

Background: Different fatty acids signal to control intracellular lipid dynamic metabolism.
Results: In skeletal muscle cells, oleic acid induces cAMP/PKA and SIRT1 Ser-434 phosphorylation and activity, causing PGC1α deacetylation and increased fatty acid oxidation.
Conclusion: Oleic acid signals to the SIRT1-PGC1α complex to increase rates of fatty acid oxidation.
Significance: Pharmacological manipulation of the oleic acid signaling pathway might be a therapeutic option for conditions of lipid dysregulation.
Keywords: Beta-oxidation, Coactivator Transcription, Cyclic AMP (cAMP), Fatty Acid, Protein Acylation, Protein Kinase A (PKA), PGC1α, SIRT1
Abstract
Fatty acids are essential components of the dynamic lipid metabolism in cells. Fatty acids can also signal to intracellular pathways to trigger a broad range of cellular responses. Oleic acid is an abundant monounsaturated omega-9 fatty acid that impinges on different biological processes, but the mechanisms of action are not completely understood. Here, we report that oleic acid stimulates the cAMP/protein kinase A pathway and activates the SIRT1-PGC1α transcriptional complex to modulate rates of fatty acid oxidation. In skeletal muscle cells, oleic acid treatment increased intracellular levels of cyclic adenosine monophosphate (cAMP) that turned on protein kinase A activity. This resulted in SIRT1 phosphorylation at Ser-434 and elevation of its catalytic deacetylase activity. A direct SIRT1 substrate is the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator 1-α (PGC1α), which became deacetylated and hyperactive after oleic acid treatment. Importantly, oleic acid, but not other long chain fatty acids such as palmitate, increased the expression of genes linked to fatty acid oxidation pathway in a SIRT1-PGC1α-dependent mechanism. As a result, oleic acid potently accelerated rates of complete fatty acid oxidation in skeletal muscle cells. These results illustrate how a single long chain fatty acid specifically controls lipid oxidation through a signaling/transcriptional pathway. Pharmacological manipulation of this lipid signaling pathway might provide therapeutic possibilities to treat metabolic diseases associated with lipid dysregulation.
Introduction
Fatty acids are central structural and functional components of lipid metabolism, forming parts of cellular membranes, lipid droplets, or lipoproteins (1–3). Fatty acids can be obtained from the diet or synthesized in cells through metabolic pathways such as de novo lipogenesis (4). Depending on the nutrient and energy conditions, fatty acids can be rapidly oxidized in peroxisomes and mitochondria to maintain the cellular bioenergetic homeostasis (5). This ability to completely oxidize fatty acids to generate energy occurs in most mammalian cells except in neurons and red blood cells that strictly depend on glucose utilization to produce sufficient quantities of ATP for survival (6). There are different types of fatty acids with specific biophysical properties based on chain length and degree of saturation, including saturated long chain fatty acids (e.g. palmitic acid), monounsaturated long chain fatty acids (e.g. oleic acid), and polyunsaturated fatty acids (e.g. docosahexaenoic acid or linoleic acid). Interestingly, whereas saturated fatty acids cause inflammation that is associated with insulin-resistant states and atherosclerosis, oleic acid and polyunsaturated omega-3 fatty acids are anti-inflammatory and protective against these metabolic diseases. In the case of omega-3 fatty acids, the mechanisms of inflammatory suppression and insulin sensitivity have been recently elucidated and involve the GPR120 receptor signaling (7). However, the molecular regulatory mechanisms by which oleic acid reduces inflammation and dyslipidemias as well as improves insulin sensitivity (8–10) are not completely understood.
The regulatory control of fatty acid oxidation rates occurs at multiple levels involving transcriptional and non-transcriptional components. For example, among the non-transcriptional mechanisms increases in intracellular malonyl-CoA (the product of the fatty acid synthesis enzyme, acetyl-CoA carboxylase) potently suppress fatty acid oxidation by inhibiting the mitochondrial fatty acid transporter, carnitine-palmitoyltransferase-1 (11–13). At the transcriptional level, several hormone nuclear receptors (e.g. peroxisome proliferator-activated receptor α (PPARα), estrogen related receptor α (ERRα)) target sets of genes encoding fatty acid oxidation enzymes (14–16). The potency of these transcription factors largely depends on the assembly of co-activators such as PGC1α, which strongly increase the ability of these hormone nuclear receptors to drive transcriptional programs (17–20). PGC1α is controlled through reversible lysine side chain hyperacetylation that depends on the enzymatic activities of the acetyltransferase General Control of Amino-acid Synthesis 5-like 2 (GCN5) and the deacetylase SIRT1 (21, 22). SIRT1 deacetylase activity is increased through different nutrient and signaling pathways, including glucose restriction, AMPK activation, and cAMP/PKA5 activation; collectively, induction of these pathways promotes fatty acid oxidation (23–26). It is unknown, however, whether lipid signaling can modulate SIRT1 and, as a consequence, increase oxidation rates of fatty acids.
In these studies, we report that oleic acid, but not other long chain fatty acids, stimulates SIRT1 deacetylase activity through the elevation of cAMP intracellular levels and PKA signaling. As a result, the transcriptional coactivator PGC1α is deacetylated and activated, leading to increases in the expression of genes linked to complete oxidation of fatty acids. Oleic acid specifically augmented rates of fatty acid oxidation in a SIRT1-PGC1α-dependent manner. These results support a model in which oleic acid promotes fatty acid oxidation through signaling/transcriptional mechanisms providing a negative feedback loop that might explain, at least in part, some of the protective effects of this fatty acid against inflammation, dyslipidemias, and insulin resistance.
EXPERIMENTAL PROCEDURES
Reagents
Oleic (O1383), palmitic (P0500), stearic (S4751), elaidic (E4637), linoleic acid (L1376), palmitoleic (P9417), H-89, and forskolin were purchased from Sigma. Oleoylethanolamide, GW9508, and EX527 were purchased from Cayman and Tocris.
SIRT1 Phosphorylation, PGC1α Acetylation, and Western Blot Analysis
SIRT1 phosphorylation and PGC1α acetylation were detected by immunoprecipitation of SIRT1 or PGC1α followed by Western blot using phospho-PKA substrate or acetyllysine antibodies (Cell Signaling Technology) as described previously (26). Briefly, C2C12 myotubes were infected with adenovirus expressing FLAG-tagged SIRT1 or PGC1α as indicated. After 48 h post-infection, cells were lysed in a buffer that contained 1% IGEPAL, 150 mm NaCl, 20 mm HEPES (pH 7.9), 10 mm sodium fluoride, 10 mm glycerol 2-phosphate, 1 mm sodium orthovanadate, and protease inhibitor mixture. SIRT1 or PGC1α was immunoprecipitated using anti-FLAG beads (Sigma), and immunocomplexes were electrophoresed on SDS-polyacrylamide gels and transferred to Immobilon-P membrane (Millipore). Membranes were incubated with primary antibodies in 5% bovine serum albumin containing 1× TBS overnight at 4 °C. The membrane was then incubated with HRP-conjugated secondary antibodies for 1 h at room temperature and visualized using a Super Signal West Dura or Super Signal West Femto Substrate (Pierce).
Quantitative Real Time PCR and Primer Sequences
Total RNA was isolated with TRIzol (Invitrogen), and 2 μg of total RNA was used for cDNA synthesis using a high capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time PCRs were performed using SYBR Green PCR Master Mix (Applied Biosystems). Experimental Ct values were normalized to 36B4, and relative mRNA expression was calculated versus 36B4 expression. MCAD cDNA was amplified with sense primer (5′-GAAGGTTGAACTCGCTAGGC-3′) and antisense primer (5′-GCTAGCTGATTGGCAATGTC-3′). PDK4 cDNA was amplified with sense primer (5′-CCGCTGTCCATGAAGCA-3′) and antisense primer (5′-GCAGAAAAGCAAAGGACGTT-3′). ERRα cDNA was amplified with sense primer (5′-CAAGAGCATCCCAGGCTT-3′) and antisense primer (5′-GCACTTCCATCCACACACTC-3′). ACO cDNA was amplified with sense primer (5′-AGAACCCATTTGCACACCTTG-3′) and antisense primer (5′-AGCGTCCGTATCTTGAGTCCT-3′). ACAD9 cDNA was amplified with sense primer (5′-TTTCCAGAGGTCAGTCAACATGA-3′) and antisense primer (5′-TGGTCAATTTTTCGAGAGTCCAC-3′). ACADSB cDNA was amplified with sense primer (5′-CCCAACCTGCTTGTCTCCTTG-3′) and antisense primer (5′-ATCCCTGGATCACCGATTTCT-3′).
Cell Culture and Adenoviral Infections
C2C12 myoblasts were cultured in 10% calf serum containing DMEM. C2C12 myoblasts were differentiated at ∼90% confluency by switching the medium to DMEM containing 2% horse serum (Invitrogen) for 72 h. Primary muscle cells were isolated and cultured from 1∼2-week-old C57BL/6 mice. To induce differentiation, isolated myoblasts were grown to 80% confluence and then switched to the differentiation medium containing 5% horse serum for 72 h (23). Differentiated C2C12 or primary myotubes were infected with adenovirus expressing FLAG-PGC1α, FLAG-SIRT1-WT, or FLAG-SIRT1-S434A for 6 h as described previously (26). After 48 h post-infection, the infected myotubes were incubated with DMEM containing 5 mm low glucose and free fatty acid-free BSA-conjugated oleic, palmitic, stearic, or elaidic acid as indicated.
Rates of Fatty Acid Oxidation
Fatty acid oxidation assays were performed as described previously (23). Briefly, differentiated myotubes were incubated with BSA-conjugated unlabeled oleic (0.3 mm), palmitic (0.3 mm) acid in 5 mm glucose and 0.5% horse serum containing DMEM. After 6 h, myotubes were incubated with [14C]oleic acid (Perkin Elmer Life Sciences) or [14C]palmitic acid (Perkin Elmer Life Sciences) for 3 h. 14C-Labeled CO2 was captured in phenylethylamine-soaked Whatman paper and measured on a scintillation counter.
Measurement of NAD+ Levels
NAD+ and NADH nucleotides were determined in cell lysates using NAD+ and NADH determination kit (Bioassay Systems). Briefly, C2C12 myotubes were lysed in 200 μl of acid extraction buffer to measure the NAD+ levels or 200 μl of alkali extraction to determine NADH levels. Then, extracts were neutralized, and the concentration of NAD+ or NADH was measured after an enzymatic cycling reaction using 5 μl of sample. NAD+/NADH ratios were calculated by comparing the ratios obtained from C2C12 myotube-derived extracts.
Measurement of cAMP Levels
Intracellular cAMP levels were measured using a nonacetylation protocol and a cAMP enzyme-linked immunoassay, according to the manufacturer's instructions (GE Healthcare). Briefly, C2C12 myoblasts were cultured and differentiated in 96-well culture plates, and then differentiated myotubes were treated with various fatty acids for 30 min in as indicated. Cells were washed and lysed, and each cAMP concentration was then measured by triplicate. cAMP concentrations were normalized to protein content of each sample by performing BCA assay.
Sirt1 Enzymatic Activity
SIRT1 enzymatic activity was measured using a fluorescent assay as described previously (26). Briefly, adenoviruses expressing either FLAG-tagged SIRT1-WT or SIRT1-S434A were used to infect C2C12 myotubes. After 48 h post-infection, cells were incubated with BSA only or BSA-conjugated oleic acid for 30 min. Cells were lysed in a buffer containing 20 mm (pH 7.5), 175 mm NaCl, 0.5% IGEPAL, 0.1% CHAPS, 10 mm sodium fluoride, 10 mm glycerol 2-phosphate, and protease inhibitor mixture. FLAG-tagged SIRT1 was immunoprecipitated using FLAG beads from whole cell extracts from oleic acid-treated myotubes, and then immunoprecipitated SIRT1 was concentrated on a 30-kDa cut-off spin column (Millipore) with buffer exchange to remove excess 3×FLAG peptide. The final eluted protein was incubated with two different concentrations (350 and 750 μm) of NAD+ and 70 μm fluorescently labeled acetylated p53 peptide for 75 min at 37 °C according to the manufacturer's instructions (Enzo Life Sciences). The reaction was terminated by adding developer solution containing nicotinamide at a final concentration of 2 mm, and then SIRT1 activity was assessed by measuring the fluorescent emission at 460 nm, following excitation at 360 nm.
PKA Activity Measurement through Fluorescent Resonance Energy Transfer (FRET) Assay
Mouse skeletal C2C12 cells were transfected with protein kinase A activity reporter (AKAR3) or Nuclear Localization Sequence (NLS)-AKAR3 cDNAs to express cytosolic or nucleic PKA activity reporter, respectively, as described previously (27). 24 h after transfection, cells were used for FRET assay. Briefly, living C2C12 cells were imaged with a CCD camera with Metamorph software (Molecular Devices) on a Zeiss Axiovert 200 m microscope (Carl Zeiss, Thornwood, NY) with a ×40/1.3 numerical aperture oil-immersion objective lens. Dual-emission recording of CFP (440/480 nm, excitation/emission) and FRET (440/535 nm, excitation/emission) were coordinated with Lambda 10-3 filter shutter wheels (Sutter Instrument Co., Novato, CA). Exposure time was 100 ms, and recording intervals were 20 s. The FRET/CFP ratio was normalized against the base-line levels.
Animal Studies
All mouse studies were performed with an approved protocol from the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee. Six-week-old C57BL/6 mice were housed on a 12:12-h light/dark cycle. Oleic acid (1 g/kg body weight, conjugated with BSA, and suspended in phosphate-buffered saline (PBS)), palmitic acid (1 g/kg body weight), or BSA were administered to mice via oral gavage. After 3 or 6 h, mice were sacrificed, skeletal muscle was rapidly harvested, and mRNA was isolated by TRIzol reagent for measurement of gene expression.
Statistics
Data are expressed as mean ± S.D. Statistical significance was evaluated by using Student's two-tailed t test. p < 0.05 was considered to be statistically significant.
RESULTS
Oleic Acid Induces PGC1α Deacetylation in a SIRT1-dependent Manner in Skeletal Muscle Cells
We have previously shown that low concentrations of glucose cause PGC1α deacetylation and promote fatty acid oxidation in skeletal muscle cells (23). However, whether changes in the concentrations of fatty acids might also affect PGC1α acetylation, thus establishing an auto-regulatory loop, was unknown. To test this, we treated skeletal muscle cells with different types of fatty acids and measured PGC1α acetylation levels. Interestingly, among the fatty acids tested only oleic acid, but not other long chain saturated or polyunsaturated fatty acids, was sufficient to robustly induce PGC1α deacetylation (Fig. 1A). In addition, oleic acid-mediated PGC1α deacetylation occurred in a dose-dependent manner (Fig. 1B). Importantly, the concentrations of non-esterified oleic acid (0.2 and 0.5 mm) fall within the range of postprandial levels of this fatty acid in mice (28) and humans (29, 30). Steady-state levels of PGC1α acetylation are dynamically controlled by endogenous SIRT1 deacetylase activity. Thus, to assess whether oleic acid-induced PGC1α deacetylation was dependent on SIRT1, we used a specific SIRT1 inhibitor (EX-527). Fig. 1C shows that oleic acid-mediated PGC1α deacetylation was completely blocked by the EX-527 inhibitor. Together, these results indicate that oleic acid, but not other long chain fatty acids, specifically induces PGC1α deacetylation in skeletal muscle cells through SIRT1.
FIGURE 1.
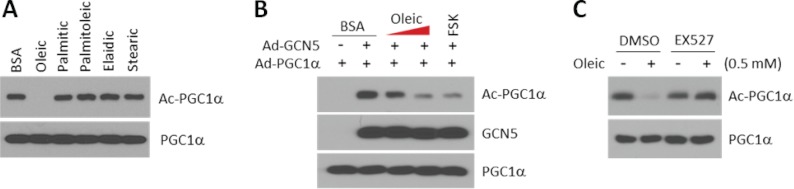
Oleic acid, but not other long chain fatty acids, induces PGC1α deacetylation through SIRT1 in skeletal muscle cells. A, oleic fatty acid deacetylates PGC1α. C2C12 myotubes were infected with adenoviruses encoding for FLAG-PGC1α. Two days after infection, cells were treated with the indicated fatty acids at 0.5 mm for 3 h. Acetylated PGC1α was detected using the acetyllysine antibody after FLAG immunoprecipation. B, oleic acid deacetylates PGC1α in a concentration-dependent manner. Indicated adenoviruses were used to infect C2C12 myotubes as indicated in A. Oleic acid (0.2 and 0.5 mm) or forskolin (FSK; 10 μm) were treated for 3 h. C, oleic acid deacetylates PGC1α in a SIRT1-dependent manner. C2C12 myotubes were treated as indicated in A, but cells were pretreated with EX-527 (2 μm) for 4 h before the addition of oleic acid. DMSO, dimethyl sulfoxide.
Oleic Acid Increases Intracellular Levels of cAMP and Activates PKA
We have recently identified that the cAMP/PKA pathway potently induces PGC1α deacetylation (26). To test whether oleic acid could affect this pathway, we measured intracellular cAMP levels. Fig. 2A shows that oleic acid treatment, but not other long chain fatty acids, caused increases in cAMP levels. In addition, we indirectly measured cAMP levels using an established FRET system based on the different cellular localization of a genetically encoded AKAR3 (31). Oleic acid induced the YFP/CFP FRET signal of AKAR or NLS-AKAR reporters (Fig. 2B). This signal induction was approximately four times less compared with a forskolin (which directly activates adenylate cyclase) control. In agreement with oleic acid induction of PKA activity, CREB phosphorylation as well as other PKA targets increased after oleic acid treatment using different concentrations and times (Fig. 2, C–E). In the same set of experiments, addition of stearic acid or other long chain fatty acids did not affect CREB phosphorylation that is consistent with its lack of effect on PGC1α acetylation levels (Fig. 2, C and E). Together, these results indicate that oleic acid treatment in skeletal muscle cells causes increases in cAMP levels and activates the PKA pathway.
FIGURE 2.

Oleic acid increases intracellular levels of cAMP and PKA phosphorylation activity. A, oleic acid increases cAMP levels. Forskolin (FSK; 10 μm) and BSA-conjugated various fatty acids (0.5 mm) were treated for 30 min either in C2C12 myotubes or myoblasts. Values represent mean ± S.D. of three independent experiments in duplicate. Statistical analysis was performed using Student's t test. *, p < 0.05 and **, p < 0.01. B, oleic acid induces PKA activity. The PKA biosensors, cytosolic AKAR3 (n = 16, upper panel), and nuclear AKAR3 (n = 17, middle panel), were expressed in C2C12 cells. Cells were treated with 0.6 mm oleic acid or 10 μm foskolin as a positive control (n = 12, lower panel) at the indicated time points. Changes in PKA FRET ratio (an indication of PKA activity) were measured. C, oleic acid but not other long chain fatty acids induces phosphorylation of PKA substrates. Indicated fatty acids were used at 0.5 mm to treat C2C12 myotubes. Phosphorylation of PKA substrates was detected using p-PKA substrate and p-CREB antibodies. D, oleic acid induces phosphorylation of PKA substrates at early time points. Experiments were performed as indicated described in C. E, concentration-dependent effects of oleic acid and stearic on p-CREB.
Oleic Acid Induces SIRT1 Phosphorylation at Ser-434
We have recently shown that activation of PKA induces SIRT1 phosphorylation at Ser-434, a residue located in the catalytic site, increasing its deacetylase activity through changes in Km and V (26). The observations that the oleic acid effects on PGC1α acetylation were dependent upon SIRT1 deacetylase activity and that oleic acid activated PKA signaling in skeletal muscle cells prompted us to test whether oleic acid could induce SIRT1 phosphorylation at Ser-434. Fig. 3A shows that among the different long chain fatty acids tested, only oleic acid was sufficient to strongly induce SIRT1 phosphorylation using an antibody that recognizes phosphorylated PKA substrates. In addition, the concentrations and times of oleic acid treatment that caused phosphorylation of SIRT1 were in the same range as the ones that promoted PKA activation (Fig. 3, B and C). Oleic acid induced SIRT1 phosphorylation in C2C12 and primary skeletal muscle myotubes (Fig. 3D). Interestingly, treatment of saturated fatty acids such as stearic and palmitic acid at different concentrations did not interfere with the ability of oleic acid to induce SIRT1 phosphorylation (Fig. 3E). The effects of oleic acid on SIRT1 phosphorylation were dependent on Ser-434 as indicated by the fact that the SIRT1 mutant (S434A) was not phosphorylated after oleic acid treatment (Fig. 3F). To test whether oleic acid induced SIRT1 phosphorylation was mediated through GPR120, a G Protein Coupled Receptor (GPCR) that binds long chain fatty acids such as omega-3, we used specific agonists of this receptor such as GW9508 or oleoylethanolamide. In contrast to oleic acid treatment, these two agonists were unable to induce SIRT1 phosphorylation (Fig. 3G), indicating that this receptor is not involved in the specific oleic acid signaling. Finally, to test whether SIRT1 phosphorylation on Ser-434 induced by oleic acid required PKA activity, we used the specific PKA inhibitor H-89 that completely blocked SIRT1 phosphorylation at this residue. As additional control, the Erk1/2 inhibitor PD98059 did not affect SIRT1 phosphorylation (Fig. 3H). The results in this section indicate that oleic acid promotes an increase in the intracellular cAMP pool, which activates PKA. The activation of PKA subsequently results in phosphorylation of SIRT1 at serine residue 434.
FIGURE 3.

SIRT1 phosphorylation at Ser-434 is induced by oleic acid treatment. A, oleic acid stimulates SIRT1 phosphorylation. C2C12 myotubes were infected with adenoviruses encoding FLAG-SIRT1 alleles. Fatty acids were used at 0.5 mm and treated for 3 h. Immunoprecipitated SIRT1 was used to detect phosphorylation using specific phospho-PKA substrate antibodies. B, time-dependent induction of SIRT1 phosphorylation by oleic acid. Experiments were performed as described in A but with the indicated times of oleic acid treatment. C, oleic acid concentration-dependent increases of SIRT1 phosphorylation. Experiments were performed as described in A, but different concentrations of oleic acid were used. D, oleic acid stimulates SIRT1 phosphorylation in primary skeletal muscle cells and C2C12 myotubes. Experiments were performed as described in A. E, saturated fatty acids do not induce SIRT1 phosphorylation. Different concentrations and combinations of fatty acids were used as indicated in the panel. F, oleic acid induces SIRT1 phosphorylation at Ser-434. C2C12 myotubes were infected with adenoviruses encoding FLAG-SIRT1 or FLAG-SIRT1-S434A and treated with oleic acid or forskolin at the indicated concentrations for 3 h. The bottom panel illustrates the quantification of SIRT1 phosphorylation. Statistical analysis was performed by Student's t test. *, p < 0.05 and **, p < 0.01. G, GPR120 agonism does not cause SIRT1 phosphorylation. C2C12 myotubes were treated with oleic acid or GPR120 agonists (oleoylethanolamide (OEA), 20 μm or GW9508, 100 μm) for 30 min, and phosphorylation of SIRT1 was analyzed. H, PKA, but not MAPK, inhibition blocks SIRT1 phosphorylation induced by oleic acid. MAPK inhibitor (PD98059, 20 μm) or PKA inhibitor (H89, 10 μm) were pretreated for 1 h before oleic acid treatment.
Oleic Acid Promotes SIRT1 Deacetylase Enzymatic Activity
Next, we assessed whether oleic acid treatment induced SIRT1 deacetylase activity through PKA-dependent phosphorylation at residue 434 using both in vitro SIRT1 deacetylase assays as well as measurements of PGC1α acetylation levels in skeletal muscle cells. In the in vitro assays, ectopically expressed SIRT1 was immunoprecipitated from cells treated with oleic acid or vehicle, and the deacetylase activity was measured using a p53-acetylated substrate. Oleic acid specifically induced the deacetylase activity of wild type SIRT1, but not mutant SIRT1 S434A, at two different concentrations of NAD+ tested (Fig. 4A). In agreement with our previous observations (26), SIRT1 S434A exhibited a slightly lower deacetylase activity, compared with wild type SIRT1, which was sensitive to NAD+ concentrations but not to oleic acid treatment. In similar sets of experiments, oleic acid specifically induced PGC1α deacetylation in muscle cells expressing wild type SIRT1, but not mutant SIRT1 S434A (Fig. 4B). Interestingly, consistent with our previous data that cAMP/PKA induces SIRT1 deacetylation activity, independent of changes in NAD+, oleic acid treatment in skeletal muscle cells did not affect NAD+ intracellular concentrations (Fig. 4C). As a positive control, we used AICAR, an AMPK activator known to increase NAD+ levels (25). These results indicate that oleic acid activates SIRT1 deacetylase through phosphorylation of SIRT1 at Ser-434.
FIGURE 4.

Oleic acid promotes SIRT1 deacetylase enzymatic activity independently of changes in NAD+ levels. A, oleic acid treatment increases SIRT1 activity. Immunoprecipitated FLAG-SIRT1 or FLAG-SIRT1 S434A from infected C2C12 myotubes were used to measure SIRT1 activity using the Fluor-de Lys® fluorimetric kit that uses acetylated p53 peptides. SIRT1 activity is represented as arbitrary fluorescence units (AFU). Values represent mean ± S.D. of one independent experiment in triplicate. B, oleic acid induces PGC1α deacetylation mediated by SIRT1. C2C12 myotubes infected with PGC1α and SIRT1 alleles were treated with oleic acid. Cellular extracts or immunoprecipitated proteins were used to detect indicated acetylated and phosphoproteins. The right panel illustrates the quantification of PGC1α acetylation. Values represent mean ± S.D. of three independent experiments. C, oleic acid does not change intracellular NAD+ levels. C2C12 myotubes treated with BSA, oleic acid (0.5 mm for 3 h), or AICAR (0.5 mm for 8 h) extracts were used to measure NAD+ and NADH levels. Values were normalized by total protein and represented as mean ± S.D. of two independent experiments in triplicate. Statistical analysis was performed by Student's t test. *, p < 0.05.
Oleic Acid Increases the PGC1α Transcriptional Activity of Genes Linked to Fatty Acid Oxidation in Vitro and in Vivo
PGC1α acetylation is a chemical enzymatic modification associated with repression of its ability to induce expression of target genes (32). We and others (23, 25, 26) have previously shown in skeletal muscle cells that SIRT1 activation induced by low concentrations of glucose, AMPK, or cAMP/PKA is sufficient to increase the expression of PGC1α target genes that are linked to the fatty acid oxidation pathway. Given that oleic acid produced similar effects as these different signals on PGC1α acetylation and in certain contexts induce PGC1α mRNA (33), we tested the ability of oleic acid to modulate PGC1α target gene expression, with particular focus on those that promote fatty acid oxidation. We first treated skeletal muscle cells with oleic, palmitic, and stearic fatty acids. Oleic acid was sufficient to significantly increase a broad range of genes associated with fatty acid oxidation that included the following: (i) transcriptional regulatory genes ERRα and NOR1; (ii) fatty acid transport genes CD36/FAT; and (iii) mitochondrial β-oxidation genes CPT1β, MCAD, and ACO. In contrast to these effects elicited by oleic acid, neither stearic nor palmitic acid caused a complete change in expression of all these genes (Fig. 5A). In some genes such as CD36 and ACAD5B, palmitic and stearic acids were sufficient to cause an increase, suggesting that these fatty acids might signal independently of cAMP/PKA.
FIGURE 5.

Oleic acid increases expression of genes linked to fatty acid oxidation in cultured muscle cells and skeletal muscle in vivo. A. Oleic acid increases gene expression of the fatty acid oxidation in C2C12 myotubes. Total RNA extracted from myotubes infected with control or PGC1α adenoviruses was used to measure the indicated genes using quantitative PCR. Indicated fatty acids were treated at 0.5 mm for 12 h. Values represent mean ± S.D. of three independent experiments in triplicate. Statistical analysis was performed using Student's t test. *, p < 0.05; **, p < 0.01, BSA versus fatty acids treatment; and #, p < 0.01, Ad-GFP versus Ad-PGC1α. B, oleic acid increases fatty acid oxidation gene expression in mouse skeletal muscle. BSA-conjugated oleic or palmitic acid (C) at 1g/kg was administered by oral gavage. Statistical analysis was performed using Student's t test (n = 4 mice). *, p < 0.05; **, p < 0.01, BSA vehicle versus fatty acid treatment. D, oleic acid induces CREB phosphorylation. Skeletal muscle from oleic or palmitic acid treated mice was homogenized and nuclear extracts were used to detect phosphorylation of CREB using specific phospho-CREB antibodies.
To assess whether oleic acid was able to induce this genetic program of fatty acid oxidation in skeletal muscle in whole animals, we administered oleic acid via oral gavage to mice and quantified changes in expression of these genes. As shown in Fig. 5, B and C, at 3 and 6 h after oleic but not palmitic acid treatment, genes linked to fatty acid oxidation were significantly increased in skeletal muscle. Consistent with oleic acid-induced PKA activation changes observed in cultured muscle cells, administration of oleic acid in mice increased CREB phosphorylation (Fig. 5D). Together, the results in this section indicate that oleic acid is sufficient to increase PKA activation and PGC1α target genes in cultures myotubes as well as skeletal muscle.
Oleic Acid Increases Fatty Acid Oxidation Genes in a SIRT1- and PGC1α-dependent Manner
Next, we performed a set of experiments to evaluate whether the oleic acid and PKA nutrient and signaling mechanisms activated fatty acid oxidation gene expression through the SIRT1-PGC1α transcriptional complex. Inhibition with SIRT1 with EX-527 attenuated the ability of oleic acid to induce fatty acid oxidation gene expression (Fig. 6A). Similarly, oleic acid effects were blocked in cells expressing the non-phosphorylated SIRT1 S434A mutant allele (Fig. 6B). These results indicate that the actions of oleic acid on gene expression linked to fatty acid oxidation are dependent on SIRT1 and, more specifically, on oleic acid-induced phosphorylation of SIRT1 S434.
FIGURE 6.

Oleic acid-mediated increases in fatty acid oxidation gene expression depend on SIRT1 and PGC1α transcriptional complex. SIRT1 inhibition by EX-527 (A) or SIRT1 S434A mutant (B) blocks the increase of fatty acid oxidation genes induced by oleic acid. Experiments were performed as described in Fig. 5A, EX-527 was treated for 13 h (added 1 h prior to oleic acid addition). Values represent mean ± S.D. of three independent experiments in triplicate. Statistical analysis was performed using Student's t test. *, p < 0.05; **, p < 0.01, BSA versus oleic acid treatment; and #, p < 0.01, Ad-GFP versus Ad-PGC1α. C, PGC1α is necessary for oleic acid-induced gene expression linked to fatty acid oxidation. C2C12 or primary myotubes were infected with adenoviruses expressing either control shRNA or PGC1α targeting shRNA for 48 h. After post infection, cells were incubated with 0.5 mm oleic acid for 12 h. Values represent mean ± S.D. of three independent experiments in triplicate. Statistical analysis was performed using Student's t test. *, p < 0.05; **, p < 0.01, BSA versus oleic acid treatment. D, effect of oleic acid is less than forskolin upon metabolic gene expression. C2C12 myotubes were incubated with oleic acid (0.5 mm) or forskolin (10 μm) for 12 h. Values represent mean ± S.D. of three independent experiments in triplicate. Statistical analysis was performed using Student's t test. *, p < 0.05.
The dependence upon PGC1α was determined through shRNA-mediated PGC1α depletion in C2C12 and primary skeletal muscle myotubes. Notably, all the oleic acid increased fatty acid oxidation gene expression was completely absent in differentiated myotubes in which PGC1α was knocked down (Fig. 6C). In agreement with oleic acid functioning through PKA, many of the same fatty acid oxidation gene targets induced by oleic acid in cultured myotubes were also induced by the PKA agonist forskolin (Fig. 6D). Collectively, these results strongly support that oleic acid signaling through PKA utilizes the transcriptional complex SIRT1-PGC1α to activate genes associated to fatty acid oxidation.
Oleic Acid Increases Rates of Fatty Acid Oxidation in a SIRT1-dependent Manner
Finally, to assess the functional consequences of the oleic acid-mediated changes in fatty acid oxidation gene expression, we measured complete rates of fatty acid oxidation in the presence and absence of oleic fatty acid. Consistent with the increases in fatty acid gene expression induced by oleic acid in skeletal muscle cells (Fig. 5), cellular fatty acid oxidation rates were markedly increased following treatment with oleic acid as measured by radioactive CO2 derived from radiolabeled [14C]palmitate or [14C]oleic acid (Fig. 7A). Ectopic expression of PGC1α synergized with oleic acid treatment to further magnify the increase in fatty acid oxidation. Importantly, these effects were both SIRT1- and PKA-dependent as the inhibitors EX-527 or H-89 and the SIRT1 S434A mutant (Fig. 7B) largely abolished oleic acid-induced lipid utilization. In contrast, palmitic acid did not substantially induce rates of fatty acid oxidation compared with vehicle controls (Fig. 7B). In short, these results indicate that oleic acid induces a signal transduction through the PKA pathway and SIRT1-PGC1α to accelerate rates of fatty acid oxidation.
FIGURE 7.

Oleic acid accelerates rates of complete fatty acid oxidation via a SIRT1-dependent mechanism. Pharmacological SIRT1 inhibition with EX527 (A) or ectopic expression of a SIRT1 S434A mutant (B) blocks rates of fatty acid oxidation induced by oleic acid. C2C12 myotubes infected with the indicated adenoviruses and treatments were used to measure fatty acid oxidation as described under “Experimental Procedures.” Values represent mean ± S.D. of two independent experiments performed in triplicate. Statistical analysis was performed using Student's t test. *, p < 0.05, BSA versus oleic acid treatment; and #, p < 0.01, Ad-GFP versus Ad-PGC1α (A) or Ad-GFP versus Ad-SIRT1-WT (B).
DISCUSSION
In these studies, we have identified a new lipid signaling/transcriptional cellular route by which oleic fatty acid communicates to increase rates of fatty acids and contribute to lipid homeostasis. This biochemical route relies upon the activation of the cAMP/PKA pathway that phosphorylates SIRT1 Ser-434 and increases its deacetylase activity. This activated SIRT1 deacetylates PGC1α and increases the expression of fatty acid oxidation genes, leading to increases in complete oxidation of fatty acids.
Specific activation of PGC1α in skeletal muscle through transgenic expression exerts strong beneficial effects and protection against age-associated diseases (34, 35). For example, lipid dysregulation and inflammation linked to aging are strongly attenuated in mice with elevated PGC1α function in skeletal muscle. These studies suggest that increasing PGC1α activity could be a valid therapeutic target to ameliorate these metabolic diseases. PGC1α functions as an scaffold protein that is recruited to specific transcription factors and potently induces expression of genes linked to oxidative metabolism (20, 36). Although not entirely impossible, it might prove difficult to identify small molecules that directly bind to and activate PGC1α. Alternatively, it might be more feasible to target pathways that lead to PGC1α activation. We and others (32, 37, 38) have shown that changes in PGC1α acetylation are linked to PGC1α transcriptional function: hyperacetylation through the GCN5 acetyl transferase is inhibitory, whereas deacetylation through the NAD+-dependent SIRT1 deacetylase is activational. In fact, increases of SIRT1 activity by different means, such as decreases of glucose concentrations, increases in AMPK or cAMP/PKA, or activators such as resveratrol, can cause deacetylation and activation of PGC1α and ultimately promote downstream target functions, including mitochondrial oxidative metabolism (23–26, 39). The study reported here suggests that oleic acid and/or small molecules that target components of this lipid signaling pathway could be used to activate PGC1α in skeletal muscle. Whereas an obvious possibility is to increase intake of oleic acid, alternative ways will need to be further explored in the future. For example, what cAMP/PKA upstream components are mediating oleic acid cellular effects? It is clear that in the case of polyunsaturated omega-3 fatty acids, direct binding to the GPCR transmembrane receptor GPR120 mediates most of the beneficial effects on inflammation and insulin resistance. It is, however, unknown whether oleic acid can bind to similar types of receptors to activate cAMP signaling but, if this was the case, agonism of this pathway in skeletal muscle would be an attractive therapeutic avenue.
Oleic acid, in complete opposition to saturated fatty acids, protects against inflammation and insulin resistance (8–10, 40). We propose here that at least one of the mechanisms by which this monounsatured ω-9 fatty acid might elicit these beneficial effects is, at least in part, through increasing lipid oxidation rates in skeletal muscle. As a result, oleic acid will help to increase clearance of excess of saturated fatty acids counteracting inflammation and insulin resistance. In addition, it is conceivable that increased rates of oxidation in skeletal muscle caused by oleic acid might also participate to boost energy expenditure and balance body weight in overnutrition states. These results might also have important therapeutic dietary implications in obesity management as indicated previously. It is, however, plausible that in addition to the effects on mitochondrial lipid oxidation other unknown cellular mechanisms could also contribute to the biological actions driven by oleic acid.
Why does there exist a big divergence in the metabolic/energetic effects of different types of fatty acids? Although at this point this is mainly speculative, it might be that a mixed diet with different type of fatty acids (saturated and unsaturated) has evolutionary advantages due to an adequate balance between energy storage and expenditure. In the same vein, unsaturated fatty acids, including oleic acid, could act as fatty acid sensors to maintain cellular and systemic levels of these lipids. Thus, oleic acid may function as a signal to trigger a negative feedback loop to deal with an excess of fatty acids and maintain lipid homeostasis. The metabolic advantage and beneficial effects triggered by oleic acid in skeletal muscle cells signaling to SIRT1-PGC1α complex and increasing levels of lipid oxidation could eventually be pharmacologically exploited. Future identification of new components in this pathway will provide therapeutic possibilities to treat diseases associated with lipid dysregulation.
Acknowledgments
We thank all members of the Puigserver laboratory for important discussions about this project.
This work was supported by the Dana-Farber Cancer Institute, the American Diabetes Association, the Department of Defense, National Institutes of Health/NIDDK Grant RO1 069966 (to P. P.) as well as National Institutes of Health Grant RO1-HL82846 and American Heart Association Grant 12EIA841007 (to Y. K. X.).
- PKA
- protein kinase A
- AKAR3
- protein kinase A activity reporter
- CREB
- cAMP-response element-binding protein
- PGC1α
- Peroxisome proliferator-activated receptor γ coactivator 1-α.
REFERENCES
- 1. Dowhan W. (1997) Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu. Rev. Biochem. 66, 199–232 [DOI] [PubMed] [Google Scholar]
- 2. Nohturfft A., Zhang S. C. (2009) Coordination of lipid metabolism in membrane biogenesis. Annu. Rev. Cell Dev. Biol. 25, 539–566 [DOI] [PubMed] [Google Scholar]
- 3. Walther T. C., Farese R. V., Jr. (2012) Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 81, 687–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Postic C., Girard J. (2008) The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab. 34, 643–648 [DOI] [PubMed] [Google Scholar]
- 5. Van den Branden C., Vamecq J. (2003) Metabolic regulation of peroxisomal and mitochondrial fatty acid oxidation. Adv. Exp. Med. Biol. 544, 307–314 [DOI] [PubMed] [Google Scholar]
- 6. Cahill G. F., Jr. (2006) Fuel metabolism in starvation. Annu. Rev. Nutr. 26, 1–22 [DOI] [PubMed] [Google Scholar]
- 7. Oh D. Y., Talukdar S., Bae E. J., Imamura T., Morinaga H., Fan W., Li P., Lu W. J., Watkins S. M., Olefsky J. M. (2010) GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tardif N., Salles J., Landrier J. F., Mothe-Satney I., Guillet C., Boue-Vaysse C., Combaret L., Giraudet C., Patrac V., Bertrand-Michel J., Migné C., Chardigny J. M., Boirie Y., Walrand S. (2011) Oleate-enriched diet improves insulin sensitivity and restores muscle protein synthesis in old rats. Clin. Nutr. 30, 799–806 [DOI] [PubMed] [Google Scholar]
- 9. Coll T., Eyre E., Rodríguez-Calvo R., Palomer X., Sánchez R. M., Merlos M., Laguna J. C., Vázquez-Carrera M. (2008) Oleate reverses palmitate-induced insulin resistance and inflammation in skeletal muscle cells. J. Biol. Chem. 283, 11107–11116 [DOI] [PubMed] [Google Scholar]
- 10. Kien C. L. (2009) Dietary interventions for metabolic syndrome: role of modifying dietary fats. Curr. Diab. Rep. 9, 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rasmussen B. B., Wolfe R. R. (1999) Regulation of fatty acid oxidation in skeletal muscle. Annu. Rev. Nutr. 19, 463–484 [DOI] [PubMed] [Google Scholar]
- 12. Schreurs M., Kuipers F., van der Leij F. R. (2010) Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes Rev. 11, 380–388 [DOI] [PubMed] [Google Scholar]
- 13. Foster D. W. (2012) Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J. Clin. Invest. 122, 1958–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sladek R., Bader J. A., Giguère V. (1997) The orphan nuclear receptor estrogen-related receptor α is a transcriptional regulator of the human medium-chain acyl coenzyme A dehydrogenase gene. Mol. Cell. Biol. 17, 5400–5409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pyper S. R., Viswakarma N., Yu S., Reddy J. K. (2010) PPARα: energy combustion, hypolipidemia, inflammation and cancer. Nucl. Recept. Signal. 8, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Narkar V. A., Downes M., Yu R. T., Embler E., Wang Y. X., Banayo E., Mihaylova M. M., Nelson M. C., Zou Y., Juguilon H., Kang H., Shaw R. J., Evans R. M. (2008) AMPK and PPARδ agonists are exercise mimetics. Cell 134, 405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Puigserver P., Spiegelman B. M. (2003) Peroxisome proliferator-activated receptor-γ coactivator 1 α (PGC-1 α): transcriptional coactivator and metabolic regulator. Endocr. Rev. 24, 78–90 [DOI] [PubMed] [Google Scholar]
- 18. Kelly D. P., Scarpulla R. C. (2004) Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 18, 357–368 [DOI] [PubMed] [Google Scholar]
- 19. Hock M. B., Kralli A. (2009) Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 71, 177–203 [DOI] [PubMed] [Google Scholar]
- 20. Scarpulla R. C., Vega R. B., Kelly D. P. (2012) Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 23, 459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodgers J. T., Lerin C., Haas W., Gygi S. P., Spiegelman B. M., Puigserver P. (2005) Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434, 113–118 [DOI] [PubMed] [Google Scholar]
- 22. Lerin C., Rodgers J. T., Kalume D. E., Kim S. H., Pandey A., Puigserver P. (2006) GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1α. Cell Metab. 3, 429–438 [DOI] [PubMed] [Google Scholar]
- 23. Gerhart-Hines Z., Rodgers J. T., Bare O., Lerin C., Kim S. H., Mostoslavsky R., Alt F. W., Wu Z., Puigserver P. (2007) Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 26, 1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fulco M., Schiltz R. L., Iezzi S., King M. T., Zhao P., Kashiwaya Y., Hoffman E., Veech R. L., Sartorelli V. (2003) Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol. Cell 12, 51–62 [DOI] [PubMed] [Google Scholar]
- 25. Cantó C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gerhart-Hines Z., Dominy J. E., Jr., Blättler S. M., Jedrychowski M. P., Banks A. S., Lim J. H., Chim H., Gygi S. P., Puigserver P. (2011) The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD(+). Mol. Cell 44, 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Arcangelis V., Liu S., Zhang D., Soto D., Xiang Y. K. (2010) Equilibrium between adenylyl cyclase and phosphodiesterase patterns adrenergic agonist dose-dependent spatiotemporal cAMP/protein kinase A activities in cardiomyocytes. Mol. Pharmacol. 78, 340–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gonçalves de Albuquerque C. F., Burth P., Younes Ibrahim M., Garcia D. G., Bozza P. T., Castro Faria Neto H. C., Castro Faria M. V. (2012) Reduced plasma nonesterified fatty acid levels and the advent of an acute lung injury in mice after intravenous or enteral oleic acid administration. Mediators Inflamm. 601032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Raatz S. K., Bibus D., Thomas W., Kris-Etherton P. (2001) Total fat intake modifies plasma fatty acid composition in humans. J. Nutr. 131, 231–234 [DOI] [PubMed] [Google Scholar]
- 30. Tholstrup T., Sandström B., Bysted A., Hølmer G. (2001) Effect of 6 dietary fatty acids on the postprandial lipid profile, plasma fatty acids, lipoprotein lipase, and cholesterol ester transfer activities in healthy young men. Am. J. Clin. Nutr. 73, 198–208 [DOI] [PubMed] [Google Scholar]
- 31. Allen M. D., Zhang J. (2006) Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem. Biophys. Res. Commun. 348, 716–721 [DOI] [PubMed] [Google Scholar]
- 32. Dominy J. E., Gerhart-Hines Z., Puigserver P. (2011) Nutrient-dependent acetylation controls basic regulatory metabolic switches and cellular reprogramming. Cold Spring Harb. Symp. Quant. Biol. 76, 203–209 [DOI] [PubMed] [Google Scholar]
- 33. Zhang P., Liu C., Zhang C., Zhang Y., Shen P., Zhang J., Zhang C. Y. (2005) Free fatty acids increase PGC-1α expression in isolated rat islets. FEBS Lett. 579, 1446–1452 [DOI] [PubMed] [Google Scholar]
- 34. Wenz T., Rossi S. G., Rotundo R. L., Spiegelman B. M., Moraes C. T. (2009) Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. U.S.A. 106, 20405–20410 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35. Boström P., Wu J., Jedrychowski M. P., Korde A., Ye L., Lo J. C., Rasbach K. A., Boström E. A., Choi J. H., Long J. Z., Kajimura S., Zingaretti M. C., Vind B. F., Tu H., Cinti S., Højlund K., Gygi S. P., Spiegelman B. M. (2012) A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481, 463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin J., Handschin C., Spiegelman B. M. (2005) Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370 [DOI] [PubMed] [Google Scholar]
- 37. Nemoto S., Fergusson M. M., Finkel T. (2005) SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem. 280, 16456–16460 [DOI] [PubMed] [Google Scholar]
- 38. Cantó C., Auwerx J. (2009) PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 20, 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baur J. A., Pearson K. J., Price N. L., Jamieson H. A., Lerin C., Kalra A., Prabhu V. V., Allard J. S., Lopez-Lluch G., Lewis K., Pistell P. J., Poosala S., Becker K. G., Boss O., Gwinn D., Wang M., Ramaswamy S., Fishbein K. W., Spencer R. G., Lakatta E. G., Le Couteur D., Shaw R. J., Navas P., Puigserver P., Ingram D. K., de Cabo R., Sinclair D. A. (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444, 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gao D., Griffiths H. R., Bailey C. J. (2009) Oleate protects against palmitate-induced insulin resistance in L6 myotubes. Br. J. Nutr. 102, 1557–1563 [DOI] [PubMed] [Google Scholar]