 expression at both the mRNA and protein levels (Fig. 2, D and E). These data suggested that BOZF1 is a transcription repressor of endogenous CDKN1A.
FIGURE 2.

BOZF1 represses transcription of CDKN1A in HEK293 cells. A, transient transcription assay. pGL2-CDKN1A-Luc and BOZF1 expression vector were transfected into HEK293 cells and analyzed for luciferase activity. Luciferase activities were normalized with co-expressed β-galactosidase activity, and data presented are the average of three independent assays. Error bars represent S.D. *, p < 0.05. B, RT-qPCR analysis of the endogenous CDKN1A mRNA of the HEK293 cells transfected with either pcDNA3.1 control or the BOZF1 expression vector. *, p < 0.05. Mean values of three independent experiments are shown. Error bars represent S.D. C, Western blot analysis of endogenous p21 expression in HEK293 cells transfected with the BOZF1 expression vector. GAPDH was used as a control. D, knockdown of endogenous BOZF1 mRNA increases CDKN1A transcription. RT-qPCR analysis of endogenous CDKN1A and BOZF1 mRNA after transient transfection of the HEK293 cells with siRNAs against BOZF1 (1–4) or negative control (N.C.) siRNA. *, p < 0.05. Mean values of three independents experiments are shown. *, p < 0.05. Error bars represent S.D. E, Western blot analysis of endogenous p21 expression in HEK293 cells transfected with siRNA against BOZF1 or a negative control siRNA. GAPDH was used as a control. Relative band intensities are indicated by number.
BOZF1 Represses Transcription Activation of the CDKN1A Promoter by Sp1
We examined which regulatory elements of the CDKN1A promoter are important in transcriptional repression by BOZF1 in HEK293 cells. BOZF1 repressed the transcription of all four promoter constructs, which had 5′-upstream regulatory regions of different lengths (bp −2308, −1462, −864, and −133 to +30 from the transcription start site). BOZF1 potently repressed transcription of the longer promoter construct containing two p53-binding elements by 80%. Transcriptional repression of the shortest promoter construct (−131 bp) was also rather potent (Fig. 3A). We also examined the functions of the C/EBPα binding site (bp −1270 to −1256), C/EBPβ binding site (bp −1928 to −1920), and retinoic acid response elements (bp −1212 to −1194) in the transcription repression by BOZF1 (Fig. 3B). Although BOZF1 repressed the promoter with intact C/EBP binding sites, it could not repress transcription of the promoter with the mutation at either one of the C/EBP binding sites and resulted in an increase in transcription. However, the mutations at retinoic acid response elements did not affect the transcription repression of CDKN1A by BOZF1 (Fig. 3B). The results suggested that the two C/EBP-binding elements are important in transcriptional repression of CDKN1A gene by BOZF1 and suggested that the binding competition between BOZF1 and C/EBPs might be important in the regulation of p21 in the tissues, such as liver, where C/EBPs are expressed.
FIGURE 3.

BOZF1 represses the transcription of the CDKN1A promoter by acting on the far distal p53-binding element and the proximal promoter. A, structures of the four CDKN1A promoter and luciferase gene fusion constructs tested and transient transcription assays. BOZF1 expression vector and pGL2-CDKN1A-Luc reporter plasmid with a variable upstream sequence were transiently co-transfected into HEK293 cells and analyzed for luciferase activity. Luciferase activities were normalized with co-expressed β-galactosidase activity, and data presented are the average of three independent assays. Error bars represent S.D. *, p < 0.05. B, transient transcription assays and structures of three additional CDKN1A promoter and luciferase gene fusion constructs designed to test the function of C/EBP- and retinoic acid receptor (RAR)/retinoid X receptor-binding elements in transcriptional regulation by BOZF1. pGL2-CDKN1A-Luc wild type (WT) or pGL2-CDKN1A-Luc mutant plasmids were co-transfected with pcDNA3 or pcDNA3-BOZF1 and analyzed as above. *, p < 0.05. Mean values of three independent experiments are shown. Error bars represent S.D. mRARE, mutated retinoic acid response element; mC/EBP, mutated C/EBP.
The data basically indicate that BOZF1 can suppress transcription by acting on the small proximal regulatory region concentrated with Sp1-binding GC boxes. Because BOZF1 can repress transcription of CDKN1A by acting on the proximal promoter with six Sp1-binding GC boxes, we investigated whether BOZF1 could suppress the transcription activation of the CDKN1A minimal promoter by Sp1. As expected, Sp1 activated the promoter, and BOZF1 repressed transcriptional activation by Sp1 (Fig. 4A). BOZF1 repressed the promoter with the deletion of Sp1-binding GC boxes 1–4 weakly (Fig. 4B). BOZF1 might repress the transcription of endogenous CDKN1A by blocking the transcription activation by Sp1 at the proximal promoter.
FIGURE 4.

BOZF1 competes with Sp1 to bind to the proximal Sp1 binding GC boxes of CDKN1A. A, transient transcription assays. pGL2-CDKN1A-Luc, −133 bp reporter, pcDNA3, pcDNA3-Sp1, and/or pcDNA3-BOZF1 plasmids were co-transfected in the various combination indicated and analyzed as described in Fig. 3. *, p < 0.05. Mean values of three independent experiments are shown. Error bars represent S.D. B, transient transcription assays. pGL2-CDKN1A-Luc (WT), pGL2-CDKN1A-Luc-ΔGC-boxes (deletion of Sp1-binding GC boxes 1–4) reporter, pcDNA3, or pcDNA3-BOZF1 plasmids were co-transfected and analyzed as above. *, p < 0.05. Mean values of three independent experiments are shown. Error bars represent S.D. C, co-immunoprecipitation and Western blot assays of molecular interaction between BOZF1 and Sp1. HEK293 cell extracts with ectopic Myc-BOZF1 expression were immunoprecipitated with anti-Myc or anti-Sp1 antibody followed by Western blot analysis with anti-Myc, anti-Sp1, and anti-GAPDH antibodies. D, oligonucleotide pulldown assays. The structure of the proximal CDKN1A promoter is shown above. Boxes, Sp1-binding GC boxes. Tsp (+1), transcription start site. E, EMSA. The 32P-labeled GC box probes were incubated with GST-ZF-BOZF1 (1.5 μg), separated by 4% nondenaturing PAGE, and analyzed by autoradiography. F, qChIP assays of Sp1 and BOZF1 binding at the endogenous CDKN1A proximal promoter in HEK293 cells. The locations of the primer binding sites are indicated by arrows in D. The ChIP antibodies used were anti-Myc for Myc-BOZF1, anti-Sp1, and control IgG. *, p < 0.05. Mean values of three independent experiments are shown. Error bars represent S.D. N.C., negative control.
BOZF1 Represses Transcription of CDKN1A by Competing with Sp1 for Binding to GC boxes 1–5/6 of the Proximal Promoter
The transcriptional repression by BOZF1 can be established by inhibiting Sp1 activity, such as DNA binding, by BOZF1-Sp1 interactions or DNA binding competition between the proteins. The co-immunoprecipitation and Western blot analyses showed that BOZF1 did not interact with Sp1, thus eliminating the possibility of transcriptional repression through Sp1-BOZF1 interaction (Fig. 4C). Because transcriptional repression is possible through DNA binding competition, we first examined whether BOZF1 could bind to the Sp1-binding GC boxes of the proximal promoter. Oligonucleotide pulldown assays using cell extracts with ectopic BOZF1 expression and EMSA with recombinant GST-ZF-BOZF1 revealed that BOZF1 directly bound to all of the Sp1-binding GC box probes (1–5/6). BOZF1 binding to GC box 3, an element critical for basal transcription and synergistic interaction with p53, was relatively weak in both experiments (Fig. 4, D and E). ChIP assays showed that BOZF1 bound to the proximal promoter of endogenous CDKN1A, and Sp1 binding was decreased by BOZF1 in vivo (Fig. 4F). These results suggested that BOZF1 binds rather extensively to the proximal six GC boxes and represses transcription of the endogenous CDKN1A by binding competition with Sp1 at the proximal promoter.
BOZF1 Interrupts p53 Binding to the Distal Promoter of CDKN1A by Protein Interactions
Another important regulator of CDKN1A transcription is p53, particularly under stress conditions (24, 25). We analyzed whether BOZF1 affects the transcriptional activation of endogenous CDKN1A through interactions with p53. We used etoposide treatment to induce p53 and observed the increased CDKN1A transcription, which was repressed by BOZF1 (Fig. 5A). In addition, ectopic p53 potently activated the transcription of pG13-Luc with 13 copies of the p53-binding element, and BOZF1 completely repressed transcriptional activation by exogenous p53 in lung cancer H1299 cells lacking p53 (supplemental Fig. 3A).
FIGURE 5.

BOZF1 represses the transcription of CDKN1A by decreasing p53 binding via protein interactions between p53 and BOZF1. A, transient transcription assays. HEK293 cells were co-transfected with pGL2-CDKN1A-Luc and pcDNA3-BOZF1 or the pcDNA3.1 control vector and cultured for 48 h. The cells were treated with etoposide (10 μm) for 24 h prior to harvest. *, p < 0.05. Mean values of three independent experiments are shown. Error bars represent S.D. B, co-immunoprecipitation and Western blot assays of protein interaction between p53 and BOZF1. HEK293 cell extracts with ectopic BOZF1-His6 expression were immunoprecipitated with anti-His tag or anti-p53 antibody. The precipitates were analyzed by Western blotting with anti-His tag, anti-p53, and anti-GAPDH antibodies. C, GST fusion protein pulldown assays. GST fusion proteins were incubated with the [35S]methionine-labeled p53 polypeptides indicated, pulled down, separated by 15% SDS-PAGE, and analyzed by autoradiography. Structures of the polypeptides used are described in supplemental Fig. 3B. D, oligonucleotide pulldown assays of BOZF1-His6 protein binding or p53 binding to the p53RE-1 and p53RE-2 sites of the CDKN1A promoter. The structure of the CDKN1A promoter is shown above. GAPDH was used as a control. Tsp (+1), transcription start site. E, qChIP assay of endogenous p53 binding at the distal p53RE-1 and -2 sites of the CDKN1A promoter in HEK293 cells. The cells were transfected with BOZF1 expression vector and fixed, and the chromatins were subjected to immunoprecipitation using the indicated antibodies: anti-Myc for BOZF1, anti-p53, and control IgG. F, qChIP assay of BOZF1-His binding at the two p53RE sites. The procedures were performed as in E. *, p < 0.05. Mean values of three independent experiments are shown. Error bars represent S.D. N.C., negative control; ab, antibody; DBD, DNA binding domain; N-Term, N terminus; C-term, C terminus; ZF, zinc finger.
We examined whether the protein interaction between p53 and BOZF1 is required for transcription repression using co-immunoprecipitation/Western blot and GST pulldown assays. BOZF1 interacted with endogenous p53 (Fig. 5B). In addition, the GST-fused POZ or zinc finger domain of BOZF1 interacted with the DNA binding domain of p53 in vitro (Fig. 5C and supplemental Fig. 3B). Thus, the repression of CDKN1A transcription by BOZF1 might involve the protein interaction between p53 and BOZF1, which might inhibit the p53 activity important in transcription activation.
The Protein Interaction between p53 and BOZF1 Inhibits the DNA Binding Activity of p53 on the Distal Promoter of CDKN1A
To test whether DNA binding competition between BOZF1 and p53 at the two distal p53-binding elements facilitates the transcription repression of CDKN1A by BOZF1, we performed EMSA analyses using recombinant zinc finger DNA binding domain from BOZF1 (GST-ZF-BOZF1). Although BOZF1 bound to the proximal Sp1-binding GC boxes (Fig. 4, D, E, and F), it did not bind the p53REs in vitro (supplemental Fig. 3C). Oligonucleotide pulldown assays of the total cell lysates using the two p53-binding elements showed that BOZF1 did not bind the two elements as in EMSA. However, ectopic BOZF1 decreased endogenous p53 binding to the p53RE-1 but not to the downstream p53RE-2 (Fig. 5D).
These data suggest that the transcription repression of the gene might be caused by the protein interaction between p53 and BOZF1, which affects the DNA binding activity of p53 to p53RE-1. Indeed, ChIP assays showed that BOZF1 reduced the DNA binding activity of p53 to p53RE-1 but not to p53RE-2 (Fig. 5E). The RT-qPCR analysis revealed that PCR amplification of the ChIP regions is detected only in a high number of PCR cycles (35 cycles), and ectopic expression of BOZF1 did not change the binding pattern of BOZF1, indicating that BOZF1 probably does not bind to the p53REs (Fig. 5F).
Furthermore, we studied the functional significance of the protein interaction between p53 and BOZF1. Western blot assays of the immunoprecipitation of HEK293 cells using anti-p53 and anti-acetyl-lysine antibodies showed a significant decrease in acetylation of p53, whereas the total amount of p53 protein remained constant (Fig. 6A). Acetylations at Lys-320 and Lys-381 of p53 were decreased significantly by the ectopic BOZF1. Because p53 is frequently acetylated by histone acetyltransferase proteins like p300 and BOZF1 decreased p53 acetylation significantly, we tested whether BOZF1 interacts with p300. BOZF1 may affect p53 acetylation by interfering with the molecular interaction between p53 and p300. The co-immunoprecipitation showed that p300 interacted with BOZF1 (Fig. 6B). The in vitro acetylation assays also showed that the acetylation of p53 by p300 was significantly decreased by the presence of BOZF1 (Fig. 6C). GST fusion protein pulldown assays revealed that the histone acetyltransferase domain of p300 (polypeptide 3) interacted directly with the POZ domain of BOZF1 (Fig. 6D). The ubiquitination assays showed that the inhibition of p53 acetylation by ectopic BOZF1 did not affect the ubiquitination of p53 (Fig. 6E). The results suggested that the inhibition of p53 acetylation may not affect p53 stability. Because acetylation of p53 is also important in DNA binding activity, BOZF1 might repress the transcription of endogenous CDKN1A through suppressed acetylation of p53 by protein interactions among BOZF1, p300, and p53.
FIGURE 6.

BOZF1 inhibits acetylation of p53 by interacting with p53 and p300. A, Western blot and immunoprecipitation (I.P.) analysis of p53 acetylation. The cell extracts were immunoprecipitated using the anti-p53 antibody (DO-1). The immunoprecipitates were analyzed for acetylation using the anti-acetyl-lysine, anti-p53, anti-GAPDH, anti-acetyl-p53 (Lys-320), and anti-acetyl-p53 (Lys-381) antibodies. B, co-immunoprecipitation and Western blot assays of protein interaction between p300 and BOZF1. HEK293 cell extracts with ectopic BOZF1-His6 expression were immunoprecipitated with anti-His tag, anti-p300 antibody, and IgG. The precipitates were analyzed by Western blotting with anti-His tag, anti-p300, and anti-GAPDH antibodies. C, in vitro acetylation assay. Full-length recombinant p53 was incubated with mock, p300, or p300 + BOZF1. Coomassie staining of the gel showed the presence and size of the proteins in the reaction mixture (right). Acetylated p53 was detected by Western blot (WB) using the anti-acetyl-lysine (Ac-K) antibody. D, GST fusion protein pulldown assays. Structures of the p300 polypeptides used are shown above. GST fusion proteins were incubated with the [35S]methionine-labeled p300 polypeptides indicated, pulled down, separated by 8% SDS-PAGE, and analyzed by autoradiography. E, ubiquitination assays of p53. H1299 p53-null cells were co-transfected with pcDNA3-p53 and pcDNA3-His-ubiquitin expression vector in the presence or absence of pcDNA3.1-BOZF1. The cell lysate pellets precipitated by MagneHis nickel particles were resolved by SDS-PAGE and analyzed by Western blotting using anti-p53 (DO-1), anti-BOZF1, and anti-GAPDH antibodies. ZnF_TAZ, Transcription Adaptor putatative Zinc finger domain; ZnF_ZZ, Zinc-binding domain; KIX, CREB and MYB interaction domain; BROMO, bromo domain; HAT, Histone Acetyltransferase domain; Ub, ubiquitin; NTA, nitrilotriacetic acid.
DISCUSSION
Some POK family proteins have been shown to be important in cell differentiation, development, and oncogenesis. In this study, we characterized a novel member of the human POK family proteins, BOZF1. BOZF1 is abundantly expressed in many types of tumor tissues, such as breast, prostate, and cervix (supplemental Fig. 4). BOZF1 stimulates cell proliferation. Biochemical analyses showed that BOZF1 appears to be a mammalian oncogenic transcription factor. We investigated the function of BOZF1 in the control of cell proliferation with particular interest in the molecular mechanism involved in the transcriptional regulation of CDKN1A, which encodes a major negative cell cycle regulator, p21. BOZF1 stimulated cell proliferation by repressing p21 expression at the transcriptional level. The transcriptional repression involved molecular interactions with p53, the inhibition of p53 DNA binding activity, and DNA binding competition with Sp1 at the proximal promoter. BOZF1 bound to the six Sp1-binding GC boxes of the proximal promoter, but it did not directly bind to the distal p53-binding elements. BOZF1 repressed transcription of the endogenous CDKN1A by interfering with Sp1 binding to the proximal promoter region containing all of the Sp1-binding GC boxes (Fig. 7, hypothetical model).
FIGURE 7.
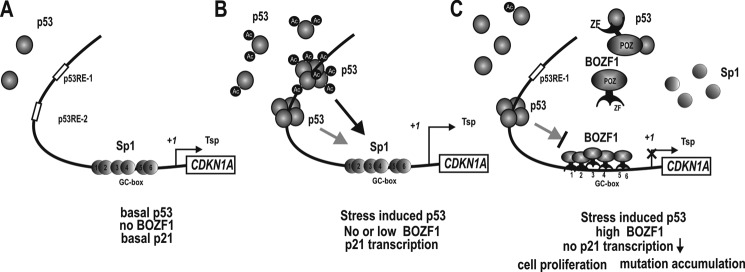
Hypothetical model of the transcriptional regulation of CDKN1A by BOZF1 under three different cellular conditions. A, transcriptional regulation of CDKN1A under normal condition and no BOZF1 expression. Constitutive Sp1 family activates transcription of CDKN1A at the basal level, and cells proliferate normally. B, transcriptional regulation of CDKN1A under stress conditions and no or low BOZF1 expression. p53 is induced and binds to the two distal p53REs. The acetylated p53 binds preferentially to p53RE-1. The p53 interacts with the Sp1 bound at the proximal promoter to synergistically activate transcription (indicated as arrows). The gray arrow indicates weaker interactions and transcription activation, and the thicker black arrow indicates a strong interaction and more potent transcription activation. C, transcriptional regulation of CDKN1A under stress conditions and high BOZF1 expression as in some cancer tissues. p53 is induced and binds to the two distal p53REs. BOZF1 represses the transcription of CDKN1A by competing with Sp1 at proximal GC boxes 1–5/6. BOZF1 bound at the proximal promoter represses the basal level of transcription and inducible transcription by blocking the interaction between p53 and Sp1. BOZF1 does not interact with Sp1. BOZF1 also represses transcription contributed by p53 at the p53REs. BOZF1 interacts with p53 and p300 and inhibits the acetylation of p53 by p300 without affecting p53 stability. The non-acetylated or partially acetylated p53 has lost the ability to bind to the p53RE-1 region but does retain DNA binding activity at the downstream p53RE-2 region. Tsp (+1), transcription start site; ZF, zinc finger.
The molecular interaction between p53 and BOZF1 resulted in the inhibition of p53 acetylation, decreased p53 DNA binding activity at the distal p53RE-1 site but not at p53RE-2. Intriguingly, the inhibition of acetylation did not affect p53 stability but appeared to affect DNA binding activity at p53RE-1. These data are consistent with a report by Luo et al. (38) indicating that acetylated p53 binds more efficiently to the p53RE-1 site.
The transcription of CDKN1A is activated through p53 and Sp1 actions at the distal and proximal elements, respectively. The protein interaction between proximal promoter-bound Sp1 and distal p53 is important in the transcriptional activation of the CDKN1A (32). Apparently, BOZF1 represses CDKN1A gene transcription through decreased Sp1 binding at the proximal promoter by binding competition at the proximal GC boxes and through inhibition of the DNA binding activity of p53 without affecting p53 stability.
Previously, we have shown that FBI-1 (ZBTB7A), ZBTB2, ZBTB5, Kr-pok, and MIZ-1 also regulate the transcription of CDKN1A (16, 43–46). FBI-1, ZBTB2, and ZBTB5 interact with Sp1 and compete with Sp1 for binding to the GC boxes of the proximal promoter. Interestingly, each POK protein has a preference for binding to a specific GC box(es) (e.g. FBI-1, GC box 3; ZBTB2, GC boxes 5/6; ZBTB5, GC boxes 5/6; Kr-pok, GC boxes 1 and 3; MIZ-1, GC boxes 3 and 5/6). In contrast, BOZF1 bound to all of the GC boxes, covering the entire proximal promoter. By competing with Sp1, BOZF1 could exclude Sp1 family transcription factors at this region. Unlike other POK proteins, such as ZBTB2 and ZBTB5, BOZF1 did not interact with Sp1 (43, 46).
At the distal p53 binding regions, FBI-1, ZBTB2, and ZBTB5 inhibit p53 activity by DNA binding competition with p53 at the p53RE-1 and/or -2 sites with preference for the p53RE-1 site (16, 43, 46). In the case of Kr-pok, the inhibition of p53 activity occurs by a different mechanism. Kr-pok does not significantly bind to the p53REs. Instead, it interacts with p53 for recruitment to the p53REs and transcriptional suppression; thus, p53 acts as a docking protein (45). Uniquely, although BOZF1 showed direct DNA binding activity at the proximal promoter GC boxes, it did not have DNA binding activity on the p53 elements, which represents a different mechanism from the other POK proteins that block transcriptional activation through p53. BOZF1 potently blocked p53 acetylation by PCAF or p300, decreased p53 binding, and inhibited transcriptional activation by p53. In particular, BOZF1 potently blocked acetylation at p53 Lys-320, a site known to be important in transcription activation of CDKN1A (40).
Accordingly, BOZF1 might play a critical role in regulating important biological processes, such as DNA repair, cell growth, differentiation, and apoptosis, by regulating the expression of p21. BOZF1 could be a novel member of the proto-oncogenic POK family of transcription factors that stimulates cell proliferation with a unique mechanism.
This work was supported by Do-Yak Program Grant 2011-0028817 (to M.-W. H.) and Midcareer Researcher Program Grant 2009-0081294) (to M.-W. H.) and in part by Medical Research Center Grant 2011-0030708) (to M.-W. H.) from the National Research Foundation of Korea, which is funded through the Korean government (Ministry of Education, Science and Technology).

This article contains a supplemental table, Figs. 1–4, and Results.
- POZ
- poxvirus and zinc finger
- POK
- POZ domain and Krüppel-like zinc finger
- BOZF-1
- BTB/POZ and zinc finger domains factor on chromosome 1
- MIZ-1
- Myc-interacting zinc finger 1
- ARF
- alternate reading frame
- CDK
- cyclin-dependent kinase
- PCAF
- p300/CBP-associated factor
- ATM
- ataxia telangiectasia mutated
- ATR
- ATM and Rad3-related
- C/EBP
- CCAAT/enhancer-binding protein
- qPCR
- quantitative PCR
- qChIP
- quantitative ChIP
- p53RE
- p53 response element
- CDKN1A
- cyclin-dependent kinase inhibitor 1A.
REFERENCES
- 1. Costoya J. A. (2007) Functional analysis of the role of POK transcriptional repressors. Brief. Funct. Genomic. Proteomic. 6, 8–18 [DOI] [PubMed] [Google Scholar]
- 2. Kelly K. F., Daniel J. M. (2006) POZ for effect-POZ-ZF transcription factors in cancer and development. Trends Cell Biol. 16, 578–587 [DOI] [PubMed] [Google Scholar]
- 3. Ye B. H., Lista F., Lo Coco F., Knowles D. M., Offit K., Chaganti R. S., Dalla-Favera R. (1993) Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science 262, 747–750 [DOI] [PubMed] [Google Scholar]
- 4. Kerckaert J. P., Deweindt C., Tilly H., Quief S., Lecocq G., Bastard C. (1993) LAZ3, a novel zinc-finger encoding gene, is disrupted by recurring chromosome 3q27 translocations in human lymphomas. Nat. Genet. 5, 66–70 [DOI] [PubMed] [Google Scholar]
- 5. Chen Z., Brand N. J., Chen A., Chen S. J., Tong J. H., Wang Z. Y., Waxman S., Zelent A. (1993) Fusion between a novel Krüppel-like zinc finger gene and the retinoic acid receptor-α locus due to a variant t(11;17) translocation associated with acute promyelocytic leukaemia. EMBO J. 12, 1161–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Melnick A., Ahmad K. F., Arai S., Polinger A., Ball H., Borden K. L., Carlile G. W., Prive G. G., Licht J. D. (2000) In-depth mutational analysis of the promyelocytic leukemia zinc finger BTB/POZ domain reveals motifs and residues required for biological and transcriptional functions. Mol. Cell. Biol. 20, 6550–6567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pessler F., Pendergrast P. S., Hernandez N. (1997) Purification and characterization of FBI-1, a cellular factor that binds to the human immunodeficiency virus type 1 inducer of short transcripts. Mol. Cell. Biol. 17, 3786–3798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee D. K., Suh D., Edenberg H. J., Hur M. W. (2002) POZ domain transcription factor, FBI-1, represses transcription of ADH5/FDH by interacting with the zinc finger and interfering with DNA binding activity of Sp1. J. Biol. Chem. 277, 26761–26768 [DOI] [PubMed] [Google Scholar]
- 9. Maeda T., Hobbs R. M., Merghoub T., Guernah I., Zelent A., Cordon-Cardo C., Teruya-Feldstein J., Pandolfi P. P. (2005) Role of the proto-oncogene Pokemon in cellular transformation and ARF repression. Nature 433, 278–285 [DOI] [PubMed] [Google Scholar]
- 10. Fujii H., Biel M. A., Zhou W., Weitzman S. A., Baylin S. B., Gabrielson E. (1998) Methylation of the HIC-1 candidate tumor suppressor gene in human breast cancer. Oncogene 16, 2159–2164 [DOI] [PubMed] [Google Scholar]
- 11. Wales M. M., Biel M. A., el Deiry W., Nelkin B. D., Issa J. P., Cavenee W. K., Kuerbitz S. J., Baylin S. B. (1995) p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat. Med. 1, 570–577 [DOI] [PubMed] [Google Scholar]
- 12. Peukert K., Staller P., Schneider A., Carmichael G., Hänel F., Eilers M. (1997) An alternative pathway for gene regulation by Myc. EMBO J. 16, 5672–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schneider A., Peukert K., Eilers M., Hänel F. (1997) Association of Myc with the zinc-finger protein Miz-1 defines a novel pathway for gene regulation by Myc. Curr. Top. Microbiol. Immunol. 224, 137–146 [DOI] [PubMed] [Google Scholar]
- 14. Phan R. T., Saito M., Basso K., Niu H., Dalla-Favera R. (2005) BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat. Immunol. 6, 1054–1060 [DOI] [PubMed] [Google Scholar]
- 15. Yeyati P. L., Shaknovich R., Boterashvili S., Li J., Ball H. J., Waxman S., Nason-Burchenal K., Dmitrovsky E., Zelent A., Licht J. D. (1999) Leukemia translocation protein PLZF inhibits cell growth and expression of cyclin A. Oncogene 18, 925–934 [DOI] [PubMed] [Google Scholar]
- 16. Choi W. I., Jeon B. N., Yun C. O., Kim P. H., Kim S. E., Choi K. Y., Kim S. H., Hur M. W. (2009) Proto-oncogene FBI-1 represses transcription of p21CIP1 by inhibition of transcription activation by p53 and Sp1. J. Biol. Chem. 284, 12633–12644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weber A., Marquardt J., Elzi D., Forster N., Starke S., Glaum A., Yamada D., Defossez P. A., Delrow J., Eisenman R. N., Christiansen H., Eilers M. (2008) Zbtb4 represses transcription of P21CIP1 and controls the cellular response to p53 activation. EMBO J. 27, 1563–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prives C., Hall P. A. (1999) The p53 pathway. J. Pathol. 187, 112–126 [DOI] [PubMed] [Google Scholar]
- 19. King K. L., Cidlowski J. A. (1998) Cell cycle regulation and apoptosis. Annu. Rev. Physiol. 60, 601–617 [DOI] [PubMed] [Google Scholar]
- 20. Lane D. P. (1992) Cancer. p53, guardian of the genome. Nature 358, 15–16 [DOI] [PubMed] [Google Scholar]
- 21. Evan G. I., Vousden K. H. (2001) Proliferation, cell cycle and apoptosis in cancer. Nature 411, 342–348 [DOI] [PubMed] [Google Scholar]
- 22. Bode A. M., Dong Z. (2004) Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 4, 793–805 [DOI] [PubMed] [Google Scholar]
- 23. Olsson A., Manzl C., Strasser A., Villunger A. (2007) How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 14, 1561–1575 [DOI] [PubMed] [Google Scholar]
- 24. el-Deiry W. S., Tokino T., Velculescu V. E., Levy D. B., Parsons R., Trent J. M., Lin D., Mercer W. E., Kinzler K. W., Vogelstein B. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 [DOI] [PubMed] [Google Scholar]
- 25. McKenzie P. P., Guichard S. M., Middlemas D. S., Ashmun R. A., Danks M. K., Harris L. C. (1999) Wild-type p53 can induce p21 and apoptosis in neuroblastoma cells but the DNA damage-induced G1 checkpoint function is attenuated. Clin. Cancer Res. 5, 4199–4207 [PubMed] [Google Scholar]
- 26. Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J. P., Sedivy J. M., Kinzler K. W., Vogelstein B. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501 [DOI] [PubMed] [Google Scholar]
- 27. Harper J. W., Adami G. R., Wei N., Keyomarsi K., Elledge S. J. (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816 [DOI] [PubMed] [Google Scholar]
- 28. van den Heuvel S., Harlow E. (1993) Distinct roles for cyclin-dependent kinases in cell cycle control. Science 262, 2050–2054 [DOI] [PubMed] [Google Scholar]
- 29. Waldman T., Kinzler K. W., Vogelstein B. (1995) p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 55, 5187–5190 [PubMed] [Google Scholar]
- 30. Gartel A. L., Tyner A. L. (1999) Transcriptional regulation of the p21WAF1/CIP1 gene. Exp. Cell Res. 246, 280–289 [DOI] [PubMed] [Google Scholar]
- 31. Gartel A. L., Goufman E., Najmabadi F., Tyner A. L. (2000) Sp1 and Sp3 activate p21WAF1/CIP1 gene transcription in the Caco-2 colon adenocarcinoma cell line. Oncogene 19, 5182–5188 [DOI] [PubMed] [Google Scholar]
- 32. Koutsodontis G., Tentes I., Papakosta P., Moustakas A., Kardassis D. (2001) Sp1 plays a critical role in the transcriptional activation of the human cyclin-dependent kinase inhibitor p21(WAF1/Cip1) gene by the p53 tumor suppressor protein. J. Biol. Chem. 276, 29116–29125 [DOI] [PubMed] [Google Scholar]
- 33. Canman C. E., Lim D. S., Cimprich K. A., Taya Y., Tamai K., Sakaguchi K., Appella E., Kastan M. B., Siliciano J. D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679 [DOI] [PubMed] [Google Scholar]
- 34. Tibbetts R. S., Brumbaugh K. M., Williams J. M., Sarkaria J. N., Cliby W. A., Shieh S. Y., Taya Y., Prives C., Abraham R. T. (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 13, 152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ito A., Lai C. H., Zhao X., Saito S., Hamilton M. H., Appella E., Yao T. P. (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 20, 1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tang Y., Luo J., Zhang W., Gu W. (2006) Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 24, 827–839 [DOI] [PubMed] [Google Scholar]
- 37. Liu L., Scolnick D. M., Trievel R. C., Zhang H. B., Marmorstein R., Halazonetis T. D., Berger S. L. (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 19, 1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luo J., Li M., Tang Y., Laszkowska M., Roeder R. G., Gu W. (2004) Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 101, 2259–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao Y., Lu S., Wu L., Chai G., Wang H., Chen Y., Sun J., Yu Y., Zhou W., Zheng Q., Wu M., Otterson G. A., Zhu W. G. (2006) Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol. Cell. Biol. 26, 2782–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chao C., Wu Z., Mazur S. J., Borges H., Rossi M., Lin T., Wang J. Y., Anderson C. W., Appella E., Xu Y. (2006) Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol. Cell. Biol. 26, 6859–6869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luo J., Nikolaev A. Y., Imai S., Chen D., Su F., Shiloh A., Guarente L., Gu W. (2001) Negative control of p53 by Sir2α promotes cell survival under stress. Cell 107, 137–148 [DOI] [PubMed] [Google Scholar]
- 42. Luo J., Su F., Chen D., Shiloh A., Gu W. (2000) Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408, 377–381 [DOI] [PubMed] [Google Scholar]
- 43. Jeon B. N., Choi W. I., Yu M. Y., Yoon A. R., Kim M. H., Yun C. O., Hur M. W. (2009) ZBTB2, a novel master regulator of the p53 pathway. J. Biol. Chem. 284, 17935–17946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee K. M., Choi W. I., Koh D. I., Kim Y. J., Jeon B. N., Yoon J. H., Lee C. E., Kim S. H., Oh J., Hur M. W. (2012) The proto-oncoprotein KR-POK represses transcriptional activation of CDKN1A by MIZ-1 through competitive binding. Oncogene 31, 1442–1458 [DOI] [PubMed] [Google Scholar]
- 45. Jeon B. N., Kim M. K., Choi W. I., Koh D. I., Hong S. Y., Kim K. S., Kim M., Yun C. O., Yoon J., Choi K. Y., Lee K. R., Nephew K. P., Hur M. W. (2012) KR-POK interacts with p53 and represses its ability to activate transcription of p21WAF1/CDKN1A. Cancer Res. 72, 1137–1148 [DOI] [PubMed] [Google Scholar]
- 46. Koh D. I., Choi W. I., Jeon B. N., Lee C. E., Yun C. O., Hur M. W. (2009) A novel POK family transcription factor, ZBTB5, represses transcription of p21CIP1 gene. J. Biol. Chem. 284, 19856–19866 [DOI] [PMC free article] [PubMed] [Google Scholar]