

Background: Dual oxidases (DUOXs) are membrane-bound ROS-generating enzymes.
Results: Conserved DUOX cysteines localized in an N-terminal domain contribute to enzymatic maturation, independent of structural stabilization.
Conclusion: Intermolecular disulfides support the interaction between DUOX enzymes and their maturation factors.
Significance: This study reflects a complex profile of protein interactions required for activity and localization of the DUOX enzymes.
Keywords: Cell Surface Protein, Chaperone Chaperonin, Cysteine-mediated Cross-linking, Disulfide, NADPH Oxidase, NOX, Protein Folding, DUOX, Maturation
Abstract
Intramolecular disulfide bond formation is promoted in oxidizing extracellular and endoplasmic reticulum compartments and often contributes to protein stability and function. DUOX1 and DUOX2 are distinguished from other members of the NOX protein family by the presence of a unique extracellular N-terminal region. These peroxidase-like domains lack the conserved cysteines that confer structural stability to mammalian peroxidases. Sequence-based structure predictions suggest that the thiol groups present are solvent-exposed on a single protein surface and are too distant to support intramolecular disulfide bond formation. To investigate the role of these thiol residues, we introduced four individual cysteine to glycine mutations in the peroxidase-like domains of both human DUOXs and purified the recombinant proteins. The mutations caused little change in the stabilities of the monomeric proteins, supporting the hypothesis that the thiol residues are solvent-exposed and not involved in disulfide bonds that are critical for structural integrity. However, the ability of the isolated hDUOX1 peroxidase-like domain to dimerize was altered, suggesting a role for these cysteines in protein-protein interactions that could facilitate homodimerization of the peroxidase-like domain or, in the full-length protein, heterodimeric interactions with a maturation protein. When full-length hDUOX1 was expressed in HEK293 cells, the mutations resulted in decreased H2O2 production that correlated with a decreased amount of the enzyme localized to the membrane surface rather than with a loss of activity or with a failure to synthesize the mutant proteins. These results support a role for the cysteine residues in intermolecular disulfide bond formation with the DUOX maturation factor DUOXA1.
Introduction
Cysteine residues often play essential roles in protein structure and function by conferring stability through disulfide bond formation, maintaining proper maturation and localization through protein-protein intermolecular interactions, or providing a thiol group for reactions with molecular substrates (1). Intramolecular disulfide bond formation, favored in oxidizing extracellular and endoplasmic reticulum (ER)2 compartments, provides structural support for native function and localization; disulfide loss can cause mistargeting or malfunction of receptors and transporters (2, 3). The mammalian peroxidases are a protein family whose rigid structure is defined by a conserved set of disulfide bonds (e.g. six in myeloperoxidase (MPO), seven in lactoperoxidase (LPO)) (4–6). These heme-containing enzymes, which catalyze the H2O2-dependent oxidation of halide and pseudohalide ions to form antimicrobial agents in phagocytes, are a critical element of the human innate immune system (7). MPO is the only member of the known mammalian peroxidases that is functional as a native homodimer linked through a disulfide bond (4, 8). This MPO oligomerization gives rise to an unusually high stability (Tm >80 °C) that is important for its physiological role. More recently, a distinct family of proteins has been identified with members that are related to the mammalian peroxidases by sequence comparisons. The NADPH oxidase (NOX)/dual oxidase (DUOX) membrane proteins are expressed in various epithelial cells and produce reactive oxygen species (ROS). In humans, two members of the NOX family of proteins, hDUOX1 and hDUOX2, contain a domain that is related by sequence to the mammalian peroxidases.
The function of the peroxidase-like domain of the two DUOX isoforms remains unclear. Although sequence identities establish a similarity to the mammalian peroxidases, the DUOX proteins lack some of the residues that in the peroxidases are known to be required for heme binding and catalytic function. Because the peroxidase-like domain is the salient difference between the DUOX enzymes and other members of the NOX family, it may be responsible for the fact that the DUOX enzymes produce H2O2, whereas the majority of the NOX family enzymes produce superoxide. Recent structural investigation of H2O2 producer NOX4 has revealed a unique large extracellular E-loop that may trap superoxide to allow conversion to H2O2, akin to the potential role of the DUOX N-terminal domain (9). Close analysis of the DUOX peroxidase-like region reveals the absence of the conserved cysteines known to confer structural stability to the mammalian peroxidases. Of the cysteine residues present in this region (seven for hDUOX11–593, six for hDUOX21–599), none corresponds by sequence alignment to a conserved disulfide pair in the mammalian peroxidases (Fig. 1A). The role of the cysteine residues that are present in the DUOX peroxidase-like region has recently been examined. Individual missense mutations of four of the cysteines in hDUOX2 (Cys351, Cys370, Cys568, Cys582) result in retention of the protein in the ER compartment, as observed by carbohydrate remodeling, indicating that these residues are critical to proper maturation (1, 10). It was speculated that the cysteines maintain a stable, N-terminal domain structure which, upon mutation, may cause misfolding and aggregation in the ER compartment. Further study demonstrated that the immature hDUOX2 is still capable of oxidant production (9). As this implies the cysteines may promote a critical protein-protein interaction rather than serving a structural function, we have modeled the N-terminal region of hDUOX2 to determine the approximate location of each of the cysteines (Fig. 1B). Modeling of the structure indicated that the thiol groups are solvent-exposed and unlikely to form intramolecular disulfide bonds due to the predicted distances between the cysteine residues.
FIGURE 1.

Comparison of sequence and structural localization of critical cysteine residues in hDUOX isoforms. A, sequence alignment of classic peroxidase domains with the hDUOX proteins. The highlighted segments shown focus on the regions encompassing the cysteine residues under study: Cys345/Cys351, Cys364/Cys370, Cys565/Cys568, Cys579/Cys582 (hDUOX1/hDUOX2). B, structural model of hDUOX21–599 produced by the SWISS-MODEL program server and PyMOL. Left, view of model structure highlighting residues investigated by point mutation in orange. Right, 45-degree y axis rotation of the model structure, illustrating residue solvent exposure and localization along one plane of the DUOX protein structure.
To obtain greater insight into the role of these cysteines in DUOX structure and function, we mutated each of four cysteine residues in both the hDUOX1 and hDUOX2 isolated peroxidase-like domains. Our experiments with these purified mutant constructs established that the mutations cause little change in the stability of the monomeric unit, in agreement with the hypothesis that these cysteine residues are solvent-exposed and do not form intramolecular disulfide bonds. However, the ability of the isolated hDUOX1 peroxidase-like domain to dimerize was affected, and the mutated full-length holoproteins did not support normal oxidant production when expressed as stable transfectants. Taken together, these findings suggest a role for these cysteines, and perhaps the surface of the domain in which they are located, in contributing to protein-protein interactions.
EXPERIMENTAL PROCEDURES
Materials, Facilities, and General Instrumentation
Sf9 cells (Invitrogen) were grown in ExCell 420TM medium (SAFC Biosciences) supplemented with glutamine (2.7 g/liter). High FiveTM cells were grown in Express FiveTM medium (Invitrogen) supplemented with glutamine (2.7 g/liter) and 10% fetal bovine serum (FBS). Both cell lines were kept in suspension at 27 °C (100 rpm) and maintained at densities between 0.5 × 106 and 2 × 106 cells/ml. Human embryonic kidney 293 (HEK) cells (ATCC CRK01573) were purchased from the American Type Culture Collection (Manassas, VA) and maintained in Dulbecco's modified Eagle's medium/Ham's nutrient mixture F-12 medium that was supplemented with 10% FBS, 100 μg/ml streptomycin, 10 mm HEPES, and 2 mm l-glutamine. Diamide, H2O2 (30% w/w), and ABTS were purchased from Sigma-Aldrich. T4 DNA ligase and restriction endonucleases were obtained from New England Biolabs. DNA sequencing was performed by Elim Biopharmaceuticals and the DNA Sequencing Facility at the University of Iowa. The entire gene insert was completely sequenced for each plasmid construct. For protein identification, mass spectra from trypsin digestion were obtained on a QSTAR-XL hybrid QqTOF mass spectrometer (Applied Biosystems). Spectrophotometric measurements were performed on a Cary 50 Bio UV-visible spectrophotometer (Varian). For experiments utilizing H2O2, concentrations were determined spectrophotometrically at 240 nm by using the molar extinction coefficient ϵ = 43.6 m−1 cm−1 (11). All experiments were performed at room temperature unless otherwise stated.
Structure Prediction
The hDUOX2 protein was truncated using the TMHMM transmembrane helix algorithm to identify the primary sequence that composes the N-terminal peroxidase-like domain (12). A structural model of hDUOX21–599 was built by sequence submission to the SWISS-MODEL program server for automatic modeling; the model with bovine LPO (Protein Data Bank ID code 3BXI) as the template was visualized with the software PyMOL (13). Heme co-factor placement in the model structure was achieved by overlay of the known bovine LPO structure and the hDUOX21–599 homology model.
Plasmid Constructs
The peroxidase-like domain constructs for hDUOX1 (residues 1–593) and hDUOX2 (residues 1–599) were described previously (14, 15). Briefly, the original source of the Homo sapiens duox11–593 gene was Quick-clone cDNA from human lung (Clontech). The gene encoding the peroxidase-like domain of hDUOX1 (residues 1–593) was PCR-amplified and inserted by ligation into pAcGP67-b (pJLM08). hduox21–599 was synthesized by GeneArt Inc. and was subcloned from the GeneArt vector into pAcGP67-b baculovirus expression vector (named pJLM025). Both plasmid constructs for hDUOX peroxidase-like domain expression encode a C-terminal His6 tag to aid protein purification.
Mutations in hduox11–593 were introduced into pJLM05 (14) by QuikChange site-directed mutagenesis (Clontech), according to the manufacturer's instructions. Primer sequences are listed in Table 1. The entire open reading frame was resequenced for each new plasmid. Each plasmid construct was subsequently digested with BamHI and EcoRI, and the gene(s) were inserted by ligation into pAcGP67-b (WT, pJLM08; C345G, pJLM072; C364G, pJLM073; C565G; pJLM074; and C579G, pJLM075). Mutations in hduox21–599 were introduced utilizing the same approach as hduox11–593; primer sequences are also listed in Table 1 (WT, pJLM025; C351G, pJLM084; C370G, pJLM085; C568G, pJLM086; and C582G, pJLM087).
TABLE 1.
Primers for mutations of hDUOX11–593 and hDUOX21–599
| Mutation | Primer | Mutation | Primer |
|---|---|---|---|
| DX1_C345G_F | 5′-catgagaaatgccagcGgccacttcc-3′ | DX2_C351G_F | 5′-gagaaatgccagcGgtcatttccggaaggtcc-3′ |
| DX1_C345G_R | 5′-ggaagtggcCgctggcatttctcatg-3′ | DX2_C351G_R | 5′-ggaccttccggaaatgacCgctggcatttctc-3′ |
| DX1_C364G_F | 5′-gctctccgggtcGgcaacagctactgg-3′ | DX2_C370G_F | 5′-cccaagctctcagggtcGgcaacaactactgg-3′ |
| DX1_C364G_R | 5′-ccagtagctgttgcCgacccggagagc-3′ | DX2_C370G_R | 5′-ccagtagttgttgcCgaccctgagagcttggg-3′ |
| DX1_C565G_F | 5′-ggcataaaggagaccccGgtccgcagccgag-3′ | DX2_C568G_F | 5′-ggtgcacccGgccctcaacctaagcagc-3′ |
| DX1_C565G_R | 5′-ctcggctgcggacCggggtctcctttatgcc-3′ | DX2_C568G_R | 5′-gctgcttaggttgagggcCgggtgcacc-3′ |
| DX1_C579G_F | 5′-gaaggcctgccagcgGgtgctccctctg-3′ | DX2_C579G_F | 5′-cggcctgccccagGgtgcacccctgac-3′ |
| DX1_C579G_R | 5′-cagagggagcacCcgctggcaggccttc-3′ | DX2_C579G_R | 5′-gtcaggggtgcacCctggggcaggccg-3′ |
The hDUOX1 and hDUOXA1 HEK293 expression constructs were generated as follows. Total RNA was extracted from well differentiated primary cultures of human airway epithelial cells and reverse transcribed using Thermoscript. The coding regions of human DUOX1 and DUOXA1 transcripts were amplified by PCR using HiFi DNA polymerase (Invitrogen) and the primers listed in Table 2. Amplification of hDUOX1 also depended on the presence of Q-solution (Qiagen; 0.5× final concentration) in the PCR buffer. The PCR products were cloned into the TOPO-TA vector (Invitrogen), sequenced, and then subcloned between the NotI and BamHI sites of the pcDNA3.1 mammalian expression vector (Invitrogen).
TABLE 2.
Primers for isolation of hDUOX1 and hDUOXA1 for HEK293 expression constructs
| Construct | Primer |
|---|---|
| hDUOX1 Forward | 5′-gcggccgccaccatgggcttctgcctggctcta-3′ |
| hDUOX1 Reverse | 5′-ggatcctctgctcaactggacagtgg-3′ |
| hDUOXA1 Forward | 5′-gcggccgccaccatggctactttgggacacacattc-3′ |
| hDUOXA1 Reverse | 5′-ggatccgcctccacggggaggaatgtta-3′ |
Generation of Recombinant Baculoviruses
Plasmids (pJLM08, pJLM025, pJLM072–75, and pJLM084–87) were co-transfected with viral baculogold DNA into Sf9 insect cells, according to the manufacturer's instructions (BD Biosciences). The resulting viruses were plaque-assayed to generate high titer recombinant baculovirus stocks amplified (two rounds of amplification in Sf9 cells) from single viral populations.
Expression and Purification of hDUOX Proteins
All hDUOX11–593 and hDUOX21–599 constructs were expressed and purified according to the previously reported procedure (14). Briefly, 1 liter of High Five cells (2.0 × 106 cells/ml) supplemented with 250 μm 5-aminolevulinic acid (Fluka) was infected with the hDUOX11–593-His6 viral stock at a multiplicity of infection of 5 for 3 days at 27 °C. After 72 h of infection, cell suspensions were concentrated and purified by nickel affinity chromatography (nickel-nitrilotriacetic acid-agarose at 4 °C, in phosphate buffer (20 mm phosphate, 400 mm NaCl, pH 8.0)). The purified proteins were stored at −20 °C, and all stock concentrations were determined in triplicate by Bradford assay (16).
Tryptophan Fluorescence Spectroscopy
The fluorescence emission spectra of DUOX11–593 Cys mutants were collected at 20 °C using a Fluorolog 3 Spectrofluorometer (Horiba Jobin Yvon), with excitation at 292 nm. Emission was monitored between 315 and 450 nm using an excitation band width of 8 nm, with slit widths set at 2.5 nm for pH 4.0 and pH 7.0 spectra. Wavelength maxima achieved for the hDUOX11–593 constructs were 341 nm (pH 4.0) and 348 nm (pH 7.0), consistent with previously published data for wild-type hDUOX11–593 (15).
Evaluation of hDUOX Secondary Structure
Far-UV circular dichroism (CD) spectra were collected on a JASCO J-815 spectropolarimeter using a cuvette path length of 1.0 mm and spectral collection in the range of 195–260 nm. All 20 °C CD experiments were conducted in 10 mm phosphate buffer, at pH 7.0 at a protein concentration of 4 μm. Temperature gradient spectra were collected at a concentration of 3 μm in 10 mm phosphate buffer, pH 7.0, over a temperature range of 20–60 °C; increasing at 2 °C/min, CD data were collected every 5 ° after 30 s of temperature stability. Raw ellipticity data were converted to mean residue ellipticity before plotting.
Creation of Stable HEK Transfectants
Mutated regions of the peroxidase-like domain of hDUOX1 were digested with restriction enzymes and inserted into the full-length hDUOX1 in pcDNA3.1(+) Zeo (from Invitrogen). Stable HEK cell lines were created as done previously (17, 18); HEK cells already stably transfected with hDUOXA1 were transfected with mutant plasmids using the Qiagen PolyFect Transfection Reagent according to the manufacturer's procedure. Stable transfectants were selected with stepwise concentrations of zeomycin (100–250 μg/ml) and cloned.
Gel Filtration Analysis of hDUOX1 and hDUOX2 Proteins
Proteins were subjected to gel filtration on a Superdex 200 10/300 column (Amersham Biosciences) equilibrated with 20 mm phosphate, 400 mm NaCl buffer, pH 8.0. Fifty microliters of each protein (40 μm) was separated at 0.5 ml/min at 4 °C and compared with gel filtration standards (Bio-Rad 151-1901) injected after reconstitution according to the manufacturer's instructions. Protein elution was monitored by the absorbance at 280 nm.
Reduction and Oxidation of hDUOX11–593
The hDUOX1 peroxidase-like domain (5 μm) was incubated with 200 mm β-mercaptoethanol (βME) in 20 mm phosphate buffer, 400 mm NaCl, pH 8.0 (4 °C) for reduction. Partial reduced protein was retained for gel analysis; reducing agent was removed from the remaining protein by sequential dilution and concentration (3×, 1:40 sample:buffer). The resulting protein was incubated at room temperature for 30 min, treated with diamide (100 μm), or untreated. All samples were loaded on SDS-PAGE for analysis.
Hydrogen Peroxidase Production by HEK Transfectants
For measurement of oxidant production, 5 × 104 wild-type or hDUOX1/hDUOXA1 cells were cultured overnight in 96-well microtiter plates. Before the assay, medium was gently removed and replaced with phosphate buffer saline (pH 7.4) containing 10 mm glucose. H2O2 production by wild-type HEK cells or hDUOX1 transfectants was measured as horseradish peroxidase-dependent (1 unit/ml) peroxidation of Amplex Red (50 μm) at 37 °C. Amplex Red peroxidation was monitored continuously at 550 nm and the amount of H2O2 produced calculated from a standard curve (0–5 nmol). The concentration of the stock H2O2 used to generate the standard curve was determined spectrophotometrically, using an extinction coefficient 43.6 m−1 cm−1 at 240 nm. In all cases, measurements of Amplex Red peroxidation were performed in triplicate.
Biosynthesis of DUOX1
Stable HEK transfectants expressing wild-type or mutant hDUOX1 with hDUOXA1 were grown in T-75 flasks and suspended in methionine-free RPMI 1640 medium with 10% dialyzed FBS and antibiotics for 1 h before biosynthetic radiolabeling, as done previously (17, 18). 25 μCi of [35S]methionine (EasyTagEXPRESS protein labeling mix, >37 TBq/mmol; Perkin-Elmer) was added, and cells were biosynthetically radiolabeled for 2 h. Cells were harvested or subjected to a chase with unlabeled methionine (100 mm) for 18 h. Cells were harvested by centrifugation and solubilized as described previously. Human DUOX1 was immunoprecipitated using a rabbit polyclonal antibody (ab106544; Abcam), and samples were separated by SDS-PAGE followed by autoradiography. Amounts of radioisotopically labeled hDUOX1 were quantitated using a PhosphorImager (Typhoon 9410; Amersham Biosciences).
RESULTS
Sequence Comparison of Peroxidase Domains Reveals That DUOX Cysteine Residues Contribute Little to Structural Stability
Cysteine residues, which are the most reactive natural amino acids due to their oxidatively susceptible thiol group, play important roles in modifying the structure and function of many proteins. Cysteine oxidation to disulfide bonds provides structural stability to many enzymes, including the mammalian peroxidases MPO and LPO, which utilize a conserved disulfide network to maintain an α-helix-rich motif. Due to the sequence and structural similarities of the DUOX peroxidase-like domain with the mammalian peroxidases, we reasoned that a comparison of the conserved structural features between these proteins might uncover important differences essential to functionality. Recently, the abundance of cysteines within the DUOX2 N-terminal region was the focus of mutagenesis experiments that suggested a role for these residues in the maturation of the DUOX proteins (1, 10, 19). To clarify the role of the cysteines, a sequence alignment analysis was performed that focused on the four cysteine residues that resulted in stalled maturation (Fig. 1A). These cysteine residues are conserved across the DUOX proteins of both lower and higher organisms, emphasizing their importance to the DUOX system. This alignment also illustrates the lack of conservation of cysteine residues between the DUOX proteins and the mammalian peroxidases, suggesting that, in contrast to MPO or LPO, these residues may not constitute a disulfide network that maintains structural integrity.
Each cysteine residue was mapped onto a structural model of the DUOX2 N-terminal region (DUOX21–599; Fig. 1B). Our previous analysis of the DUOX peroxidase-like domains (hDUOX11–593 and hDUOX21–599) showed the greatest amino acid variation to occur at the exposed extracellular face (15), a feature that may promote recognition of different interacting proteins. Each cysteine residue was found to exist as a solvent-exposed residue, situated on one face of the protein surface. The closest distance between cysteines in hDUOX2 is predicted to be 14 angstroms, between Cys351 (hDUOX1 Cys345) and Cys568 (hDUOX1 Cys565), suggesting that none of these residues is involved in disulfide bonds for structural stabilization. Due to their solvent exposure and unlikely involvement in internal disulfide bonds, we postulated that these cysteine residues may enable protein-protein interactions that are required for maturation. Two DUOX maturation factor isoforms (DUOXA1 and DUOXA2) interact specifically with their corresponding DUOX isoform (21, 22). The interaction of these two protein pairs could occur through intermolecular disulfide bonds that bridge the DUOX peroxidase-like domain and a small extracellular DUOXA domain. Interestingly, a conserved cysteine, DUOXA1/A2 Cys167, is found in both maturation factor domains (Fig. 2) (23). To explore the role(s) of the DUOX cysteine residues, they were individually mutated to glycines in both the truncated peroxidase-like domains and in the full-length DUOX proteins.
FIGURE 2.

Partial alignment of the DUOX maturation factors. The sequence alignment shown, generated by the ClustalW program, encompasses the soluble domain of DUOX maturation factors from both human (h) and mouse (m). Soluble domains were determined by the TMHMM 2.0 Server; SolhDUOXA1: 75–183 AA, SolhDUOXA2: 75–175, SolmDUOXA1: 75–181, SolmDUOXA2: 74–175. The conserved cysteine residues (Cys167) found in both DUOXA1 and DUOXA2 are highlighted in red.
Mutations of the DUOX Peroxidase-like Domain(s) Causes Significant Changes in the SDS Mobility and Oligomerization State of the Isolated Region While Structural Integrity Is Maintained
To test the role of cysteine residues in the peroxidase domain, Cys345/Cys351, Cys364/Cys370, Cys565/Cys568, and Cys579/Cys582 (hDUOX1/hDUOX2) were mutated to a glycine residue to mimic previous studies focused on hDUOX2, which suggested that membrane trafficking dysfunction was linked to these specific thiols as a result of protein misfolding (1). Each mutated DUOX protein was stably overexpressed in baculovirus and purified by nickel-nitrilotriacetic acid-agarose affinity chromatography, as reported previously for the wild-type protein (Fig. 3) (14). LC-tandem MS of the trypsin-digested hDUOX1 protein verified the presence of each mutated glycine residue. All of the purified proteins were obtained in amounts comparable with those of the wild-type domain, providing initial evidence that the cysteine residues are not essential for structural stability of the N-terminal domain. Further evidence that the mutant proteins retained overall structural integrity was provided by the absence of a notable shift in the Trp fluorescence maxima at either pH 4.0 or pH 7.0 (Fig. 3A). CD spectroscopy was performed to ascertain the relative stability of mutants versus wild-type protein under conditions of varied temperature (Fig. 3, B and C). At 20 °C, the CD profile of each mutant protein demonstrated a nearly identical CD profile to its wild-type peroxidase domain isoform. Perturbation by temperature gradient was performed for the mutation(s) that completely abolished maturation (hDUOX11–593 C579G and hDUOX21–599 C582G) and demonstrated similar profiles of unfolding and stability at the more physiologically relevant temperature of 35 °C (Fig. 3C). Heme titration of mutant constructs demonstrated no shift in the absorbance wavelength previously observed for the wild-type peroxidase-like domain, consistent with retention of a weak heme binding affinity (15).
FIGURE 3.

Structural characterization of DUOX mutants. A, tryptophan fluorescence emission spectra of DUOX11–593 cysteine mutants (C345G, diamond; C364G, circle; C565G, square; C579G, triangle), collected at 341 nm (pH 4.0, solid line) and 348 nm (pH 7.0, dashed line). B, CD spectra of hDUOX11–593 and hDUOX21–599 at 20 °C with their respective cysteine mutants compared at pH 7.0, with the spectra overlaid for comparison. C, CD spectral comparison of hDUOX11–593 versus C579G (left) and hDUOX21–599 versus C582G (right) at 35 °C, collected during temperature gradient studies. All CD experiments were conducted in 10 mm phosphate buffer. D, top, hDUOX11–593 cysteine mutants under reducing (left) and nonreducing (right) conditions. An asterisk highlights the upper proposed dimer band, seen to vary in intensity, most notably for C364G (arrow). D, bottom, hDUOX21–599 cysteine mutant set studied under reduced (left) and nonreduced (right) conditions. Obvious dimerization appears absent from wild-type and mutant proteins except for C351G (arrow).
Initial SDS-PAGE analysis of hDUOX1 protein constructs demonstrated a single band for each mutant under reducing conditions with 1.25% βME as the reductant (Fig. 3D, top, potential minor band from protein splicing observed for C364G only). Under nonreducing conditions, electrophoretic separation resulted in characteristic double band patterns composed of a high molecular mass (∼60 kDa, completely reduced protein) and low molecular mass (∼55 kDa, completely oxidized protein) band for each cysteine mutant. This effect has previously been observed in studies of human glucokinase and was linked to shifts in intramolecular disulfide bridges as a result of multiple cysteine interactions resulting in a more or less compact protein structure within the gel environment (24, 25). In our hands, each specific point mutation demonstrated a unique double band profile, perhaps due to differential disulfide bridging, though unlikely due to residue position.
Interestingly, a distinct protein at ∼120 kDa was observed under nonreducing conditions for both the wild-type and mutant hDUOX1 constructs, with the Cys364 point mutation demonstrating the least amount (Fig. 3D, top right panel, asterisk). The presumed molecular mass of this species suggests that a portion of the hDUOX1 peroxidase domain is in a homodimeric state that is susceptible to change upon reduction, implying that the state is achieved through an intermolecular disulfide interaction. The same set of conserved cysteines was mutated in the peroxidase domain of hDUOX2 to establish whether any significant differences are observed and to further clarify the recently elucidated differing nature of the two DUOX isoforms (Fig. 3D, bottom) (15). Once again, each hDUOX2 protein demonstrates a monomeric band; however, differences were noted under nonreducing conditions. No band for dimeric association was noted for WT hDUOX21–599 whereas the C351G mutation demonstrates a band suggesting dimerization and/or oligomerization (Fig. 3D, bottom right gel).
Size Exclusion Chromatography Supports Dimerization, Specific to the hDUOX1 Peroxidase Domain
To support the SDS-PAGE analysis of each DUOX peroxidase domain with a solution-based experimental approach, wild-type hDUOX11–593 was subjected to gel filtration analysis. Late eluting features, causing slight positive and negative deflections from 16 to 18 ml, were seen due to varying amount of glycerol in each sample relative to the elution buffer (26). With this technique two protein peaks were observed, suggesting multiple oligomeric states (Fig. 4). The apparent molecular masses of the peaks were determined to be 60.9 kDa for the second symmetrical peak (13.6 ml) and 133.3 kDa for the first peak at 11.9 ml. The mass of the second, evidently monomeric peak is consistent with the calculated molecular mass for hDUOX11–593 of 67.8 kDa. The first peak is close to the projected molecular mass for a dimeric hDUOX1 peroxidase domain of 135.6 kDa. The relative abundance of the dimeric species appears greater under solution phase conditions than observed via SDS-PAGE analysis, likely due to the absence of the SDS denaturant in the gel filtration study. Consistent with the polyacrylamide gel bands, the C345G, C565G, and C579G mutant proteins display slightly less dimeric protein than WT (data not shown), with C364G showing the least amount of dimeric species at a level approaching that of the fully reduced protein (Fig. 4A). hDUOX21–599 was also studied by size exclusion, to verify the unique absence of dimeric association observed with the hDUOX1 peroxidase domain. Injection of WT hDUOX21–599 (68.3 kDa) resulted in a monomeric peak eluting at the same volume as WT hDUOX11–593, consistent with their similarity in mass (Fig. 4, A and B). Low lying peaks were observed from a retention volume of 9–12.5 ml, suggestive of some nonspecific oligomerization occurring, but a significant peak for dimeric protein was not observed. SDS-PAGE analysis had suggested the presence of dimeric species by a diffuse band observed with the mutant C351G; therefore, all hDUOX21–599 mutants were also analyzed by size exclusion. Of the mutant constructs, only C351G and C582G showed unresolved peaks at 12.5 ml (Fig. 4A), within a region of oligomerization, perhaps suggesting that mutation of these cysteine residues had produced a unique minor affinity between monomeric units; however, this does not appear to be significant or favor production of a specific new protein state. To further ascertain that the dimerization observed for the hDUOX1 peroxidase domain is in fact a native, stable complex and not a product of nonspecific association, the reversibility of dimerization after disruption by reduction was studied.
FIGURE 4.

Gel filtration analysis of DUOX truncated proteins. A, comparison of hDUOX11–593 wild-type (WT), reduced (+βME), and C364G proteins (left) and hDUOX21–599 WT, C351G, and C582G (right). Arrows highlight the varying amounts of the dimeric species. B, direct comparison of hDUOX11–593 and hDUOX21–599 by size exclusion (left) and SDS-PAGE analysis (right) of hDUOX11–593 unreduced WT (lane 1) and reduced WT protein with βME (lane 2). After reductant removal, incubation at room temperature without (lane 3) or with diamide addition (lane 4) demonstrated hDUOX11–593 dimer reformation.
hDUOX11–593 Monomer/Dimer Conversion Investigated Utilizing Diamide Oxidation
Dimerization of the hDUOX1 peroxidase domain construct analyzed by both SDS-PAGE and gel filtration analysis together with the previously reported instability of the hDUOX1 construct compared with a more stable monomeric hDUOX21–599, argue for the in vivo relevance of this interaction (15). This evidence diminishes the likelihood of a nonspecific protein-protein interaction, and data collected for the C364G hDUOX11–593 mutated protein suggests dimerization occurs through a disulfide bond. To help support the specificity of this interaction and its direct association with thiol groups, a solution of purified hDUOX11–593 was fully reduced by exposure to βME (Fig. 4B, gel). In this reduced state, as shown previously, no dimer was observed. The reduced protein was dialyzed extensively to remove the reducing agent and then incubated for 30 min at room temperature, with or without diamide to determine whether oxidation can return the protein to a dimeric state. Oxidation achieved by atmospheric exposure alone demonstrated a small amount of dimer, whereas dimerization was returned to levels observed prior to reduction with the thiol-specific oxidant diamide. This supports the hypothesis that disulfide bonds result in specific dimerization of hDUOX11–593 and demonstrates that protein reduction did not disrupt dimer formation through denaturation of the overall monomeric protein structure. Review of the structural models of each DUOX N-terminal region shows that Cys364 of hDUOX1 is solvent-exposed; however, the corresponding conserved cysteine of hDUOX2 (Cys370) is partially buried and turned inward, suggesting that this cysteine may be unavailable for dimerization due to its position, in accord with our experimental results (Fig. 5).
FIGURE 5.

Overlay of DUOX1 and DUOX2 peroxidase-like domain model structures. The predicted structures of hDUOX1 (cyan) and hDUOX2 (green) N-terminal regions were overlaid to demonstrate the difference in the relative positions of the conserved cysteine residues Cys364 (red) of hDUOX1 and Cys370 (purple) of hDUOX2.
Normal and Mutant DUOX1 Expression and Activity in Stable Transfectants
To determine how the mutation of specific extracellular cysteines influences the activity and cellular fate of hDUOX1, we created stable HEK transfectants expressing wild-type and mutant hDUOX1 along with normal hDUOXA1. The agonist-dependent production of H2O2 was significantly depressed in lines expressing mutant hDUOX1 (Table 3), with the activity of C579G hDUOX1 essentially the same as that of nontransfected HEK cells.
TABLE 3.
Cysteine mutation effects on hDUOX1 system activity in stable transfectants
H2O2 production by wild-type and transfected cell lines stimulated with phorbol 12-myristate 13-acetate and ionomycin was quantitated spectrophotometrically as peroxidation of Amplex Red (n = 8). In parallel, the relative amounts of immunoreactive hDUOX1 protein in membrane-enriched fractions from HEK transfectants were assessed by immunoblotting; these data are expressed relative to levels in transfectants expressing normal hDUOX1.
| Cell line | H2O2 production | Stable protein level |
|---|---|---|
| nmol H2O2/5 × 104 cells in 2 h | % | |
| Wild-type HEK cells | 0.03 ± 0.01 | |
| DUOX1/A1 | 2.12 ± 0.22 | 100 |
| C345G/A1 | 0.16 ± 0.03 | 20.5 |
| C364G/A1 | 0.64 ± 0.10 | 40.8 |
| C565G/A1 | 0.15 ± 0.04 | 6.0 |
| C579G/A1 | 0.04 ± 0.02 | 2.2 |
Because reduced extracellular generation of H2O2 could reflect compromised production or subcellular targeting of mutant hDUOX1, as well as defective oxidase activity, we compared the biosynthesis of normal and mutant DUOX1 by pulse-chase radiolabeling (Fig. 6A). Immunoprecipitation of biosynthetically radiolabeled hDUOX1 demonstrated that all of the mutants were initially expressed at levels that exceeded ≥7-fold that of the wild-type protein (Fig. 6B). However, during the chase period, there was more rapid loss of mutant protein than of normal DUOX1, suggesting instability of the mutant proteins during processing and membrane targeting. Consistent with this interpretation, immunoblots of membrane-enriched fractions of wild-type and mutant hDUOX1/hDUOXA1 lines demonstrated reduced amounts of each of the mutants that paralleled the reduction in oxidase activity (Table 3). This observation implies that the protein fraction that reached the plasma surface was fully functional and that the effect of each mutation was to disrupt a protein association required for maturation, not to compromise structural stability or functionality of each enzyme.
FIGURE 6.
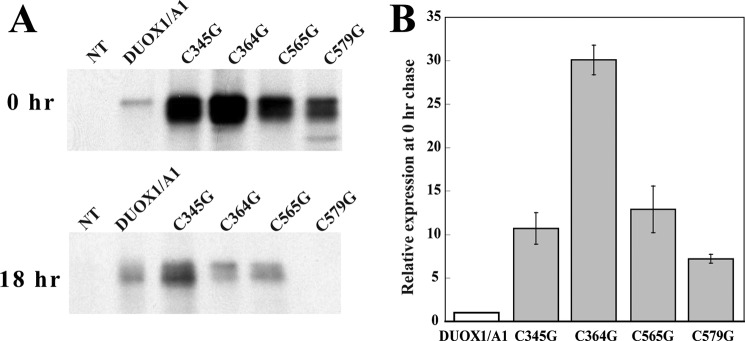
Biosynthesis and activity of hDUOX1 mutants. A, wild-type nontransfected HEK (NT) and transfected HEK cells expressing normal (hDUOX1/A1) or hDUOX1 mutants were pulse-labeled for 2 h, harvested (0 h chase), or chased (18 h) with cold methionine, and immunoprecipitated with anti-hDUOX1 antibody. Immunoprecipitates were separated by SDS-PAGE and subjected to autoradiography. DUOX1-related proteins were quantitated by a PhosphorImager. The gel is a representative of two independent experiments. B, amounts of radiolabeled mutant hDUOX1 were compared with the amount of normal hDUOX1 synthesized under the same conditions (0 h chase). Values are normalized to that for normal hDUOX1, and the data are the mean ± S.D. (error bars) for two independent pulse-chase experiments.
DISCUSSION
The absence of critical cysteine residues in enzymatic systems due to mutation has been linked to protein trafficking disorders. Specifically, thyroid dysfunction resulting in goiters is associated with missense mutations of cysteines in the thyroglobulin gene that result in its retention in the ER (27, 28). Recently, cysteine mutations in the DUOX2 protein, a ROS-generating enzyme that is highly expressed in the thyroid, have also been reported to result in failed maturation from the ER (1, 10, 19). These studies have highlighted the importance of conserved thiol residues within the hDUOX2 N-terminal peroxidase-like domain for proper targeting to the plasma membrane.
The DUOX enzymes have structural and functional features that are atypical among members of the NOX protein family. Most notably, they possess a large, extracellular domain that has sequence similarity with mammalian peroxidases, and, akin to NOX4, their stimulation yields hydrogen peroxide as the detectable oxidant (29–32). Comprehensive characterization of the two membrane-bound isoforms has been hindered by the inability to purify the full-length enzyme, a limitation circumvented in part by structural and topology modeling of important features (14, 15, 33, 34). Here, we have utilized structure prediction to identify the location of four cysteine residues previously shown to be important for DUOX2 maturation, establishing both that they are likely to be solvent-exposed and that their spatial orientation places them beyond the limits of internal disulfide bond formation (Fig. 1B). We further characterized the role of these conserved residues, not only in hDUOX2 but also in hDUOX1, through point mutations. Replacement of these specific cysteine residues did not significantly affect the structural stability of the isolated domain, as assessed through protein production levels, tryptophan fluorescence, circular dichroism, and heme binding studies.
Replacement of Cys364 with a glycine disrupted the previously unrecognized self-association of the hDUOX11–593 N-terminal region. Gel analysis and size exclusion chromatography confirmed the homodimerization of the hDUOX1-truncated construct, a behavior not shared by the hDUOX2 peroxidase-like domain (Figs. 3 and 4). Dimerization is one means by which proteins confer stabilization; therefore the observed hDUOX1 complexation is consistent with our previous demonstration that the peroxidase-like hDUOX1 domain is less stable under neutral conditions than the N-terminal region of hDUOX2 (15). Perhaps hDUOX1 dimerization is required for stability to achieve the correct structure or interaction with its maturation factor, hDUOXA1. Further analysis of the hDUOX1 dimeric state through reductive disruption and oxidative recovery of the dimeric protein with a thiol-specific oxidant, diamide, supported the conclusion that this complexation depends upon one or more disulfide bonds. Due to the sequence and structural similarities of the DUOX N-terminal region with those of the mammalian peroxidases, it is important to note that myeloperoxidase is capable of dimerizing through a disulfide bond to afford high structural stability (4, 8). Although it remains to be demonstrated in the context of the full-length DUOX proteins, it is interesting to consider that dimerization may be one factor that differentiates the DUOX isoforms. Differences in dimerization could affect localization and functionality, as homodimerization is known to be required for function in certain enzymatic systems, including caspases, metalloproteins, and serine/threonine kinases (35–41). To test the possibility that dimerization may also play a role in the type of ROS detected, hDUOX1 HEK mutant transfectants, including C364G, were assayed for both hydrogen peroxide and superoxide production; however, no significant superoxide anion was detected (data not shown).
Investigation of the localization of HEK293 transfectants, both of full-length wild-type hDUOX1/A1 and full-length mutant hDUOX1 (C345G, C364G, C565G, and C579G)/A1 demonstrated defective membrane translocation, consistent with previous investigations in which conserved cysteines in hDUOX2 were mutated (1, 10). Our findings show that mutation of these cysteines did not disrupt the stability of the peroxidase-like region. Furthermore, analysis of the full-length system demonstrated that the level of hydrogen peroxide produced by mutant constructs correlated with the amount of stably expressed protein (Table 3), suggesting that these cysteine residues did not diminish activity, but only compromised maturation. These data support a model in which direct interaction of hDUOX1 and its hDUOXA1 maturation factor occurs through contact of cysteine residue(s) in their extracellular domains (Fig. 7). This protein-protein interaction surface is then perturbed by introduction of functionally inert glycine residues in positions previously occupied by cysteine thiols. The replacement of critical cysteine residues with glycine likely compromises proper folding of hDUOX1 as well, thus enlisting ER-associated degradation of misfolded precursors. Although the fate of mutant hDUOX1 that failed to associate with hDUOXA1 was not assessed in our studies, it is conceivable that without productive interactions with hDUOXA1 in the ER, some of the mutant hDUOX1 underwent degradation in the proteasome as part of the quality control system operative during protein synthesis. Such is the fate of Y173C, a specific missense mutation underlying one genotype of inherited MPO deficiency; pharmacologic inhibition of proteasome activity in cells expressing Y173C rescues the mutant protein from degradation, although it does not restore peroxidase activity in the cell (42).
FIGURE 7.

Model of potential hDUOX1 protein and maturation factor association in vivo. Interactions of the peroxidase-like domain of hDUOX1 (green) with both a soluble extracellular region of the hDUOXA1 maturation factor (orange) and itself are demonstrated. Cys345, Cys364, Cys565, and Cys579 are highlighted, demonstrating their relative positions within the peroxidase-like domain of DUOX1. A potential disulfide bonding interaction may exist between a cysteine within the hDUOXA1 domain and a hDUOX1 cysteine (possibly Cys579). The loss of in vitro dimerization upon mutation of Cys364 within the N-terminal region of hDUOX1 suggests that this residue was responsible for self-association of the hDUOX1 peroxidase-like domain through a disulfide interaction, illustrated here in the context of the full-length protein.
We propose that the most important residue for interaction at the extracellular plane of hDUOX proteins is Cys579/Cys582 (hDUOX1/hDUOX2), as it is the only residue whose mutation completely eliminates membrane trafficking. Other cysteines may form further contact points for stability within the full-length system, either with the maturation factor, or within the DUOX protein itself at the extracellular membrane spanning loops that lead to varied levels of disturbance to maturation. Previous studies have also demonstrated loss of DUOX-thyroid peroxidase association upon both introduction of the irreversible cysteine modification agent N-ethylmaleimide or single site mutations (10, 20). This suggests DUOX-thyroid peroxidase association is dependent upon cysteine interactions to stabilize the protein complex. Findings from this study also suggest the potential for homodimeric protein association for the hDUOX1 enzyme. Although confirmation is necessary in the context of the full-length system, it is possible that a disulfide-based interaction, either sustained or transient, is required to stabilize the hDUOX1 protein. This interaction may define yet another difference between the DUOX isoforms, as it was not observed for the more stable hDUOX2 protein.
Acknowledgments
We thank Karine Reiter and the Narum laboratory for the use of their JASCO J-815 spectropolarimeter. Mass spectrometric proteomic analyses were carried out by Bio-Organic Biomedical Mass Spectrometry Resource at the University of California (director, A. L. Burlingame) supported by National Center for Research Resources Grant P41RR001614.
This work was supported by National Institutes of Health Grants DK 30297 (to P. R. O. M.), HL 090830 (to B. B.), and AI 70958 and AI 044642 (to W. M. N.). This work was also supported by the resources and use of facilities at the Iowa City Department of Veterans Affairs Medical Center, Iowa City, IA (to W. M. N.).
- ER
- endoplasmic reticulum
- ABTS
- 2,2′-azino-bis(3-ethylbenzothiazoleline-6-sulfonic acid
- βME
- β-mercaptoethanol
- DUOX
- dual oxidase
- LPO
- lactoperoxidase
- MPO
- myeloperoxidase
- NOX
- NADPH oxidase
- ROS
- superoxide + H2O2.
REFERENCES
- 1. Morand S., Agnandji D., Noel-Hudson M. S., Nicolas V., Buisson S., Macon-Lemaitre L., Gnidehou S., Kaniewski J., Ohayon R., Virion A., Dupuy C. (2004) Targeting of the dual oxidase 2 N-terminal region to the plasma membrane. J. Biol. Chem. 279, 30244–30251 [DOI] [PubMed] [Google Scholar]
- 2. Tarnow P., Schoneberg T., Krude H., Gruters A., Biebermann H. (2003) Mutationally induced disulfide bond formation within the third extracellular loop causes melanocortin 4 receptor inactivation in patients with obesity. J. Biol. Chem. 278, 48666–48673 [DOI] [PubMed] [Google Scholar]
- 3. Hänggi E., Grundschober A. F., Leuthold S., Meier P. J., St-Pierre M. V. (2006) Functional analysis of the extracellular cysteine residues in the human organic anion transporting polypeptide, OATP2B1. Mol. Pharmacol. 70, 806–817 [DOI] [PubMed] [Google Scholar]
- 4. Zeng J., Fenna R. E. (1992) X-ray crystal structure of canine myeloperoxidase at 3 Å resolution. J. Mol. Biol. 226, 185–207 [DOI] [PubMed] [Google Scholar]
- 5. Singh A. K., Singh N., Sharma S., Singh S. B., Kaur P., Bhushan A., Srinivasan A., Singh T. P. (2008) Crystal structure of lactoperoxidase at 2.4 Å resolution. J. Mol. Biol. 376, 1060–1075 [DOI] [PubMed] [Google Scholar]
- 6. Wolf S. M., Ferrari R. P., Traversa S., Biemann K. (2000) Determination of the carbohydrate composition and the disulfide bond linkages of bovine lactoperoxidase by mass spectrometry. J. Mass Spectrom. 35, 210–217 [DOI] [PubMed] [Google Scholar]
- 7. Klebanoff S. J. (2005) Myeloperoxidase: friend and foe. J. Leukoc. Biol. 77, 598–625 [DOI] [PubMed] [Google Scholar]
- 8. Banerjee S., Stampler J., Furtmüller P. G., Obinger C. (2011) Conformational and thermal stability of mature dimeric human myeloperoxidase and a recombinant monomeric form from CHO cells. Biochim. Biophys. Acta 1814, 375–387 [DOI] [PubMed] [Google Scholar]
- 9. Takac I., Schröder K., Zhang L., Lardy B., Anilkumar N., Lambeth J. D., Shah A. M., Morel F., Brandes R. P. (2011) The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 286, 13304–13313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fortunato R. S., Lima de Souza E. C., Ameziane-el Hassani R., Boufraqech M., Weyemi U., Talbot M., Lagente-Chevallier O., de Carvalho D. P., Bidart J. M., Schlumberger M., Dupuy C. (2010) Functional consequences of dual oxidase-thyroperoxidase interaction at the plasma membrane. J. Clin. Endocrinol. Metab. 95, 5403–5411 [DOI] [PubMed] [Google Scholar]
- 11. Hildebrandt A. G., Roots I., Tjoe M., Heinemeyer G. (1978) Hydrogen peroxide in hepatic microsomes. Methods Enzymol. 52, 342–350 [DOI] [PubMed] [Google Scholar]
- 12. Krogh A., Larsson B., von Heijne G., Sonnhammer E. L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 [DOI] [PubMed] [Google Scholar]
- 13. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 14. Meitzler J. L., Ortiz de Montellano P. R. (2009) Caenorhabditis elegans and human dual oxidase 1 (DUOX1) “peroxidase” domains: insights into heme binding and catalytic activity. J. Biol. Chem. 284, 18634–18643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meitzler J. L., Ortiz de Montellano P. R. (2011) Structural stability and heme binding potential of the truncated human dual oxidase 2 (DUOX2) peroxidase domain. Arch. Biochem. Biophys. 512, 197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 17. Goedken M., McCormick S., Leidal K. G., Suzuki K., Kameoka Y., Astern J. M., Huang M., Cherkasov A., Nauseef W. M. (2007) Impact of two novel mutations on the structure and function of human myeloperoxidase. J. Biol. Chem. 282, 27994–28003 [DOI] [PubMed] [Google Scholar]
- 18. Loughran N. B., Hinde S., McCormick-Hill S., Leidal K. G., Bloomberg S., Loughran S. T., O'Connor B., O'Fagáin C., Nauseef W. M., O'Connell M. J. (2012) Functional consequence of positive selection revealed through rational mutagenesis of human myeloperoxidase. Mol. Biol. Evol. 29, 2039–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ameziane-El-Hassani R., Morand S., Boucher J. L., Frapart Y. M., Apostolou D., Agnandji D., Gnidehou S., Ohayon R., Noël-Hudson M. S., Francon J., Lalaoui K., Virion A., Dupuy C. (2005) Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. J. Biol. Chem. 280, 30046–30054 [DOI] [PubMed] [Google Scholar]
- 20. Song Y., Ruf J., Lothaire P., Dequanter D., Andry G., Willemse E., Dumont J. E., Van Sande J., De Deken X. (2010) Association of duoxes with thyroid peroxidase and its regulation in thyrocytes. J. Clin. Endocrinol. Metab. 95, 375–382 [DOI] [PubMed] [Google Scholar]
- 21. Morand S., Ueyama T., Tsujibe S., Saito N., Korzeniowska A., Leto T. L. (2009) DUOX maturation factors form cell surface complexes with DUOX affecting the specificity of reactive oxygen species generation. FASEB J. 23, 1205–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luxen S., Noack D., Frausto M., Davanture S., Torbett B. E., Knaus U. G. (2009) Heterodimerization controls localization of DUOX-DUOXA NADPH oxidases in airway cells. J. Cell Sci. 122, 1238–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grasberger H., Refetoff S. (2006) Identification of the maturation factor for dual oxidase: evolution of a eukaryotic operon equivalent. J. Biol. Chem. 281, 18269–18272 [DOI] [PubMed] [Google Scholar]
- 24. Tiedge M., Krug U., Lenzen S. (1997) Modulation of human glucokinase intrinsic activity by SH reagents mirrors post-translational regulation of enzyme activity. Biochim. Biophys. Acta 1337, 175–190 [DOI] [PubMed] [Google Scholar]
- 25. Tiedge M., Richter T., Lenzen S. (2000) Importance of cysteine residues for the stability and catalytic activity of human pancreatic beta cell glucokinase. Arch. Biochem. Biophys. 375, 251–260 [DOI] [PubMed] [Google Scholar]
- 26. Kang D. C., Venkataraman P. A., Dumont M. E., Maloney P. C. (2011) Oligomeric state of the oxalate transporter, OxlT. Biochemistry 50, 8445–8453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim P. S., Hossain S. A., Park Y. N., Lee I., Yoo S. E., Arvan P. (1998) A single amino acid change in the acetylcholinesterase-like domain of thyroglobulin causes congenital goiter with hypothyroidism in the cog/cog mouse: a model of human endoplasmic reticulum storage diseases. Proc. Natl. Acad. Sci. U.S.A. 95, 9909–9913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hishinuma A., Takamatsu J., Ohyama Y., Yokozawa T., Kanno Y., Kuma K., Yoshida S., Matsuura N., Ieiri T. (1999) Two novel cysteine substitutions (C1263R and C1995S) of thyroglobulin cause a defect in intracellular transport of thyroglobulin in patients with congenital goiter and the variant type of adenomatous goiter. J. Clin. Endocrinol. Metab. 84, 1438–1444 [DOI] [PubMed] [Google Scholar]
- 29. De Deken X., Wang D., Many M. C., Costagliola S., Libert F., Vassart G., Dumont J. E., Miot F. (2000) Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J. Biol. Chem. 275, 23227–23233 [DOI] [PubMed] [Google Scholar]
- 30. Martyn K. D., Frederick L. M., von Loehneysen K., Dinauer M. C., Knaus U. G. (2006) Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 18, 69–82 [DOI] [PubMed] [Google Scholar]
- 31. Serrander L., Cartier L., Bedard K., Banfi B., Lardy B., Plastre O., Sienkiewicz A., Fórró L., Schlegel W., Krause K. H. (2007) NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 406, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Geiszt M., Witta J., Baffi J., Lekstrom K., Leto T. L. (2003) Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 17, 1502–1504 [DOI] [PubMed] [Google Scholar]
- 33. Edens W. A., Sharling L., Cheng G., Shapira R., Kinkade J. M., Lee T., Edens H. A., Tang X., Sullards C., Flaherty D. B., Benian G. M., Lambeth J. D. (2001) Tyrosine cross-linking of extracellular matrix is catalyzed by DUOX, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J. Cell Biol. 154, 879–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leto T. L., Morand S., Hurt D., Ueyama T. (2009) Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox Signal. 11, 2607–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Riedl S. J., Fuentes-Prior P., Renatus M., Kairies N., Krapp S., Huber R., Salvesen G. S., Bode W. (2001) Structural basis for the activation of human procaspase-7. Proc. Natl. Acad. Sci. U.S.A. 98, 14790–14795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen M., Orozco A., Spencer D. M., Wang J. (2002) Activation of initiator caspases through a stable dimeric intermediate. J. Biol. Chem. 277, 50761–50767 [DOI] [PubMed] [Google Scholar]
- 37. Renatus M., Stennicke H. R., Scott F. L., Liddington R. C., Salvesen G. S. (2001) Dimer formation drives the activation of the cell death protease caspase 9. Proc. Natl. Acad. Sci. U.S.A. 98, 14250–14255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koo B. H., Kim Y. H., Han J. H., Kim D. S. (2012) Dimerization of matrix metalloproteinase-2 (MMP-2): Functional implication in MMP-2 activation. J. Biol. Chem. 287, 22643–22653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Günther V., Davis A. M., Georgiev O., Schaffner W. (2012) A conserved cysteine cluster, essential for transcriptional activity, mediates homodimerization of human metal-responsive transcription factor-1 (MTF-1). Biochim. Biophys. Acta 1823, 476–483 [DOI] [PubMed] [Google Scholar]
- 40. Constantinescu Aruxandei D., Makbul C., Koturenkiene A., Lüdemann M. B., Herrmann C. (2011) Dimerization-induced folding of MST1 SARAH and the influence of the intrinsically unstructured inhibitory domain: low thermodynamic stability of monomer. Biochemistry 50, 10990–11000 [DOI] [PubMed] [Google Scholar]
- 41. Singh M., Kumar P., Karthikeyan S. (2011) Structural basis for pH dependent monomer-dimer transition of 3,4-dihydroxy 2-butanone-4-phosphate synthase domain from Mycobacterium tuberculosis. J. Struct. Biol. 174, 374–384 [DOI] [PubMed] [Google Scholar]
- 42. DeLeo F. R., Goedken M., McCormick S. J., Nauseef W. M. (1998) A novel form of hereditary myeloperoxidase deficiency linked to endoplasmic reticulum/proteasome degradation. J. Clin. Invest. 101, 2900–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]