

Background: Type III polyketide synthase is hypothesized to produce quinolones, but no such enzyme has been identified so far.
Results: QNS, a type III PKS from Aegle marmelos synthesizes diketide 4-hydroxy-1-methyl-2H-quinolone using a unique substrate binding site.
Conclusion: QNS is a novel 4-hydroxy-1-methyl-2H-quinolone synthase.
Significance: This is the first report of a gene involved in quinolone biosynthesis from plants.
Keywords: Acetyl Coenzyme A, Drug Design, Flavonoids, Molecular Biology, Natural Product Biosynthesis, Plant Molecular Biology, Acridone, Aegle marmelos, Quinolone, Type III PKS
Abstract
Quinolone alkaloids, found abundantly in the roots of bael (Aegle marmelos), possess various biological activities and have recently gained attention as potential lead molecules for novel drug designing. Here, we report the characterization of a novel Type III polyketide synthase, quinolone synthase (QNS), from A. marmelos that is involved in the biosynthesis of quinolone alkaloid. Using homology-based structural modeling, we identify two crucial amino acid residues (Ser-132 and Ala-133) at the putative QNS active site. Substitution of Ser-132 to Thr and Ala-133 to Ser apparently constricted the active site cavity resulting in production of naringenin chalcone from p-coumaroyl-CoA. Measurement of steady-state kinetic parameters demonstrates that the catalytic efficiency of QNS was severalfold higher for larger acyl-coenzymeA substrates as compared with smaller precursors. Our mutagenic studies suggest that this protein might have evolved from an evolutionarily related member of chalcone synthase superfamily by mere substitution of two active site residues. The identification and characterization of QNS offers a promising target for gene manipulation studies toward the production of novel alkaloid scaffolds.
Introduction
Quinolones are anthranilic acid-derived alkaloids that are largely or completely restricted to plants of Rutaceae (1). Some of these naturally occurring quinolones have profound medicinal properties, whereas others have served as lead structures and provided inspiration for the design of synthetic quinolones as useful drugs (2, 3). Quinoline alkaloids such as quinine, chloroquin, mefloquine, and amodiaquine are used as efficient drugs for the treatment of malaria (4). Skimmianine is a potential quinolone alkaloid derived from the roots of many rutacean plants and is reported to possess various biological activities such as inhibitory effect on spontaneous motor function (5), cardiovascular effect (6), antiplatelet aggregation activity (7), cytotoxic in HeLa cell line (8), and also as antagonists of 5-hydroxytryptamine receptors (9). Because of these properties, quinolone biosynthesis is a subject of great interest. Recently, a report by Abe et al. (10) demonstrated the ability of benzalacetone synthase (BAS),2 a type III PKS to accept non-physiological substrate N-methylanthraniloyl CoA and catalyze formation of 4-hydroxy-2(1H)-quinolones. However, until now, no type III PKS dedicated to biosynthesis of quinolone alkaloid has been identified. In this report, we characterize a novel type III PKS from Aegle marmelos Corr. and demonstrate its remarkable ability to catalyze formation of quinolones scaffold. Strikingly, A. marmelos Correa belongs to the Rutaceae family, which is widely recognized for the diversity of the associated quinolone alkaloid (11, 12).
Type III polyketide synthases (PKS) are homodimeric enzymes where the single active site in each monomer iteratively catalyzes the priming, extension, and cyclization reactions to generate the polyketide product. Despite their structural simplicity, type III PKS produces a variety of compounds, including pyrones, resorcinols, acridones, chalcones, stilbenes, and phloroglucinols (13, 14). The diversity observed with type III PKS arises from its ability to accept variety of acyl-CoA thioesters for priming the reaction, its capability to extend this starter with variable number of extender units and also perform different cyclizations of the poly-β-keto linear polyketide intermediate. Although type III PKS have been studied in plants for decades, genome sequencing projects has resulted in identification of type III PKS homologues from several bacteria, fungi, and lower eukaryotes (15). Interestingly, the first report of a type III PKS accepting long chain acyl-CoA substrates was reported from a bacterium, Mycobacterium tuberculosis (16).
In this report, we describe cloning and characterization of a novel type III PKS from A. marmelos, which is named quinolone synthase (QNS) on the basis of its activity. The reaction involves decarboxylative condensation of malonyl-CoA with N-methylanthraniloyl-CoA to form an intermediate, which spontaneously cyclize by amide formation to yield 4-hydroxy-2(1H)-quinolone. Kinetic analysis indicated that the catalytic efficiency of QNS protein to accept larger acyl-CoA substrate is severalfold higher than that for smaller substrates. Molecular modeling studies suggested that QNS might have emerged by the gain of function (by the substitution of simply two active site residues) mutation from a structurally homologous CHS type III protein. The catalytic and structural importance of active site residues, as predicted by our structural model, was investigated by performing site-directed mutagenesis. The modeling and mutagenesis studies provide an insight into the structural mechanism for the enzyme that could be used to generate pharmaceutically important products.
EXPERIMENTAL PROCEDURES
Materials
[2-14C]Malonyl-CoA (58.40 mCi/mmol) was purchased from PerkinElmer Life Sciences, and nonradioactive acyl-CoA starter substrates such as malonyl-CoA, n-butyryl-CoA, isovaleryl-CoA, n-hexanoyl-CoA, benzoyl CoA, naringenin, 4-hydroxy 1-methyl quinolone, and p-hydroxy-benzalacetone were purchased from Sigma. p-Coumaroyl-CoA, feruloyl-CoA, and N-methylanthraniloyl-CoA were purchased from TransMIT.
Cloning, Expression, and Purification of QNS
5′ and 3′ RACE experiments using gene-specific primers were performed for obtaining the full-length cDNA of QNS from RNA prepared using First Choice RNA Ligase Mediated RACE kit (Ambion) as per manufacturer's protocol. For 3′ RACE, 5′-TAGTGGGTCAGGCTCTTTTTGC-3′ and 5′-GCATCCATTGAGCGTCCATTGT-3′ and for 5′ RACE, 5′-AATGAACTGTTAGACCCGCCTC-3′ and 5′-CCATCAGAATCAGGCAGAAGCG-3′ were used along with adaptor specific primers provided with the kit. Full-length QNS was isolated using forward primer, 5′-ATGGTAACCATGGAGGAGATTAGA-3′ and reverse primer, 5′-TCAAGCTTCGATGGGGACACTGCG-3′. Using the full-length information, QNS was also amplified from genomic DNA using the same primers for full-length QNS cDNA amplification. The QNS was expressed in the prokaryotic system as a fusion protein with His6 tag using the expression vector of the series pET32 (Novagen). QNS was in-frame cloned into the EcoRI site of pET32b vector and was transformed into competent Escherichia coli BL21(DE3) cells. The inducibility was monitored at regular interval of 1 h in presence of 0.4 mm isopropyl-1-thio-β-galactopyranoside and confirmed by Western blotting using anti-His tag antibody. For the purification of protein, the transformed BL21(DE3) cells were grown to A600 = 0.5 and induced in 0.4 mm isopropyl-1-thio-β-galactopyranoside at 28 °C for 6 h. The bacterial pellet was then resuspended in protein buffer A (50 mm KPO4 buffer, pH 8.0, 100 mm NaCl, 0.7 mg/ml lysosyme, 40 mm imidazole) and sonicated in ice at 50% duty cycle at intermediate power. The lysate was analyzed by SDS-PAGE and subjected to nickel-nitrilotriacetic acid-agarose column chromatography (Invitrogen). After washing with protein buffer B (50 mm KPO4 buffer, pH 7.9, 500 mm NaCl, and 40 mm imidazole), QNS was eluted with protein buffer C (15 mm KPO4 buffer, pH 7.5, 150 mm imidazole, and 10% glycerol) and analyzed by SDS-PAGE.
Site-directed Mutagenesis
A. marmelos QNS mutants MSD1 and MSD2 were constructed using the QuikChange site-directed mutagenesis kit (Stratagene) and a pair of primers as follows (mutated codons are underlined): MSD1 (S132T/A133S) (5′-CTCATTTTCTGCACAACTTCAGGCGTCGACATG-3′ and 5′-CATGTCGACGCCTGAAGTTGTGCAGAAAATGAG-3′)) and MSD2 (V265F in addition to S132T/A133S) (5′)-GAGGCGGGTCTAACATTTCATTTGAAGAAAGAC-3′ and 5′-GTCTTTCTTCAAATGAAATGTTAGACCCGCCTC- 3′). Mutagenesis was confirmed by automated DNA sequencing. The mutant proteins were expressed and tried to purify analogous to the wild-type QNS protein. The mutants expressed as inclusion bodies and could not be expressed in soluble form when similar conditions as wild-type QNS protein were used. The expression of mutant protein was modulated to soluble form by inducing cultures with 0.4 mm isopropyl-1-thio-β-d-galactopyranoside concentration at 18 °C for 24 h.
Enzyme Assay and Product Characterization Using Mass Spectrometry
For the in vitro enzyme assay, the standard reaction conditions involved 200 μm starter molecule (n-butyryl-CoA, isovaleryl-CoA, n-hexanoyl-CoA, benzoyl-CoA, cinnamoyl-CoA, p-coumaroyl-CoA, feruloyl-CoA, and N-methylanthraniloyl-CoA) and 100 μm malonyl-CoA (inclusive of 9.12 μm [2-14C]malonyl-CoA (58.40 mCi/mmol)). Reactions were carried out with 45 μg of protein at 30 °C for 60 min and quenched with 5% acetic acid. The products were extracted with 2 × 300 μl of ethyl acetate and dried under vacuum. Radiolabeled products were resolved on silica gel 60 F254 TLC plates (Merck) in ethyl acetate/hexanes/acetic acid (63:27:5, v/v/v). Polyketide products were characterized using nanospray electrospray ionization MS (API QSTAR Pulsar i MS/MS; Applied Biosystems).
Determination of the Kinetic Parameters
A standard reaction contained 100 mm potassium phosphate buffer (pH 7.0), 200 μm malonyl-CoA, N-methylanthraniloyl-CoA (0.5–200 μm), and 40 μg of QNS in a total volume of 100 μl for determination of kinetic values for N-methylanthraniloyl-CoA, and 100 mm potassium phosphate buffer (pH 7.0), 200 μm malonyl-CoA, varied starter concentrations (2.5–200 μm), and 40 μg of QNS in a total volume of 100 μl for determination of kinetic values for all other starter CoAs. After the reaction mixture had been preincubated at 37 °C for 2 min, reactions were initiated by adding the substrate and continued for 20 min. The reactions were stopped with 5 μl of 5% acetic acid, and the material in the mixture was extracted with 2× ethyl acetate. The organic layer was collected and evaporated. The residual materials were dissolved in 20 μl of methanol for Radio-TLC analysis. The kinetic parameters were calculated for formation of major product of the enzyme reactions.
Homology Modeling and Docking Studies
Structural modeling was done based on the homology using the SWISS-MODEL workspace (17–19), which is an integrated web-based modeling expert system. To get the suitable template for QNS, we did Blastp search (www.ncbi.nlm.nih.gov/blast) against the Protein Data Bank using an E value cut-off: 0.001. Based on the result Medicago sativa CHS (Protein Data Bank code 1BI5, chain A; resolution, 1.56; 74% identity) was selected as template. Calculation of cavity volumes (Connolly's surface volumes) was then performed with the CASTP program (available online) (20). To find out the preferred substrates for the enzyme, we tried to dock substrate molecules like p-coumaroyl-CoA and N-methylanthraniloyl-CoA to the QNS wild-type and mutant proteins. For docking studies, HEX (version 4.5, 21) was used with default parameters. The substrates were obtained from PubChem database.
RESULTS
Sequence Analysis and Homology Modeling
The full-length cDNA of ∼1200 bp (Fig. 1A) was obtained from young leaves of A. marmelos via amplification through RNA Ligase Mediates RACE experiments (the details of the method are described under “Experimental Procedures”). To study the genomic organization of QNS, the full-length gene was also isolated and cloned from A. marmelos genomic DNA, which was ∼1305 bp in length (GenBank ID, JX679860) (Fig. 1B). The sequencing results showed that the coding region of QNS is interrupted by one intervening sequence of 127 bp (Fig. 1C) as reported in almost all plant-specific type III PKS genes (22–24) that is located between the first and second nucleotides of Cys-60-encoding triplet (numbering of M. sativa CHS2). The primary cDNA sequence of A. marmelos QNS (GenBank ID JX679859) exhibits 60–75% identity to CHS superfamily enzymes from other plant species, including M. sativa CHS and Gerbera hybrida 2-PS. The closest homologue to QNS is chalcone synthase from Dictamus albus (81% amino acid identity) and acridone synthase (ACS) from Ruta graveolens (79% amino acid identity). An alignment was made, and a phylogenetic tree was constructed with others type III PKS in the database, including the highly homologous ones to study the evolutionary relationship of QNS (Fig. 2, A and B). Phylogenetic analysis revealed that QNS groups with non-chalcone forming PKS and cluster with R. graveolens ACS (Fig. 2B). Comparative analysis of protein sequences of plants PKS superfamily of enzymes have previously led to the clustering of these proteins based on their function and not based on species-specific nature (25, 26). A. marmelos QNS maintains an almost identical CoA binding site with the reported type III PKS and a conserved catalytic triad of Cys-164, His-303, and Asn-336 (numbering in M. sativa CHS) (Table 1). In addition, the active site residues, including Leu-214, Ile-254, Gly-256, Thr-197, Leu-263, and Ser-338 of M. sativa CHS are well conserved in QNS (Table 1). Strikingly, A. marmelos QNS lacks active site Phe-265, the second “gatekeeper Phe” in type III PKS of plant and bacterial origin and is replaced by Val-265. Additionally the active site residues, Thr-132 and Ser-133 of M. sativa CHS are replaced by Ser-132 and Ala-133 residues (Table 1). Similar type of T132S, S133A, and F265V substitutions are present in both ACS isoforms from R. graveolens and are reported to play key role in starter substrate specificity (27).
FIGURE 1.

Amplification of QNS from A. marmelos leaves. A and B, PCR amplification of QNS from cDNA ad genomic DNA, respectively. 1, 1-kb DNA ladder; 2, PCR product; 3, control reaction. C, organization of intron in QNS gene. The arrow indicates the predicted intron position. The diagrammatic representation of QNS genome organization was also shown below.
FIGURE 2.

Alignment and phylogenetic analysis of QNS with other type III PKS. A, alignment of QNS. The alignment was generated by CLUSTALW. AmQNS, A. marmelos quinolone synthase; RgACS, R. graveolens acridone synthase; GhPYS, Gerbera hybrida 2-pyrone synthase; MsCHS, M. sativa CHS2; RpBAS, R. palmatum benzalacetone synthase. The amino acids of the catalytic Cys-His-Asn triad are marked by bold rectangular boxes. The residues at positions Thr-132, Ser-133, and Phe-265 corresponding to M. sativa CHS (marked in thin rectangular boxes) in QNS have been shown to be lie near or line the active site cavity and Thr-132, Ser-133 are crucial for the binding of bulky starter substrate. The stars below each line of alignment indicates conserved sites. B, phylogenetic analysis of QNS with other type III PKS. The dendrogram was generated for A. marmelos QNS with other plant PKS. The accession numbers are given in brackets. E. coli ketoacyl synthase III (FabH) was used as an out group. CHS, chalcone synthase; VPS, valerophenone synthase; ACS, acridone synthase; ALS, aleosone synthase; PYS, pyrone synthase; CURS, curcuminoid synthase; BLS, benzalacetone synthase; PSS, pinosylvin synthase; STS, stilbene synthase.
TABLE 1.
Comparison of active site cavity residues of crystallized M. sativa CHS (Protein Data Bank code 1BI5) and A. marmelos QNS
The varied residues in QNS are underlined.
| CHS M. sativa | QNS A. marmelos |
|---|---|
| Initiation pocket | |
| Leucine 214 | Leucine 214 |
| Isoleucine 254 | Isoleucine 254 |
| Glycine 256 | Glycine 256 |
| Phenylalanine 265 | Valine 265 |
| Elongation pocket | |
| Threonine 132 | Serine 132 |
| Serine 133 | Alanine 133 |
| Threonine 194 | Threonine 194 |
| Valine 196 | Proline 196 |
| Threonine 197 | Threonine 197 |
| Glycine 216 | Alanine 216 |
| Leucine 263 | Leucine 263 |
| Serine 338 | Serine 338 |
| Catalytic triad | |
| Cysteine 164 | Cysteine 164 |
| Histidine 303 | Histidine 303 |
| Asparagine 336 | Asparagine 336 |
Structure-based homology modeling and docking studies were carried out to get an insight into the role of active site residues in QNS substrate binding activity. Homology models of QNS wild-type and mutants, MSD1 (S132T/A133S), and MSD2 (S132T/A133S/V265F) were generated based on M. sativa CHS (Protein Data Bank code 1BI5) as template (Fig. 3). The homology model of A. marmelos QNS wild-type showed that it has the same overall fold as M. sativa CHS, with cavity volume estimated to be 1196 Å3, which is much larger than that of M. sativa CHS (1019 Å3). This also suggested that the active site cavity of QNS is easily large enough to accommodate bulkier starter substrate than CHS. The three residues Ser-132, Ala-133, and Val-265 lie near or line the active cavity. The mutant MSD1 and MSD2 homology models shows remarkable constriction in the active site cavity with a volume estimated to be 1188 and 1168 Å3, respectively (Fig. 3). We therefore predicted that the steric bulk from three active site residues substitution, S132T, A133S, and V265F, in mutants significantly affect the active site volume and might hinder the binding of bulkier starter substrates in mutants. To improve our predictability for MSD1 and MSD2 mutants, N-methylanthraniloyl-CoA and p-coumaroyl-CoA starter substrates were docked in the wild-type QNS and mutant substrate binding pocket. As expected, N-methylanthraniloyl-CoA and p-coumaroyl CoA starter substrates were finely docked into the active site cavities of QNS (Fig. 4). However, N-methylanthraniloyl-CoA, owing to steric clashes with both the substituted side chains residues, failed to dock in the active site pocket of both mutants, MSD1 and MSD2. Interestingly, p-coumaroyl-CoA finely docked in the substrate-binding pocket of the mutant proteins. Thus homology modeling studies suggest the crucial role of Ser-132 and Ala-133 in allowing binding of bulkier substrate as N-methylanthraniloyl-CoA (Fig. 4).
FIGURE 3.

Homology modeling and comparison of the active site cavities of A. marmelos QNS wild-type with mutant (MSD1 and MSD2) proteins. Some of the crucial active site residues along with the catalytic triad Cys, His, and Asn are shown here.
FIGURE 4.

Docking studies of QNS wild-type and mutant proteins, MSD1 and MSD2 with ligands. A, QNS WT with N-methylanthraniloyl-CoA; B, QNS WT with p-coumaroyl-CoA; C and D, MSD1 (S132T/A133S) and MSD2 (S132T/A133S/V265F) with p-coumaroyl-CoA. Substrates were shown in yellow. Some of the crucial active site residues along with the catalytic triad Cys, His, and Asn were shown in red.
Wide Starter Substrate Specificity and Characterization of Novel Products
Due to the interesting results obtained by comparative modeling studies, we proceeded to biochemically characterize the QNS protein. The ability of QNS to accept various starter CoA with [2-14C]malonyl-CoA as extender was investigated using Radio-TLC (Fig. 5). Several significant bands appear on the TLC plate, clearly indicating that QNS protein is able to accept various aromatic and small as well as bulky aliphatic substrates. Analogous broad starter substrate specificity has also been reported for other plant type III PKS (13). In support of our homology modeling and substrate binding studies of QNS that showed highest similarity with ACS, our radio-TLC analysis revealed a significant amount of product formation in the N-methylanthraniloyl-CoA-primed reaction. We decided to unambiguously characterize the nature of the product formed using nanospray electrospray ionization MS.
FIGURE 5.

TLC-based analysis of radiolabeled products of QNS wild-type. The overall reaction was carried out by QNS with different substrate. Reactions were carried out for 2 h using [2-14C]malonyl-CoA as an extender unit along with different starter substrates. The starter substrates used in the assay are indicated above each lane. Chemical structures of the major products obtained by N-methylanthraniloyl-CoA and p-coumaroyl-CoA primed reactions are indicated next to the radioactive bands. Because the Rf value of 1,3-dihydroxy-N-methylacridone was not known, it was not shown.
The major product shows Rf value similar to 4-hydroxy-1-methyl-2(1H)-quinolone standard. On MS analysis of the product, two molecular ion peak, (M-H)− at m/z 174.08 and (M-H)− at m/z 240.09 were observed. The structural elucidation of QNS reaction products was performed by using tandem mass spectrometry. Fig. 6A shows the fragmentation pattern for the molecular ion (M-H)− m/z 174.08 (obtained for a), which yielded a fragment of m/z 159.05 corresponding to (C9H5NO2) (with the loss of −CH3 group), and m/z 131.05 corresponding to (MCO2-H)− ion. This fragmentation pattern has been reported for the standard 4-hydroxy-1- methyl-2(1H)-quinolone also; which confirmed the formation of quinolone scaffold by QNS. Interestingly, the molecular ion peak (M-H)− at m/z 240.09 (obtained for b) yielded a fragment of m/z 225.06 corresponding to (C13H8NO3) (with the loss of −CH3 group) and m/z 197.07 corresponding to (MCO2-H)− ion (Fig. 6B), which were similar to the fragmentation pattern reported for 1,3-dihydroxy-N-methylacridone. The spectroscopic data (electrospray ionization MS and UV) of the compounds were characteristic of those of the quinolone and acridone and completely identical with those of authentic compounds and those cited in literatures. Thus, QNS may be a bifunctional enzyme capable of producing both quinolone and acridone in vitro. Because BAS is the only enzyme reported so far to produce quinolone scaffold whose physiological substrate is p-coumaroyl-CoA, we also tried to characterize the products biosynthesized by QNS protein with p-coumaroyl-CoA. Interestingly, the nanospray electrospray ionization MS analysis of the p-coumaroyl-CoA primed reaction showed a major molecular ion peak, (M-H)− at m/z 161.08 equivalent to p-hydroxybenzalacetone (C10H10O2). Fig. 6C shows the fragmentation pattern for the molecular ion (M-H)− m/z 161.08 (obtained for c), which yielded a fragment of m/z 146.05 corresponding to (C9H7O2) (with the loss of −CH3 group) and m/z 119.06 corresponding to the (MCO2-H)− ion. The products obtained with other substrates were similarly characterized by nanospray MS and determined to be triketide and tetraketide pyrones. With some notable exceptions (28, 29), the exclusive formation of pyrones by plant PKS seems to be an indicator that the starter CoA ester in the reaction is not the physiologically relevant substrate for the enzyme under assay (30). These findings suggest that pyrones produced by QNS in vitro might be derailment products due to non-physiological substrates or suboptimal reaction conditions.
FIGURE 6.
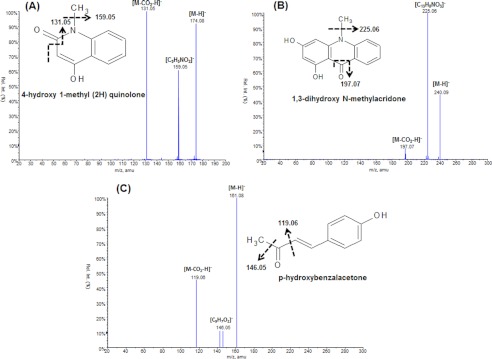
Identification of QNS wild-type products. The molecular mass obtained for each peak by nanospray mass spectrometric analysis is indicated below the structures for the expected products. A and B illustrate the fragmentation pattern observed by tandem mass spectrometry for N-methylanthraniloyl-CoA-primed products, 4-hydroxy-1-methyl-2(1H)-quinolone (M-H)− m/z 174.08 (A) and 1,3-dihydroxy-N-methylacridone (M-H)− m/z 240.09 (B). C illustrates the fragmentation pattern observed by tandem mass spectrometry for p-coumaroyl-CoA-primed product, p-hydroxybenzalacetone (M-H)− at m/z 161.08. Fragment ions are indicated above the corresponding peaks. The structure of the molecule with the expected fragmentation profile is drawn next to its MS/MS spectra. Rel. Int., Relative Intensity.
Transformation of QNS to CHS by Site-directed Mutagenesis
From the sequence comparison and docking studies, it was observed that residues Ser-132 and Ala-133 line the substrate binding pocket of wild-type QNS (Fig. 3). Two mutant enzymes, a doublet (MSD1), in which the residues Ser-132 and Ala-133 were mutated to Thr-132 and Ser-133 and a triplet (MSD2), in which three residues, Ser-132, Ala-133 and Val-265 were mutated to Thr, Ser and Phe respectively were generated to gain an insight into the functional role of these residues. (MSD1 and MSD2 mutations were similar as generated for homology modeling studies.) QNS double and triple mutant homology model showed remarkable constriction in the active site cavity compared with wild-type QNS (Fig. 2). The steric hindrance was most probably due to the substitution of methyl group for hydrogen and charge substitution by the added hydroxyl group, thus occluding the chance of binding of bulky starter substrate in the substrate binding pocket of mutant enzyme. To validate our prediction, the QNS S132T/A133S and S132T/A133S/V265F mutants was heterologously overexpressed in E. coli as a recombinant protein with a His tag at the N terminus as in the case of wild-type QNS and purified to homogeneity by a nickel-chelate column. QNS S132T/A133S mutant (MSD1) exhibited chalcone-forming activity and performed sequential condensations of three malonyl-CoA units with p-coumaroyl-CoA to form tetraketide naringenin chalcone. The S132T/A133S/V265F triple mutant (MSD2), on the other hand, showed no product formation using p-coumaroyl-CoA as starter unit (Fig. 7). The production of naringenin chalcone by MSD1 was confirmed using authentic standard (Rf value, 0.8; retention time, 31.7). In support of our docking studies, none of the mutants formed products with N-methylanthraniloyl-CoA. Thus, the two amino acid substitution, S132T and A133S, which were predicted to line the QNS substrate binding pocket in the mutant MSD1, considerably shifted the starter substrate preference to p-coumaroyl-CoA and hence transformed the enzyme completely to a functional CHS producing naringenin (Fig. 7). Our results correlate with the reported transformation of ACS to CHS by triple mutation (27). The triple ACS mutant (S132T/A133S/V265F) retained ∼35% ACS activity with an overall 10-fold increase in specific CHS activity. Interestingly, although the double mutant protein (S132T/A133S) displayed 100% CHS activity, the triple mutant showed no QNS/ACS/CHS activity, emphasizing a rather different enzyme mechanism.
FIGURE 7.
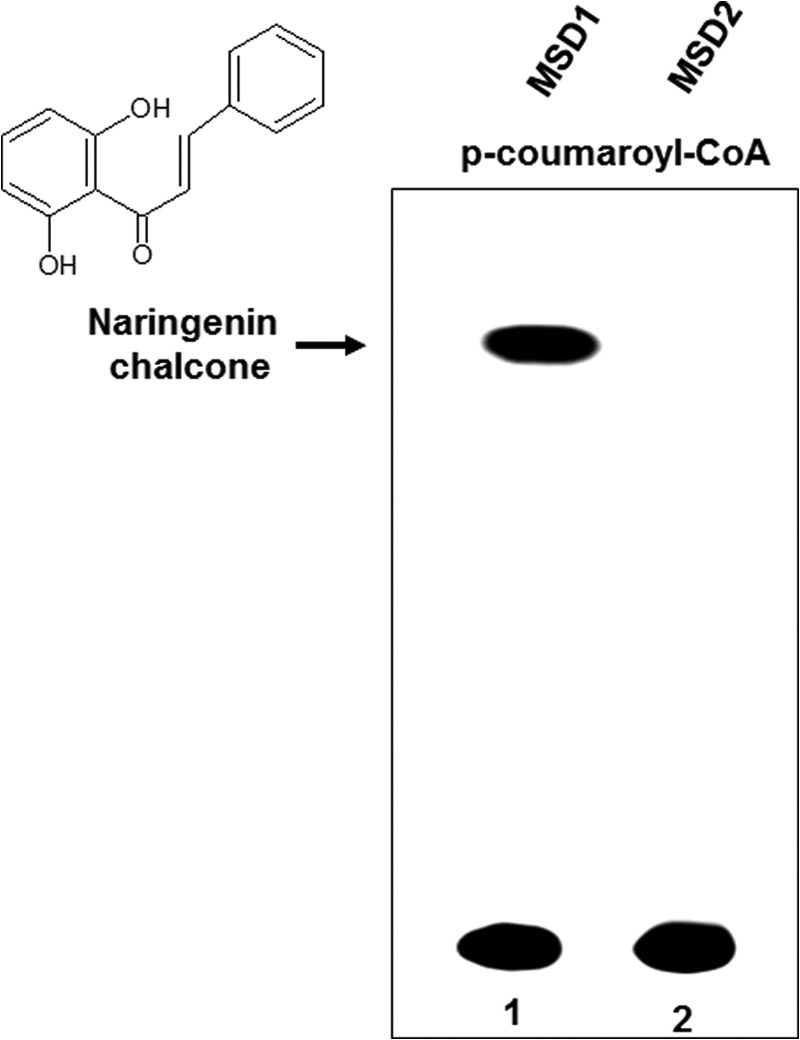
Comparative TLC of radiolabeled products of QNS mutants, MSD1 (S132T/A133S) and MSD2 (S132T/A133S/V265F) of p-coumaroyl-CoA-primed reactions. [2-14C]Malonyl-CoA was used as an extender unit in both the reactions. Chemical structure of the major product obtained is indicated next to the radioactive band.
Characterization of Kinetic Parameters of QNS
The starter unit specificity of QNS was investigated by performing steady state kinetic analysis using various CoA substrates. Type III PKS are generally known to possess broad substrate tolerance; we therefore performed kinetic studies with several different acyl-CoAs, the result of which was summarized in Table 2. Even though QNS produced aromatic products only with N-methylanthraniloyl-CoA and p-coumaroyl-CoA starter substrates, detailed kinetic studies were carried out with various starter CoAs, the result of which was summarized in Table 2. QNS displayed poor substrate affinity toward shorter aliphatic CoA thioesters and the benzoyl-, feruloyl-derived aromatic CoA-thioesters as reflected in the Km values (Table 2). Hexanoyl-CoA and cinnamoyl-CoA were found to be efficient starter molecules for QNS, with kcat/Km values comparable with that of the physiological substrate. As reflected by the Km values, the affinity of QNS for larger N-methylanthraniloyl-CoA starter was found to be higher compared with p-coumaroyl-CoA starter units. It can also be observed that the kcat/Km values for the biosynthesis of 4-hydroxy-1-methyl-quinolone are several folds higher than that observed for p-hydroxybenzalacetone (Table 2). For the formation of 4-hydroxy-1-methyl-quinolone, Km = 2.93 μm, kcat = 3.78 min−1, kcat/Km = 21,501.71 m−1 s−1 for N-methylanthraniloyl-CoA and Km = 21.3 μm, kcat = 2.56 min−1, kcat/Km = 2003.13 m−1 s−1) for extender malonyl-CoA. For p-hydroxybenzalacetone formation, the Km = 3.62 μm, kcat = 0.23 min−1, kcat/Km = 1,058.93 m−1 s−1 for p-coumaroyl-CoA, and Km = 26.7 μm, kcat = 0.93 min−1, kcat/Km = 580.52 m−1 s−1 for extender malonyl-CoA. N-Methylanthraniloyl-CoA is clearly the most preferred substrate in vitro and could also be the most preferred physiological starter substrate for QNS. To test whether the double mutant, MSD1, is biochemically relevant or not, we carried out a detailed kinetic studies for the double mutant (triple mutant, MSD2 is found to be biochemically non-functional and excluded from kinetic studies). As the double mutant showed activity only with p-coumaroyl-CoA, the same strater unit was used for kinetic analysis. For the mutant (MSD1) chalcone-forming reaction, the kinetic parameters for naringenin chalcone formation were Km = 3.43 μm, kcat = 2.14 min−1, kcat/Km = 10,398 m−1 s−1) for p-coumaroyl-CoA and Km = 23.6 μm, kcat = 1.6 min−1, kcat/Km = 1129.94 m−1 s−1) for extender malonyl-CoA. The mutant enzyme thus exhibited 10-fold increase in kcat/Km for p-coumaroyl-CoA as compared with the wild type QNS protein. Interestingly, MSD1 mutant showed rather narrow substrate tolerance and did not accept diverse starter substrates other than benzoyl-CoA.
TABLE 2.
Steady state kinetic parameters for QNS protein with different starter units
Results are means (n = 3) with S.E. values < 15%. ND, not determined.
| Starter CoAs | kcat | Km | kcat/Km |
|---|---|---|---|
| min−1 | μm | s−1 m−1 | |
| Butryl-CoA | ND | ND | ND |
| Isovaleryl-CoA | 1.8 | 7.9 | 3797.47 |
| Hexanoyl-CoA | 2.53 | 4.2 | 10,039.68 |
| Benzoyl-CoA | 0.5 | 10.25 | 813.01 |
| Cinnamoyl-CoA | 2.9 | 5.2 | 9294.87 |
| Feruloyl-CoA | 1.12 | 9.14 | 2042.30 |
| p-Coumaroyl-CoA | 0.23 | 3.62 | 1058.93 |
| N-Methylanthraniloyl- CoA | 3.78 | 2.93 | 21,501.71 |
DISCUSSION
The chemical diversity of alkaloids in the Rutaceae family has led to the isolation of various pharmacologically active natural products (31). Anthranilic acid has been postulated to be a key intermediate in the biosynthesis of various rutacean alkaloids, viz. quinolone and acridone alkaloids. Naturally occurring quinolones have profound medicinal properties and have served as lead structures for the design of synthetic quinolones as useful drugs (2). Although a number of functionally diverse type III PKS have been identified and characterized (13), no gene encoding for an enzyme catalyzing the formation of quinolone has yet been identified. Here, we report the identification of a novel type III PKS, QNS capable of catalyzing quinolone alkaloid biosynthesis from a rutacean member, A. marmelos. The overall reactions catalyzed by QNS protein is summarized in Fig. 8. In its native form, the QNS catalyzes the condensation of N-methylanthraniloyl-CoA with malonyl-CoA to form quinolone scaffold (major product, 89%) and acridone scaffold (minor product, 11%). QNS also shows significant BAS (benzalacetone) side activities in assays with p-coumaroyl-CoA. QNS also displays promiscuous substrate specificity in vitro by accepting both aliphatic and aromatic CoAs. Interestingly, CHS and other type III PKS, except ACS, do not accept the shorter and the bulkier N-methylanthraniloyl-CoA as a starter unit. Although the M. sativa CHS F215S mutant has been shown to accept N-methylanthraniloyl-CoA to produce an unnatural novel tetraketide lactone after three condensations with malonyl-CoA (14), it does not catalyze biosynthesis of acridone or quinolone.
FIGURE 8.
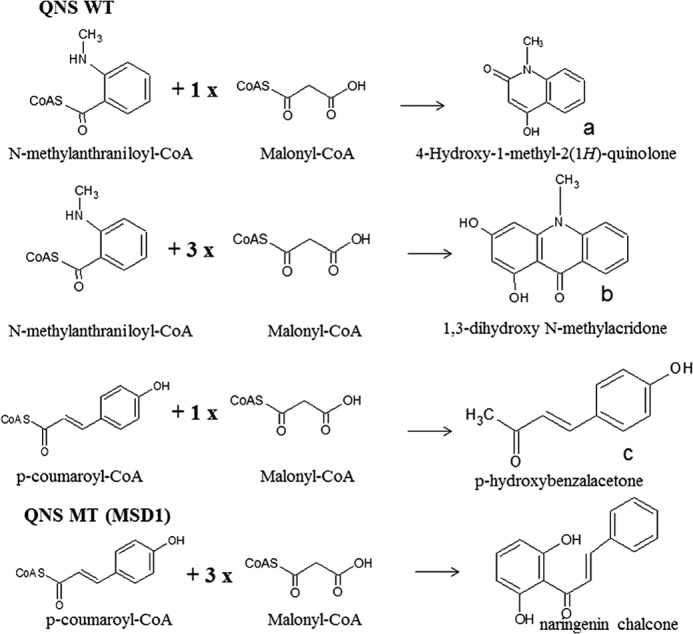
Overall reactions catalyzed by A. marmelos QNS wild-type and mutant, MSD1.
R. graveolens ACS is the only type III PKS reported to be involved in rutacean alkaloid (acridone) biosynthesis (32). In ACS, the residues Ser-132, Ala-133, and Val-265 (numbering in M. sativa CHS) have been proposed to play a critical role for the selection of N-methylanthraniloyl-CoA as a starter substrate and for the formation of the acridone alkaloid. Indeed, an ACS triple mutant (S132T/A133S/V265F) has been shown to yield an enzyme that was functionally identical with CHS (27). A similar type of active site residue substitution in QNS prompted us to look into the role of these residues in substrate binding activity. Homology model and mutant studies of QNS showed that Ser-132 and Ala-133 residues to be involved in substrate binding, whereas Val-265 is occluded from starter substrate binding. These amino acids line the active site cavity of QNS and are not conserved in other members of the PKS family other than ACS. In accordance with our docking studies the functional behavior of QNS was dramatically altered in the mutant, MSD1, where S132T/A133S substitution in the active site cavity transformed QNS completely into CHS. Substitution of Ser-132 to Thr and Ala-133 to Ser had dual effects on enzyme activity. First, the double mutant completely lost the ability to accept N-methylanthraniloyl-CoA. Secondly, the mutant showed an overall increase in polyketide chain elongation. This could be explained by the constriction of the active site cavity by the introduction of methyl group in the Thr residue, thus excluding larger substrates in active site pocket. In Aloe arborescens pentaketide chromone synthase, a point mutation, M207G, expanded the volume of cavity, thereby converting pentaketide chromone synthase into octaketide synthase (33). Compared with the substrate cavity of wild-type QNS, the size of the accessible pocket in MSD1 and MSD2 is smaller and does not allow the binding of bulky starter substrate such as N-methylanthraniloyl-CoA, thereby preventing the formation of quinolone or acridone. The docking studies with double and triple mutant confirm the complete exclusion of N-methylanthraniloyl-CoA from substrate binding pocket. Although the double mutant protein (MSD1) displayed 100% CHS activity with p-coumaroyl-CoA, showing no QNS side activity with N-methylanthraniloyl-CoA, the triple mutant purified enzyme (MSD2), when incubated with both starter CoAs, was not able to produce any significant bands in the radio-TLC. It was rather surprising because similar triple mutant generated for acridone synthase were reported to be 100% functional producing naringenin chalcone with p-coumaroyl-CoA (27). The mutations, in particular, in MSD1 must have significantly altered the space and/or charge restrictions of the active site pocket, and the difference in geometry caused the change in the starter substrate preference. The shape and volume of the active site cavity greatly influence the substrate specificity and control the final length of the polyketide (34). Replacement of the S132T and A133S may open the gate to a buried pocket, into which the triketide intermediate is redirected during the elongation process of the diketide. It was thus confirmed that the residues S132T and A133S in A. marmelos QNS are indeed involved in the formation of quinolone and that the preference toward increased polyketide chain extension at the diketide stage in the mutant was due to the absence of these residues. A combination of three mutations (T197L/G256L/S338I) converted M. sativa CHS into 2-pyrone synthase by reducing the volume of the active site cavity from 923 to 274 Å3 (34). The T135L point mutation not only changed the product specificity but also the substrate specificity of BPS, thus transforming it into a functional phenylpyrone synthase (35).
BAS, a member of the type III PKS superfamily from Rheum palmatum, which catalyzes a one-step decarboxylative condensation of p-coumaroyl-CoA with malonyl-CoA to produce p-hydroxybenzalacetone is the only reported enzyme capable of enzymatic synthesis of quinolone derivatives (10). Because quinolone alkaloids have never been isolated from R. palmatum (Polygonaceae) and are completely or largely restricted to Rutaceae, the physiological role of BAS in quinolone alkaloid biosynthesis still remains obscure. The identified QNS from A. marmelos, a rutacean member rich in quinolone and acridone alkaloid derivatives, is a more physiologically relevant novel potential type III PKS catalyzing quinolone scaffold biosynthesis. As mentioned previously, QNS double mutant (S132T/A133S) yield an enzyme that was functionally identical with CHS, whereas triple mutant fail to produce quinolone or acridone or naringenin. However, two of the mentioned residues, Ser-132 and Ala-133 are not conserved in R. palmatum BAS that accepted the anthraniloyl starter; instead, it was S132L/V265F. One of the most unique features of R. palmatum BAS is that it lacks active site Phe-215; the conserved residues Ile214Leu. Interestingly, the diketide-forming activity of the BAS enzyme is reported to be derived from the characteristic substitution of the active site gatekeeper Phe-215 by Leu (10). A BAS I214L/L215F mutant exhibited chalcone-forming activity, which also supported a hypothesis that the absence of Phe-215 in BAS accounts for the interruption of the polyketide chain elongation at the diketide stage (36). Even though QNS also showed considerable benzalacetone side activity with p-coumaroyl-CoA, Phe-215 is well conserved in QNS similar to all other type III PKS.
In addition to the diketide condensation, wild-type QNS shows a minor tetraketide condensation leading to the formation of acridone such as ACS. ACS and CHS catalyze similar condensation reactions up to the tetraketide stage, but subtle differences exist in the modes of cyclization. The final bridging of rings in acridone biosynthesis must formally precede from a non-aromatic “trione” intermediate (37) proceeds through nucleophilic substitution followed by dehydration. In contrast, CHS releases the chalcone as the immediate product, which in the absence of chalcone isomerase, cyclizes non-stereoselectively to the flavanone naringenin. Although QNS is also capable of similar type of condensation reaction up to the tetraketide stage releasing acridone scaffold, under most of the experimental tests conducted, it favors interruption of the chain extension at the diketide stage, releasing quinolone scaffold. QNS preference toward single condensation is well shown with p-coumaroyl substrate also, by catalyzing a one-step decarboxylative condensation with malonyl-CoA to produce a diketide benzalacetone. The BAS is reported to carry out only one condensation with N-methylanthraniloyl-CoA and methylmalonyl-CoA to produce quinolone scaffold, even though it can easily carry out two condensation reaction with p-coumaroyl-CoA (10). The results support a different type of reaction mechanism for QNS.
Kinetic characterization of QNS and MSD1 demonstrated that the S132T/A133S mutation results in increased affinity for the starter substrate, p-coumaroyl-CoA, and increased turnover number resulting in an overall increased catalytic efficiencies (kcat/Km). Steady-state enzyme kinetics for the formation of 4-hydroxy-1,3-dimethyl-2(1H)-quinolone by reported BAS is Km of 23.7 μm and kcat of 1.48 min−1 for N-methylanthraniloyl-CoA, whereas for QNS these are 2.93 μm and 3.8 min−1, respectively. For the formation of benzalacetone, the normal product of the BAS enzyme is Km of 10.0 μm and kcat of 1.79 for p-coumaroyl-CoA (25) and for QNS, 3.62 μm and 0.23 min−1, respectively. The severalfold increase in affinity of QNS for N-methylanthraniloyl-CoA and overall increased catalytic efficiency for quinolone formation in comparison with BAS suggests its physiological preference toward quinolone biosynthesis.
In conclusion, we have characterized a novel type III PKS protein that catalyzes the formation of quinolones using bulky acyl-CoA substrates. Structural and mutational studies have suggested that QNS might have emerged by the gain of function mutation (by the substitution of simply two active site residues) from a structurally homologous CHS type III protein. The functional equivalence of mutant QNS to CHS proteins rather presents a fascinating evolutionary link. Furthermore, recent structure-based engineering of functionally divergent type III PKS has significantly expanded the catalytic repertoire of the enzymes and the product diversity (38–40). The identified PKS (QNS) due to its unusual substrate specificity and with its ability to produce quinolone scaffold can be utilized for the production of chemically and structurally distinct unnatural novel alkaloids.
This work was supported by Department of Science and Technology, New Delhi, India, and a senior research fellowship from Council of Scientific and Industrial Research (New Delhi, India).
- BAS
- benzalacetone synthase
- PKS
- polyketide synthase(s)
- QNS
- quinolone synthase
- RACE
- rapid amplification of cDNA ends
- ACS
- acridone synthase.
REFERENCES
- 1. Michael J. P. (2008) Quinoline, quinazoline and acridone alkaloids. Nat. Prod. Rep. 25, 166–187 [DOI] [PubMed] [Google Scholar]
- 2. Heeb S., Fletcher M. P., Chhabra S. R., Diggle S. P., Williams P., Cámara M. (2011) Quinolones: from antibiotics to autoinducers. FEMS Microbiol. Rev. 35, 247–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Robert A., Meunier B. (1998) Is alkylation the main mechanism of action of the antimalarial drug artemisinin? Chem. Soc. Rev. 27, 273–274 [Google Scholar]
- 4. Alhaider A. A., Abdelkader M. A., Lien E. J. (1985) Design, synthesis, and pharmacological activities of 2-substituted 4-phenyl quinolines as potential antidepressant drugs. J. Med. Chem. 28, 1394–1398 [DOI] [PubMed] [Google Scholar]
- 5. Cheng J. T. (1986) Effect of skimmianine on animal behavior. Arch. Int. Pharmacodyn. Ther. 281, 35–43 [PubMed] [Google Scholar]
- 6. Cheng J. T., Chang S. S., Chen I. S. (1990) Cardiovascular effect of skimmianine in rats. Arch. Int. Pharmacodyn. Ther. 306, 65–74 [PubMed] [Google Scholar]
- 7. Chen K. S., Chang Y. L., Teng C. M., Chen C. F., Wu Y. C. (2000) Furoquinolines with antiplatelet aggregation activity from leaves of Melicope confusa. Planta Med. 66, 80–81 [DOI] [PubMed] [Google Scholar]
- 8. Jansen O., Akhmedjanova V., Angenot L., Balansard G., Chariot A., Ollivier E., Tits M., Frédérich M. (2006) Screening of 14 alkaloids isolated from Haplophyllum A Juss. for their cytotoxic properties. J. Ethnopharmacol. 105, 241–245 [DOI] [PubMed] [Google Scholar]
- 9. Cheng J. T., Chang T. K., Chen I. S. (1994) Skimmianine and related furoquinolines function as antagonists of 5-hydroxytryptamine receptors in animals. J. Auton. Pharmacol. 14, 365–374 [DOI] [PubMed] [Google Scholar]
- 10. Abe I., Abe T., Wanibuchi K., Noguchi H. (2006) Enzymatic formation of quinolone alkaloids by a plant type III polyketide synthase. Org. Lett. 8, 6063–6065 [DOI] [PubMed] [Google Scholar]
- 11. Nugroho A. E., Riyanto S., Sukari M. A., Maeyamad K. (2010) Effects of skimmianine, a quinolone alkaloid of Aegle marmelos Correa roots, on the histamine release from rat mast cells. J. Basic Appl. Sci. 6, 141–148 [Google Scholar]
- 12. Riyanto S., Sukari M. A., Rahmani M., Ee G. C. L., Yap Y. H. T., Aimi N., Kitajima M. (2001) Alkaloids from Aegle marmelos (Rutaceae). Malays. J. Analy. Sci. 7, 463–465 [Google Scholar]
- 13. Austin M. B., Noel J. P. (2003) The chalcone synthase superfamily of type III polyketide synthases. Nat. Prod. Rep. 20, 79–110 [DOI] [PubMed] [Google Scholar]
- 14. Jez J. M., Bowman M. E., Noel J. P. (2002) Expanding the biosynthetic repertoire of plant type III polyketide synthases by altering starter molecule specificity. Proc. Natl. Acad. Sci. U.S.A. 99, 5319–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ghosh R., Chhabra A., Phatale P. A., Samrat S. K., Sharma J., Gosain A., Mohanty D., Saran S., Gokhale R. S. (2008) Dissecting the functional role of polyketide synthases in Dictyostelium discoideum: biosynthesis of the differentiation regulating factor 4-methyl-5-pentylbenzene-1,3-diol. J. Biol. Chem. 283, 11348–11354 [DOI] [PubMed] [Google Scholar]
- 16. Saxena P., Yadav G., Mohanty D., Gokhale R. S. (2003) A new family of type III polyketide synthases in Mycobacterium tuberculosis. J. Biol. Chem. 278, 44780–44790 [DOI] [PubMed] [Google Scholar]
- 17. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 18. Kiefer F., Arnold K., Künzli M., Bordoli L., Schwede T. (2009) The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 37, D387–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peitsch M. C. (1995) Protein modeling by E-mail Bio/Technology. Nat. Biotechnol. 13, 658–660 [Google Scholar]
- 20. Liang J., Edelsbrunner H., Woodward C. (1998) Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. 7, 1884–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ritchie D. W., Kemp G. J. (2000) Protein docking using spherical polar Fourier correlations. Proteins 39, 178–194 [PubMed] [Google Scholar]
- 22. Durbin M. L., McCaig B., Clegg M. T. (2000) Molecular evolution of the chalcone synthase multigene family in the morning glory genome. Plant Mol. Biol. 42, 79–92 [PubMed] [Google Scholar]
- 23. Schroder J. (1997) A family of plant-specific polyketide synthases: Facts and predictions. Trends Plant Sci. 2, 373–378 [Google Scholar]
- 24. Zheng D., Schröder G., Schröder J., Hrazdina G. (2001) Molecular and biochemical characterization of three aromatic polyketide synthase genes from Rubus idaeus. Plant Mol. Biol. 46, 1–15 [DOI] [PubMed] [Google Scholar]
- 25. Abe I., Takahashi Y., Morita H., Noguchi H. (2001) Benzalacetone synthase. A novel polyketide synthase that plays a crucial role in the biosynthesis of phenylbutanones in Rheum palmatum. Eur. J. Biochem. 268, 3354–3359 [DOI] [PubMed] [Google Scholar]
- 26. Resmi M. S., Soniya E. V. (2012) Molecular cloning and differential expressions of two cDNA encoding Type III polyketide synthase in different tissues of Curcuma longa L. Gene 491, 278–283 [DOI] [PubMed] [Google Scholar]
- 27. Lukacin R., Schreiner S., Matern U. (2001) Transformation of acridone synthase to chalcone synthase. FEBS Lett. 508, 413–417 [DOI] [PubMed] [Google Scholar]
- 28. Akiyama T., Shibuya M., Liu H. M., Ebizuka Y. (1999) p-Coumaroyl triaceticacid synthase, a new homologue of chalcone synthase, from Hydrangea macrophylla var. thunbergii. Eur. J. Biochem. 263, 834–839 [DOI] [PubMed] [Google Scholar]
- 29. Eckermann S., Schroder G., Schmidt J., Strack D., Edrada R. A., Helariutta Y., Elomaa P., Kotilainen M., Kilpelainen I., Proksch P., Teeri T. H., Schroder J. (1998) New pathway to polyketides in plants. Nature 396, 387–390 [Google Scholar]
- 30. Samappito S., Page J. E., Schmidt J., De-Eknamkul W., Kutchan T. M. (2003) Aromatic and pyrone polyketides synthesized by a stilbene synthase from Rheum tataricum. Phytochemistry 62, 313–323 [DOI] [PubMed] [Google Scholar]
- 31. Tillequin F. (2007) Rutaceous alkaloids as models for the design of novel antitumor drugs. Phytochem. Rev. 6, 65–79 [Google Scholar]
- 32. Junghanns K. T., Kneusel R. E., Baumert A., Maier W., Gröger D., Matern U. (1995) Molecular cloning and heterologous expression of acridone synthase from elicited Ruta graveolens L. cell suspension cultures. Plant Mol. Biol. 27, 681–692 [DOI] [PubMed] [Google Scholar]
- 33. Morita H., Kondo S., Oguro S., Noguchi H., Sugio S., Abe I., Kohno T. (2007) Structural insight into chain-length control and product specificity of pentaketide chromone synthase from Aloe arborescens. Chem. Biol. 14, 359–369 [DOI] [PubMed] [Google Scholar]
- 34. Jez J. M., Austin M. B., Ferrer J., Bowman M. E., Schröder J., Noel J. P. (2000) Structural control of polyketide formation in plant-specific polyketide synthases. Chem. Biol. 7, 919–930 [DOI] [PubMed] [Google Scholar]
- 35. Klundt T., Bocola M., Lütge M., Beuerle T., Liu B., Beerhues L. (2009) A Single Amino Acid Substitution Converts Benzophenone Synthase into Phenylpyrone Synthase. J. Biol. Chem. 284, 30957–30964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abe I., Sano Y., Takahashi Y., Noguchi H. (2003) Site-directed mutagenesis of benzalacetone synthase. The role of the Phe215 in plant type III polyketide synthases. J. Biol. Chem. 278, 25218–25226 [DOI] [PubMed] [Google Scholar]
- 37. Dewick P. M. (1997) Medicinal Natural Products. A Biosynthetic Approach, pp. 123–124, Wiley, New York [Google Scholar]
- 38. Abe I., Watanabe T., Morita H., Kohno T., Noguchi H. (2006) Engineered biosynthesis of plant polyketides: manipulation of chalcone synthase. Org. Lett. 8, 499–502 [DOI] [PubMed] [Google Scholar]
- 39. Shi S. P., Wanibuchi K., Morita H., Endo K., Noguchi H., Abe I. (2009) Enzymatic formation of unnatural novel chalcone, stilbene, and benzophenone scaffolds by plant type III polyketide synthase. Org. Lett. 11, 551–554 [DOI] [PubMed] [Google Scholar]
- 40. Morita H., Yamashita M., Shi S. P., Wakimoto T., Kondo S., Kato R., Sugio S., Kohno T., Abe I. (2011) Synthesis of unnatural alkaloid scaffolds by exploiting plant polyketide synthase. Proc. Natl. Acad. Sci. U.S.A. 108, 13504–13509 [DOI] [PMC free article] [PubMed] [Google Scholar]