

Background: Telomerase is an essential enzyme for the immortalization of stem and cancer cells.
Results: Dyrk2-associated E3 ligase targets telomerase reverse transcriptase, a catalytic subunit of telomerase.
Conclusion: Dyrk2-E3 ligase is necessary to negatively regulate telomerase activity.
Significance: Learning how telomerase is regulated is crucial for understanding telomerase regulatory mechanism in cancer and stem cells.
Keywords: Breast Cancer, Protein Degradation, Telomerase, Telomeres, Ubiquitination, Dyrk2, E3 Ligase, TERT, Ubiquitination
Abstract
Telomerase maintains the telomere, a specialized chromosomal end structure that is essential for genomic stability and cell immortalization. Telomerase is not active in most somatic cells, but its reactivation is one of the hallmarks of cancer. In this study, we found that dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 2 (Dyrk2) negatively regulates telomerase activity. Dyrk2 phosphorylates TERT protein, a catalytic subunit of telomerase. Phosphorylated TERT is then associated with the EDD-DDB1-VprBP E3 ligase complex for subsequent ubiquitin-mediated TERT protein degradation. During the cell cycle, Dyrk2 interacts with TERT at the G2/M phase and induces degradation. In contrast, depletion of endogenous Dyrk2 disrupts the cell cycle-dependent regulation of TERT and elicits the constitutive activation of telomerase. Similarly, a Dyrk2 nonsense mutation identified in breast cancer compromises ubiquitination-mediated TERT protein degradation. Our findings suggest the novel molecular mechanism of kinase-associated telomerase regulation.
Introduction
In stem and regenerating cells, maintenance of genomic stability is essential for self-renewal and the subsequent transmission of accurate genetic information to progenitor cells. In eukaryotes, telomerase overcomes the end-replication problem by adding the telomeric repeat sequence (TTAGGG) to the ends of chromosomes (1, 2). In somatic cells, the absence of telomerase results in the gradual loss of telomeric repeats with every cell division, which is referred to as a telomere crisis. These cells typically undergo growth arrest and cellular senescence (3–5). However, genetic and epigenetic defects in oncogenic pathways can induce abnormal cell proliferation, resulting in cancer (6, 7). Importantly, the re-activation of telomerase in most cancers prevents a telomere crisis and leads to cellular immortality.
Telomerase is composed of two subunits: telomerase RNA template (TERC),2 an RNA template, and telomerase reverse transcriptase (TERT) (8). The results of several studies suggest that telomerase activity is modulated at various steps, including TERC biosynthesis, TERT transcription, TERT protein modification, telomerase holoenzyme formation, and telomere-associated proteins (9–17). Recent studies have shown that Wnt/β-catenin signaling participates in transactivating TERT in cancer and stem cells (18, 19), highlighting the importance of the regulatory mechanism of TERT in controlling telomerase activity. Although telomerase's pivotal roles in cancer have been extensively studied, its regulatory mechanism in such pathological conditions is still unclear. In this study, we determined an important telomerase regulatory mechanism. We found that the dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 2 (Dyrk2)-associated E3 ligase complex is involved in degradation of TERT protein and subsequent inhibition of telomerase activity.
EXPERIMENTAL PROCEDURES
Mammalian Cell Culture
HeLa, 293T, and MCF-7 cells were maintained with Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS). Mouse embryonic stem cells were maintained in DMEM, 20% FBS, and leukemia inhibitory factor (1000 units per ml) on feeders that were isolated from 13.5-day-old mouse embryos. HeLa cells that stably expressed either FLAG-TERT or HA-Dyrk2 were established by transduction with 3FLAG-TERT-pMGIB and 3HA-Dyrk2-pWZL retroviruses.
Constructs
Full-length or fragments of TERT, Dyrk2, and FOXO3A were cloned into HA-tagged pcDNA3.1, FLAG-tagged pcDNA3.1, HA-tagged pWZL (retrovirus), FLAG-tagged pMIGB (retrovirus), and pGEX-6T vectors. Myc-tagged EDD, Myc-tagged VprBP, S-protein/FLAG/streptavidin-binding protein-tagged DDB1, Cul4A, and Roc1 constructs were provided by J. Chen (The University of Texas M.D. Anderson Cancer Center, Houston, TX) and R. Maddika (Centre for DNA Fingerprinting and Diagnostics, Hyderabad, India). The Dyrk2 K178R and TERT S457A mutants were generated via polymerase chain reaction (PCR). Dyrk2 shRNA lenti-viral plasmids (Sigma) and retroviral plasmids (pMGIB and pWZL) were packaged into 293T cells with standard protocols.
Small Scale Kinase Screening
23 genes encoding kinases in various signaling pathways were subcloned into pcDNA3.1 and pCS2 expression vector. Then, HeLa cells stably expressing FLAG-TERT were transiently transfected with each construct. 36 h after transfection, cell lysates were analyzed for immunoblotting with anti-FLAG antibody (M2; Sigma).
Pulse-chase Labeling Assay
HeLa-S3 cells that stably expressed FLAG-TERT were washed and pre-incubated for 1 h with 10% Met- and Cys-free DMEM in FBS. The medium was then replaced with fresh Met- and Cys-free medium containing 1.48 × 106 Bq of [35S]Met or -Cys, and the cells were incubated for 1 h before being washed and incubated in complete medium for each time point. Two milligrams of cell lysates were immunoprecipitated with anti-FLAG-agarose (Sigma). The samples were subjected to 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), followed by autoradiography.
Immunoblotting and Immunoprecipitation
Whole cell lysates were prepared using Nonidet P-40 lysis buffer (0.5% Nonidet P-40, 1.5 mm MgCl2, 25 mm HEPES, 150 mm KCl, 10% glycerol, 1 mm phenylmethanesulfonyl fluoride (PMSF), 12.7 mm benzamidine HCl, 0.2 mm aprotinin, 0.5 mm leupeptin, and 0.1 mm pepstatin A) for 20 min at 4 °C, followed by centrifugation (14,000 rpm, 10 min). Supernatant fractions were denatured with 5× SDS sample buffer (200 mm Tris-Cl, pH 6.8; 40% glycerol; 8% SDS, 200 mm dithiothreitol; and 0.08% bromphenol blue) at 95 °C for 5 min, followed by SDS-PAGE. For immunoblot blocking and antibody incubation, 0.1% nonfat dry milk in Tris-buffered saline with Tween-20 (25 mm Tris-HCl, pH 8.0; 125 mm NaCl; and 0.5% Tween-20) was used. Super Signal West Pico and Femto (Pierce Biotechnology) reagents were used to detect horseradish peroxidase-conjugated secondary antibodies. For immunoprecipitation, cells on 10-cm plates were lysed with 0.3 ml of Nonidet P-40 lysis buffer for 20 min at 4 °C, followed by spinning at 14,000 rpm for 10 min. One hundred microliters of extract (10% of input) were saved for later immunoblotting. The cell lysates were incubated with 20 μl of HA-agarose or FLAG-magnetic beads at 4 °C overnight. Immunoprecipitates were washed with Nonidet P-40 lysis buffer four times, eluted using SDS sample buffer, and analyzed by immunoblotting. The following antibodies were used: anti-Dyrk2 (Abcam), FLAG (M2, Sigma), HA (12CA5, HA-7, and 3F10), Myc (9E10), Brg-1 (Santa Cruz Biotechnology), BAF57 (Santa Cruz Biotechnology), PCNA (Santa Cruz Biotechnology), TPP1 (Bethyl), TRF2 (Santa Cruz Biotechnology), and tubulin (Sigma).
Telomerase Rapid Amplification Protocol (TRAP) Assay
Cells (293T or MCF-7) were lysed with 100 μl of Nonidet P-40 lysis buffer containing 1 mm PMSF, 12.7 mm benzamidine HCl, 0.2 mm aprotinin, 0.5 mm leupeptin, 0.1 mm pepstatin A, and 400 units of RNase inhibitor. The cell lysates were analyzed using the TRAP reaction assay with the TRAPEZE telomerase detection kit (Chemicon) or real-time PCR (20).
Cell Synchronization
HeLa cells with FLAG-tagged TERT- and HA-tagged Dyrk2 were synchronized using a double thymidine block. Cells were harvested, washed with phosphate-buffered saline, and fixed with ice-cold 70% ethanol. Cells were then treated with 5 μg/μl RNase A and 50 μg/μl propidium iodide for 30 min at 37 °C and analyzed on a C6 flow cytometer (Accuri).
Transfection
HeLa S3 and 293T cells were transfected with plasmids and siRNAs using Lipofectamine 2000 (Invitrogen) in accordance with the manufacturer's protocol. Control siRNA, VprBP siRNAs (siRNA1, GGAGGGAAUUGUCGAGAAU; siRNA2, CCACAGAAUUUGUUGCGCA; siRNA3, GGAAUGACACUGUGCGCUU; siRNA4, CGGAGUUGGAGGAGGACGA), and Dyrk2 siRNAs (siRNA1, CAAAUGGGCUUACAACAGU; siRNA2, UCACGUGGCUUACAGGUAU; siRNA3, GGUGCUAUCACAUCUAUAU; siRNA4, GGCCUACGAUCACAAAGUC) were purchased from Dharmacon.
Semiquantitative PCR
One microgram of RNA extracted from each cell was used to generate cDNA samples (Superscript II; Invitrogen). Next, cDNA was analyzed using standard PCR. The sequence information for each primer is available upon request.
GST Pull-down Assay
GST-enhanced green fluorescent protein (EGFP; control) and Dyrk2 proteins were purified from an Escherichia coli BL21 strain using a standard procedure. The purified protein (0.1 μg) was incubated with HeLa-FLAG-TERT cell lysates for 30 min and precipitated using glutathione-Sepharose 4B beads (GE Healthcare). Then precipitates were analyzed using IB with an anti-FLAG antibody.
Ubiquitination Assay
For in vivo ubiquitination assay, HeLa S3 cells were transiently co-transfected with pCMV-HA-tagged UBC and 3FLAG-tagged TERT-pcDNA3.1, and subsequently immunoprecipitated with HA-agarose (HA-7). The precipitates were then immunoblotted with an anti-FLAG antibody. In vitro ubiquitination reactions were performed at 30 °C for 8 h in 30 μl of ubiquitination reaction buffer (40 mm Tris-HCl, pH 7.6, 2 mm dithiothreitol, 5 mm MgCl2, 0.1 m NaCl, 2 mm ATP) containing 100 μm ubiquitin, 20 nm E1 (UBE1), 100 nm UbcH5b (all from Boston Biochem), and EDVP E3 ligase components (50 ng each of EDD, DDB1, VprBP, and Dyrk2). A FLAG-TERT substrate was generated using a TNT-coupled reticulocyte lysate system (Promega). After the ubiquitination reaction, samples were boiled in an SDS-PAGE loading buffer. Ubiquitination of TERT was monitored using IB with anti-ubiquitin and anti-FLAG antibodies.
In Vitro Kinase Assay
Wild-type and mutant (S457A) TERT expressed in bacterial cells (BL21) were purified using glutathione-Sepharose 4B beads. FLAG-tagged Dyrk2 expressed in 293T cells was immunoprecipitated using anti-FLAG-agarose and used as a kinase source. The Dyrk2 kinase and TERT substrate were incubated in a kinase reaction buffer (10 mm HEPES, pH 7.5, 50 mm NaCl, 10 nm MgCl2, 10 mm MnCl2, 1 mm EGTA, 1 mm dithiothreitol, 10 mm NaF, 10 μCi [γ-32P]) for 60 min at 30 °C. Kinase reactions were resolved using SDS-PAGE and autoradiography.
RESULTS
Dyrk2-induced TERT Ubiquitination and Degradation
To understand the regulatory mechanism of telomerase, we first compared the level of TERT mRNA transcript with telomerase activity in various cells, including human embryonic stem cells, mouse embryonic stem cells, mouse embryonic fibroblasts, and HeLa cells. Interestingly, we observed that the level of the TERT mRNA transcript was not correlated with telomerase activity in several cell lines (supplemental Fig. S1), indicating the existence of an additional layer of telomerase activity regulation beyond TERT transcription. Among several interpretations of this discrepancy between TERT transcription and telomerase activity, we hypothesized that post-transcriptional TERT regulation determines telomerase activity. Then, we measured TERT protein half-life using a pulse-chase labeling assay. HeLa cells stably expressing FLAG-tagged TERT (HeLa-FLAG-TERT) were metabolically labeled with [35S]methioinine and immunoprecipitated with FLAG antibody for subsequent autoradiography. We found that the half-life of TERT protein was ∼2.1 h (Fig. 1, A and B), suggesting that telomerase activity might be determined by TERT protein stability.
FIGURE 1.

TERT protein down-regulation by Dyrk2-associated E3 ligase complex. A and B, instability of TERT protein. In HeLa cells that stably expressed FLAG-TERT (HeLa-TERT), total translated proteins were labeled with [35S]Met and chased for each time point (0, 2, 4, and 6 h). Cell lysates were immunoprecipitated (FLAG) and analyzed using autoradiography (A). Solid arrow, full-length of TERT; empty arrows, cleaved TERT fragments. The half-life (T1/2) of TERT protein was calculated using an ImageJ analysis (B). The plot was derived from the intensity of the TERT protein on autoradiography (A). n = 3; error bars, S.D. C, Dyrk2 down-regulation of TERT protein expression. HeLa (GFP-expressing control) and HeLa-TERT cells were transiently transfected with three plasmids (Dyrk2, Dyrk3, and FOXO3A; 24 h) and harvested to quantify TERT protein level using IB (FLAG). FOXO3A was used as a negative control. The expression of HA-Dyrk2, Dyrk3, and HA-FOXO3A was confirmed using IB (HA). D, Dyrk2 down-regulation of TERT protein expression in a dose-dependent manner. HeLa-TERT cells were transfected with HA-Dyrk2 at different concentrations (0.5, 1, and 2 μg of plasmids) and then analyzed using IB. Brg-1 and BAF57 were used as negative controls. E, Dyrk2 inhibition of telomerase activity. 293T and HeLa cells were transiently transfected (24 h) with Dyrk2 for TRAP assays. An RNase A-treated sample of cell lysates (lane 5) was used as a negative control. F, HeLa-TERT cells were transfected with Dyrk2 siRNA (36 h) and analyzed using IB (TERT and Dyrk2). G, HeLa-TERT cells were stably transduced with shRNA against Dyrk2 (clones #3 and #4) and analyzed using IB (lanes 2 and 4). Dyrk2 depletion-induced up-regulation of TERT protein was rescued by shRNA nontargetable Dyrk2-pMGIB retroviral infection (lanes 3 and 5). H and I, increased TERT protein stability by Dyrk2 depletion. HeLa-TERT stably expressing shGFP or shDyrk2 cells was treated with cycloheximide (CHX) at each time point. Then cell lysates were analyzed for IB (H) and quantified using ImageJ (I).
Protein stability is generally controlled by the ubiquitin-mediated proteasome pathway (21). Specific post-translational modifications of a target protein are recognized by the E1-E3 ubiquitination cascade, which initiates proteasome-mediated degradation of the target protein (22). Of the various types of post-translational modifications, we focused on kinase-induced target protein phosphorylation and ubiquitination. To identify kinases involved in down-regulation of TERT protein, we performed small-scale kinase screening. Briefly, we transiently overexpressed plasmids encoding individual kinases in HeLa-FLAG-TERT cells, and analyzed the level of TERT protein using immunoblotting (IB) for FLAG-TERT. We found that a member of Dyrk family proteins significantly downregulated TERT (Fig. 1C). Dyrk proteins are categorized into two groups: 1) Dyrk1A and Dyrk1B, and 2) Dyrk2, Dyrk3, and Dyrk4 (23). We observed that Dyrk1A did not down-regulate TERT protein level, but Dyrk2 and Dyrk3 did, indicating substrate specificity among the Dyrk proteins. Additionally, ectopic expression of Dyrk2 decreased the level of TERT protein in a dose-dependent manner (Fig. 1D). Next, we examined whether Dyrk2-induced down-regulation of TERT protein is independent of transcriptional down-regulation of TERT. Indeed, ectopic expression of Dyrk2 did not down-regulate endogenous TERT transcripts in HeLa cells. To further analyze the effects of Dyrk2 on TERT transcription, we established TERT knock-in mouse embryonic stem cells (mESCs) that harbor tdTomato fluorescent protein in place of TERT allele (TERTtdTomato/+). tdTomato expression represents the transcriptional level of TERT. Consistently, Dyrk2 expression did not affect endogenous TERT promoter activity (supplemental Fig. S2). These data suggest that Dyrk2 down-regulates TERT protein at post-transcriptional level. Next, we sought to determine whether Dyrk2-mediated TERT inhibition affects telomerase activity. We ectopically expressed Dyrk2 in 293T and HeLa cells, and performed the TRAP assay. Similar to IB results, the overexpression of Dyrk2 decreased telomerase activity. To compensate Dyrk2 gain-of-function analysis, we also employed Dyrk2 knockdown approaches. Depletion of endogenous Dyrk2 using either small interfering RNA (siRNA) or short hairpin RNA (shRNA) increased the level of TERT protein in HeLa-FLAG-TERT cells (Fig. 1F). Moreover, Dyrk2 knockdown (shRNA)-induced TERT stabilization was reverted by introducing shRNA non-targetable Dyrk2, confirming the specific on-target effects of Dyrk2 shRNA on TERT stabilization (Fig. 1G). Additionally, we examined whether Dyrk2 depletion affects TERT protein half-life using cycloheximide (CHX) treatment in HeLa-FLAG-TERT cells (shGFP control versus shDyrk2). We found that Dyrk2 depletion increased TERT protein half-life in HeLa cells (Fig. 1, H and I). These data suggest that Dyrk2 specifically destabilizes TERT protein and subsequently inhibits telomerase activity.
Dyrk2 Binds to TERT and Phosphorylates Its Serine Residue
We next explored the molecular mechanism of Dyrk2-mediated TERT protein down-regulation. First, we tested whether Dyrk2 is associated with TERT. Coimmunoprecipitation (co-IP) of ectopically expressed TERT and Dyrk2 exhibited the reciprocal protein interaction in 293T cells (Fig. 2A). Additionally, a glutathione S-transferase (GST) pull-down assay of GST-Dyrk2 from HeLa-FLAG-TERT cell lysates showed that TERT protein bound directly to Dyrk2 (Fig. 2B). Moreover, we examined the endogenous interaction between TERT and Dyrk2. Due to absence of antibody detecting endogenous TERT protein (24), we utilized triple-HA epitope knockin mESCs (TERTHA/+) for co-IP assays. By gene targeting in mESCs, HA epitope was inserted into start codon of TERT to express HA-TERT fusion protein (25). Indeed, endogenous TERT protein is associated with Dyrk2 in mESCs (Fig. 2C). To further analyze TERT-Dyrk2 protein interaction, we performed binding domain mapping using truncated mutants of TERT and Dyrk2. We observed that the reverse transcriptase (RT) domain and C-terminal regions in TERT and the kinase domain in Dyrk2 are responsible for TERT-Dyrk2 interaction (Fig. 2, D and E). These data suggest that TERT protein is physically associated with Dyrk2 protein.
FIGURE 2.
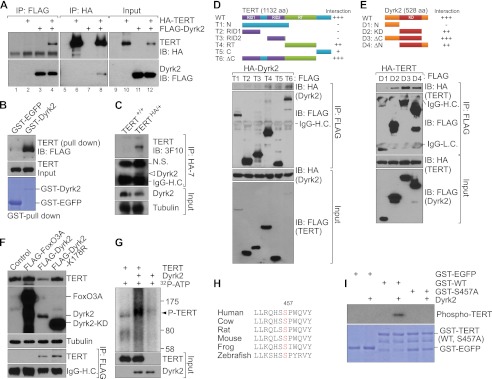
Dyrk2 binds to TERT and phosphorylates its serine residue. A, binding of Dyrk2 to TERT. 293T cells were transiently transfected with FLAG-TERT and HA-Dyrk2 plasmids. Twenty-four hours later, 293T cell lysates were analyzed using reciprocal IP with either anti-HA or anti-FLAG antibodies. Dyrk2-TERT interaction was detected (lane 4, upper panel; lane 8, lower panel). B, direct interaction of Dyrk2 with TERT. Purified GST-EGFP (control) and GST-Dyrk2 proteins were used for a GST pull-down assay by incubating each protein with HeLa-TERT cell lysates. The resulting GST-protein complex was resolved using IB (FLAG). C, association of endogenous TERT with Dyrk2. Endogenous TERT-Dyrk2 interaction was analyzed using IP (HA) and IB (Dyrk2) with parental (TERT+/+) and TERT knockin (TERTHA/+) mouse embryonic stem cells. D and E, binding domain mapping of the TERT-Dyrk2 interaction. D, various FLAG-TERT fragments and full-length HA-Dyrk2 plasmids were cotransfected into 293T cells. After 36 h, each lysate of these cells was analyzed using IP (FLAG) and IB (HA). Of note is that T4, T5, and T6 deletion mutants interacted with Dyrk2, indicating that a C-terminal domain including the RT domain is necessary for TERT-Dyrk2 interaction. RID1 and 2, RNA-interacting domain 1 and 2; N, N terminus; C, C terminus; ΔC, C terminus-deleted mutant. E, each FLAG-Dyrk2 fragment with full-length HA-tagged TERT (HA-TERT) was cotransfected into 293T cells. Lysates of these cells were analyzed using IP (FLAG) and IB (HA). D1 fragment lacking a kinase domain failed to interact with TERT protein. Also, deletion of the C terminus in Dyrk2 was dispensable for TERT interaction, indicating that the Dyrk2 kinase domain is sufficient for TERT interaction. N, N terminus; KD, kinase-dead; ΔC, ΔC terminus-deleted mutant; ΔN, N terminus-deleted mutant. F, kinase activity of Dyrk2 is not required for TERT-Dyrk2 interaction but is necessary for TERT protein degradation. In HeLa cells stably expressing HA-TERT, the plasmids FOXO3A, Dyrk2, and Dyrk2-kinase dead mutant were transiently transfected (24 h) for assessment of TERT protein expression using IB (HA) and TERT association with Dyrk2 (IP, FLAG; IB, HA). Of note, whereas the KD mutant still bound to TERT (lanes 4, lower panel), it did not down-regulate TERT protein expression (lane 4, top panel). G, Dyrk2 phosphorylation of TERT protein. In vitro transcribed and translated TERT (substrate) and Dyrk2 (kinase) were incubated with [32P]ATP. Dyrk2 phosphorylated TERT (lane 2, arrowhead). H, evolutionary conservation of the Dyrk2 consensus substrate sequence (R/KXX(X)S/TP) in TERT in vertebrates. Serine (red): candidate phosphorylation site for Dyrk2. I, phosphorylation of TERT at Ser457 by Dyrk2. GST-EGFP (control), wild-type TERT, and S457A (mutant) TERT fragments were incubated with Dyrk2 for in vitro kinase assay. GST-EGFP served as a negative control for the kinase reaction. Wild-type TERT was phosphorylated by Dyrk2 (lane 4), but the S457A mutant was not (lane 6).
Dyrk2 belongs to a serine/threonine protein kinase family (26), implying that Dyrk2 might phosphorylate TERT protein and lead to degradation. To address this, we examined whether Dyrk2's kinase activity is required for TERT protein destabilization. A point mutation (K178R) in the kinase domain of Dyrk2 results in loss of its kinase activity (27). We ectopically expressed wild-type or K178R Dyrk2 in HeLa-FLAG-TERT cells and performed IB for TERT. Intriguingly, a K178R kinase-inactive mutant of Dyrk2 failed to induce TERT protein degradation (Fig. 2F), suggesting that Dyrk2-induced TERT phosphorylation is required for TERT degradation. Nonetheless, co-IP assays showed that Dyrk2 K178R can still bind to TERT (Fig. 2F). Next, we examined whether Dyrk2 phosphorylates TERT protein using in vitro kinase assay. As shown in Fig. 2G, Dyrk2 specifically phosphorylated TERT protein. Then, we located the evolutionarily conserved Dyrk2 consensus phosphorylation sequence (RKXX[X]S/TP) (28) at Ser457 in TERT's RNA-interacting domain 2 (Fig. 2H). To verify Ser457 as a Dyrk2 target serine residue, we utilized a site-specific TERT mutant (Ser to Ala at 457) as a substrate for kinase assay. Indeed, S457A mutant was not phosphorylated by Dyrk2, while wild-type TERT was phosphorylated (Fig. 2I). These data showed that Dyrk2 specifically phosphorylated TERT at Ser457. These results suggested that Dyrk2 induces TERT phosphorylation, which is required for TERT protein down-regulation.
The Dyrk2-EDVP E3 Ligase Complex Ubiquitinates TERT for Degradation
Previously, it was suggested that Dyrk2 acts as a scaffolding protein for ubiquitination substrates and the E3 ligase complex EDD, DDB1, and VprBP (EDVP) (29). Thus, we hypothesized that Dyrk2 and its associated EDVP E3 ligase complex are involved in Dyrk2-mediated TERT protein down-regulation. First we questioned whether Dyrk2 induces ubiquitination of TERT for protein degradation. To address this, we performed in vivo ubiquitination assays. Indeed, in HeLa cells, ectopic expression of Dyrk2 moderately increased TERT ubiquitination (Fig. 3A), suggesting that Dyrk2 down-regulates TERT protein via ubiquitination. Next, we examined whether the EDVP E3 ligase complex is also associated with TERT protein. In the EDVP E3 ligase complex, 1) EDD functions as a homologous to the E6-AP C terminus (HECT) E3 ligase (30), 2) DDB1 plays a dual role as a linker between the cullin E3 ligase and a substrate receptor (31), and 3) VprBP, also known as a DDB1-cullin-associated factor, serves as a substrate receptor for target protein ubiquitination (32). As expected, in co-IP experiments, we found that all components of the EDVP E3 ligase interacted with the TERT protein in HeLa cells (Fig. 3, B and C). Interestingly, similar to Dyrk2's effects on telomerase, ectopic expression of DDB1 or VprBP decreased telomerase activity. However, EDD had little effect on telomerase activity (Fig. 4, D and E). Given that only the DDB1-VprBP module specifies the target recognition by physical binding, EDD alone may be insufficient for destabilization of TERT protein. Conversely, depletion of VprBP using siRNA increased TERT protein (Fig. 3F), suggesting that VprBP functions as a substrate receptor for TERT in EDVP E3 ligase complexes. Importantly, we found that TERT-EDD and TERT-VprBP interactions were impaired upon depletion of Dyrk2 (Fig. 3G), supporting the previously identified role of Dyrk2 as a scaffolding protein between substrates and the EDVP E3 ligase (29). Also, in vitro ubiquitination assays showed that Dyrk2 was indispensable for EDVP-mediated TERT ubiquitination (Fig. 3H). These data suggest that the Dyrk2-EDVP E3 ligase complex ubiquitinates TERT.
FIGURE 3.
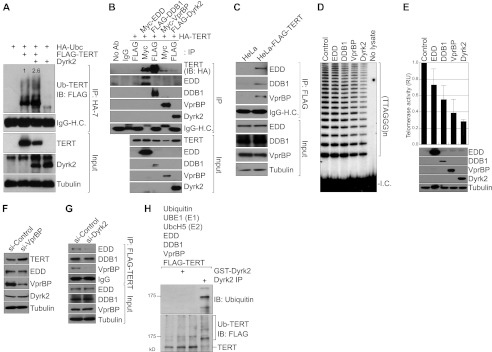
The Dyrk2-EDVP E3 ligase complex ubiquitinates TERT for degradation. A, Dyrk2-increased TERT ubiquitination. HeLa cells were transiently transfected with UBC, TERT, and Dyrk2 plasmids. After 36 h, cells were collected for IP (HA-tagged UBC) and IB (FLAG-TERT) analysis. Dyrk2 down-regulated the total level of TERT protein expression (lanes 2 and 3, input), and increased TERT ubiquitination (lanes 2 and 3, IP). Blots were normalized by TERT input and quantified using ImageJ. B, association of the EDVP E3 ligase complex with TERT. 293T cells transfected with HA-TERT, Myc-tagged EDD, FLAG-tagged DDB1, Myc-tagged VprBP, or FLAG-tagged Dyrk2 were analyzed using IP (FLAG or Myc) and IB (HA). Of note is that each component of the EDVP complex bound to TERT protein (lanes 4–7, top panel). C, association of TERT with endogenous EDVP E3 ligase components. HeLa-TERT cell lysates were analyzed using IP and IB. D and E, inhibition of telomerase activity by the EDVP E3 ligase complex. 293T cells transiently transfected with each EDVP component were analyzed for telomerase activity using TRAP assays (D). To quantify the effects of Dyrk2-EDVP on telomerase activity, the TRAP assay results were quantified using the ImageJ software program (E) and plotted (n = 3). The error bars indicate S.D. RU, relative unit. F, VprBP depletion stabilizes TERT protein. HeLa-TERT cells transfected with VprBP siRNA were analyzed using IB. Of note is that knockdown of VprBP expression increased the level of TERT protein, indicating an endogenous function of VprBP in TERT destabilization. G, mediation of the TERT-EDVP E3 ligase complex by Dyrk2. HeLa-TERT cells were transfected with a control siRNA (si-Control) or Dyrk2 siRNA (si-Dyrk2), and analyzed using IP (FLAG) and IB. Under Dyrk2-depleted conditions, both EDD and VprBP exhibited decreased binding to TERT. Of note is that DDB1-TERT interaction was not affected by Dyrk2 shRNA, indicating an EDVP-independent DDB1 association with TERT. H, requirement of Dyrk2 for EDVP E3 ligase-mediated TERT ubiquitination. For in vitro ubiquitination assays of TERT, E1, E2, and E3 components, and Dyrk2 were incubated with TERT protein. Next, ubiquitinated TERT was analyzed using IB (FLAG and ubiquitin). In the presence of Dyrk2, TERT was ubiquitinated (lane 4). Of note, bacterially expressed GST-Dyrk2 failed to ubiquitinate TERT because of a lack of post-translational modification of GST-Dyrk2 activity (tyrosine phosphorylation in the activation loop of Dyrk2; lane 2).
FIGURE 4.

Cell cycle-dependent TERT regulation by Dyrk2. A, cell cycle of HeLa (GFP-expressing control cells) and HeLa-FLAG-TERT + HA-Dyrk2 cells was arrested by treatment with thymidine (2 mm, 24 h), serum starvation (48 h), or nocodazole (100 μm, 14 h). Cell lysates from each condition were analyzed by co-immunoprecipitation (HA) and immunoblotting (FLAG) assays. TERT protein level was inversely correlated with the TERT-Dyrk2 association (see lanes 3 and 5). Blots were quantified using ImageJ. B—D, HeLa-FLAG-TERT + HA-Dyrk2 cells were synchronized using thymidine double block (200 nm, more than 17 h each) and released for cell cycle progression. At each time point (0, 2, 4, 6, and 10 h), cells were analyzed using flow cytometry (B) and co-immunoprecipitation (HA)-immunoblotting (FLAG) assays (C), physical association between TERT and Dyrk2 during the cell cycle was quantified and plotted (ImageJ) (D) (n = 2). When the TERT protein level decreased during the G2/M phase, the TERT-Dyrk2 association increased. TERT:Dyrk2/TERT indicates the normalized TERT-Dyrk2 interaction by total TERT protein level (asterisk). E and F, HeLa-TERT cells transiently transfected with siControl or siDyrk2 were synchronized by thymidine double block and released for cell cycle progression. At different time points (0, 2, 4, 6, and 8 h), HeLa-TERT cells were analyzed using immunoblotting assays for TERT protein (E). TERT protein levels were quantified using ImageJ and plotted (n = 3) (F). Compared with the control (lanes 2–8), Dyrk2-depleted HeLa-TERT cells exhibited an overall increase in TERT protein levels (lanes 10–14). Error bars, S.D.; NC: nocodazole treatment (100 μm, 14 h). G, hyperactivation of endogenous telomerase activity by Dyrk2 knockdown. MCF-7 cells were stably transduced with lentivirus expressing shGFP (control) or shDyrk2. shDyrk2 non-targetable Dyrk2 retrovirus was also stably transduced for reconstitution assay (lane 3). Cell lysates were prepared for TRAP using real-time PCR. n = 3; Student's t test; error bars, S.D.
Cell Cycle-dependent Association of Dyrk2 and TERT
Telomerase activity is only detectable in self-renewing cells, including stem cells, regenerating cells, and germ cells (2, 33), but its reactivation is strongly associated with cell immortalization in human cancer (34, 35), suggesting that telomerase activity should be tightly controlled during cellular and tissue homeostasis. Therefore, we hypothesized that Dyrk2 controls TERT protein under normal physiological conditions, such as the cell cycle. Because telomere synthesis mainly occurs in the S phase of cell cycle (36), Dyrk2 likely plays a role in eliminating the TERT protein after the S phase. Prior to analyzing Dyrk2's effects on TERT, we determined whether Dyrk2 affected the cell cycle. In our experimental setting, we observed that Dyrk2 overexpression or knockdown did not affect cell cycle (supplemental Fig. S3), which rules out the effects of Dyrk2-induced cell cycle arrest on TERT protein regulation.
Because the event that Dyrk2 binds to and phosphorylates TERT seems to be a prerequisite for TERT protein ubiquitination and degradation, the TERT-Dyrk2 physical interaction may be a key event in regulating telomerase activity during the cell cycle. To address this, we induced cell cycle arrest at each stage using a specific treatment: thymidine for 24 h, serum starvation for 48 h, and nocodazole for 14 h in HeLa cells; we then analyzed the interaction between TERT and Dyrk2 using co-IP assays. Intriguingly, we found that the TERT-Dyrk2 protein interaction mainly occurred in G2/M phase-arrested cells. Moreover, the TERT-Dyrk2 interaction was correlated with a decrease in TERT protein levels (Fig. 4A, lane 5). Conversely, TERT weakly interacted with Dyrk2 in S phase-arrested cells with the increase in TERT protein levels (Fig. 4A, lane 3), indicating the inversely proportional relationship between TERT protein levels and TERT-Dyrk2 interaction. These results suggest that Dyrk2 conditionally binds to TERT protein only in the G2/M phase of the cell cycle for ubiquitin-mediated TERT protein degradation. To further validate these results, we analyzed TERT-Dyrk2 interaction in each cell cycle stage of synchronized HeLa-FLAG-TERT cells (Fig. 4B). Consistent with the results using cell cycle-arrested HeLa cells, the synchronized HeLa cells displayed an increase in the TERT-Dyrk2 association in the G2/M phase, with a decreased level of TERT protein (Fig. 4C, lane 5). This was also manifested by quantitative analysis showing the increased TERT-Dyrk2 interaction at G2/M phase (Fig. 4D). These results recapitulate that TERT-Dyrk2 interaction is a prerequisite for TERT protein degradation and strongly suggest that the dynamic association of Dyrk2 with TERT is an essential biological event that negatively modulates telomerase activity in a cell-cycle-dependent manner.
Next, we validated our observation that Dyrk2 specifically destabilizes TERT protein during the G2/M phase of the cell cycle via a Dyrk2 loss-of-function approach. Using shRNA-mediated depletion of endogenous Dyrk2, we determined whether Dyrk2 depletion compromises cell cycle-dependent regulation of TERT protein stability. Indeed, knockdown of endogenous Dyrk2 disrupted G2/M phase-specific down-regulation of TERT protein, resulting in the constitutive up-regulation of TERT protein, independently of cell cycle (Fig. 4, E and F). Of note, nocodazole treatment decreased TERT protein levels (Fig. 4E, lane 7), as also shown in Fig. 4A (lane 5) and Fig. 4C (lane 6). However, Dyrk2 knockdown abolished the effects of nocodazole-induced TERT down-regulation (Fig. 4E, compare lane 7 and 14; F), indicating that Dyrk2 is necessary to down-regulate TERT protein at the G2/M phase of the cell cycle. Although it is evident that Dyrk2 targets TERT protein in a cell cycle-dependent manner, our studies utilized cells that ectopically expressed TERT. To validate the effects of Dyrk2 on endogenous telomerase activity, we examined whether depletion of endogenous Dyrk2 enhances endogenous telomerase activity in MCF-7 cells. shRNA-mediated Dyrk2 knockdown markedly increased telomerase activity. By contrast, introduction of shRNA non-targetable Dyrk2 reverted telomerase activity (Fig. 4G), verifying the specific effects of Dyrk2 on telomerase activity. These results suggest that Dyrk2 is a key molecule in controlling TERT protein during the cell cycle.
Dyrk2 Nonsense Mutation Compromises TERT Protein Destabilization
Telomerase reactivation is observed in 90% of human cancers (35). Having determined that Dyrk2 negatively regulates telomerase activity via ubiquitin-mediated protein degradation, we reasoned that Dyrk2-mediated telomerase regulation is associated with cancer. To evaluate this, we determined whether any components of the Dyrk2-EDVP E3 ligase complex were transcriptionally suppressed or genetically inactivated in human cancer using the Oncomine database (www.oncomine.org). We found no down-regulation or genetic inactivation of EDVP components, but we observed that Dyrk2 is somatically mutated in human breast cancer from the analysis of the COSMIC database (www.sanger.ac.uk/genetics/CGP/cosmic/). These Dyrk2 mutations were initially identified in genome-wide analyses of various cancers (37–39) (supplemental Fig. S4). Of the Dyrk2 somatic mutations, which are mostly missense mutations in Dyrk2's kinase domain, we focused on the Dyrk2 S471X nonsense mutation (X stands for the stop codon generated by the frameshift mutation) in human breast cancer cells. Dyrk2-S471X mutant lacks the Kelch motif, which may be responsible for interaction with E3 ubiquitin ligase (Fig. 5A) (40, 41). Therefore, the Dyrk2-S471X nonsense mutation likely comprises an endogenous telomerase degradation mechanism. To test this, we determined whether Dyrk2 S471X down-regulates TERT protein in HeLa-FLAG-TERT cells. Dyrk2-S471X mutant failed to decrease TERT protein levels, whereas wild-type Dyrk2 induced TERT protein degradation (Fig. 5B). Consistent with the protein level of TERT, TRAP assays also showed that ectopic expression of Dyrk2 S471X mutant was unable to inhibit telomerase activity (Fig. 5C). Because the Kelch motif is missing in Dyrk2 S471X mutant, Dyrk2 S471X's inability to target TERT protein may be resulted from the loss of interaction with EDVP E3 ligase components. Thus, we evaluated the interaction between Dyrk2-S471X mutant and EDVP E3 ligase components. Indeed, S471X mutant no longer bound to the EDD and VprBP proteins (Fig. 5D). These results suggest that Dyrk2 mutant fails to target TERT protein and leads to the constitutive hyperactivation of telomerase.
FIGURE 5.

Dyrk2 mutant fails to induce TERT de-stabilization. A, three-dimensional structure of Dyrk2 protein. DOI: 10.2210/pdb3k2l/pdb. The protein kinase domain (blue) is based on PFAM (PF00069). Y382 is in the activation loop of the kinase domain for autophosphorylation. The S471X nonsense mutation results in the partial loss (two α-helices and two β-sheets) of the kinase domain (red). B, Dyrk2 S471X had no effects on TERT protein down-regulation. HeLa-TERT cells were transiently transfected with Dyrk2 (wild-type) or S471X mutant. Twenty-four hours after transfection, cells were analyzed for TERT quantification using an immunoblotting assay. (FLAG). Blots were normalized by tubulin and quantified using ImageJ. C, Dyrk2 S471X mutant fails to inhibit telomerase activity. 293T cells were transiently transfected with wild-type or S471X Dyrk2 plasmids and analyzed for telomerase activity using a TRAP assay. I.C.: internal control. Ladders indicate telomeric repeats (TTAGGG), as represented by six-base pair increments. Autoradiography was quantified using ImageJ. D, S471X nonsense mutation of Dyrk2 disrupts interaction with EDVP E3 ligase components. HeLa cells were transiently transfected with wild-type or S471X mutant Dyrk2 plasmids and analyzed for protein interaction by co-immunoprecipitation (EDD and VprBP) and immunoblotting assays.
DISCUSSION
In this study, we identified an novel regulatory mechanism of telomerase. Dyrk2 and its associated EDVP E3 ligase complex specifically target TERT protein via ubiquitination-mediated degradation. Importantly, the genetic mutation in Dyrk2 identified in breast cancer cells fails to elicit TERT protein degradation, implying that Dyrk2 mutation-induced telomerase deregulation may contribute to tumorigenesis (Fig. 6).
FIGURE 6.

Illustration of Dyrk2-mediated telomerase regulation. A, in normal cells expressing telomerase, Dyrk2 phosphorylates TERT protein (i). Then, phosphorylated TERT is recognized by the Dyrk2-associated EDD-DDB1-VprBP E3 ligase complex (ii) for subsequent ubiquitination and protein degradation of TERT. In contrast, Dyrk2 mutation (S471X) fails to recruit the EDVP-E3 ligase to TERT protein, which did not induce TERT ubiquitination and degradation. Of note, the upstream regulatory mechanism of Dyrk2 is still ambiguous. B, cell cycle-dependent TERT regulation by Dyrk2-E3 ligase. Stable TERT protein is associated with TERC, a RNA template of telomerase, and synthesizes a telomere repeat sequence at the early S phase of the cell cycle. During the G2/M phase, the Dyrk2-E3 ligase targets TERT protein for degradation and inhibits telomerase activity.
Telomerase activity is strictly controlled at multiple levels, including TERC biosynthesis, TERT transcription, telomerase holoenzyme formation, telomerase recruitment to telomere, and telomere-associated proteins. Telomere elongation is coupled with DNA replication and cell cycle progression (36). In studies in budding yeast, cell cycle-dependent telomere elongation was regulated by various safeguards: (i) telomerase recruitment to the telomere by Cdk1-induced Cdc13 phosphorylation in yeast, (ii) changes in the telomere's chromosomal structure, and (iii) telomere end processing (9, 42–44). Accumulating evidence indicates that various cell cycle-related proteins are regulated at the protein level by kinase-mediated ubiquitination and degradation at different cell cycle checkpoints (45, 46). In addition, transcriptional activation of TERT by Wnt/β-catenin signaling is required for telomerase activity in cancer and stem cells (18, 19). These studies suggest that telomerase activity is tightly controlled to allow full activation of telomerase only in specific phases of the cell cycle. We found that the Dyrk2-associated E3 ubiquitin ligase complex negatively regulates telomerase in the G2/M phase. However, Dyrk2 depletion stabilizes TERT protein, resulting in constitutive activation of telomerase, regardless of the cell cycle checkpoint. Importantly, Dyrk2 knockdown also reverses the effects of nocodazole on TERT protein down-regulation (see Fig. 4). These findings underline the essential function of Dyrk2-E3 ligase complex in regulating TERT protein during cell cycle.
The EDVP components DDB1 and VprBP are also part of the Cul4A-Roc1 E3 ligase complex (29). In this complex, DDB1 functions as a linker protein between Cul4-Roc1 and VprBP (47). DDB1 is relatively larger than the substrate receptors for other proteins in the cullin family, implying that it is versatile in protein-protein interactions (31). Additionally, this suggests the various roles of DDB1 in diverse physiological events. In agreement with this, we found that TERT was also associated with Cul4A and Roc1 E3 ligase (supplemental Fig. S5). Nonetheless, Cul4A and Roc1 failed to destabilize TERT protein. Instead, Cul4A slightly enhanced telomerase activity in HeLa cells (supplemental Fig. S6), suggesting that Cul4-mediated TERT ubiquitination might provide an additional layer of telomerase activity regulation via a nondegradation mechanism.
Previous studies suggested that TERT protein is ubiquitinated by several E3 ligases, including MKRN1, CHIP, and HDM2 (48–50). Although these studies suggest the effects of ubiquitination of TERT on regulating telomerase activity, their pathological relevance remains elusive. In our study, we found that Dyrk2 mutation (S471X) in breast cancer disrupts the interaction between Dyrk2 and the EDVP E3 ligase complex, which leads to interruption of Dyrk2-mediated TERT protein degradation. Dyrk2 phosphorylates p53 at serine 46 and induces apoptosis upon DNA damage (27). Therefore, Dyrk2 may play a key role in cellular homeostasis through two different mechanisms: (i) eliminating irreparable DNA-damaged cells through p53-induced cell death and (ii) limiting telomerase activity in specific self-renewal cells and during the S phase of the cell cycle. Thus, a truncated mutation in Dyrk2 may compromise both the p53-mediated DNA damage response and cell cycle-dependent telomerase regulation, which may contribute to tumorigenesis. Thus, it will be necessary to further examine the pathological association of telomerase deregulation with Dyrk2's genetic inactivation in human cancers. Additionally, employing genetically engineered mouse models for Dyrk2 knock-out will address the function of Dyrk2 in regulating p53 and telomerase in vivo.
The Dyrk2 level remains constant during the cell cycle, without intracellular re-distribution (data not shown). These results suggest that the unknown upstream regulator(s) may control Dyrk2-TERT interaction. Previously, it was shown that Dyrk2 phosphorylation by ATM controls nuclear stabilization of Dyrk2 (51), implying possible connection of telomerase regulation with DNA damage response via Dyrk2. Therefore, it will be interesting to identify the upstream regulator of Dyrk2 in telomerase regulation. Additionally, it is also necessary to evaluate the effects of Dyrk2 on telomerase's non-telomeric functions in stem cells and cancer (25, 52–57) (Fig. 6A). Taken together, our findings suggest that Dyrk2-associated E3 ligase complex induces TERT destabilization, and propose a novel molecular mechanism of telomerase homeostasis.
Acknowledgments
We thank Pierre McCrea, Lei Li, and Junjie Chen for helpful comments on the manuscript and Junjie Chen and Subbareddy Maddika for providing reagents. The DNA Analysis Core Facility was supported by the National Institutes of Health through M.D. Anderson's Cancer Center Support Grant, CA016672.
This work was supported by the University Cancer Foundation (IRG-08-061-01), a Center for Stem Cell and Developmental Biology Transformative Pilot Grant (M.D. Anderson), an Institutional Research Grant (M.D. Anderson), a New Faculty Award (M.D. Anderson Cancer Center Support Grant), and a Metastasis Research Center Grant (M.D. Anderson).

This article contains supplemental Figs. S1—S6.
- TERC
- telomerase RNA template
- Dyrk2
- dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 2
- TERT
- telomerase reverse transcriptase
- TRAP
- telomerase rapid amplification protocol
- EDVP
- EDD, DDB1, and VprBP complex.
REFERENCES
- 1. Cech T. R. (2004) Beginning to understand the end of the chromosome. Cell 116, 273–279 [DOI] [PubMed] [Google Scholar]
- 2. Negrini S., Gorgoulis V. G., Halazonetis T. D. (2010) Genomic instability–an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228 [DOI] [PubMed] [Google Scholar]
- 3. Bodnar A. G., Ouellette M., Frolkis M., Holt S. E., Chiu C. P., Morin G. B., Harley C. B., Shay J. W., Lichtsteiner S., Wright W. E. (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349–352 [DOI] [PubMed] [Google Scholar]
- 4. Deng Y., Chan S. S., Chang S. (2008) Telomere dysfunction and tumour suppression: the senescence connection. Nat. Rev. Cancer 8, 450–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rhyu M. S. (1995) Telomeres, telomerase, and immortality. J. Natl. Cancer Inst. 87, 884–894 [DOI] [PubMed] [Google Scholar]
- 6. Hastie N. D., Dempster M., Dunlop M. G., Thompson A. M., Green D. K., Allshire R. C. (1990) Telomere reduction in human colorectal carcinoma and with ageing. Nature 346, 866–868 [DOI] [PubMed] [Google Scholar]
- 7. Maser R. S., DePinho R. A. (2002) Connecting chromosomes, crisis, and cancer. Science 297, 565–569 [DOI] [PubMed] [Google Scholar]
- 8. Feng J., Funk W. D., Wang S. S., Weinrich S. L., Avilion A. A., Chiu C. P., Adams R. R., Chang E., Allsopp R. C., Yu J. (1995) The RNA component of human telomerase. Science 269, 1236–1241 [DOI] [PubMed] [Google Scholar]
- 9. Blackburn E. H. (2000) The end of the (DNA) line. Nat. Struct. Biol. 7, 847–850 [DOI] [PubMed] [Google Scholar]
- 10. Podlevsky J. D., Chen J. J. (2012) It all comes together at the ends: telomerase structure, function, and biogenesis. Mutat. Res. 730, 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McEachern M. J., Krauskopf A., Blackburn E. H. (2000) Telomeres and their control. Annu. Rev. Genet. 34, 331–358 [DOI] [PubMed] [Google Scholar]
- 12. Martínez P., Blasco M. A. (2011) Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 11, 161–176 [DOI] [PubMed] [Google Scholar]
- 13. de Lange T. (2005) Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100–2110 [DOI] [PubMed] [Google Scholar]
- 14. Zhou X. Z., Huang P., Shi R., Lee T. H., Lu G., Zhang Z., Bronson R., Lu K. P. (2011) The telomerase inhibitor PinX1 is a major haploinsufficient tumor suppressor essential for chromosome stability in mice. J. Clin. Invest. 121, 1266–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou J., Monson E. K., Teng S. C., Schulz V. P., Zakian V. A. (2000) Pif1p helicase, a catalytic inhibitor of telomerase in yeast. Science 289, 771–774 [DOI] [PubMed] [Google Scholar]
- 16. Venteicher A. S., Abreu E. B., Meng Z., McCann K. E., Terns R. M., Veenstra T. D., Terns M. P., Artandi S. E. (2009) A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323, 644–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pfingsten J. S., Goodrich K. J., Taabazuing C., Ouenzar F., Chartrand P., Cech T. R. (2012) Mutually exclusive binding of telomerase RNA and DNA by Ku alters telomerase recruitment model. Cell 148, 922–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoffmeyer K., Raggioli A., Rudloff S., Anton R., Hierholzer A., Del Valle I., Hein K., Vogt R., Kemler R. (2012) Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Science 336, 1549–1554 [DOI] [PubMed] [Google Scholar]
- 19. Zhang Y., Toh L., Lau P., Wang X. (2012) Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/β-catenin pathway in human cancer. J. Biol. Chem. 287, 32494–32511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herbert B. S., Hochreiter A. E., Wright W. E., Shay J. W. (2006) Nonradioactive detection of telomerase activity using the telomeric repeat amplification protocol. Nat. Protoc. 1, 1583–1590 [DOI] [PubMed] [Google Scholar]
- 21. Kornitzer D., Ciechanover A. (2000) Modes of regulation of ubiquitin-mediated protein degradation. J. Cell Physiol. 182, 1–11 [DOI] [PubMed] [Google Scholar]
- 22. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Becker W., Joost H. G. (1999) Structural and functional characteristics of Dyrk, a novel subfamily of protein kinases with dual specificity. Prog. Nucleic Acids Res. Mol. Biol. 62, 1–17 [DOI] [PubMed] [Google Scholar]
- 24. Wu Y. L., Dudognon C., Nguyen E., Hillion J., Pendino F., Tarkanyi I., Aradi J., Lanotte M., Tong J. H., Chen G. Q., Ségal-Bendirdjian E. (2006) Immunodetection of human telomerase reverse-transcriptase (hTERT) re-appraised: nucleolin and telomerase cross paths. J. Cell Sci. 119, 2797–2806 [DOI] [PubMed] [Google Scholar]
- 25. Park J. I., Venteicher A. S., Hong J. Y., Choi J., Jun S., Shkreli M., Chang W., Meng Z., Cheung P., Ji H., McLaughlin M., Veenstra T. D., Nusse R., McCrea P. D., Artandi S. E. (2009) Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 460, 66–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aranda S., Laguna A., de la Luna S. (2011) DYRK family of protein kinases: evolutionary relationships, biochemical properties, and functional roles. FASEB J. 25, 449–462 [DOI] [PubMed] [Google Scholar]
- 27. Taira N., Nihira K., Yamaguchi T., Miki Y., Yoshida K. (2007) DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell 25, 725–738 [DOI] [PubMed] [Google Scholar]
- 28. Campbell L. E., Proud C. G. (2002) Differing substrate specificities of members of the DYRK family of arginine-directed protein kinases. FEBS Lett. 510, 31–36 [DOI] [PubMed] [Google Scholar]
- 29. Maddika S., Chen J. (2009) Protein kinase DYRK2 is a scaffold that facilitates assembly of an E3 ligase. Nat. Cell Biol. 11, 409–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Callaghan M. J., Russell A. J., Woollatt E., Sutherland G. R., Sutherland R. L., Watts C. K. (1998) Identification of a human HECT family protein with homology to the Drosophila tumor suppressor gene hyperplastic discs. Oncogene 17, 3479–3491 [DOI] [PubMed] [Google Scholar]
- 31. He Y. J., McCall C. M., Hu J., Zeng Y., Xiong Y. (2006) DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 20, 2949–2954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Angers S., Li T., Yi X., MacCoss M. J., Moon R. T., Zheng N. (2006) Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 443, 590–593 [DOI] [PubMed] [Google Scholar]
- 33. Branzei D., Foiani M. (2010) Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 11, 208–219 [DOI] [PubMed] [Google Scholar]
- 34. Kim N. W., Piatyszek M. A., Prowse K. R., Harley C. B., West M. D., Ho P. L., Coviello G. M., Wright W. E., Weinrich S. L., Shay J. W. (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 35. Shay J. W., Bacchetti S. (1997) A survey of telomerase activity in human cancer. Eur. J. Cancer 33, 787–791 [DOI] [PubMed] [Google Scholar]
- 36. Marcand S., Brevet V., Mann C., Gilson E. (2000) Cell cycle restriction of telomere elongation. Curr. Biol. 10, 487–490 [DOI] [PubMed] [Google Scholar]
- 37. Greenman C., Stephens P., Smith R., Dalgliesh G. L., Hunter C., Bignell G., Davies H., Teague J., Butler A., Stevens C., Edkins S., O'Meara S., Vastrik I., Schmidt E. E., Avis T., Barthorpe S., Bhamra G., Buck G., Choudhury B., Clements J., Cole J., Dicks E., Forbes S., Gray K., Halliday K., Harrison R., Hills K., Hinton J., Jenkinson A., Jones D., Menzies A., Mironenko T., Perry J., Raine K., Richardson D., Shepherd R., Small A., Tofts C., Varian J., Webb T., West S., Widaa S., Yates A., Cahill D. P., Louis D. N., Goldstraw P., Nicholson A. G., Brasseur F., Looijenga L., Weber B. L., Chiew Y. E., DeFazio A., Greaves M. F., Green A. R., Campbell P., Birney E., Easton D. F., Chenevix-Trench G., Tan M. H., Khoo S. K., Teh B. T., Yuen S. T., Leung S. Y., Wooster R., Futreal P. A., Stratton M. R. (2007) Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stephens P., Edkins S., Davies H., Greenman C., Cox C., Hunter C., Bignell G., Teague J., Smith R., Stevens C., O'Meara S., Parker A., Tarpey P., Avis T., Barthorpe A., Brackenbury L., Buck G., Butler A., Clements J., Cole J., Dicks E., Edwards K., Forbes S., Gorton M., Gray K., Halliday K., Harrison R., Hills K., Hinton J., Jones D., Kosmidou V., Laman R., Lugg R., Menzies A., Perry J., Petty R., Raine K., Shepherd R., Small A., Solomon H., Stephens Y., Tofts C., Varian J., Webb A., West S., Widaa S., Yates A., Brasseur F., Cooper C. S., Flanagan A. M., Green A., Knowles M., Leung S. Y., Looijenga L. H., Malkowicz B., Pierotti M. A., Teh B., Yuen S. T., Nicholson A. G., Lakhani S., Easton D. F., Weber B. L., Stratton M. R., Futreal P. A., Wooster R. (2005) A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat. Genet. 37, 590–592 [DOI] [PubMed] [Google Scholar]
- 39. Bonifaci N., Górski B., Masoj B., Wokolorczyk D., Jakubowska A., Dbniak T., Berenguer A., Serra Musach J., Brunet J., Dopazo J., Narod S. A., Lubiński J., Lázaro C., Cybulski C., Pujana M. A. (2010) Exploring the link between germline and somatic genetic alterations in breast carcinogenesis. PLoS One 5, e14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cullinan S. B., Gordan J. D., Jin J., Harper J. W., Diehl J. A. (2004) The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell Biol. 24, 8477–8486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu L., Wei Y., Reboul J., Vaglio P., Shin T. H., Vidal M., Elledge S. J., Harper J. W. (2003) BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature 425, 316–321 [DOI] [PubMed] [Google Scholar]
- 42. de Lange T. (2009) How telomeres solve the end-protection problem. Science 326, 948–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li S., Makovets S., Matsuguchi T., Blethrow J. D., Shokat K. M., Blackburn E. H. (2009) Cdk1-dependent phosphorylation of Cdc13 coordinates telomere elongation during cell-cycle progression. Cell 136, 50–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O'Sullivan R. J., Karlseder J. (2010) Telomeres: protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 11, 171–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nakayama K. I., Nakayama K. (2006) Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381 [DOI] [PubMed] [Google Scholar]
- 46. Reinhardt H. C., Yaffe M. B. (2009) Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 21, 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang J., Chen J. (2008) VprBP targets Merlin to the Roc1-Cul4A-DDB1 E3 ligase complex for degradation. Oncogene 27, 4056–4064 [DOI] [PubMed] [Google Scholar]
- 48. Kim J. H., Park S. M., Kang M. R., Oh S. Y., Lee T. H., Muller M. T., Chung I. K. (2005) Ubiquitin ligase MKRN1 modulates telomere length homeostasis through a proteolysis of hTERT. Genes Dev. 19, 776–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee J. H., Khadka P., Baek S. H., Chung I. K. (2010) CHIP promotes human telomerase reverse transcriptase degradation and negatively regulates telomerase activity. J. Biol. Chem. 285, 42033–42045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oh W., Lee E. W., Lee D., Yang M. R., Ko A., Yoon C. H., Lee H. W., Bae Y. S., Choi C. Y., Song J. (2010) Hdm2 negatively regulates telomerase activity by functioning as an E3 ligase of hTERT. Oncogene 29, 4101–4112 [DOI] [PubMed] [Google Scholar]
- 51. Taira N., Yamamoto H., Yamaguchi T., Miki Y., Yoshida K. (2010) ATM augments nuclear stabilization of DYRK2 by inhibiting MDM2 in the apoptotic response to DNA damage. J. Biol. Chem. 285, 4909–4919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Artandi S. E., Alson S., Tietze M. K., Sharpless N. E., Ye S., Greenberg R. A., Castrillon D. H., Horner J. W., Weiler S. R., Carrasco R. D., DePinho R. A. (2002) Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc. Natl. Acad. Sci. U.S.A. 99, 8191–8196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. González-Suárez E., Flores J. M., Blasco M. A. (2002) Cooperation between p53 mutation and high telomerase transgenic expression in spontaneous cancer development. Mol. Cell Biol. 22, 7291–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stewart S. A., Hahn W. C., O'Connor B. F., Banner E. N., Lundberg A. S., Modha P., Mizuno H., Brooks M. W., Fleming M., Zimonjic D. B., Popescu N. C., Weinberg R. A. (2002) Telomerase contributes to tumorigenesis by a telomere length-independent mechanism. Proc. Natl. Acad. Sci. U.S.A. 99, 12606–12611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Armstrong L., Saretzki G., Peters H., Wappler I., Evans J., Hole N., von Zglinicki T., Lako M. (2005) Overexpression of telomerase confers growth advantage, stress resistance, and enhanced differentiation of ESCs toward the hematopoietic lineage. Stem Cells 23, 516–529 [DOI] [PubMed] [Google Scholar]
- 56. Imamura S., Uchiyama J., Koshimizu E., Hanai J., Raftopoulou C., Murphey R. D., Bayliss P. E., Imai Y., Burns C. E., Masutomi K., Gagos S., Zon L. I., Roberts T. M., Kishi S. (2008) A non-canonical function of zebrafish telomerase reverse transcriptase is required for developmental hematopoiesis. PLoS One 3, e3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang C., Przyborski S., Cooke M. J., Zhang X., Stewart R., Anyfantis G., Atkinson S. P., Saretzki G., Armstrong L., Lako M. (2008) A key role for telomerase reverse transcriptase unit in modulating human embryonic stem cell proliferation, cell cycle dynamics, and in vitro differentiation. Stem Cells 26, 850–863 [DOI] [PubMed] [Google Scholar]