

Background: The Hsp90 chaperoning machine is an exciting therapeutic target for cancer treatment.
Results: We report that the natural product gedunin inactivates the Hsp90 co-chaperone p23 in vitro and in vivo. The lethal effect of gedunin-p23 complex on cancer cells is amplified by caspase-7-mediated cleavage of the co-chaperone, leading to apoptotic cell death.
Conclusions: Gelduin binds directly to p23 leading to inactivation of the Hsp90 machine and selective destabilization of steroid receptors.
Significance: Gedunin is a promising compound to develop anti-cancer therapeutics.
Keywords: Hsp90, Molecular Chaperone, Protein Drug Interactions, Protein Kinases, Steroid Hormone Receptor, Cancer Therapy, Co-chaperone p23
Abstract
Pharmacological inhibition of Hsp90 is an exciting option for cancer therapy. The clinical efficacy of Hsp90 inhibitors is, however, less than expected. Binding of the co-chaperone p23 to Hsp90 and induced overexpression of anti-apoptotic proteins Hsp70 and Hsp27 are thought to contribute to this outcome. Herein, we report that the natural product gedunin may provide a new alternative to inactivate the Hsp90 machine. We show that gedunin directly binds to p23 and inactivates it, without overexpression of Hsp27 and relatively modest induction of Hsp70. Using molecular docking and mutational analysis, we mapped the gedunin-binding site on p23. Functional analysis shows that gedunin inhibits the p23 chaperoning activity, blocks its cellular interaction with Hsp90, and interferes with p23-mediated gene regulation. Cell treatment with gedunin leads to cancer cell death by apoptosis through inactivation of p23 and activation of caspase 7, which cleaves p23 at the C terminus. These results provide important insight into the molecular mechanism of action of this promising lead compound.
Introduction
Hsp90 is essential for normal cellular homeostasis, but it is also known to play important roles in several pathological conditions (1). Hsp90 works in concert with a number of chaperones and co-chaperones to modulate the conformation of Hsp90 and its client proteins. Over 20 co-chaperone proteins, such as p23, Cdc37, HIP, HOP, PP5, UNC45A, and immunophilins (FKBP51, FKBP52, and Cyp40), have been shown to regulate the function of the Hsp90 protein-folding machine (2–4). These co-chaperones are thought to provide Hsp90 selectivity toward different client proteins. For example, Cdc37 is thought to recruit the Hsp90 machine to kinases (5–7), and p23 is essential for the activation of steroid hormone receptors (6, 8, 9).
The Hsp90 machine has also been implicated as a target to treat neurodegenerative and cardiovascular diseases in addition to cancer. Hsp90 maintains the functional stability of aggregation-prone neuronal proteins and prevents the accumulation of toxic aggregates (10). Similarly, the normal and patho-physiological conditions of the cardiovascular system are highly dependent on Hsp90 machinery. Endothelial nitric oxide synthase (11), vascular endothelial growth factor (VEGF) receptor, and focal adhesion kinase (12) require Hsp90 for their functional activity.
Hsp90 and its co-chaperones are, however, most extensively studied in tumorigenesis, wherein the Hsp90 machine is required for the proper functioning of regulatory proteins that contribute to the hallmarks of cancer (13). More than 15 Hsp90 inhibitors are currently in clinical trials for cancer treatment worldwide (13, 14). Inhibitors of Hsp90, such as the ansamycin antibiotic geldanamycin and its derivatives, as well as purine-scaffold derivatives, are effective in treating different tumor types, including but not limited to melanoma, multiple myeloma, breast, and prostate cancers (3, 15). These drugs bind to and inactivate the Hsp90 N-terminal ATP-binding site, causing proteasomal degradation of Hsp90-dependent client proteins (13, 15, 16). Unfortunately, these inhibitors also induce overexpression of Hsp70 and Hsp27, which are anti-apoptotic proteins that diminish the efficacy of Hsp90 inhibitors (17). Thus, new methods are needed to inhibit the Hsp90 chaperone machinery without inducing the heat shock response. Great efforts have been put forth to develop compounds that target other domains of Hsp90, such as the C-terminal ATP binding pocket (18, 19). However, alternative strategies to block the Hsp90 machine, including inactivation of Hsp90 co-chaperones such as FKBP52, are beginning to be investigated (20).
Gedunin is a tetranortriterpenoid natural product (Fig. 3A) that is isolated from the Meliacae family of medicinal plants and has been used for the treatment of malaria and other infectious diseases in traditional Indian medicine. Recent reports have shown that gedunin exhibits anti-proliferative activity against tumor cells from prostate, ovarian, and colon origins (21–23). Connectivity map analysis established that gedunin is an inhibitor of the Hsp90 machine (24) via a yet unknown mechanism, which is distinct from that of ansamycin analogues (23). Gedunin failed to compete with geldanamycin in a fluorescence polarization assay using purified Hsp90, but it showed a synergistic effect with 17-AAG2 in inhibiting growth of LNCaP cells and AR signaling (23). Extensive chemical modification of gedunin led to the conclusion that Michael acceptor properties of gedunin are not required for its inhibitory activity, and consequently, gedunin represents a promising scaffold for further development (25), and its molecular mechanism of action needs further characterization.
FIGURE 3.

Mapping gedunin-binding site on p23. A, 7-oxo-gedunin has improved efficiency in inhibiting PR chaperoning in vitro. Hormone binding activity of PR after reconstitution using RRL without or with 10 μm 17AAG, gedunin (Gd), 7-carbamate gedunin (Gd-3f), or 7-oxo-gedunin (Gd-4). B, PR protein complex analysis. Samples from A were subjected to SDS-PAGE (10% gel) and Coomassie Blue staining. Heavy (HC) and light (LC) chains of the PR antibody are indicated along with PR, Hsp90, and Hsp70. The level of p23 was assessed by Western blotting using JJ3 antibody (bottom). C and D, gedunin and 7-oxo-gedunin docked into the p23 crystal structure (32). E, docking of both parent gedunin and 7-oxo-gedunin on p23. p23 residues within reasonable distance to establish bonds with gedunin are shown. F, importance of the residues Thr-90, Ala-94, and Lys-95 in gedunin binding was confirmed by mutational analysis. Experiments were repeated three times (see also Table. 1). G, purification of wild-type and p23 point mutants. 12% SDS-PAGE was loaded with 1 μg of each purified protein and gel was stained with Coomassie Blue.
In this study, we sought to identify the molecular target of gedunin. Using a biotin-gedunin conjugate we show that gedunin specifically binds to p23 both in vitro and in vivo and interferes with the passive (Hsp90-independent) chaperoning activity of p23 in vitro. We also show that upon gedunin binding, the conformation of p23 is altered making it more amenable to proteolysis by caspase-7. Using molecular docking and mutational analysis, we identified and validated specific residues of p23 that appear essential for gedunin binding. Treatment of breast and cervical carcinoma cell lines with gedunin induces caspase-dependent cleavage of p23 and apoptotic cell death. This cytotoxic effect of gedunin was specific for tumor cells, and normal cells were relatively unaffected. In addition, gedunin perturbs the ligand-dependent nuclear localization of the glucocorticoid receptor and affects p23-mediated regulation of gene expression. The results below support a role for gedunin as a mediator of apoptotic cancer cell death through a p23-dependent mechanism.
EXPERIMENTAL PROCEDURES
Progesterone Receptor (PR) Reconstitution Assay
Purified PR was adsorbed onto PR22 antibody-protein A-Sepharose resin beads and was assembled into complexes as described previously (26). Briefly, about 0.05 μm PR was incubated with 1 μm each of Hsp90β, Hsp70, Hop, Hsp40 (Ydj), and p23 in reaction buffer (20 mm Tris/HCl, pH 7.5, 5 mm MgCl2, 2 mm DTT, 0.01% Nonidet P-40, 50 mm KCl, and 5 mm ATP). After incubation for 30 min at 30 °C, 0.1 μm [3H]progesterone (American Radiolabeled Chemicals, Inc., (catalog no. ART 0063) was added. Samples were incubated on ice for 3 h at 4 °C. Complexes were then washed three times with 1 ml of reaction buffer and assessed for bound progesterone by liquid scintillation using a Microbeta plate reader (PerkinElmer Life Sciences) and for composition of protein complexes by SDS-PAGE (10% gel) and Coomassie Blue staining.
Pulldown of Biotin-Gedunin Interacting Chaperones
1 μm of purified Hsp90β, Hsp70, HOP, Hsp40 (Ydj), and p23 was incubated with 1 μm biotin-gedunin conjugate (or biotin control) in 500 μl of buffer A (20 mm Tris/HCl, pH 7.8, 50 mm KCl) for 45 min, shaking slowly at room temperature. This was followed by addition of 250 μl of 1% Nonidet P-40 in 20 mm Tris/HCl, pH 7.8, and a 60-μl slurry of NeutrAvidin-agarose resin (Thermo Scientific, catalog no. 29200). Samples were further incubated for 30 min shaking slowly at room temperature. Samples were washed three times with 1 ml of buffer A and analyzed by SDS-PAGE (10% gel) and Western blotting.
Pulldown of Biotin-Gedunin Protein Complexes from Cells
HeLa-PRB cells were plated on a 10-cm dish and treated at 60% confluency with 40 μm biotin-gedunin conjugate (or biotin control) for 15 h. Cells were lysed in buffer B (20 mm Tris/HCl, pH 7.8, 50 mm KCl, 2 mm DTT, 0.1% Nonidet P-40, 10 mm NaF, 1 mm NaVO4, 2 mm β-glycerophosphate, 2 mm sodium pyrophosphate) supplemented with a protease inhibitor mixture (Roche Applied Science, catalog no. 11 836 170 001). 500 μg of clarified cell lysate was incubated with a 200-μl slurry of pre-blocked NeutrAvidin-agarose resin for 3 h at 4 °C followed by three washes with 1 ml of buffer B. Protein complexes were resolved on SDS-PAGE (15% gel) and analyzed by Western blotting.
Gedunin-p23 Docking Studies
All molecular modeling studies were performed in Sybyl version 7.2; docking studies were carried out using Surflex-Dock, and all images were generated in PyMOL. The binding site ProtoMol was generated using the residues identified in (8) as a guide, although Surflex default parameters for threshold and bloat were used. Gedunin and 7-oxo-gedunin were minimized, and partial charges were assigned (Gasteiger Marsili). Each compound was then independently docked with 10 different starting conformations into the p23 ProtoMol allowing for rotation of bonds and sampling of 20 different conformations per fragment; all other Surflex docking parameters were left at default. Docking scores were then analyzed, and the overall best conformations were presented herein.
Protein Expression and Purification
Human Hsp90β was expressed in Sf9 cells and purified as described previously. Hsp70, Hsp40 (Ydj), Hop, and p23 were expressed and purified as described previously (26, 27).
p23 Passive Chaperoning Assay
Thermal aggregation of citrate synthase (Sigma, catalog no. C 3260) was measured as described previously (28). 0.5 μm citrate synthase was incubated alone or in combination with 2.5 μm p23 in 40 mm HEPES, pH 7.5, equilibrated to 41 °C. Light scattering during thermal aggregation of citrate synthase was measured using a Jasco FP-750 spectrofluorometer.
Site-directed Mutagenesis
We used the QuickChange® XL site-directed mutagenesis kit (Agilent Technologies, catalog no. 200522-5) to introduce specific mutations such as T90A, K95A, and T90A/K95A/A94D in pET23a-hp23. All mutant clones were verified by DNA sequencing (McLAB).
Cell Culture and siRNA Knockdown
HeLa-PRB and the breast cancer Hs578T cells at 30–40% confluence in 6-well plates (Corning, catalog no. 3516) were transfected with 100 nm siRNA against p23 using DharmaFECT duo transfection reagent according to the manufacturer's protocol. The nonspecific control VIII was used as negative control to assess off-target effects. Cells were harvested at 96 h, and cell lysates were made. 10 μg of protein lysate were analyzed by Western blotting using the following specific antibodies: homemade antibodies HMGR and PR6 for GR and PRB, respectively; AR from Santa Cruz Biotechnology (catalog no. sc-816); CDK4 from Santa Cruz Biotechnology (catalog no. sc-260); Raf-A from Santa Cruz Biotechnology (catalog no. sc-8408); Chk1 from Santa Cruz Biotechnology (catalog no. sc-8408); and β-actin from Santa Cruz Biotechnology (catalog no. sc-477786).
Mouse Embryonic Fibroblast (MEF) Cultures
The wild-type and corresponding p23 gene disruption mouse lines were developed in the Charles Miller laboratory (9). Cells were plated on a 6-well plate as triplicate samples. When cells were about 50% confluent, they were treated with serial gedunin concentrations or DMSO control. After 24 h of treatment, cells were collected and counted.
Cytosol Preparation, Immunoprecipitation, and Western Blotting
Cells were lysed in buffer C (20 mm Tris/HCl, pH 7.5, 25 mm KCl, 2 mm DTT, 20 mm sodium molybdate, 2 mm ATP, pH 7.6, 2 mm MgCl2, 0.1% Nonidet P-40 supplemented with ATP regeneration system and protease inhibitor mixture) (Roche Applied Science catalog no. 11 836 170 001). After centrifugation at 16,000 × g for 10 min, clarified lysates (∼250 μg of protein) were incubated with antibody to Hsp90 (H90.10) or murine IgG control for 2 h at 4 °C. Protein-A-Sepharose (Pierce, catalog no. 17-0963-03) resin beads were then added to the lysate and incubated for 1.5 h at 4 °C. The immunoprecipitates were washed three times with 1 ml of buffer C. Bound proteins were eluted with SDS sample buffer, resolved by SDS-PAGE (10% gel), and transferred to PVDF membranes. Proteins were then detected by Western blotting with antibodies against Hsp90 (H90.10), p23 (JJ3), or Hop (F5). PARP antibody was a generous gift by Dr. Scott Kaufmann (Mayo Clinic, MN).
Thioflavin-T Binding
Triplicate samples of 25 μm p23 were treated with 50 μm celastrol and 150 μm gedunin for 1 h at 37 °C. 5 μm thioflavin-T was then added followed by analysis of samples for enhancement of thioflavin-T fluorescence using a Safire-Tecan plate reader at the excitation wavelength of 450 nm following emission from 470 to 500 nm as described previously (8).
Immunocytochemisty and Fluorescence Microscopy
HeLa-PRB cells were grown in 24-well plates (Corning, catalog no. 3337) on micro-cover glasses (Electron Microscopy Sciences) to about 50% confluency in MEM, 1× (Cellgro, catalog no. 10-010-CV) medium supplemented with 10% fetal bovine serum. Cells were treated with 30 μm gedunin (or DMSO control) for 2.5 h followed by addition of 150 nm dexamethasone (or ethanol control) for another 1 h. Cells were fixed with 0.1 m PIPES, pH 6.95, 1 mm EGTA, pH 8.0, 3 mm MgSO4, 3% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked with 10% fetal bovine serum with 5% glycerol, and stored at 4 °C. Primary and secondary antibodies were prepared in the blocking buffer.
p23-regulated Gene Analysis
MCF7 cells were grown to 50% confluence on 10-cm culture dishes (Falcon, catalog no. 353003). Cells were treated with 30 μm gedunin or DMSO control for about 20 h. Cells were harvested, and reverse transcriptase PCR was done using a two-step RT-PCR kit (Qiagen, catalog no. 205920). We used the same primers as in Ref. 29. β-Actin was used as an internal control.
Cell Proliferation Assay
To monitor proliferation, cells were grown to 50% confluency on 96-well tissue culture plates (Corning, catalog no. 3599) followed by treatment with gedunin or DMSO control. Cell proliferation was measured using The CellTiter 96® AQueous One Solution Cell Proliferation Assay reagent (Promega, catalog no. G3580).
Compounds
Gedunin was from Gaia Chemical Corp. (catalog no. L4000); dihydrocelastrol was from Gaia Chemical Corp. (catalog no. C2310); 17-AAG was from ChemieTek (catalog no. 75747-14-7); and biotin was from Fisher (catalog no. BP232-1). Gedunin semisynthetic derivatives, 7-carbamate (Gd-3f) and 7-oxo-gedunin (Gd-4) were made in the Brian Blagg laboratory. Biotin-gedunin conjugate was synthesized and purified by Dr. Abdul Fauq (Chemical Synthesis Core, Mayo Clinic, Jacksonville, FL), and Z-VAD-fmk was from Bachem (catalog no. N1510).
RESULTS
Gedunin Selectively Destabilizes Steroid Receptors but Not Signaling Kinase Clients of Hsp90
We compared the effects of increasing concentrations of gedunin and the prototypical Hsp90 inhibitor 17-AAG on Hsp90 client protein stability in HeLa-PRB cells that stably express the B isoform of the progesterone receptor (PRB) as well as in the breast cancer cell lines Hs578T and MCF7 (Fig. 1, A and C). As expected, 17-AAG induced a broad and dose-dependent degradation of Hsp90 client proteins such as signaling kinases, CDK4 and Chk1, and steroid receptors, PRB, GR, and AR. However, gedunin selectively caused a dramatic dose-dependent degradation of steroid receptors PRB and GR without significantly affecting the cellular levels CDK4, Raf-A, and Chk1. Although Hsp90 itself is required for both steroid receptors and kinases, earlier reports have showed that steroid receptors require co-chaperone p23 for their assembly and intracellular stability (8), and signaling kinases require Cdc37 (30). The data in Fig. 1 therefore suggest that gedunin might affect p23 function without affecting the function of Cdc37. Importantly, unlike 17-AAG, gedunin treatment induced relatively little overexpression of Hsp70 in all of the cell lines tested (Fig. 1). Furthermore, contrary to its induction by 17-AAG, gedunin treatment decreased the level of Hsp27 protein in Hs578T, MCF7, and HeLa-PRB cell lines. Taken together, these findings suggest that gedunin selectively inactivates a portion of the Hsp90 machine by a mechanism distinct from that of other Hsp90 inhibitors, such as 17-AAG, that target the ATP-binding site in the Hsp90 N terminus. Because Hsp70 and Hsp27 are known apoptosis inhibitors (17) and thought to contribute to the low efficacy of Hsp90 inhibitors observed in the clinic, gedunin appears to represent a novel leading compound for further optimization, and its mechanism of action was investigated in more detail.
FIGURE 1.

Gedunin induces selective degradation of steroid hormone receptors. Cervical cancer HeLa-PRB (A), human breast cancer Hs578T (B), and MCF7 (C) cell lines were treated with increasing concentrations of gedunin or 17-AAG. Levels of Hsp90, Hsp70, Hsp27, as well as the indicated Hsp90 client proteins were assessed by Western blotting using specific antibodies. β-Actin was used as loading control. PRB, progesterone receptor B isoform; GR, glucocorticoid receptor; AR, androgen receptor; CDK4, cyclin-dependent kinase 4; Chk1, checkpoint kinase 1. For each cell line, the images shown are representative of several experiments.
Gedunin Inhibits PR Chaperoning in Vitro
To determine the mechanism of action for gedunin, we used the in vitro PR reconstitution assay as a model system. This assay uses rabbit reticulocyte lysate (RRL) as a source of molecular chaperones, and it has been fundamental in furthering our understanding of how geldanamycin and related compounds inhibit Hsp90-dependent chaperoning (31). The assay directly measures the ability of molecular chaperones to refold the heat-denatured PR to its hormone-binding state. We thus used the hormone binding activity of the PR as a readout of the functional integrity of molecular chaperones and tested whether gedunin treatment affects the recovery of hormone binding activity of PR after heat denaturation. As shown in Fig. 2A, gedunin inhibits PR reconstitution in RRL in a dose-dependent manner, albeit with lower potency as compared with 17-AAG. Upon further examination of the Hsp90 heteroprotein complexes by SDS-PAGE and Coomassie Blue staining, it was determined that gedunin reduced the levels of p23 and Hsp90 in the PR complex (Fig. 2B). Because p23 is difficult to detect in 10% SDS-PAGE stained with Coomassie Blue, we used Western blot analysis to assess the level of p23 upon gedunin treatment (Fig. 2B, lower panel).
FIGURE 2.

Gedunin inhibits PR chaperoning in vitro and specifically binds to p23. PR hormone binding activity was reconstituted using RRL (A) and the 5P-system containing the five purified chaperones Hsp90, Hsp70, Hsp40 (Ydj), HOP, and p23 (C) in the presence of various concentrations of gedunin (Gd). 17-AAG was used as positive controls. B, samples from A were used for analysis of protein complexes by SDS-PAGE and Coomassie Blue staining (top). The level of p23 was assessed by Western blotting using JJ3 antibody (bottom). WB, Western blot; HC, heavy chain; LC, light chain. D, structures of gedunin, deacetyl-gedunin, and the biotin-gedunin conjugate. E, comparison of the inhibitory effect of gedunin, deacetyl-gedunin, and the biotin-gedunin conjugate on the PR chaperoning using the 5P-system. Binding of [3H]progesterone is expressed as a percentage of the DMSO control. Experiments were reproduced three times in duplicate. F, mixture of Hsp90, Hsp70, Hsp40 (Ydj), Hop, and p23 was incubated with gedunin-biotin conjugate for 45 min at room temperature, and then NeutrAvidin-agarose resin was added. After further incubation for 30 min, resin was washed, and bound proteins were eluted with sample buffer and detected by Western blotting using specific antibodies. G, full-length purified p23 or the C-terminal deletion mutant p23C35 was incubated with gedunin-biotin conjugate and then with NeutrAvidin agarose resin as in F. Each experiment was repeated at least four times. H, unlike celastrol, gedunin does not cause p23 to form amyloid-like fibrils in vitro. 25 μm purified full-length p23 was treated with 2-fold molar excess of celastrol or 6-fold excess of gedunin for 1 h at 37 °C. 5 μm thioflavin-T was then added followed by analysis of samples for enhancement of thioflavin-T (ThT) fluorescence emission. Thioflavin-T binding was monitored using 96-well plates and the Safire-TECAN microplate reader at the excitation wavelength of 450 nm following emission from 470 to 500 nm.
To further our understanding of the mechanism by which gedunin inhibits PR chaperoning, the effect of gedunin on the hormone binding activity of PR was evaluated using the five purified chaperone systems containing Hsp90, Hsp70, Hsp40 (Ydj), Hop, and p23 (5P-system). These chaperones constitute the minimum combination required to preserve the hormone binding of PR following mild heat treatment (26). The 5P-system has been shown to mimic the RRL in chaperoning PR. As seen in Fig. 2C, gedunin efficiently inhibited the recovery of the PR hormone binding activity in this purified system in a concentration-dependent manner. Analysis of protein complexes showed that, as in the case of RRL, gedunin decreased the levels of p23 and Hsp90 in PR complexes (data not shown), which lead to lower hormone binding. These data strongly suggest that the molecular target of gedunin is present among the five essential components of the Hsp90 chaperoning machine, namely Hsp90, Hsp70, Hsp40 (Ydj), Hop, and p23, that are necessary for the folding of steroid receptors.
Gedunin Specifically Targets the Co-chaperone p23
To identify the molecular target of gedunin among the mixture of the above five molecular chaperones, deacetylgedunin was conjugated with a biotin moiety (Fig. 2D). Deacetylgedunin and the biotin-gedunin conjugate inhibit PR reconstitution when tested in the 5P-system (Fig. 2E), suggesting that the biological activity of gedunin was preserved after biotin conjugation, and the biotin-gedunin conjugate could be used as a powerful tool to identify the molecular target of gedunin. We used NeutrAvidin-agarose beads to pull down gedunin and its interacting proteins. The mixture of five purified protein chaperones was incubated with the gedunin-biotin conjugate for 45 min at room temperature before gedunin-interacting proteins were captured with the NeutrAvidin-agarose beads. After multiple washes, protein complexes were analyzed by Western blot using specific antibodies to Hsp90, Hsp70, Hsp40 (Ydj), Hop, and p23. Surprisingly, only p23 showed specific pulldown above background with the biotin-gedunin conjugate (Fig. 2F). Further analysis using only purified p23 in pulldown experiments confirmed that p23 directly interacts with the biotin-gedunin conjugate (Fig. 2G). Interestingly, deletion of the C-terminal 35 amino acids of p23 (p23C35) abolished gedunin binding in pulldown experiments (Fig. 2G), indicating that the flexible C-terminal tail of p23 is critical for p23 interaction with the gedunin-biotin conjugate. Previous work has shown that p23C35 fragment binds Hsp90 and preserves more than 80% of its chaperoning activity when tested in the 5P-system. Furthermore, we have previously shown that wild-type p23 and p23C35 bind to a structurally related compound celastrol, which induces fibrillization of wild-type p23 and p23C35 (8). We therefore tested whether gedunin, like celastrol, could cause fibrillization of wild-type p23. As shown in Fig. 2H, celastrol efficiently causes wild-type p23 to bind thioflavin-T dye, indicating fibril formation. In contrast, gedunin was unable to induce p23 fibrillization at concentrations 6× in molar excess of celastrol. Therefore, despite the fact that gedunin and celastrol share structural similarities, their binding to p23 elicits different outcomes.
Interestingly, modification of the parent gedunin at position C7 of the α,β-unsaturated ketone moiety with an oxygen atom resulted in a compound that was ∼2-fold more potent than gedunin (Fig. 3, A and B) in inhibiting chaperoning of PR in a cell-free system. This conclusion was further supported by the analysis of PR heteroprotein complexes using SDS-PAGE and Coomassie Blue staining showing that 7-oxygedunin (Gd-4) is more efficient in reducing the levels of Hsp90 and p23 co-immunoprecipitated with PR (Fig. 3B).
Mapping the Gedunin-binding Site on p23
To identify the binding site of gedunin on p23, we used the available structure of p23C35 (Protein Data Bank code 1EJF) (32) to perform in silico docking experiments using the residues identified by NMR to be important in celastrol binding to p23 (8). We identified a potential binding site on the surface of p23 that could reasonably accommodate gedunin binding (Fig. 3C). This ligand-binding model is relatively superficial, but this could be attributed to the lack of the p23 C terminus in the available p23 crystal structure. The residues Thr-90 and Lys-95 appear to mediate hydrogen bonds, whereas Ala-94 provides hydrophobic interactions with the cyclohexenone ring of gedunin (Fig. 3C). Interestingly, 7-oxogeduinin docked into this binding pocket in a slightly different orientation and manifested a 10-fold increase in calculated relative binding energy as compared with gedunin (Table 1). This can be reasoned by the fact that the predicted hydrogen bonds between p23 and gedunin/7-oxo-gedunin are maintained at shorter distances with 7-oxo-gedunin (bond lengths, 3.9 and 3.3 Å, respectively) than the corresponding hydrogen bonds with parent gedunin (bond lengths, 5.6 and 4.9 Å, respectively) (Fig. 3, D and E). This observation correlates with the higher efficacy of 7-oxo-gedunin in inhibiting PR chaperoning in vitro (Fig. 3A).
TABLE 1.
Gedunin-p23 binding energy scores
Table represents the relative binding energy scores (in log units) for 7-oxo-gedunin, parent gedunin, and celastrol.
| Compound | Relative binding energy |
|---|---|
| 7-Oxo-gedunin | 3.38 |
| Gedunin | 2.67 |
| Celastrol | 2.47 |
To test our model, p23 binding to gedunin was analyzed with T90A, K95A, and T90A/K95A/A94D mutations. The three mutant proteins showed similar characteristics during the entire purification procedure, and the purity of each mutant is shown on an SDS-PAGE stained with Coomassie Blue in Fig. 3G. Individual mutations of Thr-90 and Lys-95 to alanine (T90A and K95A, respectively) or the combination of these mutations with Ala-94 to aspartic acid mutation (T90A/K95A/A94D) dramatically reduced gedunin binding, demonstrating that these amino acids are crucial for gedunin binding (Fig. 3F). Because the C-terminal 50 amino acids are unstructured and thus not visible in the available crystal structure of the p23 dimer (32), we believe that the identified binding site of gedunin on p23 could only be partially resolved by our docking studies. This is further supported by the fact that the C-terminal tail of 35 amino acids is required for stabilizing the gedunin-p23 interaction as suggested by the loss of gedunin binding with p23C35 in pulldown experiments (Fig. 2G).
p23 Binds to Gedunin in the Cell
To verify whether p23 is a target of gedunin inside the cell, HeLa-PRB cells were treated with the biotin-gedunin conjugate or biotin alone as a control for 15 h. Cells were harvested, and lysates were incubated with NeutrAvidin-agarose resin for 3 h at 4 °C. As shown in Fig. 4A, only the biotin-gedunin conjugate efficiently pulled down p23. Importantly, p23 is competed from the resin when the lysate was incubated with free gedunin or excess dihydrocelastrol, confirming that this interaction is specific. Interestingly, biotin-gedunin conjugate induced cleavage of p23 (Fig. 4A, input) (discussed below).
FIGURE 4.

Gedunin targets p23 in cells. A, HeLa-PRB cells were treated with 40 μm of the biotin-gedunin conjugate or biotin as control for 15 h. Left panel, cytosols were made and incubated with NeutrAvidin-agarose resin with or without free gedunin (40 μm) or free dihydrocelastrol (400 μm). Right panel, cytosols used in the left panel show that biotin-gedunin conjugate treatment caused cleavage of p23 to generate a smaller form of about 18 kDa. B and C, gedunin treatment mimics p23 silencing. Breast cancer Hs578T (A) and HeLa-PRB (B) cells were cultured in 6-well plates then treated with 100 nm of p23 or control siRNA for 96 h or with the indicated concentrations of gedunin and 17-AAG for 24 h. Cytosols were made, and levels of p23, Hsp90, and Hsp90 client proteins (GR, PRB AR, CDK4, Raf-A, and ChK1) were assessed by Western blotting. D, mouse embryonic fibroblasts derived from p23 knock-out mouse are resistant to gedunin treatment. In concentration-dependent manner, gedunin induces cell death of p23+/+ MEFs (black bars) but not that of p23−/− MEFs (open bars).
Because gedunin binding inhibits PR reconstitution in vitro, it was predicted that gedunin treatment would mimic the lack of p23 within the cell. To test this hypothesis, the effects of gedunin were compared with silencing of p23 with siRNA, and the profile of Hsp90 client proteins was assessed by Western blot analysis. Both treatments resulted in similar destabilization of GR and PRB in breast cancer Hs578T and HeLa-PRB cells without significantly affecting the levels of kinases CDK4, Raf-A, and ChK1 (Fig. 4, B and C). Interestingly, AR, which interacts with Cdc37 (33), is neither destabilized by gedunin treatment nor by p23 silencing in HeLa-PRB cells. A similar finding was reported upon celastrol treatment, which leads to the inactivation of p23 by inducing fibrillization (8). Together, these data demonstrate that gedunin targets p23 in Hs578T and HeLa-PRB cells. This interpretation was further supported by the observation that MEFs derived from p23 null mice were resistant to gedunin treatment when compared with parental p23+/+ cells (Fig. 4D).
Gedunin Affects p23 Functions in Vitro and in Vivo
The effect of gedunin on the function of p23 using both in vitro and in vivo approaches was further evaluated. p23 has been shown to possess an Hsp90-independent passive chaperoning activity, which prevents heat-induced aggregation of citrate synthase (28). Gedunin blocked, although modestly, the ability of p23 to prevent thermal aggregation of citrate synthase upon heating at 41 °C (Fig. 5A). This modest effect could be explained by a reduced p23 binding to gedunin at 41 °C.
FIGURE 5.
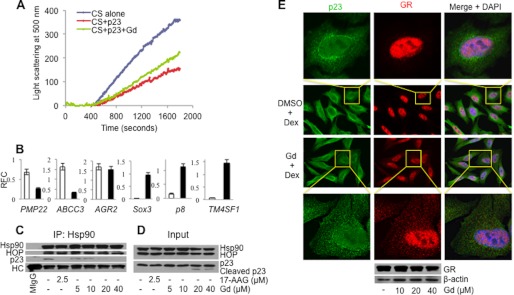
Gedunin affects p23 function in vitro and in vivo. A, gedunin (Gd) inhibits the ability of p23 to chaperone citrate synthase (CS) in vitro. B, gedunin interferes with p23 regulation of gene expression. MCF7 cells were treated with 30 μm gedunin for 24 h, and the expression of the indicated genes was assessed by RT-PCR. Data were quantified using ImageJ software. Open bars represent DMSO-treated samples, and black bars represent gedunin-treated samples. Data represents S.E. of duplicate experiments. RFC, relative fold change. C, gedunin destabilizes the p23-Hsp90 complex in HeLa-PRB cells. Cells were treated with increasing concentrations of gedunin for 24 h. 2.5 μm of 17-AAG was used as positive control and DMSO as a negative control. Cell lysates were made, and Hsp90 was immunoprecipitated (IP) using the H90.10 antibody. MIgG is the murine IgG antibody incubated with cell lysate. Protein complexes were then analyzed by Western blotting using antibodies against Hsp90 (H90.10), HOP (F5), or p23 (JJ3). HC, heavy chain of H90.10 and murine IgG. D, cell lysates used in C were analyzed for Hsp90, HOP, and p23. p23 cleavage can be seen with higher (20 and 40 μm) concentrations of gedunin treatment but not with 17-AAG treatment or DMSO control. E, gedunin (Gd) treatment disrupts p23 perinuclear localization and GR nucleo-cytoplasmic trafficking (upper panel). Gedunin-treated cells were analyzed by immunocytochemistry using p23 and GR-specific antibodies. Cell images obtained at ×100 magnification are shown in center panels. The stability of GR was assessed after 3 h of gedunin treatment using Western blotting (lower panel). Dex, dexamethasone.
Furthermore, p23 has been established as a modulator of gene expression at the promoter level. Using the yeast system as well as breast cancer cell line MCF7, Garabedian and co-workers have demonstrated that p23 modulates ER signaling (34–36). Overexpression of p23 enhances the expression of genes that are regulated by direct binding of ER to the estrogen-response element, without affecting genes that are regulated by ER via indirect interaction with their promoters and other transcription factors (36). Genome-wide analysis showed that small increases in p23 levels amplify estradiol-induced ER-binding sites and that ER tends to bind more with ER-induced genes (37). Overexpression of p23 in MCF7 cells enhances expression of the genes PMP22, ABCC3, and AGR2, although down-regulation of the genes p8, TM4SF1, and Sox3 (29) occurs. We treated MCF7 cells with gedunin and analyzed the effect of gedunin treatment on these genes using RT-PCR. As shown in Fig. 5B, gedunin reversed the expression of these genes. The genes PMP22 and ABCC3 were down-regulated upon gedunin treatment. AGR2 expression, however, did not show any significant change upon gedunin treatment. The genes p8, TM4SF1, and Sox3 were reciprocally up-regulated after gedunin treatment. These data strongly suggest that gedunin interferes with p23-mediated gene regulation.
Gedunin Destabilizes the p23-Hsp90 Complex and Disrupts GR Nuclear Localization
Analysis of protein complexes from PR reconstitution with RRL indicated that gedunin causes a dose-dependent decrease of p23 levels in PR complexes (Fis. 2B and 3B). This translates into a decrease of Hsp90 levels in PR complexes because p23 stabilizes the Hsp90-PR interaction (31). Importantly, gedunin-induced destabilization of the p23-Hsp90 complex is observed in cells. As shown in Fig. 5C, immunoprecipitation of Hsp90 complexes from cells treated with gedunin resulted in a concentration-dependent loss of p23. In contrast, the level of another co-chaperone, Hop, did not change (Fig. 5C), demonstrating that gedunin effect is specific to p23-Hsp90 interactions. Interestingly, treatment of cells with gedunin also induced a dose-dependent cleavage of p23 (Fig. 5D) (see below).
In addition to its role in maintaining the stable (ligand binding competent) oligomeric complexes of steroid receptors such as GR, the co-chaperone p23 is an important cofactor for the nucleo-cytoplasmic trafficking of GR. p23, along with Hsp90, Hsp70, FKBP51 and FKBP52, forms a heterocomplex with GR that interacts with importin-β nuclear pore machinery, thus facilitating movement of GR into the nuclear compartment (38). p23 has also been shown to interact with the nuclear pore complex protein, Nup62, in an Hsp90-independent manner (38). The integrity of p23 function is necessary for the hormone binding and transcriptional activity of GR (9, 31). It was therefore hypothesized that inhibition of p23 function by gedunin treatment would block the nucleo-cytoplasmic trafficking of GR. To investigate this hypothesis, HeLa-PRB cells were treated with dexamethasone either in combination with gedunin or DMSO (control) for 3 h before assessing the nucleo-cytoplasmic distribution of GR by immunofluorescence microscopy (Fig. 5E, upper panel). As compared with DMSO control, GR was markedly retained in the cytoplasm of gedunin-treated cells. We confirmed by Western blotting that GR was not degraded after 3 h of gedunin treatment (Fig. 5E, lower panel). Although no significant impact on the nucleo-cytoplasmic distribution of p23 was observed in gedunin-treated cells, a clear disruption of classical perinuclear localization of p23 in HeLa-PRB cells was noted. Together, these data demonstrate that gedunin blocks p23 function and prevents the efficient nucleo-cytoplasmic trafficking of GR.
Gedunin Induces Apoptotic Death of Cancer Cells and Caspase-dependent Cleavage of p23
Gedunin has been reported to induce cancer cell death in prostate, ovarian, and colon cancer cell lines (21–23). Data in Fig. 6A show that gedunin also induces cancer cell death in cervical (HeLa-PRB) and breast carcinoma (MDA-MB-231, MDA-MB-453, Hs578T, T47D, and MCF7) cell lines in a dose-dependent manner. Comparison of cell growth of cancer versus immortalized normal cell lines showed that gedunin selectively killed cancer cells. Adult-derived normal cells, Hs578Bst cells, and human mammary epithelial cells (HME) were remarkably resistant to gedunin treatment (Fig. 6B). Apoptosis mediated by activation of caspase-7 leading to cleavage of PARP seems to be the death pathway triggered by gedunin in HeLa-PRB and MCF7 cells (Fig. 6, C and D).
FIGURE 6.

Gedunin induces apoptotic death of various human cancer cell lines. A, human breast cancer cell lines MDA-MB-231, T47D, Hs578T, MDA-MB-453, and MCF7 and human cervical cancer cell line HeLa-PRB cells were treated with the indicated concentrations of gedunin (Gd) for 72 h. Cell proliferation was assessed by MTS assay at 24 h (circle), 48 h (square), and 72 h (triangle). Data represent S.E. of quadruplicate samples for each gedunin concentration normalized to DMSO control. Experiment was repeated three times. B, human breast cancer cell line (Hs578T), nontransformed immortalized normal breast cells (Hs578Bst), and immortalized normal HME cells were treated with 12.5 μm gedunin concentration for the indicated time. Cell proliferation was assessed by MTS assay at 0, 24, 48, 72, and 96 h. Data represent S.E. of quadruplicate samples for each time point normalized to 0-h control. Experiment was repeated three times. HeLa-PRB (C) and MCF7 (D) cells were treated with increasing concentrations of 17-AAG or gedunin. DMSO was used as a control. Cell lysates were analyzed for PARP cleavage by Western blotting. β-Actin was used as a loading control.
It has been reported that p23 is cleaved by caspases during endoplasmic reticulum stress response (39, 40) and during apoptotic cell death induced by several stimuli, including the commonly used chemotherapeutic agents such as anthracyclines, cytarabine, etoposide (41), and tunicamycin (42). Cleavage of p23 is reported to occur at the C-terminal Asp-142 residue, and caspase-7 is the most effective executioner caspase catalyzing this reaction (41–43).
To verify whether gedunin-induced cleavage of p23 is a caspase-dependent phenomenon, the pan-caspase inhibitor Z-VAD-fmk was used to prevent gedunin-induced proteolysis of p23. As shown in Fig. 7A, co-treatment of cells with gedunin and Z-VAD-fmk completely blocked the cleavage of both caspase-7 and p23. This indicates that gedunin-mediated activation of caspase-7 (by yet unknown mechanism) subsequently triggers cleavage of p23. Furthermore, gedunin-induced PARP degradation was completely abrogated by co-treatment with Z-VAD-fmk. Cell survival analysis however reflected only partial rescue from apoptotic cell death (Fig. 7B) in spite of no biochemical evidence of apoptosis in gedunin-Z-VAD-fmk co-treated cells (Fig. 7A). This suggests that gedunin binding to p23 exerts other toxic events that contribute to cell death. As shown in Figs. 1 and 7A, gedunin treatment causes cellular destabilization of GR. However, inhibition of gedunin-induced p23 cleavage with Z-VAD-fmk failed to prevent the degradation of GR (Fig. 7A), suggesting that gedunin-bound p23 (although not cleaved) is inactive and therefore unable to maintain chaperoning and stability of intracellular GR.
FIGURE 7.
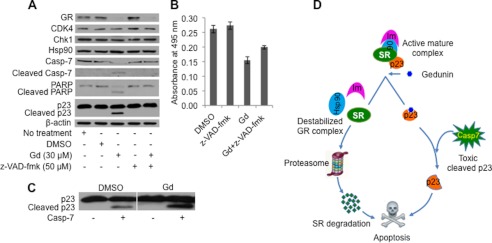
Gedunin induces cleavage of p23 in caspase-7-dependent manner. A, gedunin induces p23 cleavage, caspase-7 activation, and PARP degradation. The pan-caspase inhibitor Z-VAD-fmk blocks p23 cleavage but does not prevent gedunin-induced GR degradation. B, MTS assay showing partial rescue of HeLa-PRB cells from 30 μm gedunin-induced apoptotic cell death when co-treated with 50 μm Z-VAD-fmk. Data represent absorbance at 495 nm. The standard deviation of quadruplet samples is shown here. Experiment was repeated three times. C, gedunin binding sensitizes p23 to purified caspase-7. Data were reproduced by two independent experiments. D, model for gedunin effects. Binding of gedunin causes a destabilization of steroid receptor (SR) chaperone complexes leading to proteasomal degradation of steroid receptors. p23 bound to gedunin is cleaved more efficiently by the activated caspase-7, which promotes apoptotic cell death.
Interestingly, the p23-bound gedunin is more prone to caspase hydrolysis. As shown in Fig. 7C, purified caspase-7 hydrolyzes p23 more efficiently in the presence of gedunin as compared with apo-p23. This is additional evidence that gedunin directly binds to p23 and changes its conformation to better expose the Asp-142 cleavage site to caspase-7.
DISCUSSION
In this study, we showed that the natural product gedunin inactivates Hsp90 chaperoning machinery by targeting the co-chaperone p23 in vivo and in vitro. Using a biotin-gedunin conjugate, we pulled down p23 from cell lysates as well as purified p23 in vitro, thus demonstrating for the first time a direct interaction between gedunin and p23. Using computational modeling studies, we showed that gedunin can be successfully docked into the p23 structure, and three amino acids (Thr-90, Ala-94, and Lys-95) possibly mediate noncovalent interactions with gedunin. The crystal structure of p23 dimer reported by Weaver et al. (32) showed that the surface of p23 molecule exhibits a solvent-accessible cavity surrounded by polar amino acids, whereas the floor of the cavity contains nonpolar hydrophobic amino acids, which appear to provide a ligand-binding site on the surface of p23. Utilizing molecular modeling and docking software, we observed that gedunin docks into this ligand-binding site, forms hydrogen bonds with Thr-90 and Lys-95, and maintains hydrophobic interactions with Ala-94. Mutagenesis studies confirmed the validity of the proposed model and demonstrated gedunin-p23 interactions at an atomic level. In addition, it appears that the C-terminal 35 amino acids are essential for gedunin-p23 binding, which suggests that the unstructured C-terminal tail stabilizes gedunin binding to the identified pocket. The ligand-binding cleft, along with other amino acids in this vicinity, compose the Hsp90-binding site on the surface of p23 (32, 44), and gedunin binding to p23 appears to alter the topology of this region thus disrupting the p23-Hsp90 interaction. Data in Fig. 5C support this hypothesis and explain why complexes of Hsp90 with client proteins (such as GR and PR) that require p23 are destabilized upon treatment with gedunin. It is well established that inactivation of the Hsp90 machine by small molecules targeting the N-terminal ATP-binding site of Hsp90 causes degradation of intracellular steroid receptors GR, PR, and ER as well as signaling protein kinases by the ubiquitin-proteasome machinery. In contrast, gedunin causes only a minimal destabilization of protein kinase clients of Hsp90. This is in agreement with previous work showing that p23 is critical for steroid receptor complex assembly but not for the assembly of protein kinase complexes, which instead require Cdc37 (6, 27). A recent comprehensive report has confirmed the requirement of Cdc37 for the kinase clients of Hsp90 (30).
Death of cancer but not nontransformed immortalized cells was observed after gedunin treatment (Fig. 6A), and the fact that inhibition of gedunin-induced p23 cleavage failed to completely rescue dying cells (Fig. 7B) and to prevent degradation of GR indicates that the gedunin-p23 complex is toxic to cancer cells. An earlier report by Woo et al. (42) showed that overexpression of cleaved p23, as opposed to wild-type p23, is toxic to HEK 293 cells. Together, these data suggest that gedunin exerts a dual cytotoxic effect by binding to p23 and also by triggering caspase-7-mediated cleavage of p23, both of which ultimately kill cancer cells (Fig. 7D). Our data, however, indicate that the primary cause of gedunin-induced cell death is the binding of gedunin to p23 and its inactivation (Fig. 7D). This is further supported by the fact that a high concentration of gedunin kills MEFs derived from p23 wild-type mice; however, MEFs from p23−/− mice were relatively unaffected. Perhaps cancer cells living with and depending on p23 are more sensitive to a sudden loss of p23 function upon gedunin treatment as compared with normal cells. Indeed, lower doses of gedunin kill cancer cell line Hs578T over time without significantly affecting nontransformed immortalized cells lines Hs578Bst and HME. At the molecular level, cancer cells seem to respond differently to the lack of p23. Indeed, an earlier report (9) shows that the level of GR in MEFs does not decrease significantly in p23 null cell, but in cancer cells the lack of p23 causes dramatic GR degradation (Figs. 1, 4, and 7). This may also be the case for other prosurvival protein clients of Hsp90 that are dependent on p23 such as other steroid receptors and telomerase (45). Therefore, a thorough comparative study on gedunin's effects on normal versus cancer cells warrants further investigation.
Targeting p23 may also improve the efficacy of Hsp90 inhibitors as p23 has been shown to protect Hsp90 against its inhibitors targeting the N-terminal ATP binding domain (46). Indeed, expression of p23 correlates with breast tumor grades, and it is highly overexpressed in malignant metastatic tissues. Importantly, microarray analysis using breast cancer cell line MCF7 showed that overexpression of p23 changes the expression levels of several genes involved in metastasis and drug resistance (29), and p23 is required for optimal cell mobility and invasion (36). Recent global analysis of p23 functions by Echtenkamp et al. (47) suggests an extensive cellular network through which p23 modulates a wide range of physiological functions in a manner dependent and independent of Hsp90. Their data also showed that p23 interactors are primarily distributed outside the endoplasmic reticulum. We thus propose that gedunin can be used as a pharmacological probe to further dissect the biological consequences of disrupting p23 interactions in vivo. In fact, data from our experiments showed that the perinuclear localization of p23 was altered after gedunin treatment (Fig. 5E), and it will be interesting to test if there is any alteration of p23 localization and function at the level of cellular organelles.
The chaperoning activity of p23 is being extended to include nuclear activities such as GR mobility (48). p23 has also been established as a modulator of gene expression at the promoter level. Although there are some discrepancies regarding how p23 modulates the transcription regions, this co-chaperone is clearly important for assembling and/or disassembling of steroid receptor-DNA complexes. p23 is recruited to promoters of GR-regulated genes and inhibits GR transcriptional activity by disassembling transcriptional regulatory complexes (49). In a recent report, Freeman and co-workers (50) have further shown that p23 initiates the dissociation of protein-DNA complexes and the acetyltransferase GCN5 prolongs this dissociated state. Other groups, however, have shown that p23 stabilizes GR binding to promoters (51) and p23 stimulates ER transcriptional activity (36). Furthermore, genome-wide analysis showed that overexpression of p23 leads to a large increase in a number of promoter sites bound by ER but also a p23-dependent eviction of ER in a small number of promoter sites (37). Gene expression analysis presented in this report therefore supports the idea that gedunin could be used as a pharmacological tool to probe the regulatory functions of p23 at the promoter level.
Data in this study support our previous findings with celastrol, which shares structural similarity to gedunin and was also identified in the same screen as an Hsp90 pathway inhibitor (23). Similar to gedunin, celastrol inactivates p23 function and destabilizes steroid receptors without affecting protein kinase clients of Hsp90 (8). A comparison of gedunin and celastrol demonstrated that, unlike celastrol, gedunin does not induce fibrillization of p23 in vitro (Fig. 2H). Thus, contrary to the drastic conformational changes of p23 induced by celastrol (8), gedunin binding to p23 seems to cause more subtle conformational changes, which are significant enough to sensitize p23 to caspase-7-mediated proteolysis (Fig. 7C). The rationale for these differences is still unclear; however, one possible reason could be due to the fact that celastrol may form covalent bonds with the cysteines of p23, as apposed to the noncovalent interactions between gedunin and p23. Celastrol has been shown to react with cysteine residues of Cdc37 (52), with the N-terminal domain of Hsp90 (53), and it interferes with the inducible nitric-oxide synthase, topoisomerase II, NF-κB, potassium channels, and the proteasome activity (54–59). It also induces the heat shock response by activating the transcription factor Hsf1 leading to overexpression of Hsp70 (60), and it activates antioxidant proteins in yeast and mammalian cells (61). Furthermore, because gedunin and 7-oxo-gedunin show 10- and 100-fold higher binding scores, respectively, as compared with celastrol (Table. 1), we suggest that gedunin may be a better lead compound to develop the first generation of anti-cancer therapeutics against the co-chaperone p23.
Acknowledgment
We thank Dr. Kang Won Jang for insightful discussion.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM102443-01 (to A. C.). This work was also supported by Scientist Development Grant 0930019N from the American Heart Association (to A. C.).
This article was selected as a Paper of the Week.
- 17-AAG
- 17-allylamino-17-demethoxy-geldanamycin
- ER
- estrogen receptor
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- PR
- progesterone receptor
- GR
- glucocorticoid receptor
- AR
- androgen receptor
- RRL
- rabbit reticulocyte lysate
- PARP
- poly(ADP-ribose) polymerase
- HME
- human mammary epithelial
- MTS
- [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt]
- MEF
- mouse embryonic fibroblast.
REFERENCES
- 1. Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 2. Wegele H., Müller L., Buchner J. (2004) Hsp70 and Hsp90–a relay team for protein folding. Rev. Physiol. Biochem. Pharmacol. 151, 1–44 [DOI] [PubMed] [Google Scholar]
- 3. Whitesell L., Lindquist S. L. (2005) HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772 [DOI] [PubMed] [Google Scholar]
- 4. Johnson J. L. (2012) Evolution and function of diverse Hsp90 homologs and co-chaperone proteins. Biochim. Biophys. Acta 1823, 607–613 [DOI] [PubMed] [Google Scholar]
- 5. Pearl L. H. (2005) Hsp90 and Cdc37 – a chaperone cancer conspiracy. Curr. Opin. Genet. Dev. 15, 55–61 [DOI] [PubMed] [Google Scholar]
- 6. Felts S. J., Karnitz L. M., Toft D. O. (2007) Functioning of the Hsp90 machine in chaperoning checkpoint kinase I (Chk1) and the progesterone receptor (PR). Cell Stress Chaperones 12, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vaughan C. K., Gohlke U., Sobott F., Good V. M., Ali M. M., Prodromou C., Robinson C. V., Saibil H. R., Pearl L. H. (2006) Structure of an Hsp90-Cdc37-Cdk4 complex. Mol. Cell 23, 697–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chadli A., Felts S. J., Wang Q., Sullivan W. P., Botuyan M. V., Fauq A., Ramirez-Alvarado M., Mer G. (2010) Celastrol inhibits Hsp90 chaperoning of steroid receptors by inducing fibrillization of the co-chaperone p23. J. Biol. Chem. 285, 4224–4231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grad I., McKee T. A., Ludwig S. M., Hoyle G. W., Ruiz P., Wurst W., Floss T., Miller C. A., 3rd, Picard D. (2006) The Hsp90 cochaperone p23 is essential for perinatal survival. Mol. Cell. Biol. 26, 8976–8983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Voisine C., Pedersen J. S., Morimoto R. I. (2010) Chaperone networks: tipping the balance in protein folding diseases. Neurobiol. Dis. 40, 12–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. García-Cardeña G., Fan R., Shah V., Sorrentino R., Cirino G., Papapetropoulos A., Sessa W. C. (1998) Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 392, 821–824 [DOI] [PubMed] [Google Scholar]
- 12. Le Boeuf F., Houle F., Huot J. (2004) Regulation of vascular endothelial growth factor receptor 2-mediated phosphorylation of focal adhesion kinase by heat shock protein 90 and Src kinase activities. J. Biol. Chem. 279, 39175–39185 [DOI] [PubMed] [Google Scholar]
- 13. Trepel J., Mollapour M., Giaccone G., Neckers L. (2010) Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 10, 537–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim Y. S., Alarcon S. V., Lee S., Lee M. J., Giaccone G., Neckers L., Trepel J. B. (2009) Update on Hsp90 inhibitors in clinical trial. Curr. Top. Med. Chem. 9, 1479–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taldone T., Gozman A., Maharaj R., Chiosis G. (2008) Targeting Hsp90: small-molecule inhibitors and their clinical development. Curr. Opin. Pharmacol. 8, 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pearl L. H., Prodromou C., Workman P. (2008) The Hsp90 molecular chaperone: an open and shut case for treatment. Biochem. J. 410, 439–453 [DOI] [PubMed] [Google Scholar]
- 17. Lanneau D., Brunet M., Frisan E., Solary E., Fontenay M., Garrido C. (2008) Heat shock proteins: essential proteins for apoptosis regulation. J. Cell. Mol. Med. 12, 743–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marcu M. G., Chadli A., Bouhouche I., Catelli M., Neckers L. M. (2000) The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J. Biol. Chem. 275, 37181–37186 [DOI] [PubMed] [Google Scholar]
- 19. Burlison J. A., Avila C., Vielhauer G., Lubbers D. J., Holzbeierlein J., Blagg B. S. (2008) Development of novobiocin analogues that manifest anti-proliferative activity against several cancer cell lines. J. Org. Chem. 73, 2130–2137 [DOI] [PubMed] [Google Scholar]
- 20. De Leon J. T., Iwai A., Feau C., Garcia Y., Balsiger H. A., Storer C. L., Suro R. M., Garza K. M., Lee S., Kim Y. S., Chen Y., Ning Y. M., Riggs D. L., Fletterick R. J., Guy R. K., Trepel J. B., Neckers L. M., Cox M. B. (2011) Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc. Natl. Acad. Sci. U.S.A. 108, 11878–11883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uddin S. J., Nahar L., Shilpi J. A., Shoeb M., Borkowski T., Gibbons S., Middleton M., Byres M., Sarker S. D. (2007) Gedunin, a limonoid from Xylocarpus granatum, inhibits the growth of CaCo-2 colon cancer cell line in vitro. Phytother. Res. 21, 757–761 [DOI] [PubMed] [Google Scholar]
- 22. Kamath S. G., Chen N., Xiong Y., Wenham R., Apte S., Humphrey M., Cragun J., Lancaster J. M. (2009) Gedunin, a novel natural substance, inhibits ovarian cancer cell proliferation. Int. J. Gynecol. Cancer 19, 1564–1569 [DOI] [PubMed] [Google Scholar]
- 23. Hieronymus H., Lamb J., Ross K. N., Peng X. P., Clement C., Rodina A., Nieto M., Du J., Stegmaier K., Raj S. M., Maloney K. N., Clardy J., Hahn W. C., Chiosis G., Golub T. R. (2006) Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 10, 321–330 [DOI] [PubMed] [Google Scholar]
- 24. Lamb J., Crawford E. D., Peck D., Modell J. W., Blat I. C., Wrobel M. J., Lerner J., Brunet J. P., Subramanian A., Ross K. N., Reich M., Hieronymus H., Wei G., Armstrong S. A., Haggarty S. J., Clemons P. A., Wei R., Carr S. A., Lander E. S., Golub T. R. (2006) The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 [DOI] [PubMed] [Google Scholar]
- 25. Brandt G. E., Schmidt M. D., Prisinzano T. E., Blagg B. S. (2008) Gedunin, a novel hsp90 inhibitor: semisynthesis of derivatives and preliminary structure-activity relationships. J. Med. Chem. 51, 6495–6502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kosano H., Stensgard B., Charlesworth M. C., McMahon N., Toft D. (1998) The assembly of progesterone receptor-hsp90 complexes using purified proteins. J. Biol. Chem. 273, 32973–32979 [DOI] [PubMed] [Google Scholar]
- 27. Arlander S. J., Felts S. J., Wagner J. M., Stensgard B., Toft D. O., Karnitz L. M. (2006) Chaperoning checkpoint kinase 1 (Chk1), an Hsp90 client, with purified chaperones. J. Biol. Chem. 281, 2989–2998 [DOI] [PubMed] [Google Scholar]
- 28. Bose S., Weikl T., Bügl H., Buchner J. (1996) Chaperone function of Hsp90-associated proteins. Science 274, 1715–1717 [DOI] [PubMed] [Google Scholar]
- 29. Simpson N. E., Lambert W. M., Watkins R., Giashuddin S., Huang S. J., Oxelmark E., Arju R., Hochman T., Goldberg J. D., Schneider R. J., Reiz L. F., Soares F. A., Logan S. K., Garabedian M. J. (2010) High levels of Hsp90 cochaperone p23 promote tumor progression and poor prognosis in breast cancer by increasing lymph node metastases and drug resistance. Cancer Res. 70, 8446–8456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taipale M., Krykbaeva I., Koeva M., Kayatekin C., Westover K. D., Karras G. I., Lindquist S. (2012) Quantitative analysis of hsp90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pratt W. B., Toft D. O. (2003) Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp. Biol. Med. 228, 111–133 [DOI] [PubMed] [Google Scholar]
- 32. Weaver A. J., Sullivan W. P., Felts S. J., Owen B. A., Toft D. O. (2000) Crystal structure and activity of human p23, a heat shock protein 90 co-chaperone. J. Biol. Chem. 275, 23045–23052 [DOI] [PubMed] [Google Scholar]
- 33. Rao J., Lee P., Benzeno S., Cardozo C., Albertus J., Robins D. M., Caplan A. J. (2001) Functional interaction of human Cdc37 with the androgen receptor but not with the glucocorticoid receptor. J. Biol. Chem. 276, 5814–5820 [DOI] [PubMed] [Google Scholar]
- 34. Knoblauch R., Garabedian M. J. (1999) Role for Hsp90-associated cochaperone p23 in estrogen receptor signal transduction. Mol. Cell. Biol. 19, 3748–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oxelmark E., Knoblauch R., Arnal S., Su L. F., Schapira M., Garabedian M. J. (2003) Genetic dissection of p23, an Hsp90 cochaperone, reveals a distinct surface involved in estrogen receptor signaling. J. Biol. Chem. 278, 36547–36555 [DOI] [PubMed] [Google Scholar]
- 36. Oxelmark E., Roth J. M., Brooks P. C., Braunstein S. E., Schneider R. J., Garabedian M. J. (2006) The cochaperone p23 differentially regulates estrogen receptor target genes and promotes tumor cell adhesion and invasion. Mol. Cell. Biol. 26, 5205–5213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simpson N. E., Gertz J., Imberg K., Myers R. M., Garabedian M. J. (2012) Research resource: enhanced genome-wide occupancy of estrogen receptor α by the cochaperone p23 in breast cancer cells. Mol. Endocrinol. 26, 194–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Echeverría P. C., Mazaira G., Erlejman A., Gomez-Sanchez C., Piwien Pilipuk G., Galigniana M. D. (2009) Nuclear import of the glucocorticoid receptor-hsp90 complex through the nuclear pore complex is mediated by its interaction with Nup62 and importin β. Mol. Cell. Biol. 29, 4788–4797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rao R. V., Niazi K., Mollahan P., Mao X., Crippen D., Poksay K. S., Chen S., Bredesen D. E. (2006) Coupling endoplasmic reticulum stress to the cell-death program: a novel HSP90-independent role for the small chaperone protein p23. Cell Death Differ. 13, 415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Poksay K. S., Banwait S., Crippen D., Mao X., Bredesen D. E., Rao R. V. (2012) The small chaperone protein p23 and its cleaved product p19 in cellular stress. J. Mol. Neurosci. 46, 303–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gausdal G., Gjertsen B. T., Fladmark K. E., Demol H., Vandekerckhove J., Døskeland S. O. (2004) Caspase-dependent, geldanamycin-enhanced cleavage of co-chaperone p23 in leukemic apoptosis. Leukemia 18, 1989–1996 [DOI] [PubMed] [Google Scholar]
- 42. Woo S. H., An S., Lee H. C., Jin H. O., Seo S. K., Yoo D. H., Lee K. H., Rhee C. H., Choi E. J., Hong S. I., Park I. C. (2009) A truncated form of p23 down-regulates telomerase activity via disruption of Hsp90 function. J. Biol. Chem. 284, 30871–30880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walsh J. G., Cullen S. P., Sheridan C., Lüthi A. U., Gerner C., Martin S. J. (2008) Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. U.S.A. 105, 12815–12819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ali M. M., Roe S. M., Vaughan C. K., Meyer P., Panaretou B., Piper P. W., Prodromou C., Pearl L. H. (2006) Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 440, 1013–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Toogun O. A., Zeiger W., Freeman B. C. (2007) The p23 molecular chaperone promotes functional telomerase complexes through DNA dissociation. Proc. Natl. Acad. Sci. U.S.A. 104, 5765–5770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Forafonov F., Toogun O. A., Grad I., Suslova E., Freeman B. C., Picard D. (2008) p23/Sba1p protects against Hsp90 inhibitors independently of its intrinsic chaperone activity. Mol. Cell. Biol. 28, 3446–3456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Echtenkamp F. J., Zelin E., Oxelmark E., Woo J. I., Andrews B. J., Garabedian M., Freeman B. C. (2011) Global functional map of the p23 molecular chaperone reveals an extensive cellular network. Mol. Cell 43, 229–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Elbi C., Walker D. A., Romero G., Sullivan W. P., Toft D. O., Hager G. L., DeFranco D. B. (2004) Molecular chaperones function as steroid receptor nuclear mobility factors. Proc. Natl. Acad. Sci. U.S.A. 101, 2876–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Freeman B. C., Yamamoto K. R. (2002) Disassembly of transcriptional regulatory complexes by molecular chaperones. Science 296, 2232–2235 [DOI] [PubMed] [Google Scholar]
- 50. Zelin E., Zhang Y., Toogun O. A., Zhong S., Freeman B. C. (2012) The p23 molecular chaperone and GCN5 acetylase jointly modulate protein-DNA dynamics and open chromatin status. Mol. Cell 48, 459–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stavreva D. A., Müller W. G., Hager G. L., Smith C. L., McNally J. G. (2004) Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Mol. Cell. Biol. 24, 2682–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sreeramulu S., Gande S. L., Göbel M., Schwalbe H. (2009) Molecular mechanism of inhibition of the human protein complex Hsp90-Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew Chem. Int. Ed. Engl. 48, 5853–5855 [DOI] [PubMed] [Google Scholar]
- 53. Zhang T., Hamza A., Cao X., Wang B., Yu S., Zhan C. G., Sun D. (2008) A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 7, 162–170 [DOI] [PubMed] [Google Scholar]
- 54. Jin H. Z., Hwang B. Y., Kim H. S., Lee J. H., Kim Y. H., Lee J. J. (2002) Anti-inflammatory constituents of Celastrus orbiculatus inhibit the NF-κB activation and NO production. J. Nat. Prod. 65, 89–91 [DOI] [PubMed] [Google Scholar]
- 55. Nagase M., Oto J., Sugiyama S., Yube K., Takaishi Y., Sakato N. (2003) Apoptosis induction in HL-60 cells and inhibition of topoisomerase II by triterpene celastrol. Biosci. Biotechnol. Biochem. 67, 1883–1887 [DOI] [PubMed] [Google Scholar]
- 56. Zhang D. H., Marconi A., Xu L. M., Yang C. X., Sun G. W., Feng X. L., Ling C. Q., Qin W. Z., Uzan G., d'Alessio P. (2006) Tripterine inhibits the expression of adhesion molecules in activated endothelial cells. J. Leukocyte Biol. 80, 309–319 [DOI] [PubMed] [Google Scholar]
- 57. Sethi G., Ahn K. S., Pandey M. K., Aggarwal B. B. (2007) Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-κB-regulated gene products and TAK1-mediated NF-κB activation. Blood 109, 2727–2735 [DOI] [PubMed] [Google Scholar]
- 58. Sun H., Liu X., Xiong Q., Shikano S., Li M. (2006) Chronic inhibition of cardiac Kir2.1 and HERG potassium channels by celastrol with dual effects on both ion conductivity and protein trafficking. J. Biol. Chem. 281, 5877–5884 [DOI] [PubMed] [Google Scholar]
- 59. Yang H., Chen D., Cui Q. C., Yuan X., Dou Q. P. (2006) Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 66, 4758–4765 [DOI] [PubMed] [Google Scholar]
- 60. Westerheide S. D., Bosman J. D., Mbadugha B. N., Kawahara T. L., Matsumoto G., Kim S., Gu W., Devlin J. P., Silverman R. B., Morimoto R. I. (2004) Celastrols as inducers of the heat shock response and cytoprotection. J. Biol. Chem. 279, 56053–56060 [DOI] [PubMed] [Google Scholar]
- 61. Trott A., West J. D., Klaić L., Westerheide S. D., Silverman R. B., Morimoto R. I., Morano K. A. (2008) Activation of heat shock and antioxidant responses by the natural product celastrol: Transcriptional signatures of a thiol-targeted molecule. Mol. Biol. Cell 19, 1104–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]