

Background: ATM acts as a master controller of the DSB response by phosphorylating numerous DSB response proteins, some of which, including histone H2AX, function in homologous recombination (HR).
Results: HR is not impaired in mouse ATM-null ES cells.
Conclusion: ATM is dispensable for HR, including H2AX-dependent HR.
Significance: This study helps clarify the perception that ATM has a general role in HR, including that controlled by H2AX.
Keywords: Cancer, DNA Damage Response, DNA Repair, Genomic Instability, Phosphorylation Enzymes, ATM, DSB Repair, MDC1, Histone H2AX, Homologous Recombination
Abstract
Ataxia telangiectasia mutated (ATM) is activated upon DNA double strand breaks (DSBs) and phosphorylates numerous DSB response proteins, including histone H2AX on serine 139 (Ser-139) to form γ-H2AX. Through interaction with MDC1, γ-H2AX promotes DSB repair by homologous recombination (HR). H2AX Ser-139 can also be phosphorylated by DNA-dependent protein kinase catalytic subunit and ataxia telangiectasia- and Rad3-related kinase. Thus, we tested whether ATM functions in HR, particularly that controlled by γ-H2AX, by comparing HR occurring at the euchromatic ROSA26 locus between mouse embryonic stem cells lacking either ATM, H2AX, or both. We show here that loss of ATM does not impair HR, including H2AX-dependent HR, but confers sensitivity to inhibition of poly(ADP-ribose) polymerases. Loss of ATM or H2AX has independent contributions to cellular sensitivity to ionizing radiation. The ATM-independent HR function of H2AX requires both Ser-139 phosphorylation and γ-H2AX/MDC1 interaction. Our data suggest that ATM is dispensable for HR, including that controlled by H2AX, in the context of euchromatin, excluding the implication of such an HR function in genomic instability, hypersensitivity to DNA damage, and poly(ADP-ribose) polymerase inhibition associated with ATM deficiency.
Introduction
DNA double strand breaks (DSBs)4 are among the most dangerous DNA lesions that can lead to cell death or genomic instability. Once they occur, proper DSB repair is required for cell survival and maintenance of genome integrity. DSBs can arise during normal cellular activities, such as metabolism and DNA replication, by exposure to exogenous DNA-damaging agents, including ionizing radiation (IR) and various chemotherapeutic drugs, or as a programmed event during lymphocyte development and meiosis. DSBs are repaired by two major DSB repair mechanisms: homologous recombination (HR) and non-homologous end joining (NHEJ). In somatic cells, HR operates primarily in the S and G2 phases of the cell cycle during which neighboring sister chromatids are available as a repair template for potentially error-free HR, termed sister chromatid recombination (1, 2). HR defects often result in “chromatid-type” aberrations, including chromatid radial structures and chromatid breaks, and are associated with many human cancers (3).
To ensure proper DSB repair, the DSB response has evolved to coordinate DSB repair with other cellular activities, such as cell cycle control, transcription repression, and programmed cell death (4). Ataxia telangiectasia mutated (ATM), a member of the phosphoinositide 3-kinase-related protein kinase family that also includes DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and ataxia telangiectasia- and Rad3-related kinase (ATR), serves as a master controller of the DSB response (5). Upon DSBs, ATM is activated to phosphorylate a host of DSB response proteins, including histone H2A variant H2AX, MDC1, BRCA1, and NBS1 (5). Rapid phosphorylation of H2AX on serine 139 (Ser-139) of the C-terminal SQEY motif expands over a large chromatin domain flanking DSBs (6) and initiates a series of downstream reactions that include protein recruitment and post-translational protein modifications on this chromatin domain (4). Cells lacking ATM or H2AX exhibit DSB repair defects, genomic instability, radiosensitivity, and checkpoint dysfunction (5, 7). However, although defective HR has been identified as a molecular mechanism causing genomic instability in H2AX−/− cells (8–11), it is unclear whether this is true in ATM−/− cells.
Ser-139 phosphorylation of H2AX and subsequent interaction of γ-H2AX with MDC1 are essential for H2AX-dependent HR (10, 12). As the major kinase for H2AX Ser-139 phosphorylation in a variety of cell lines (13–15), ATM may be responsible for this H2AX HR function. However, whether ATM has a function in HR independent of H2AX is not clear. Several lines of evidence support a role for ATM in HR. 1) Besides H2AX and MDC1, many HR factors, including BRCA1 and NBS1, are ATM substrates (5). Phosphorylation of these proteins by ATM may regulate HR. 2) Because poly(ADP-ribose) polymerase (PARP) inhibition selectively kill cells defective for HR (16), synthetic lethality caused by combined deficiency of ATM and PARP1 or PARP2 may be due to synergistic impacts on ATM-dependent HR and repair function of PARP1/2 (17, 18). In fact, cells carrying homozygous ATM kinase-dead mutations display HR defects and elevated sensitivity to PARP inhibition (19, 20). 3) Small molecule inhibitors of ATM and siRNA-mediated ATM depletion reduce HR in human cells (21, 22). 4) In proliferating cells, although the majority (∼85%) of IR-induced DSBs are likely repaired by NHEJ with fast kinetics in an ATM-independent manner, repair of the remaining ∼15% of IR-induced DSBs is dependent upon ATM and has slower repair kinetics that may reflect either NHEJ-mediated repair in heterochromatin or a possible HR-directed postreplication repair process (23–26). Unlike proliferating cells, Purkinje neurons require ATM for repairing the majority of IR-induced DSBs likely by NHEJ (27). 5) ATM drives the ATM-to-ATR switch that may promote HR (28). Despite this strong supposition of ATM HR function, there is evidence contradicting it. For example, high levels of spontaneous, unrepaired DSBs in ATM−/− cells are mainly chromosome breaks, which generally represent a deficiency in repairing prereplication DNA lesions by NHEJ (11, 29). In contrast, DNA lesions in H2AX−/− cells are equally divided between chromosome and chromatid breaks (11, 29). Occurrence of chromatid breaks is at least in part due to inactivation of H2AX-dependent HR/sister chromatid recombination (10). In this regard, the difference in genomic abnormalities between ATM−/− cells and H2AX−/− cells implies that H2AX HR function may be independent of ATM. In addition, it has been suggested that ATM is dispensable for HR in human embryonic stem cells and neural progenitor cells (30).
ATM and H2AX synergistically suppress genomic instability in mammalian cells (29). This synergistic impact may be partly mediated by their independent functions in HR. In this study, using ATM and H2AX double deficient mouse embryonic stem (ES) cells carrying a green fluorescent protein (GFP)-based HR reporter, we directly tested whether ATM has separable HR functions that are either H2AX-dependent or H2AX-independent. Because at least three possible fractions of γ-H2AX can be generated in response to DSBs, each by ATM, DNA-PKcs, and ATR, respectively, we investigated whether the ATM-dependent γ-H2AX fraction is responsible for H2AX-dependent HR. Surprisingly, we found that ATM is dispensable for HR and not even required for HR controlled by γ-H2AX.
EXPERIMENTAL PROCEDURES
Plasmids, Antibodies, and Small Molecule Inhibitors
The ROSA26 targeting vector for the HR reporter and pcDNA3β-based expression vectors with the hygromycin-resistant (HygR) marker for hemagglutinin (HA)-tagged human H2AX, mouse MDC1 tandem BRCT domain (MDC1 BRCT), H2AX mutants, and MDC1 BRCT K1554M mutant were described previously (10, 12, 31). Antibodies used in this study include anti-HA tag (sc-805; 1:200), anti-p53 (sc-6243; 1:200), and anti-Chk1 (sc-8408; 1:200) from Santa Cruz Biotechnology; anti-phospho-p53 Ser-15 (9284; 1:1000) and anti-phospho-Chk1 Ser-345 (2348; 1:1000) from Cell Signaling Technology; anti-ATM (ab78; 1:2000) and anti-Mre11 (ab397; 1:5000) from Abcam; and anti-γ-H2AX (JBW301; 1:2500), anti-histone H2A (07-146; 1:1000), and anti-histone H4 (07-108; 1:2000) from Millipore. Rabbit polyclonal anti-histone H2AX antibody (1:2000) was described before (8). Small molecule inhibitors include KU55933 from Calbiochem, KU60019 and NU7441 from Tocris, olaparib from Selleck, caffeine from Sigma, and VE821 from Axon MedChem.
Cell Lines, Cell Culture, and Transfection
Mouse ATMflox/floxH2AXneo/neo ES cells were generated previously (29) and grown in ES medium on either mouse embryonic fibroblast feeder cells or gelatinized plates. The ATMflox/floxH2AXneo/neo ES cells carrying an intact single copy HR reporter at the ROSA26 locus of ES cells were similarly generated as described previously (10). Adeno-Cre infection to generate isogenic ES reporter clones lacking either ATM or H2AX and transfection of mouse ES cells using Lipofectamine 2000 (Invitrogen) were performed as described before (10). Mouse ES cells stably expressing H2AX were generated as described previously (10).
Western Blotting
To analyze non-histone proteins, cells were lysed using radioimmune precipitation assay buffer. To analyze histones, cells were first lysed using cytolysis buffer (10 mm Hepes, pH7.9, 50 mm NaCl, 0.25 m sucrose, 0.1 mm EDTA, 0.5% Triton X-100). Histones were then acid-extracted from pellets of cell lysates as described previously (10), resolved by SDS-PAGE, and analyzed by Western blotting. To analyze the effect of small molecule inhibitors of ATM, DNA-PKcs, and ATR on IR or hydroxyurea (HU)-induced phosphorylation of histone H2AX and Chk1, cells were pretreated with drugs for 30 min at the doses indicated, irradiated with 10 Gy of IR or treated with 5 mm HU for 30 min, recovered for 30 min, and lysed for Western blotting.
Southern Analysis
Genomic DNA was extracted from ∼5 × 106 cells using the Puregene DNA Isolation kit (Gentra Systems). Southern blotting analysis for correct gene targeting was performed on 5 μg of genomic DNA using a ROSA26 probe and a GFP probe as described before (10).
Survival Competition Assay for IR and Drug Sensitivity
The survival competition assay was based upon a multicolor competition assay described previously with some modifications (32, 33). Specifically, mouse BRCA1flox/mut ES cells (34) expressing GFP were generated as reference cells and mixed with GFP− sample ES cells or GFP− control ES cells in a fixed ratio. Mixed cells were cultured for 1 day and treated the next day with either drugs for 18–20 h or IR at different doses. After culturing for another 5 days, percentages of viable GFP+ cells for each treatment were determined by flow cytometry using a BD Biosciences SORP LSR II analyzer. Percentages of viable GFP− cells were derived and normalized with percentages of viable GFP− cells for the untreated set at 100%. Relative survival of each sample was then determined as percentage of control with relative survival of control normalized to 100%.
I-SceI-induced HR Assay
Analysis of I-SceI-induced GFP+ frequencies in mouse HR reporter ES cells was performed as described previously (10). In co-transfection experiments, each expression plasmid and the I-SceI plasmid were used in a 2:3 ratio. Transfection efficiencies were measured by parallel transfection of wild-type (WT) GFP expression vector at an amount that was one-tenth of that of total DNA. If drug treatment was required, drugs were added at 5 h post-transfection, replaced with fresh addition at 24 h, and removed at 48 h. I-SceI-induced GFP+ was measured at 72 or 96 h by flow cytometry using a BD Biosciences SORP LSR II analyzer. Relative HR of tested cells was determined as percentage of control with relative HR of control normalized to 100%. Statistical comparisons between two unpaired populations and between paired samples were analyzed by Student's two-tailed unpaired t test (unknown variance) and two-tailed paired t test, respectively.
RESULTS
Loss of ATM Does Not Exacerbate HR Deficiency in Mouse H2AX−/− ES Cells
To define the role of ATM in HR, including that controlled by H2AX, we targeted an HR reporter into the ROSA26 locus of mouse ATMflox/floxH2AXneo/neo ES cells (29). Similar to the HR reporter described previously (10), this HR reporter (Fig. 1A) contains two inactivated GFP copies, one (TrGFP) truncated at the 5′-end and the other (I-SceI-GFP) interrupted with an 18-bp recognition site for the rare cutting endonuclease I-SceI (35). Using TrGFP as a template, HR-directed repair of a site-specific chromosomal DSB induced by I-SceI cutting generates a WT GFP copy and thereby GFP+ cells (Fig. 1A). The frequency of I-SceI-induced GFP+ cells reflects HR efficiency.
FIGURE 1.

Loss of ATM does not exacerbate HR deficiency in mouse H2AX−/− ES cells. A, the HR reporter. Repair of an I-SceI-induced DSB by HR between sister chromatids generates WT GFP. B, relative HR of multiple isogenic ES clones containing the HR reporter. Relative HR of one A+H+ clone serves as control and is normalized to 100%. Symbols represent the mean of at least two independent experiments, each with triplicates. Error bars indicate S.E. Statistical significance was determined by Student's two-tailed unpaired t test (unknown variance): p < 0.0002 between A+H+ and others and p < 0.04 between A+H− and A−H−. C and D, effect of KU55933 and KU60019 on HR in isogenic A+H+, A+H−, and A−H− reporter ES cells. I-SceI-induced HR assays were performed on A+H+, A+H−, and A−H− reporter cells transiently transfected with the I-SceI plasmid. Transfected cells were treated with 10 μm KU55933 (C) or 5 and 10 μm KU60019 (D) with DMSO as the mock control for 42 h starting at 5 h post-transfection, and I-SceI-induced GFP+ cells were quantified by flow cytometry 96 h post-transfection. Relative HR of A+H+ cells treated with DMSO serves as control and is set to 100%. Bars represent the mean of three independent experiments, each with triplicates. Error bars indicate S.E. Statistical significance was determined by paired t test between DMSO and ATM inhibitors: with KU55933 (C), p < 0.0004 in A+H+, not significant (p > 0.05) in A+H−, and p < 0.03 in A−H−, and with 5 μm KU60019 (D), p < 0.0006 in A+H+, p < 0.04 in A+H−, and p < 0.01 in A−H− and with 10 μm KU60019, p < 0.0008 in A+H+, p < 0.04 in A+H−, and p < 0.02 in A−H−.
Following gene targeting, ATMflox/floxH2AXneo/neo ES reporter clones containing a single copy of the HR reporter at the ROSA26 locus were established and identified by Southern blot (supplemental Fig. S1). These clones exhibit a low frequency of spontaneous GFP+ cells (background) and a high frequency of I-SceI-induced GFP+ cells (supplemental Fig. S2A and data not shown). Using adenovirus encoding Cre-recombinase to delete ATM or H2AX through flanking loxP sites (29), we generated several isogenic ATM+/+H2AX+/+ (A+H+), ATM+/+H2AX−/− (A+H−), and ATM−/−H2AX−/− (A−H−) reporter clones from one representative ATMflox/floxH2AXneo/neo parental HR reporter clone. ATM deletion seemed to be always accompanied with H2AX deletion; as a result, we were unable to generate ATM−/−H2AX+/+ (A−H+) clones. Cells lacking either ATM or H2AX were identified by Western blot (supplemental Fig. S2B).
I-SceI-induced HR assays were performed on isogenic A+H+, A+H−, and A−H− reporter clones (four clones for each genotype). To correct for interexperimental variations, percentages of I-SceI-induced GFP+ cells from one A+H+ reporter clone in each independent experiment were normalized to 100% as relative HR of control, and relative HR of other samples was then determined as percentage of control. Consistent with a previous observation (10), relative HR levels in the A+H− clones were about 20% of those of the A+H+ clones, indicating that deletion of H2AX reduces HR by ∼5-fold (Fig. 1B). Surprisingly, HR in the A−H− clones was not reduced at all and even slightly increased in comparison with the A+H− clones, suggesting that ATM either has no function in I-SceI-induced HR or has an HR function that is dependent upon H2AX.
We also asked whether ATM inhibition by small molecule ATM inhibitors affects HR in H2AX+/+ and H2AX−/− cells. To address this question, A+H+ and A+H− reporter cells along with A−H− reporter cells as control were treated with KU55933 and the improved ATM inhibitor KU60019 (36) at 5 or 10 μm in I-SceI-induced HR assays. Both ATM inhibitors reduced the HR levels in A+H+ and A+H− cells (Fig. 1, C and D). Unexpectedly, both ATM inhibitors also caused a reduction in HR in A−H− cells (Fig. 1, C and D), suggesting an off-target effect of the ATM inhibitors on HR. Although this off-target effect limits the use of ATM inhibitors in determining whether ATM has an HR function that is dependent upon H2AX, both ATM inhibitors reduced HR proportionally more in A+H+ cells than in A−H− cells (Fig. 1, C and D), indicating that a portion of HR reduction may be specific to ATM kinase inactivation by the inhibitors. It is possible that catalytically inactive ATM may interfere with HR as proposed recently (19, 20, 37, 38) even if fully functional ATM is not involved in HR.
HR Is Not Impaired in Mouse ATM−/− ES Cells
To further determine whether ATM has no HR function or an HR function that is dependent upon H2AX, A+H− and A−H− reporter cells were transiently transfected with HA-tagged H2AX, and HR of these cells was analyzed. In comparison with the S139A mutant or the empty vector (EV) control, WT H2AX restored HR in A+H− cells, indicating that the HR function of H2AX is dependent upon Ser-139 phosphorylation (Fig. 2A). Unexpectedly, WT H2AX alone (versus S139A or the EV control) rescued HR in A−H− cells as efficiently as, if not more than, that in A+H− cells (Fig. 2A), suggesting that ATM is dispensable for HR, including that controlled by γ-H2AX. Protein levels of HA-tagged WT H2AX and S139A were similar among transfected cells (Fig. 2A). To confirm that these observations are not limited to this particular set of A+H− and A−H− reporter clones, additional A+H− and A−H− reporter clones were tested, and similar results were observed (data not shown). Expression of WT H2AX in A+H+ cells did not significantly alter HR (Fig. 2A and data not shown).
FIGURE 2.
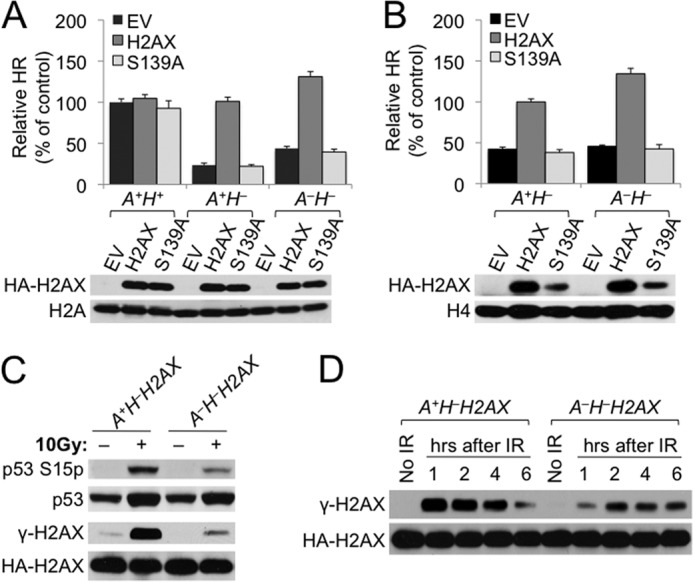
Loss of ATM does not impair HR. A, relative HR of each of the ES cell lines transiently transfected with H2AX expression plasmids. Relative HR of A+H+ cells transfected with EV serves as control. Bars represent the mean of three independent experiments, each with triplicates. Error bars indicate S.E. Statistical significance was determined by Student's two-tailed paired t test: between H2AX and other samples, not significant (p > 0.05) in A+H+ cells, p < 0.0002 in A+H− cells, and p < 0.0005 in A−H− cells; between A−H− cells transfected with H2AX and A+H− cells transfected with H2AX, p < 0.0001; and between A−H− cells transfected with H2AX and A+H+ cells with EV, p < 0.00005. H2AX expression plasmids, i.e. the EV control, H2AX, and S139A, for transient transfection and genotypes of transfected ES cells are indicated. H2AX protein levels after transient transfection of H2AX alleles in reporter ES cell lines are shown under the chart. Exogenous HA-tagged WT H2AX and S139A mutant detected by anti-HA antibody are indicated. Histone H2A serves as the loading control. B, relative HR of reconstituted ES cell lines stably expressing different H2AX alleles as indicated. Relative HR of A+H− cells stably expressing WT H2AX serves as control. Bars represent the mean of three independent experiments, each with triplicates. Error bars indicate S.E. Statistical significance was determined by Student's two-tailed paired t test: between H2AX and other samples, p < 0.001 in both A+H− cells (i.e. A+H−H2AX versus A+H−EV) and A−H− cells (i.e. A−H−H2AX versus A−H−EV); between A+H− and A−H− cells, p < 0.0001 (i.e. A−H−H2AX versus A+H−H2AX), p < 0.00001 (i.e. A−H−H2AX versus A+H−EV), and p < 0.00001 (i.e. A−H−EV versus A+H−H2AX). Steady-state HA-tagged H2AX protein levels recognized by anti-HA antibody in each of the reconstituted ES cell lines are shown under the chart with histone H4 as the loading control. C and D, ablation of IR-induced γ-H2AX formation in ATM−/− ES cells. A+H−H2AX and A−H−H2AX ES cells were treated with 10 Gy of IR, and extracts were prepared after 30 min (C) or at different time points over 6 h as indicated (D) and immunoblotted. HA-tagged H2AX protein was detected by anti-HA antibody.
Similar to transient rescue, stable expression of WT H2AX in comparison with S139A or the EV control restored HR in both A+H− and A−H− cells (Fig. 2B). In other words, the HR level in A−H− reporter cells stably expressing ectopic H2AX (i.e. A−H−H2AX cells, which are equivalent to A−H+ cells) was no less than that in A+H− reporter cells stably expressing ectopic H2AX (i.e. A+H−H2AX cells, which are equivalent to A+H+ cells). Taken together, this suggests that 1) ATM is dispensable for promoting HR, including H2AX-dependent HR, and 2) ATM-independent HR function of H2AX also requires Ser-139 phosphorylation. Of note, the abundance of H2AX S139A protein in stable cell lines was lower than that of WT H2AX for reasons unknown (Fig. 2B).
In many cell lines, ATM is the primary phosphoinositide 3-kinase-related protein kinase that phosphorylates H2AX Ser-139 (13–15). However, the fact that H2AX HR function requires Ser-139 phosphorylation but not ATM raises a possibility that ATM is not the major kinase for H2AX Ser-139 phosphorylation in mouse ES cells upon DSBs, and DSB-induced γ-H2AX formation is therefore little affected in ATM−/− ES cells. To test this, A+H−H2AX cells and A−H−H2AX cells were treated with 10 Gy of IR and analyzed for Ser-139 phosphorylation of H2AX upon IR treatment. Because ATM phosphorylates p53 on Ser-15 in response to DSBs (39, 40), we also analyzed and compared IR-induced p53 Ser-15 phosphorylation between A+H−H2AX and A−H−H2AX cells. In comparison with A+H−H2AX cells, A−H−H2AX cells had only a residual level of phosphorylated p53 and γ-H2AX at 30 min or over 6 h (Fig. 2, C and D), indicating that ATM remains the major kinase for H2AX Ser-139 phosphorylation and p53 Ser-15 phosphorylation in mouse ES reporter cells upon induction of DSBs. This also suggests that the remaining fraction of γ-H2AX generated in ATM−/− ES cells is sufficient for H2AX HR function. In addition, IR-induced H2AX phosphorylation is delayed in A−H−H2AX cells as compared with A+H−H2AX cells (Fig. 2D).
Two other phosphoinositide 3-kinase-related protein kinases, DNA-PKcs and ATR, have been shown to compensate for ATM for H2AX phosphorylation (13–15, 41). We asked which kinase is responsible for IR-induced H2AX phosphorylation in ATM−/− ES cells. Addressing this question may provide a clue about the kinase responsible for the ATM-independent HR function of γ-H2AX. Thus, A+H−H2AX (i.e. ATM+/+) and A−H−H2AX (i.e. ATM−/−) reporter ES cells were pretreated with small molecule inhibitors of DNA-PKcs, ATR, and ATM and irradiated at 10 Gy or treated with 5 mm HU, and Ser-139 phosphorylation of H2AX was then determined.
In A+H−H2AX cells, the ATM inhibitor KU60019 (5 μm) greatly reduced IR-induced H2AX phosphorylation, whereas the DNA-PKcs inhibitor NU7441 (5 μm) did not (Fig. 3A, upper panel), again indicating the primary role of ATM in phosphorylating H2AX in response to IR in mouse ES cells. In contrast, NU7441 abrogated nearly all of IR-induced γ-H2AX formation in A−H−H2AX cells (Fig. 3, A and B), suggesting that DNA-PKcs may compensate for ATM in IR-induced H2AX phosphorylation when ATM is absent. Of note, KU60019 also reduced γ-H2AX in A−H−H2AX cells (Fig. 3A, lower panel). This is consistent with the off-target effect of ATM inhibitors revealed previously (Fig. 1, C and D).
FIGURE 3.
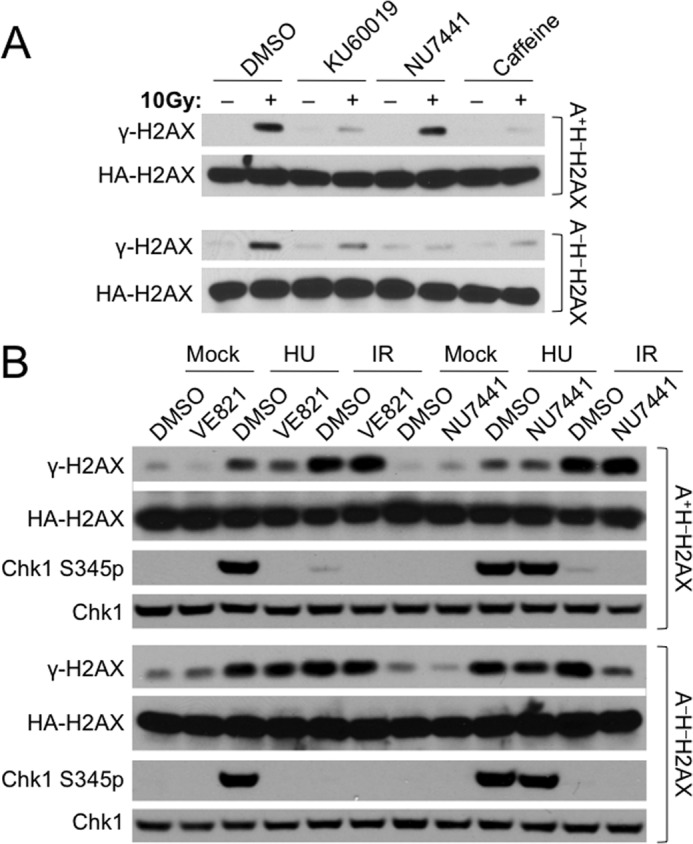
Inhibition of DNA-PKcs nearly abrogates IR-induced γ-H2AX formation in ATM−/− ES cells. A, effect of KU60019, NU7441, or caffeine on IR-induced γ-H2AX formation in A+H−H2AX (upper panel) and A−H−H2AX ES cells (lower panel) as indicated. Cells were pretreated with DMSO, 5 μm KU60019, 5 μm NU7441, or 4 mm caffeine for 30 min and irradiated with 10 Gy of IR. Histone extracts were prepared after 30 min and immunoblotted. HA-tagged H2AX protein was detected by anti-HA antibody. B, effect of VE821 and NU7441 on IR-induced γ-H2AX formation and HU-induced Chk1 Ser-345 phosphorylation in A+H−H2AX (upper panel) and A−H−H2AX ES cells (lower panel) as indicated. Cells were pretreated with DMSO, 5 μm VE821, and 5 μm NU7441 for 30 min and then exposed to 10 Gy of IR, 5 mm HU, or mock treatment. Extracts were prepared after 30 min and immunoblotted. HA-tagged H2AX protein was recognized by anti-HA antibody.
Although the ATR inhibitor caffeine (4 mm) nearly ablated IR-induced H2AX phosphorylation in both A+H−H2AX and A−H−H2AX cells, whether or not this effect is due to ATR inhibition is not clear because caffeine also targets other kinases, including ATM. Thus, we used the newly developed, potent ATR inhibitor VE821 to assess the effect of ATR inhibition on IR-induced H2AX phosphorylation (42). VE821 (5 μm) indeed specifically inhibited HU-induced ATR-mediated Chk1 Ser-345 phosphorylation in both A+H−H2AX and A−H−H2AX cells (Fig. 3B) but had little effect on IR-induced H2AX phosphorylation in both cells (Fig. 3B), indicating little involvement of ATR in phosphorylating H2AX in response to IR. Interestingly, VE821 reduced HU-induced H2AX phosphorylation only slightly in A+H−H2AX cells and little in A−H−H2AX cells (Fig. 3B). It is possible that ATM or DNA-PKcs may efficiently compensate for ATR in HU-induced H2AX phosphorylation in the absence of ATR.
ATM/H2AX Deficiency Has Independent Contributions to Cellular Sensitivity to IR
ATM and H2AX have independent functions in maintaining genomic instability (29). Here, we found that H2AX-dependent HR is not dependent upon ATM. Thus, we asked whether ATM/H2AX deficiency affects cellular sensitivity to DNA damage independently of each other. Using survival competition assays, we first analyzed IR sensitivity of A+H− and A−H− cells. Consistent with previous observations (8, 9, 44), H2AX deletion rendered cells sensitivity to IR (1, 2.5, and 5 Gy) (Fig. 4A). This sensitivity may be partly attributable to the HR defect caused by H2AX deficiency. ATM deletion in H2AX−/− cells further sensitized cells to IR (Fig. 4A), suggesting that ATM has an H2AX-independent function, loss of which renders cells sensitive to IR.
FIGURE 4.
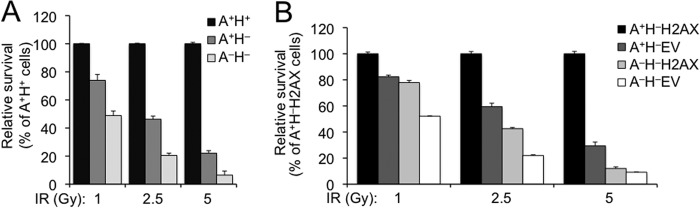
Loss of ATM and H2AX has independent contributions to IR sensitivity. A, IR sensitivity of mouse ES cells lacking H2AX or both ATM and H2AX. Relative survival as compared with A+H+ control sample was determined after 5-day culture following treatment with IR at 1, 2.5, and 5 Gy. Bars represent the mean of triplicates. Error bars indicate S.E. Statistical significance was determined by paired t test: at 1 Gy, p < 0.02 between A+H+ and A+H− and p < 0.005 between A−H− and others; at 2.5 Gy, p < 0.001 between A+H+ and others and p < 0.01 between A+H− and A−H−; and at 5 Gy, p < 0.001 between A+H+ and others and not significant (p > 0.05) between A+H− and A−H−. B, IR sensitivity of reconstituted mouse ES cells lacking ATM, H2AX, or both. Relative survival as compared with A+H−H2AX control sample was determined after 5-day culture following treatment with IR at 1, 2.5, and 5 Gy. Bars represent the mean of triplicates. Error bars indicate S.E. Statistical significance was determined by paired t test: at 1 Gy, p < 0.004 except not significant between A+H−EV and A−H−H2AX; at 2.5 Gy, p < 0.004 except that p = 0.03 between A+H−EV and A−H−H2AX; and at 5 Gy, p < 0.04 between A+H−H2AX versus A+H−EV, p < 0.02 between A+H−EV and A−H−H2AX, not significant between A−H−H2AX and A−H−EV, and p < 0.01 between every other two samples.
To determine whether IR sensitivity caused by H2AX deficiency is dependent upon ATM, we then used reconstituted A+H− and A−H− reporter ES cell lines stably expressing either ectopic H2AX (i.e. A+H−H2AX cells and A−H−H2AX cells) or the EV control (i.e. A+H−EV cells and A−H−EV cells) to analyze the impacts of either ATM deficiency, H2AX deficiency, or both on IR sensitivity. After exposure to IR, relative survival of A+H−EV and A−H−H2AX cells was lower than that of control A+H−H2AX cells (Fig. 4B). This again confirms that loss of either ATM or H2AX alone renders cells sensitive to IR. In addition, relative survival of A−H−H2AX cells was less than that of A+H−EV cells, suggesting that ATM deficiency has a greater effect on IR sensitivity than H2AX deficiency. Furthermore, relative survival of A−H−EV cells was lower than that of A−H−H2AX cells (Fig. 4B), indicating that IR sensitivity caused by H2AX deficiency is at least in part independent of ATM. We also noted that the difference in relative survival between A−H−H2AX and A−H−EV cells at 5 Gy was quite small (Fig. 4B). It is likely that 5 Gy of IR induces additional toxicity not specific to ATM or H2AX deficiency, thus masking IR sensitivity specific to H2AX deficiency.
Loss of ATM or H2AX Renders Cells Sensitive to the PARP Inhibitor Olaparib
ATM or H2AX deficiency in combination with PARP1 or PARP2 deficiency causes embryonic lethality (17, 18, 45). It is unclear whether ATM/H2AX deficiency contributes to this synthetic lethality through the same molecular mechanism. In addition, although PARP inhibition selectively kill cells defective for HR (16), it is not known whether HR-proficient ATM−/− ES cells and HR-deficient H2AX−/− ES cells have different sensitivity to PARP inhibition. Using survival competition assays, we analyzed the following two groups of cells for their sensitivity to the PARP inhibitor olaparib at 2.5 μm: 1) A+H− and A−H− with A+H+ as control and 2) A+H−EV, A−H−H2AX, and A−H−EV with A+H−H2AX as control. With olaparib exposure, relative survival of A+H− cells was reduced compared with A+H+ cells (Fig. 5A), and ectopic expression of H2AX improved survival of A+H− cells (A+H−H2AX versus A+H−EV; Fig. 5B), indicating that loss of H2AX renders cells sensitive to PARP inhibition. This is consistent with the fact that cells defective for HR are hypersensitive to PARP inhibition. Loss of ATM also renders cells hypersensitive to olaparib (comparing A+H− versus A−H− cells in Fig. 5A and A+H−H2AX versus A−H−H2AX and A+H−EV versus A−H−EV in Fig. 5B). In particular, such effect in H2AX−/− cells suggests that loss of ATM confers PARP inhibition sensitivity at least in part independently of H2AX.
FIGURE 5.
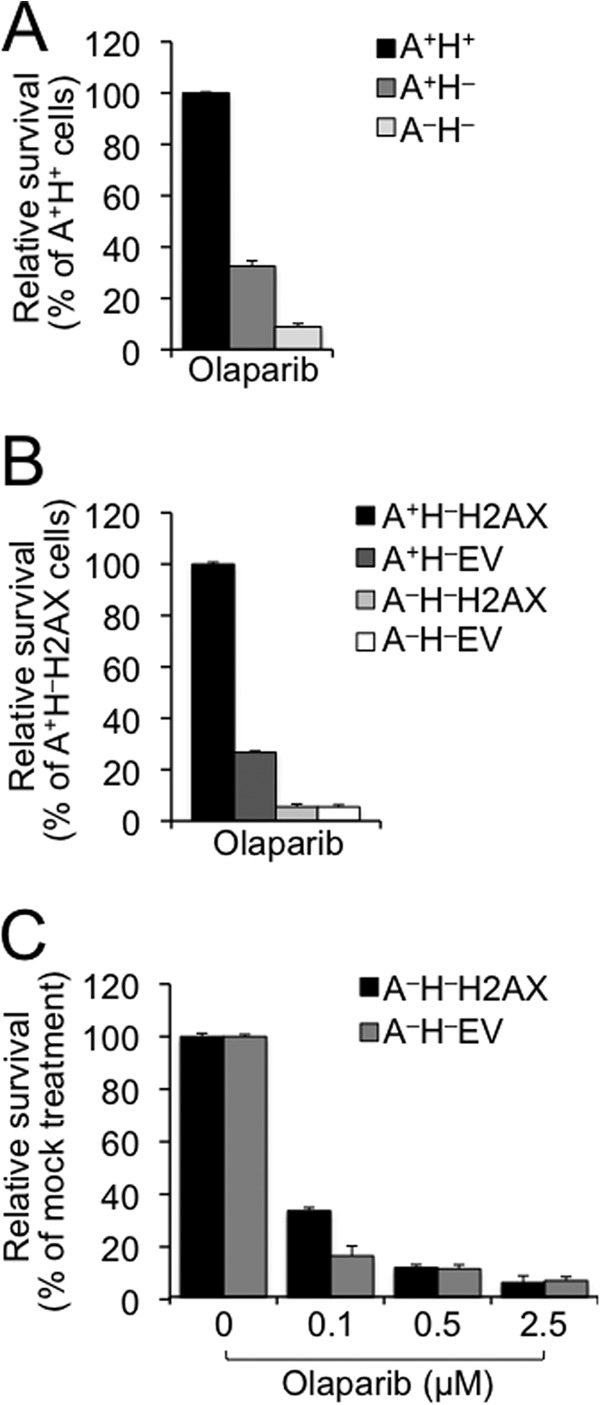
Loss of ATM or H2AX confers sensitivity to PARP inhibition. A, olaparib sensitivity of H2AX−/− ES cells in the presence or absence of ATM. Relative survival as compared with A+H+ control sample was determined after 5-day culture following treatment with 2.5 μm olaparib. Bars represent the mean of triplicates. Error bars indicate S.E. Statistical significance was determined by Student's two-tailed paired t test: p < 0.003 between A+H+ and other samples and p < 0.005 between A+H− and A−H−. B, olaparib sensitivity of mouse ES cells lacking ATM, H2AX, or both. Relative survival was determined as compared with A+H−H2AX control sample after 5-day culture following treatment with 2.5 μm olaparib. Bars represent the mean of triplicates. Error bars indicate S.E. Statistical significance was determined by paired t test between every two samples: p < 0.0007 except not significant (p > 0.05) between A−H−H2AX and A−H−EV. C, olaparib dose response of mouse A−H− ES cells reconstituted with WT H2AX or the EV control. Relative survival as compared with mock treatment (control) was determined after 5-day culture following treatment with olaparib at different doses as indicated. Bars represent the mean of triplicates. Error bars indicate S.E. Statistical significance was determined by paired t test between A−H−H2AX and A−H−EV: p < 0.01 at 0.1 μm olaparib and not significant at 0.5 and 2.5 μm.
In addition, loss of ATM (i.e. A+H−H2AX versus A−H−H2AX) appeared to confer higher sensitivity to olaparib than loss of H2AX (i.e. A+H−H2AX versus A+H−EV) (Fig. 5B). Although the effect of ATM deficiency on sensitivity to PARP inhibition is consistent with the observed synthetic lethality of combined ATM and PARP deficiency (17, 18), the normal HR level in ATM−/− cells suggests that an HR-independent function of ATM may be responsible for this synthetic lethality.
To determine whether loss of H2AX confers sensitivity to olaparib in an ATM-dependent manner, we compared relative survival between A−H−H2AX and A−H−EV cells. When exposed to 2.5 μm olaparib, these two cell lines have similar survival (Fig. 5B), seemingly suggesting that the contribution of H2AX deficiency to the olaparib sensitivity is ATM-dependent. However, 2.5 μm olaparib may induce additional toxicity that is not specific to H2AX loss, thus masking the possible contribution of H2AX deficiency to olaparib sensitivity in ATM−/− cells. To address this, lower doses of olaparib were administered, and relative survival of A−H−H2AX and A−H−EV cells was measured again. Although relative survival between these cells was similar in response to olaparib at 0.5 and 2.5 μm, A−H−H2AX cells were more sensitive to 0.1 μm olaparib than A−H−EV cells (Fig. 5C), indicating that PARP inhibition sensitivity caused by H2AX loss is at least in part ATM-independent. Together, these data suggest that ATM deficiency and H2AX deficiency have independent contributions to PARP inhibition sensitivity. The ATM-independent hypersensitivity associated with H2AX loss is likely a result of inactivation of H2AX-dependent HR.
ATM-independent H2AX HR Function Is Mediated by MDC1
H2AX Ser-139 phosphorylation is a critical event in the DSB response and essential for H2AX-dependent HR in both ATM+/+ cells and ATM−/− cells. Additional post-translational modifications of H2AX may occur for H2AX to fully function in the DSB response and DSB repair. In fact, using mass spectrometry, we have previously identified several new IR-induced modifications of H2AX (e.g. Lys-5 and Lys-36 acetylation and Thr-101 phosphorylation) in addition to constitutive Thr-120 and Ser-121 phosphorylation (31).
To determine whether these H2AX modifications and the well known Lys-119 ubiquitylation as well as other potential modifications, including Ser-1 and Tyr-142 phosphorylation, Lys-9 acetylation, and Lys-74, Lys-75, and Arg-76 methylation, are involved in H2AX HR function in ATM−/− cells, A−H− HR reporter cells were transfected with either of the H2AX mutants in which post-translational modification was abolished by specific mutation, and HR was measured. Like WT H2AX, most of these mutants, including S1A, K5R, K9R, K36R, K75R/R76Q, T101A, T120A/S121V, T136V, and K118R/K119R, efficiently restored HR in A−H− cells in which HR is defective (Fig. 6A). In contrast, Y142A, similar to S139A, did not restore HR, whereas Y142W did (Fig. 6A). It is known that phosphorylated SQEY motif of H2AX mediates the interaction of γ-H2AX with MDC1 through its tandem BRCT repeats (46, 47). Tyr-142 to Ala or Ser-139 to Ala mutation, but not Tyr-142 to Trp, disrupts the interaction of MDC1 with γ-H2AX (46, 47). Hence, ATM-independent H2AX HR function is mediated by MDC1. Similar protein levels of these H2AX mutants were confirmed by Western blot (Fig. 6A), excluding any impact by different protein levels of H2AX mutants on HR.
FIGURE 6.
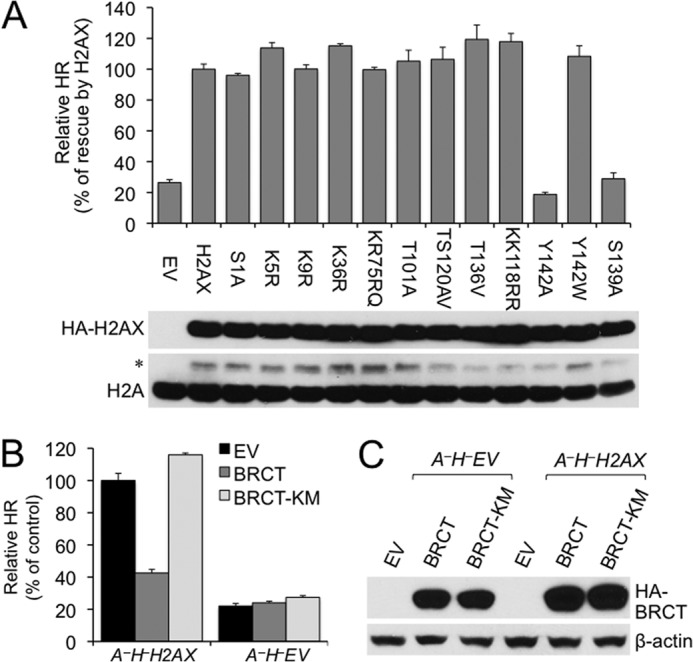
Disruption of the γ-H2AX/MDC1 interaction abolishes H2AX-dependent HR in ATM−/− ES cells. A, effect of H2AX mutations on H2AX-dependent HR in ATM−/− cells. A−H− ES cells were transiently transfected with expression plasmids for WT and mutant H2AX as indicated. Relative HR of A−H− cells transfected with WT H2AX serves as control. Bars represent the mean of triplicates. Error bars indicate S.E. Statistical significance was determined by paired t test between H2AX and other samples: not significant (p > 0.05) except that p < 0.004 between H2AX and EV, Y142A, or S139A and p < 0.002 between Y142A and Y142W. H2AX protein levels after transient transfection of H2AX alleles are shown under the chart. Exogenous HA-tagged WT and mutant H2AX recognized by anti-HA antibody are indicated with histone H2A as the loading control. * indicates exogenous HA-tagged H2AX recognized by anti-H2A antibody. B, effect of MDC1 BRCT on H2AX-dependent HR in ATM−/− cells. I-SceI-induced HR assays were performed on A−H−H2AX and A−H−EV ES reporter cells transiently transfected with EV and expression plasmids for HA-tagged mouse MDC1 BRCT or MDC1 BRCT K1554M (KM). Relative HR of A−H−H2AX cells transfected with EV serves as control. Bars represent the mean of triplicates. Error bars indicate S.E. Statistical significance was determined by paired t test between “BRCT” and others: p < 0.002 in A−H−H2AX and not significant (p > 0.05) in A−H−EV. C, protein levels of MDC1 BRCT and BRCT K1554M produced in I-SceI-induced HR assays and detected by anti-HA antibody. β-Actin serves as a loading control.
Because of its strong binding affinity with phosphorylated SQEY motif of γ-H2AX, MDC1 BRCT has been used as a potent MDC1 dominant negative to specifically block the interaction of MDC1 with γ-H2AX (12, 47). To further test whether MDC1 mediates ATM-independent H2AX HR function, MDC1 BRCT was used to disrupt the interaction between γ-H2AX and MDC1 in ATM−/− cells, and HR was analyzed. Indeed, by comparison with the EV control or the MDC1 BRCT K1554M mutant, which does not bind γ-H2AX (47), overexpression of MDC1 BRCT reduced HR by about 2.5-fold in A−H−H2AX reporter cells but not in A−H−EV reporter cells (Fig. 6B). This further confirms that MDC1 mediates ATM-independent H2AX HR function. Protein levels of MDC1 BRCT were similar among transfected cells as confirmed by Western blot (Fig. 6C).
DISCUSSION
ATM deficiency does not cause any reduction in I-SceI-induced HR in either H2AX+/+ or H2AX−/− cells, indicating that ATM is genetically dispensable for HR, including H2AX-dependent HR. However, a role for ATM in HR cannot be completely ruled out. Unlike I-SceI-induced DSBs, physiologic and pathologic DSBs, such as those induced by IR, radiomimetic drugs, and oxidative stress, have complex DSB ends that require additional processing before end resection for HR (48). ATM may have a role in this end processing and thereby in HR. The I-SceI-based reporter system is not designed for detecting such a role. In addition, DSBs in heterochromatin activate ATM signaling in part to enhance heterochromatin relaxation, thus facilitating DSB repair by HR or NHEJ in this highly compact, transcriptionally inert chromatin environment (24). However, the HR we measured in this study occurs at the euchromatic ROSA26 locus (49). Barring that, it is surprising that ATM as a master regulator of the DSB response has no function in HR, including that controlled by γ-H2AX, in euchromatin. Two models are proposed to explain this observation. 1) ATM has no HR function, and a different kinase is required for the formation of the HR-performing γ-H2AX fraction (Fig. 7A, Model 1). 2) ATM has an HR function that can be efficiently compensated by other kinases in the absence of ATM (Fig. 7B, Model 2).
FIGURE 7.
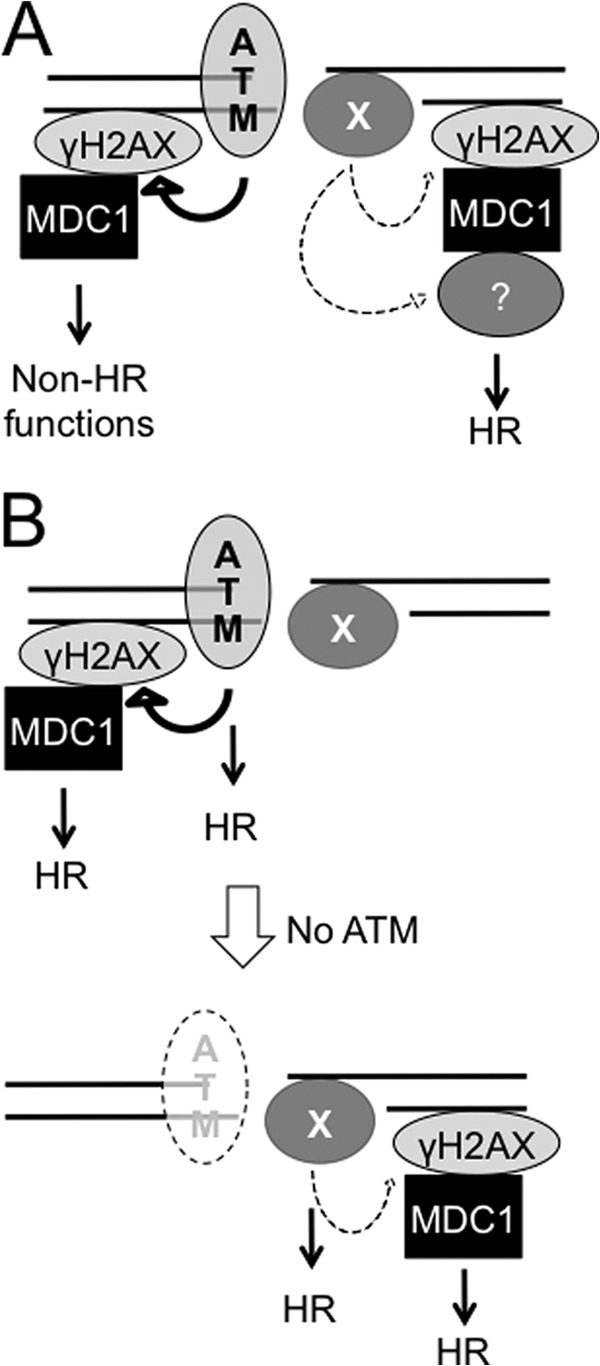
Models depicting the possible action of ATM in HR, including H2AX-dependent HR. A, ATM has no HR function, and a different kinase phosphorylates H2AX and promotes H2AX-dependent HR (Model 1). B, ATM has an HR function that can be efficiently compensated by other kinase in the absence of ATM (Model 2).
In Model 1, a further validation would be identification of the kinase that promotes H2AX-dependent HR. DNA-PKcs and ATR are among candidate kinases possibly performing this function. Also, H2AX can be phosphorylated by c-Jun N-terminal kinase (JNK), the mitogen-activated protein kinase (MAPK) p38, and mammalian STE20-like kinase 1 (MST1) during apoptosis regulation (50–52). It has yet to be determined which kinase is responsible for H2AX-dependent HR. Nevertheless, DSB-induced H2AX phosphorylation mediated by this kinase appears less efficient and covers a short range of chromatin near DSB ends. This is different from ATM-mediated H2AX phosphorylation, which expands over a much larger region of chromatin flanking DSBs (6, 15). In addition, different γ-H2AX fractions generated by different kinases may coexist on chromatin surrounding DSBs. A mechanism is needed to distinguish one fraction from the other so that the HR-performing γ-H2AX fraction can be recognized among different γ-H2AX fractions for H2AX-dependent HR. Two possible mechanisms may be at play. First, HR-performing γ-H2AX is generated at a specific time for its HR function. For example, if ATR is the kinase responsible for H2AX-dependent HR, ATR activation after the ATM-to-ATR switch (28) may initiate ATR-dependent γ-H2AX formation near DSB ends, and this temporally controlled γ-H2AX formation then mediates H2AX-dependent HR. Second, the factor mediating H2AX HR function is activated only by the kinase responsible for H2AX-dependent HR. This factor could be MDC1 or factors downstream of MDC1. Activation of this factor, possibly by phosphorylation, specifies the kinase that promotes H2AX HR function. H2AX Ser-139 phosphorylation, which is required for H2AX-dependent HR, plays no part in this decision.
In Model 2, ATM would normally perform two separable HR functions that are either H2AX-dependent or H2AX-independent. However, in the absence of ATM, these functions can be efficiently executed by substitute kinase(s) and therefore exhibit no HR deficiency. As a result, we did not observe any HR impairment associated with ATM deficiency in either H2AX+/+ or H2AX−/− cells in this study. One approach to test Model 2 is to block compensating pathways mediated by such a substitute kinase in H2AX+/+ and H2AX−/− cells and then determine whether loss of ATM affects HR, including H2AX-dependent HR, in this context. Because ATM, DNA-PKcs, and ATR have redundant functions in the DNA damage response (43, 53, 54), it is possible that DNA-PKcs or ATR may function as a substitute kinase for ATM in HR when ATM is absent. Other kinases that may have potential to compensate for ATM loss in HR also have yet to be identified.
In ATM−/− cells, DSB-induced γ-H2AX formation still occurs but with lower efficiency. Mapping of γ-H2AX chromatin domain by chromatin immunoprecipitation (ChIP) reveals that γ-H2AX formation near a site-specific DSB is less expansive or intense in ATM−/− cells than in ATM+/+ cells (15). Unexpectedly, HR in ATM−/− cells remains as efficient as, if not more than, that in ATM+/+ cells. This suggests that in either model the HR-performing γ-H2AX fraction is located near DSB ends and comprises a small amount of γ-H2AX, which is sufficient to fully execute H2AX-dependent HR. This γ-H2AX fraction requires MDC1 interaction for its action in HR. Therefore, in ATM+/+ ES cells, it is possible that the γ-H2AX domain near DSB ends, not the one distal to DSBs, mediates H2AX-dependent HR through the MDC1 interaction.
ATM and H2AX have synergistic functions in suppression of genomic instability through their independent functions (29). Here, we show that loss of these independent functions together leads to severe hypersensitivity of ATM/H2AX double deficient cells to DNA damage and PARP inhibition. Although defects in DSB repair are believed to be a major contributing factor to this hypersensitivity, ATM-deficient cells are not defective for HR. Therefore, loss of ATM may cause defects in other DSB repair pathways, such as NHEJ; these defects in turn lead to this hypersensitivity. To maintain genome stability, this ATM-dependent alternative repair pathway along with ATM-independent HR may be required to properly repair DSBs, at least those arising in euchromatin.
As PARP inhibition selectively kills cells defective for HR (16), it is consistent that H2AX−/− cells, which are defective for HR, are sensitive to PARP inhibitors, and combined PARP1 and H2AX deficiency causes embryonic lethality (45). In contrast, as loss of ATM does not impair HR, HR defects do not appear to be a contributing factor to the molecular mechanism of synthetic lethality imposed by combined loss of ATM and PARP1/2. However, as discussed above, we cannot rule out a role for ATM in HR-directed repair of DSBs with complex ends or in heterochromatin and thus a possible contribution of such a function, when defective, to this synthetic lethality. Nevertheless, synthetic lethality caused by PARP1/2 inhibition and HR defects has provided a promising strategy for development of targeted cancer therapy (16). Elucidation of the underlying mechanism that does not involve HR defects may be beneficial to the development of PARP inhibitors into targeted cancer therapy for HR-proficient cancer.
Acknowledgments
We thank Drs. Nicholas Willis, Yosef Shiloh, and Ralph Scully for valuable comments and critique.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM073894.

This article contains supplemental Figs. S1 and S2.
- DSB
- double strand break
- HR
- homologous recombination
- NHEJ
- non-homologous end joining
- EV
- empty vector
- IR
- ionizing radiation
- HU
- hydroxyurea
- ATM
- ataxia telangiectasia mutated
- DNA-PKcs
- DNA-dependent protein kinase catalytic subunit
- ATR
- ataxia telangiectasia- and Rad3-related kinase
- PARP
- poly(ADP-ribose) polymerase
- BRCT
- BRCA1 C terminus
- Gy
- grays.
REFERENCES
- 1. Johnson R. D., Jasin M. (2000) Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 19, 3398–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kadyk L. C., Hartwell L. H. (1992) Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 132, 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Helleday T. (2011) The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 5, 387–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ciccia A., Elledge S. J. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shiloh Y. (2003) ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3, 155–168 [DOI] [PubMed] [Google Scholar]
- 6. Rogakou E. P., Boon C., Redon C., Bonner W. M. (1999) Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146, 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fernandez-Capetillo O., Lee A., Nussenzweig M., Nussenzweig A. (2004) H2AX: the histone guardian of the genome. DNA Repair 3, 959–967 [DOI] [PubMed] [Google Scholar]
- 8. Bassing C. H., Chua K. F., Sekiguchi J., Suh H., Whitlow S. R., Fleming J. C., Monroe B. C., Ciccone D. N., Yan C., Vlasakova K., Livingston D. M., Ferguson D. O., Scully R., Alt F. W. (2002) Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. U.S.A. 99, 8173–8178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Celeste A., Petersen S., Romanienko P. J., Fernandez-Capetillo O., Chen H. T., Sedelnikova O. A., Reina-San-Martin B., Coppola V., Meffre E., Difilippantonio M. J., Redon C., Pilch D. R., Olaru A., Eckhaus M., Camerini-Otero R. D., Tessarollo L., Livak F., Manova K., Bonner W. M., Nussenzweig M. C., Nussenzweig A. (2002) Genomic instability in mice lacking histone H2AX. Science 296, 922–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xie A., Puget N., Shim I., Odate S., Jarzyna I., Bassing C. H., Alt F. W., Scully R. (2004) Control of sister chromatid recombination by histone H2AX. Mol. Cell 16, 1017–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Franco S., Gostissa M., Zha S., Lombard D. B., Murphy M. M., Zarrin A. A., Yan C., Tepsuporn S., Morales J. C., Adams M. M., Lou Z., Bassing C. H., Manis J. P., Chen J., Carpenter P. B., Alt F. W. (2006) H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol. Cell 21, 201–214 [DOI] [PubMed] [Google Scholar]
- 12. Xie A., Hartlerode A., Stucki M., Odate S., Puget N., Kwok A., Nagaraju G., Yan C., Alt F. W., Chen J., Jackson S. P., Scully R. (2007) Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol. Cell 28, 1045–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burma S., Chen B. P., Murphy M., Kurimasa A., Chen D. J. (2001) ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 276, 42462–42467 [DOI] [PubMed] [Google Scholar]
- 14. Stiff T., O'Driscoll M., Rief N., Iwabuchi K., Löbrich M., Jeggo P. A. (2004) ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 64, 2390–2396 [DOI] [PubMed] [Google Scholar]
- 15. Savic V., Yin B., Maas N. L., Bredemeyer A. L., Carpenter A. C., Helmink B. A., Yang-Iott K. S., Sleckman B. P., Bassing C. H. (2009) Formation of dynamic γ-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol. Cell 34, 298–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martin S. A., Lord C. J., Ashworth A. (2008) DNA repair deficiency as a therapeutic target in cancer. Curr. Opin. Genet. Dev. 18, 80–86 [DOI] [PubMed] [Google Scholar]
- 17. Ménisser-de Murcia J., Mark M., Wendling O., Wynshaw-Boris A., de Murcia G. (2001) Early embryonic lethality in PARP-1 Atm double-mutant mice suggests a functional synergy in cell proliferation during development. Mol. Cell. Biol. 21, 1828–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huber A., Bai P., de Murcia J. M., de Murcia G. (2004) PARP-1, PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA Repair 3, 1103–1108 [DOI] [PubMed] [Google Scholar]
- 19. Daniel J. A., Pellegrini M., Lee B.-S., Guo Z., Filsuf D., Belkina N. V., You Z., Paull T. T., Sleckman B. P., Feigenbaum L., Nussenzweig A. (2012) Loss of ATM kinase activity leads to embryonic lethality in mice. J. Cell Biol. 198, 295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yamamoto K., Wang Y., Jiang W., Liu X., Dubois R. L., Lin C.-S., Ludwig T., Bakkenist C. J., Zha S. (2012) Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. J. Cell Biol. 198, 305–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Köcher S., Rieckmann T., Rohaly G., Mansour W. Y., Dikomey E., Dornreiter I., Dahm-Daphi J. (2012) Radiation-induced double-strand breaks require ATM but not Artemis for homologous recombination during S-phase. Nucleic Acids Res. 40, 8336–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Serrano M. A., Li Z., Dangeti M., Musich P. R., Patrick S., Roginskaya M., Cartwright B., Zou Y. (July 16, 2012) DNA-PK, ATM and ATR collaboratively regulate p53-RPA interaction to facilitate homologous recombination DNA repair. Oncogene 10.1038/onc.2012.257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beucher A., Birraux J., Tchouandong L., Barton O., Shibata A., Conrad S., Goodarzi A. A., Krempler A., Jeggo P. A., Löbrich M. (2009) ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 28, 3413–3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goodarzi A. A., Jeggo P. A. (2012) The heterochromatic barrier to DNA double strand break repair: how to get the entry visa. Int. J. Mol. Sci. 13, 11844–11860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goodarzi A. A., Noon A. T., Deckbar D., Ziv Y., Shiloh Y., Löbrich M., Jeggo P. A. (2008) ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 31, 167–177 [DOI] [PubMed] [Google Scholar]
- 26. Shibata A., Conrad S., Birraux J., Geuting V., Barton O., Ismail A., Kakarougkas A., Meek K., Taucher-Scholz G., Löbrich M., Jeggo P. A. (2011) Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 30, 1079–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dar I., Yosha G., Elfassy R., Galron R., Wang Z.-Q., Shiloh Y., Barzilai A. (2011) Investigation of the functional link between ATM and NBS1 in the DNA damage response in the mouse cerebellum. J. Biol. Chem. 286, 15361–15376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shiotani B., Zou L. (2009) Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 33, 547–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zha S., Sekiguchi J., Brush J. W., Bassing C. H., Alt F. W. (2008) Complementary functions of ATM and H2AX in development and suppression of genomic instability. Proc. Natl. Acad. Sci. U.S.A. 105, 9302–9306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adams B. R., Golding S. E., Rao R. R., Valerie K. (2010) Dynamic dependence on ATR and ATM for double-strand break repair in human embryonic stem cells and neural descendants. PLoS One 5, e10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xie A., Odate S., Chandramouly G., Scully R. (2010) H2AX post-translational modifications in the ionizing radiation response and homologous recombination. Cell Cycle 9, 3602–3610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scully R., Ganesan S., Vlasakova K., Chen J., Socolovsky M., Livingston D. M. (1999) Genetic analysis of BRCA1 function in a defined tumor cell line. Mol. Cell 4, 1093–1099 [DOI] [PubMed] [Google Scholar]
- 33. Smogorzewska A., Matsuoka S., Vinciguerra P., McDonald E. R., 3rd, Hurov K. E., Luo J., Ballif B. A., Gygi S. P., Hofmann K., D'Andrea A. D., Elledge S. J. (2007) Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129, 289–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Farmer H., McCabe N., Lord C. J., Tutt A. N., Johnson D. A., Richardson T. B., Santarosa M., Dillon K. J., Hickson I., Knights C., Martin N. M., Jackson S. P., Smith G. C., Ashworth A. (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 [DOI] [PubMed] [Google Scholar]
- 35. Rouet P., Smih F., Jasin M. (1994) Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 14, 8096–8106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Golding S. E., Rosenberg E., Valerie N., Hussaini I., Frigerio M., Cockcroft X. F., Chong W. Y., Hummersone M., Rigoreau L., Menear K. A., O'Connor M. J., Povirk L. F., van Meter T., Valerie K. (2009) Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 8, 2894–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Choi S., Gamper A. M., White J. S., Bakkenist C. J. (2010) Inhibition of ATM kinase activity does not phenocopy ATM protein disruption: implications for the clinical utility of ATM kinase inhibitors. Cell Cycle 9, 4052–4057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. White J. S., Choi S., Bakkenist C. J. (2010) Transient ATM kinase inhibition disrupts DNA damage-induced sister chromatid exchange. Sci. Signal. 3, ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Banin S., Moyal L., Shieh S., Taya Y., Anderson C. W., Chessa L., Smorodinsky N. I., Prives C., Reiss Y., Shiloh Y., Ziv Y. (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674–1677 [DOI] [PubMed] [Google Scholar]
- 40. Canman C. E., Lim D. S., Cimprich K. A., Taya Y., Tamai K., Sakaguchi K., Appella E., Kastan M. B., Siliciano J. D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679 [DOI] [PubMed] [Google Scholar]
- 41. Ward I. M., Chen J. (2001) Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J. Biol. Chem. 276, 47759–47762 [DOI] [PubMed] [Google Scholar]
- 42. Reaper P. M., Griffiths M. R., Long J. M., Charrier J.-D., Maccormick S., Charlton P. A., Golec J. M., Pollard J. R. (2011) Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 7, 428–430 [DOI] [PubMed] [Google Scholar]
- 43. Neal J. A., Meek K. (2011) Choosing the right path: does DNA-PK help make the decision? Mutat. Res. 711, 73–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang J., Gong Z., Chen J. (2011) MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J. Cell Biol. 193, 267–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Orsburn B., Escudero B., Prakash M., Gesheva S., Liu G., Huso D. L., Franco S. (2010) Differential requirement for H2AX and 53BP1 in organismal development and genome maintenance in the absence of poly(ADP)ribosyl polymerase 1. Mol. Cell. Biol. 30, 2341–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee M. S., Edwards R. A., Thede G. L., Glover J. N. (2005) Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the γ-H2AX histone tail. J. Biol. Chem. 280, 32053–32056 [DOI] [PubMed] [Google Scholar]
- 47. Stucki M., Clapperton J. A., Mohammad D., Yaffe M. B., Smerdon S. J., Jackson S. P. (2005) MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226 [DOI] [PubMed] [Google Scholar]
- 48. Lieber M. R. (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Soriano P. (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70–71 [DOI] [PubMed] [Google Scholar]
- 50. Lu C., Zhu F., Cho Y.-Y., Tang F., Zykova T., Ma W. Y., Bode A. M., Dong Z. (2006) Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 23, 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu C., Shi Y., Wang Z., Song Z., Zhu M., Cai Q., Chen T. (2008) Serum starvation induces H2AX phosphorylation to regulate apoptosis via p38 MAPK pathway. FEBS Lett. 582, 2703–2708 [DOI] [PubMed] [Google Scholar]
- 52. Wen W., Zhu F., Zhang J., Keum Y.-S., Zykova T., Yao K., Peng C., Zheng D., Cho Y.-Y., Ma W. Y., Bode A. M., Dong Z. (2010) MST1 promotes apoptosis through phosphorylation of histone H2AX. J. Biol. Chem. 285, 39108–39116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cimprich K. A., Cortez D. (2008) ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Falck J., Coates J., Jackson S. P. (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434, 605–611 [DOI] [PubMed] [Google Scholar]