

Abstract
Biological membranes are essential for cell viability. Their functional characteristics strongly depend on their protein content, which consists of transmembrane (integral) and peripherally associated membrane proteins. Both integral and peripheral inner membrane proteins mediate a plethora of biological processes. Whereas transmembrane proteins have characteristic hydrophobic stretches and can be predicted using bioinformatics approaches, peripheral inner membrane proteins are hydrophilic, exist in equilibria with soluble pools, and carry no discernible membrane targeting signals. We experimentally determined the cytoplasmic peripheral inner membrane proteome of the model organism Escherichia coli using a multidisciplinary approach. Initially, we extensively re-annotated the theoretical proteome regarding subcellular localization using literature searches, manual curation, and multi-combinatorial bioinformatics searches of the available databases. Next we used sequential biochemical fractionations coupled to direct identification of individual proteins and protein complexes using high resolution mass spectrometry. We determined that the proposed cytoplasmic peripheral inner membrane proteome occupies a previously unsuspected ∼19% of the basic E. coli BL21(DE3) proteome, and the detected peripheral inner membrane proteome occupies ∼25% of the estimated expressed proteome of this cell grown in LB medium to mid-log phase. This value might increase when fleeting interactions, not studied here, are taken into account. Several proteins previously regarded as exclusively cytoplasmic bind membranes avidly. Many of these proteins are organized in functional or/and structural oligomeric complexes that bind to the membrane with multiple interactions. Identified proteins cover the full spectrum of biological activities, and more than half of them are essential. Our data suggest that the cytoplasmic proteome displays remarkably dynamic and extensive communication with biological membrane surfaces that we are only beginning to decipher.
An in-depth understanding of cellular proteomes requires knowledge of protein subcellular topology, assembly in macromolecular complexes, and modification and degradation of poplypeptides. Escherichia coli, a model organism for many such studies, is by far the best studied. The genomes of strain K-12 derivatives MG1655 and W3110 have been sequenced (1, 2), and >75% of their genes have been functionally assigned (3). Almost 90% of the K-12 proteome has been identified experimentally, and >73% of its proteins have known structures (4, 5). Moreover, the genomes of another 38 E. coli strains have been determined (see EcoliWiki for details).
In E. coli, like in all Gram-negative bacteria, the bacterial cell envelope comprises the plasma or inner membrane and the outer membrane, which are separated by the periplasmic space. The inner membrane encloses the cytoplasm and is a dynamic substructure. It harbors a wide variety of proteins that function in vital cell processes such as the trafficking of ions, molecules, and macromolecules; cell division; environmental sensing; lipid, polysaccharide, and peptidoglycan biosynthesis; and metabolism. Inner membrane proteins either fully span the lipid bilayer using one or more hydrophobic transmembrane helices (integral) or are bound either directly to phospholipid components or via protein–protein interactions to the surface of the membrane (peripheral) (6) (Fig. 1A). Peripheral inner membrane proteins exist on either side of the membrane and may be recruited in membrane-associated complexes on demand (7). Peripheral inner membrane proteins on the cytoplasmic side constitute a sub-proteome of central importance because of their interaction with the cytoplasmic proteome, the nucleoid, and most of the cell's metabolism. Thanks to their soluble character and the nature of their interactions with the membrane (mostly electrostatic and moderate hydrophobic interactions (7)), peripheral inner membrane proteins can be extracted using high salt concentrations, extreme pH levels, or chaotropes without disrupting the lipid bilayer (8–11). In contrast, the solubilization of integral proteins requires amphiphilic detergents in order to displace the membrane phospholipids and maintain them as soluble in aqueous solutions (12).
Fig. 1.

Bioinformatics and experimental workflow for characterizing peripheral inner membrane proteins. A, schematic representation of the subcellular localization of the E. coli inner membrane peripherome. Protein topology assignment is based on the cellular compartment: A, cytoplasmic; B, integral inner membrane proteins; F1, peripheral inner membrane proteome; r, ribosome. B, schematic diagram for PIM protein annotation. 130 cytoplasmic and PIM E. coli K-12 proteins were downloaded from Uniprot (November 2010) (81) and EchoLOCATION (23). A set of bioinformatics tools was used to predict topologies and features of the unassigned and differently assigned proteins and to further validate existing protein annotations (see supplemental text). For the annotation of additional peripheral membrane proteins, the literature was extensively searched. Additional, other E. coli K-12 databases containing gene ontology annotations (84, 85) and protein homologies through BLAST (44) were employed. Homologues of curated E. coli K-12 proteins were identified in E. coli BL21(DE3) (supplemental Table S1A). C, preparation strategy for detecting the E. coli inner membrane peripherome via nanoLC-MS/MS. Inverted membrane vesicles (IMVs) were isolated and washed extensively with the indicated chemical agents to extract cytoplasmic and PIM proteins (“IMVs washed”), and then their surface was trypsinized (gray arrow). Following digestion, soluble peptides were analyzed via nanoLC-MS/MS. D, protein enrichment at different sample preparation conditions. Top: Relative percentage of proteins detected with the proteolysis approach. Proteins are classified here in three major categories: cytoplasmic (A), ribosomal (r), and peripheral (F1). The bar graphs indicate the percentage of proteins in each category relative to the proteins in other categories at a given sample preparation condition. Bottom: Heat maps showing relative quantities of individual proteins at different sample preparation conditions. Perseus (version 1.2.0.16), a part of the MaxQuant bioinformatics platform, was used for the construction of the heat map (86). A top-three label-free quantitative method was employed (27). Individual protein values across the various treatments are given in supplemental Table S3B.
Unlike the cytoplasmic proteome of E. coli, which has been extensively characterized (13), its membrane sub-proteome is still poorly defined. Of 1133 predicted integral inner membrane proteins, only half were experimentally identified through proteomics approaches (14). These figures are constantly being re-evaluated,2 but most protein identifications appear robust. In contrast to integral inner membrane proteins, bioinformatics prediction of peripheral inner membrane proteins is currently not possible because they are not known to possess any specific features. Despite the occasional designation of partner proteins identified as peripheral in studies that target inner membrane complexes (15–21), no systematic effort has been undertaken to analyze the peripheral inner membrane proteome.
Here we have used a multi-pronged strategy employing bioinformatics, biochemistry, proteomics, and complexomics to systematically determine the peripheral inner membrane proteome of E. coli. We focus exclusively on the peripheral inner membrane proteome that faces the cytoplasm, referred to hereinafter as PIM,1 and do not analyze peripheral inner membrane proteins residing on the periplasm. Manually curated and re-evaluated topology of the E. coli K-12 proteome was extrapolated to the non-K-12 strain BL21(DE3) (95% proteome homology to K-12) (22). By combining various biochemical treatments, we determined experimentally that several cytoplasmic proteins are also novel PIM proteins, and many of them participate in protein complexes associated with the membrane. Collectively, we demonstrate that a significant, previously unsuspected percentage of the expressed polypeptides constitute the PIM proteome.
RESULTS
Curation of the Theoretical Inner Membrane Peripherome of E. coli BL21(DE3)
The PIM sub-proteome is largely elusive. The complete absence of bioinformatic predictor tools for PIM proteins renders experimental approaches, such as proteomics, essential. To curate the currently available PIM proteome, we performed an exhaustive in-depth analysis combining data from the literature, a variety of bioinformatics tools (Fig. 1B), and topological information for the E. coli K-12 proteins available in public databases such as EchoLOCATION (23), Uniprot (5), and EcoCyc (3). We then extrapolated the results to the E. coli BL21(DE3) strain (supplemental Table S1A). The nomenclature of protein classes used here was based on that of EchoLOCATION (Fig. 1A).
A total of 138 K-12 proteins were annotated as PIM (indicated with “F1” in supplemental Table S1A) in EchoLOCATION and Uniprot, but this number was subsequently reduced to 123 after manual validation (supplemental Table S1B). These corresponded to 133 homologues in BL21(DE3). PIM proteins of strain BL21(DE3) that were curated for the first time in the present study (138 proteins, indicated with “F1*” in supplemental Table S1A) were derived mainly from a thorough search of the E. coli literature, comparison to homologues from other organisms, and a multi-pronged iterative curation approach (supplemental Table S1C). We reexamined any proposed topology classification for cytoplasmic or integral inner membrane proteins using several bioinformatic predictors (SignalP, TatP, LipoP, TMHMM, Phobius, Protscale, SOSUI, and AmphipaSeek) and homology (BLAST) tools. The results were subsequently refined using the available databases (EcoGene, EcoCyc) and the literature (supplemental Table S1A). The collective use of these approaches increased the total number of curated PIM proteins to 278.
PIM Proteins Identified via Membrane Proteolysis
To experimentally identify PIM proteins, we used inverted inner membrane vesicles (IMVs), in which the cytoplasmic face of the membrane is exposed and accessible and the periplasmic side faces the sealed lumen (24). The surfaces of intact IMVs were treated with trypsin, and soluble tryptic peptides were analyzed via nanoLC-MS/MS. Multiple technical and biological repeats were performed to ensure statistical robustness (supplemental Table S3A). This method is highly specific for surface-attached proteins, because trypsin (27 kDa) cannot penetrate IMVs, and lumenally trapped cytoplasmic proteins are therefore neither trypsined nor analyzed here (supplemental Fig. S1A). Two major challenges were the discrimination of PIM from non-specifically bound cytoplasmic proteins and overcoming “peptide noise” derived from abundant membrane-bound ribosomes.
In the untreated IMVs, ∼70% of identified proteins corresponded to cytoplasmic and ribosomal polypeptides (Fig. 1D), indicating that these are present on the IMV surface. We systematically probed the degree of their extraction from the membrane through the consecutive use of chemical agents that are traditionally used to characterize peripherally attached proteins (Fig. 1C) and to remove unspecifically bound cytoplasmic proteins (25, 26). Such treatments included KCl, EDTA, Na2CO3 at pH 11, and urea combined with mild sonication and sucrose density centrifugations (see supplemental Materials and Methods) (8–11). These treatments did not functionally compromise IMVs, which remained fully competent for protein translocation (supplemental Fig. S1B). Finally, the IMVs were treated with low levels of the non-ionic detergent n-dodecyl β-D-maltoside (DDM) (0.1–2.0 critical micellar concentration (CMC)) (supplemental Fig. S1C) to disrupt hydrophobic protein–protein interactions.
The chemical agents used gradually removed cytoplasmic, ribosomal, and known PIM proteins from the membrane periphery, as indicated by the significant decrease of the total number of soluble proteins identified (Fig. 1D) and by the decrease of their relative amounts across consecutive treatments (supplemental Table S3). Compared with the untreated IMVs, new proteins were also identified across treatments, indicating that their identification was obstructed by contaminating polypeptides residing on the membrane surface.
Assignment Criteria for PIM Proteins
The relative protein amounts during the multiple consecutive treatments described above (Fig. 1D) were determined using label-free quantitation (27). Some known peripheral proteins such as DhsA and DhsB (of the succinate dehydrogenase heterotetramer), SecA (the motor of the Sec translocase), and the FtsE component of the cell division ring remained membrane associated even after extensive treatments. All of these proteins were highly abundant and were detected in almost all technical repeats of a given sample preparation condition (columns F–H and I–M in supplemental Table S3B). In contrast, the intensity of some other subunits of these complexes decreased upon IMV treatment. For example, SecB was detected at low abundance only in the untreated IMV samples and was removed completely upon treatment with KCl and EDTA. Another example was FtsZ, which was gradually removed during the various treatments as indicated by its abundance values (∼90-fold decrease) and detection rate (found in all repeats in the untreated IMVs and in only 1 out of the 13 repeats in the DDM-treated IMVs). Remarkably, several proteins classified previously as cytoplasmic, suchas HemG/HemH/YbbO (involved in heme biosynthesis), DeoB/DeoC/DeoD (involved in the synthesis of nucleoside catabolic enzymes), and AccA/AccD (of the acetyl coenzyme A carboxylase), survived several IMV treatments (supplemental Table S3B), indicating that they are avid membrane binders and could therefore be considered legitimate PIM proteins.
In order to assign a cytoplasmic protein as PIM in a systematic fashion, a number of criteria were applied (supplemental Fig. S2). For each sample preparation condition (not treated, salt and then DDM, etc.), a threshold was set based on the confidence with which a protein was detected (how many times a protein was detected in technical repeats). The threshold was higher for the untreated IMVs (which were anticipated to contain nonspecific bound proteins, meaning their detection would be random) and lower for the treated and DDM-treated IMVs (nonspecific bound proteins would have been removed by the various treating agents). Proteins that fulfilled this criterion were directly annotated as peripheral (supplemental Fig. S2). For example, HemG and YbbO were detected in all sample preparation conditions, but they fulfilled the criteria only for the washed IMVs (and DDM-washed IMVs) (supplemental Table S3B). If these thresholds were not met, then the abundance of a protein in the cell was taken into account. Low-abundance proteins (<500 copies per cell) were assigned as peripheral even if they were detected in fewer repeats (in more than three) in the IMV fraction (i.e. YggL, which has ∼400 molecules per cell (supplemental Table S1) and was detected in six repeats in the untreated IMVs only), whereas high abundance proteins were considered as lower confidence PIM proteins if they had the same detection score, because it could not be excluded that their detection was due to nonspecific, low-affinity associations (i.e. AmpM with ∼2000 molecules per cell that was detected in four repeats in the untreated IMVs only). A protein that did not meet any of the above criteria but which was part of a known membrane-associated protein complex (3), for which a minimum of one peripheral protein subunit was detected in this study, was also considered peripheral. In this way we reduced the possibility of false positive identifications. Therefore, proteins such as CspC (∼379,000 molecules per cell), which was identified in two repeats, were not assigned as peripheral proteins.
In conclusion, this analysis proposes that a total of 169 of the cytoplasmic proteins identified on IMVs are novel PIM proteins.
PIM Protein Complexes
Our analysis suggested that several of the PIM proteins might associate with the membrane as part of complexes rather than directly. Given the interfacial location of PIM proteins and the dynamic equilibria with the cytoplasmic proteome pool, it was of interest to determine interacting partners. Based on interactomics (28, 29) and gel-based complexome (15–19, 21, 30, 31) data, >90% of the known and PIM proteins curated in this study have known interactors (supplemental Table S6) (32), but these are not necessarily membrane-associated proteins.
In order to experimentally extract stable PIM complexes that employ either electrostatic or hydrophobic interactions, the IMVs were treated with mild methods: either KCl (1 m) or DDM (1–2 times CMC), or a sequential combination of both (supplemental Fig. S3). Extracted complexes were separated first via size-exclusion chromatography and then via N-PAGE (Fig. 2). Proteins belonging to the same complex co-elute in size-exclusion chromatography (low resolution) and should co-migrate in N-PAGE (high resolution). Bands of complexes visible with Coomassie staining were isolated, and their protein constituents were identified. In total, 730 proteins were detected, with ∼14% of these identified as PIM proteins (supplemental Table S1A).
Fig. 2.

Workflow for the analysis of peripherome complexes. For the isolation of PIM complexes, the IMVs were treated independently, either with 2 m KCl or 0.34 mm DDM (2 times CMC) or sequentially, with KCl first and then DDM. Extracted native complexes were fractionated using size-exclusion chromatography. Fractions were pooled in six groups (as indicated) and further separated via N-PAGE. Bands of interest were excised, trypsinized, and analyzed via nanoLC-MS/MS.
Some proteins were found to be associated with more than one complex. This could have resulted from the dissociation of the native complexes caused by the experimental conditions we used, giving rise to “artificial” complexes with different combination of subunits. To decipher meaningful interactions and exclude false identifications, we pruned our data as follows: (a) The experimental mass of each complex was determined within a 10% error using mass markers (supplemental Table S4). (b) We downloaded subunit stoichiometry and theoretical masses of all known E. coli K-12 complexes curated in EcoCyc (supplemental Table S7) (3). (c) Complex identities were assigned when at least two of the subunits were identified in the same band. (d) Based on the known subunit stoichiometry of a complex, possible subunit combinations delimited by the experimental mass range were calculated. (e) Only abundant polypeptides were included. A total of 34 heteromultimeric complexes were thus identified experimentally (supplemental Table S4, supplemental Fig. S4).
Out of the proteins identified through this approach, almost 50% were annotated as cytoplasmic in the available databases, and 6% of those are known to participate in heteromultimers (supplemental Table S7). By applying our criteria for the complexes identified (supplemental Fig. S4) and by taking into account partner proteins identified in the membrane proteolysis experiments, we were able to propose 56 novel PIM proteins using this approach, 11 of which participate in 9 PIM heteromultimers.
Collectively, we propose that E. coli BL21(DE3) has at least 503 PIM proteins (Fig. 3A). Of these, 353 were identified via our membrane proteolysis and N-PAGE approaches. Based on EcoCyc data, at least 102 of these PIM proteins are expected to participate in heteromultimeric complexes (supplemental Table S7). Several of these complexes are schematically shown in Fig. 4.
Fig. 3.

Features of the peripherome. A, combined protein identifications of PIM and protein complexes from two approaches. The proposed total E. coli peripherome is composed of at least 503 PIM proteins (47% from membrane proteolysis, 16% from complexomics, and 37% from the literature and re-annotation). In this study, a total of 342 peripheral proteins were identified by our approaches; 106 of them are known to participate in 63 PIM complexes. A total of 34 heteromultimeric complexes were identified experimentally. B, abundance of PIM proteins. The abundance of the experimentally detected proteins (colored circles, supplemental Table S1A, Fig. 4) is depicted using quantitation values previously determined for E. coli K-12. The right-hand y-axis represents the percentage of detection and expresses the number of times a protein has been identified relative to the number of experimental repeats. Peripheral proteins are shown with green circles, and ribosomal and integral membrane proteins are denoted by blue and yellow circles, respectively. The abundance distribution of 1783 proteins available in the literature is plotted on the background (gray area) (33–35). C, functional assignment of PIM proteins. Distribution of PIM proteins based on the MultiFun cell function classification system (87). Gene ontology information of individual proteins was collected from EcoCyc and Uniprot (3, 5). Most of the PIM proteins are involved in metabolism (i.e. energy metabolism, electron transport, amino acid biosynthesis) and in information transfer (protein-, DNA-, RNA-related) processes of the cell (supplemental Table S7).
Fig. 4.
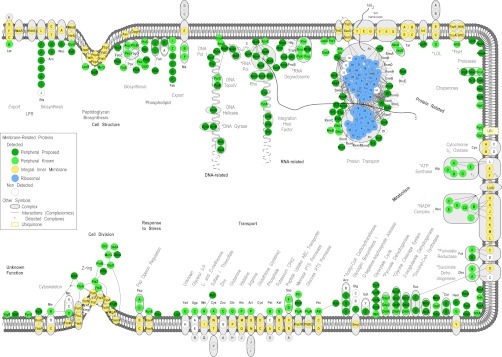
Bird's-eye view of the E. coli peripherome. A schematic longitudinal section of an E. coli dividing cell is shown. A section of the inner membrane is depicted; the rest of the cell envelope is omitted for simplicity. Based on MultiFun classification, PIM proteins (in green circles) participate in nine cellular processes (indicated with bold letters): cell structure; DNA-, RNA-, and protein related; metabolism; transport; response to stress; and cell division. Sub-categories of these processes are indicated with bold gray letters. The proteins are grouped according to known complexes (encircled in gray color schemes) and pathways, together with their integral inner membrane partners (yellow circles) where known (3). Protein–protein interactions derived from tandem-affinity purification experiments (88) are also mapped (where available) and depicted with connecting lines. For visualization purposes, the protein–protein interactions were integrated and loaded in Cytoscape (3). The interactions shown are not necessarily simultaneous and might have derived from the capture of different holoenzyme variants with different subunits. Proteins drawn in the peptidoglycan and phospholipid biosynthesis and cell division are based on known interactions and were downloaded only from IntAct (88). Proteins of the ribosome, arranged in the 50S and the 30S subunits, are drawn in the membrane's vicinity (blue circles). Following translation in the ribosome, proteins (indicated with a thick gray scribbled line) are recognized co- or post-translationally by the ribosome-bound signal recognition particle or the SecA and/or SecB chaperones, respectively, and are destined for the membrane for secretion through the Sec translocase (89). A number of proteins, previously annotated as cytoplasmic, are shown to associate with the inner membrane and are proposed as new PIM proteins. These are represented with dark green circles, whereas previously known and curated (in this study) peripheral proteins are featured with light green circles. For a comprehensive view of the mapped complexes, some periplasmic proteins are also drawn (where available) on the membrane outer surface. Complexes identified are indicated with a red star. Proteins with no functional annotations are indicated as “Unknown Function.” For the complete list, annotation, and description of protein complexes identified, see supplemental Table S4.
PIM Protein Abundance
An unexpectedly large number of cytoplasmic proteins were proposed as novel PIM proteins. Could it be that their binding on membranes is nonspecific and related only to their cellular abundance? The distribution of abundance values for 1834 E. coli proteins available in the literature (33–35) was plotted (gray color on the background) against the number of times they have been experimentally identified here (Fig. 3B). Proteins were classified as those annotated previously and in this study as peripheral (261 proteins, PIM known) and as those identified experimentally and proposed in this study to be novel PIM proteins (225 proteins, PIM proposed). Ribosomal and integral inner membrane proteins identified were also included for reference purposes. The identification of proteins as PIM by our approach does not correlate with high cellular abundance; they span the entire range of concentrations. Some of them exist in fewer than 100 copies per cell (e.g. RadA, which is involved in DNA repair, LonH of the peptidase S16 family, and HemH, which is involved in heme biosynthesis).
Bioinformatic Search for PIM Protein Features
We next searched for common features that would reflect a protein's ability to associate with the inner membrane peripherally. Two characteristic features that helped us discriminate PIM from purely cytoplasmic proteins were amphipathic helices and hydrophobic patches that can promote interactions with membrane lipids and membrane-embedded proteins. Amphipathic helices are known to be used for peripheral protein interaction with membranes. For example, MinD, an ATPase that regulates the division site in bacteria, contains an amphipathic helix at its C-terminal region that mediates its direct interaction with membrane lipids (36–38). GlpD is a membrane enzyme that penetrates the lipid monolayer with a basic amphipathic helix formed within the protein's body (39). PtgA, an enzyme of the sugar phosphotransferase system, adopts an amphitrophic helical conformation that allows it to interact with membrane lipids to exert its function (40, 41). The amino acid sequences of cytoplasmic and previously curated PIM proteins were searched for their ability to form amphipathic α-helices. Our analysis showed that all proteins were able to form at least one amphipathic α-helix, and no difference was observed between the number (∼4) and length (∼17 amino acids) of such elements per protein for the two protein categories (not shown).
Hydrophobic region probabilities were analyzed using Phobius (42). As expected, the transmembrane helices of inner membrane proteins exhibited high probabilities (close to 1). One-third of PIM and cytoplasmic proteins have weak hydrophobic patches that could be hydrophobic-interaction elements. However, no significant differences were seen between cytoplasmic and PIM proteins (supplemental Fig. S5).
We concluded that no discernible features can be proposed at this stage to identify cytoplasmic proteins that have the tendency to become PIM.
Functional Analysis of PIM Proteins
PIM proteins are involved in a wide variety of cellular interactions (supplemental Table S6; Fig. 4) that encompass all of the main cellular processes (supplemental Table S6; Figs. 3C, 4). A small number of these proteins still have unknown functions (e.g. YifE and YjgR) (supplemental Table S7). Functional assignments indicate that the majority of the PIM proteins identified in this study (∼30%) belong to metabolic processes involved in cell division and cell wall biosynthesis, protein trafficking and processing, energy conversions, membrane transport, small molecule metabolism, and nucleic-acid-related processes ranging from DNA replication and transcription to RNA translation and turnover. Functional assignment of the complexes that the PIM proteins participate in shows that most of them are engaged in RNA-related processes and the transport of molecules. Many of these processes are schematically shown in Fig. 4.
To investigate whether PIM proteins known from the literature or curated in this study are indispensable to the cell's life, protein sequences of essential genes were downloaded from DEG (43), and a multiblast query was performed in which essential proteins were the target database and PIM proteins were the query (44). Protein homologues with identities > 20 and e-values ≤ 0.001 were accepted. Our results indicated that 44% of PIM proteins are homologous to at least one essential protein.
We next searched how many of the PIM proteins are conserved among bacteria and pathogenic bacteria. The complete proteomes of 25 bacteria and 22 pathogenic bacteria (supplemental Table S8) were downloaded from Uniprot and used to create blast databases. Our PIM protein sequences were searched against these databases. A protein was accepted as conserved in bacteria if it had a homologue sequence (criteria same as above) in at least 16 strains. Our analysis showed that >60% of the PIM proteins are found throughout the bacteria, including pathogens, and approximately half of them are highly conserved (found in 20 strains).
DISCUSSION
We present a systematic analysis of the poorly characterized bacterial PIM proteome. Our analysis relied on experimental proteomics data, as current in silico approaches cannot predict the cell topology of cytoplasmic proteins that also associate with membranes. Proteomics was coupled to complete re-annotation of E. coli protein subcellular localization. We determined that the E. coli BL21(DE3) proteome comprises a total of at least 503 PIM proteins accounting for ∼17% of the basic proteome and a remarkable ∼27% of the estimated expressed proteome of this cell grown in LB medium to mid-log phase. Our analysis doubled the number of PIM proteins available in Uniprot and EchoLOCATION.
PIM protein interactions with the membrane appear to be multiple and complex. They associate with the lipid bilayer non-covalently and reversibly without penetrating the hydrocarbon hydrophobic core (7). Moreover, the indirect association of soluble components with the membrane may also occur through interactions with PIM proteins physically attached to the membrane. Though these components are not directly associated with the membrane, we propose that they should also be considered as PIM proteins. For example, FtsE, which is involved in cell division, interacts directly with the integral inner membrane FtsX, but also with FtsZ, which in turn recruits other known division proteins that do not interact directly with the membrane (Fig. 4) (45, 46). Some PIM proteins form associations with the membrane that are easily abrogated. These can be weak electrostatic interactions with lipid head groups (i.e. PspA) or with other proteins (e.g. SecB, HflD, and NuoEFG). Another example is AccA and AccD subunits of acetyl-CoA carboxylase, a metabolic complex that catalyzes the synthesis of fatty acids. In E. coli, the complex is ascribed to the cytoplasm (5), whereas in B. subtilis evidence has been provided regarding its membrane proximity (47), consistent with systematic anecdotal evidence for the co-fractionation of these enzymes with membranes.3 Here, we identified AccA, AccC, and AccD as PIM proteins that associate tightly with membranes electrostatically and can be removed by high ionic strength (supplemental Table S3B). These proteins were also identified in the size-exclusion chromatography/N-PAGE approach as subunits of acetyl-CoA carboxylase sub-complexes (supplemental Table S4) (in E. coli, the acetyl-CoA carboxylase complex is known to be rather unstable (48), which explains why some subunits eluded detection before). Most important, the organization of these proteins in membrane-associated complexes suggested a possible function, although unknown at present. One obvious possibility is that such complexes might facilitate metabolic channeling of small molecule substrates (49, 50).
In other cases, interactions with the membrane surface are extremely tight and resistant to several chemical agents. These are suggestive of a multitude of interactions, almost certainly with proteinaceous receptors (e.g. SecA on SecYEG, SRP on FtsY, SeqA on DnaA). Some of these interactions (e.g. MinD, MlaB) can be disrupted only with the use of non-ionic detergent, indicating the importance of strong hydrophobic interactions. This tight interaction behavior is reminiscent of that of integral membrane proteins. Clearly, the biochemical means traditionally used for assigning PIM protein status (8–11, 25) are inadequate when annotating these tight interactions and should be revised.
Membrane proteomics studies tend to consider cytoplasmic proteins that co-purify with membrane as “contaminants” (26, 51–54), as it is difficult to conclude whether these represent real interacting partners or nonspecific associations. Especially for novel proteins of unknown subcellular localization and/or function, more biological, biochemical, and cell biological evidence is required. In this context, the purity of inner membranes is crucial. Many of the PIM proteins we identified participate in multimeric complexes and/or have known interactors. The combination of mild membrane treatment (to leave complexes intact) with an orthogonal complexome purification approach identified several PIM complexes comprising abundant proteins (e.g. ATP synthase, NADH dehydrogenase, and succinate dehydrogenase). A future application of our PIM complexomics pipeline will be to identify unknown complexes and discover how these change in various growth regimes.
Our approach relied heavily on subcellular fractionation. This is a way of eliminating contaminants and detecting low-copy-number proteins. Its inherent limitation is that topological information for proteins in the real cellular context is lost, as are unstable interactions. Also lost are the dynamics of complex formation and the kinetics of membrane-surface occupancy. In order to overcome these limitations, future approaches should employ tools such as monitoring green-fluorescent-protein-tagged proteins in the cell. Given the dual location of PIM proteins in both the cytoplasm and the membrane periphery, advanced kinetic- and FRET-based studies together with single molecule approaches and Total internal reflection fluorescence-based high resolution microscopy will have to be used to strengthen signals from membrane-associated interactions and distinguish them from an extensive cytoplasmic labeling background. Transient or weak interactions may be stabilized using chemical cross-linking prior to MS-based identification (55–57).
The dynamic aspect of the peripherome is particularly fascinating, as it suggests that the membrane can act as a “temporary storage pool” to remove factors from cytoplasmic circulation and thus reduce their effective concentration or deliver them only upon external stimulation. This is the case with transcription factors NadR (58), RpoE, and PutA (59) detected here, as well as BglG, which was not identified (60). Similarly, LacI, identified here as associating strongly with the membrane, is normally ascribed to a cytoplasmic location but was consistently found here in slightly overexpressed conditions as a membrane-associated tetramer (supplemental Fig. S3A, band K10). These transcription factors may be stored until needed—for example, when the relevant small molecule ligand enters the cell. Likewise, the RNA degradosome component RNE remains bound to the membrane during cell growth and is released and becomes active only in the late stationary phase (61, 62). Another example of dynamic behavior is that of PspA, which either forms oligomeric ring structures on the membrane surface thought to be ion leakage sites (63) or interacts as a monomer with other Psp proteins in solution and at the membrane (64).
Here, we focused only on stable complexes, unambiguously annotated. As a result, we estimate that the PIM interactome is more complex. Many cytoplasmic proteins are expected to form fleeting interactions with the membrane or with PIM proteins attached to the membrane, as discussed earlier. Rather than static pair-wise interactions, the physiological interactions in the cell are likely to be multidimensional and might even involve “moonlighting” proteins (65) (supplemental Fig. S6). Thus the same PIM protein could be a partner in multiple different complexes, as in the cases of MukB (66, 67), MurG (68), RpoE (69, 70), MreB (71–74), PyrG (75), and GlpD (19, 76, 77). To make things more complex, the same protein can have strikingly different structural behaviors and functions, such as the actin-like protein MreB (detected in all our experiments), which can determine cell polarity in bacteria (78), function as a structural filament, interact with elongation factor EF-Tu to define cell morphology (79), and also interact with RNA polymerase to initiate chromosome segregation (74).
In conclusion, a remarkable number of cytoplasmic proteins interact with the bacterial inner membrane. The PIM proteome acts as a dynamic and extensive liaison that connects the inner membrane with most cell processes. Our pipeline is applicable to investigations of peripheromes in other cells such as pathogenic bacteria. We expect future studies to elaborate on these networks of cytoplasmic-membrane cross talk, identify the role and dynamics of PIM proteins of unknown function, and determine the way in which they exert their central role in the cell's biology.
MATERIALS AND METHODS
Chemicals and Reagents
Chemicals were obtained from Sigma Aldrich, Fisher Scientific, and Bioline-Applichem (Athens, Greece). Iodoacetamide was acquired from Merck, and trypsin (sequencing grade) from Roche Applied Science. Acetonitrile (LC-MS grade) was obtained from Fisher Scientific.
Membrane Preparation
Membrane Proteolysis
IMVs (10 μg protein) were reduced with a 10-fold molar excess of tris(2-carboxyethyl)phosphine for 30 min at room temperature and alkylated with iodoacetamide (5 mM) for 45 min in the dark. Trypsin was added at a ratio of 1:200 (enzyme:protein), and samples were digested overnight at 37 °C. The samples were diluted 2-fold with deionized water and acidified with 1 μl trifluoroacetic acid. Following ultracentrifugation (200,000 × g, 4 °C, 30 min, Sorvall WX Ultra Series 80, TH641), the supernatant was desalted using StageTips (C18) according to a standard protocol (Thermo Scientific). The peptide mixtures were dried in a SpeedVac concentrator (Savant ISS110, Thermo Scientific) and reconstituted in 0.5% formic acid prior to nanoLC-MS/MS analysis.
Size Exclusion Chromatography and N-PAGE
IMVs (14 mg of protein in 2 ml of IMVs) were treated with different chemicals (indicated in supplemental Materials and Methods). Supernatants from such treatments were collected after centrifugation (200,000 × g, Sorvall WX Ultra Series 80, TH641, 4 °C, 45 min). The total protein concentration was measured using a bicinchoninic acid assay (protein assay kit, Pierce). Routinely, ∼0.5 to 1.5 mg of PIM proteins are extracted from 14 mg of IMVs. The samples were loaded on an analytical high-resolution pre-packed column (Superdex HR200 10/30) on an HPLC system (Shimadzu, Asteriadis S.A., Thessaloniki, Greece). Fractions were collected at 30-s intervals (0.8 ml) at 25 °C. Based on their chromatographic profile, the aliquots were pooled in six major fractions (supplemental Fig. S3), concentrated to a 20 μl volume using Amicon (Amicon Ultra, 0.5 ml, 30K c.o.), and loaded on N-PAGE (7% acrylamide). High-molecular-weight native markers were used (Amersham Biosciences, GE Healthcare). The gel was run at 8 mA for 16 h at 4 °C. Proteins were stained with blue silver solution (10% phosphoric acid, 10% ammonium sulfate, Coomassie brilliant blue G-250 (Bioline-Applichem, Athens, Greece), 20% methanol, 3 h) (80). Gel bands were excised, and samples were reduced with 10 mm dithiothreitol at 56 °C for 1 h and carboxyamidomethylated with 55 mm iodoacetamide in the dark at room temperature for 1 h. Samples were digested with modified trypsin (100 ng per sample) at 37 °C for 15 h. Proteolysis was terminated by acidification of the reaction mixture with glacial acetic acid (2 μl). The peptide mixtures were dried in a SpeedVac Concentrator (Savant ISS110, Thermo Scientific) and re-constituted in 0.5% formic acid prior to nanoLC-MS/MS analysis.
Calculation of Mass of Protein Complexes
The distance that the molecular weight native markers migrated on each N-PAGE gel was used to fit the exponential equation y = y0ekx, in which x is the distance measured from the top of a gel to the center of each marker band and y is the molecular weight of each marker. The respective coefficient values deduced for the KCl/DDM-, KCl-, and DDM-treated samples were as follows: y0, 876.9, 862.9, and 929.5; k, −0.616, −0.1603, and −0.3371. These values were used to calculate the molecular weight of individual bands via curve fitting; a 10% error was applied (supplemental Table S4).
NanoLC-MS/MS Analysis
The nanoLC-MS/MS analysis was performed on an EASY-nLC system (Proxeon, software version 2.7.6 #1) coupled with an LTQ-Orbitrap XL ETD (Thermo Scientific, Bremen, Germany) through a nanoES ion source (Proxeon). Data were acquired with Xcalibur software (LTQ Tune 2.5.5 sp1, Thermo Scientific). Prior to the analysis, the mass spectrometer was calibrated with a standard ESI positive ion calibration solution of caffeine (Sigma), l-methionyl-arginyol-phenylalanylalanine acetate H2O (MRFA, Research Plus, Barnegat, NJ), and perfluoroalkyl triazine (Ultramark 1621, Alfa Aesar, Ward Hill, MA). Samples were reconstituted in 0.5% formic acid, and the tryptic peptide mixture was separated on a reversed-phase column (Reprosil Pur C18 AQ, particle size = 3 μm, pore size = 120 Å (Dr. Maisch, AnaLab, Athens, Greece), fused silica emitters 100 mm long with a 75 μm internal diameter (Proxeon, Rigas Labs S.A., Thessaloniki, Greece)) packed in-house using a pressurized (35 to 40 bars of helium) packing bomb (Loader kit SP035, Proxeon). The nanoLC flow rate was 300 nl min−1. The LC mobile phase consisted of 0.5% formic acid in water (A) and 0.5% formic acid in acetonitrile (B). A multi-step gradient was employed, from 5% to 13% B in 10 min, to 30% B in 120 min, to 95% B in 3 min. After the gradient had been held at 95% B for 7 min, the mobile phase was re-equilibrated at initial gradient conditions. The MS was operated with a spray voltage of 2300 V, a capillary voltage of 35 V, a tube lens voltage of 140 V, and a capillary temperature of 180 °C. A survey scan was acquired in the range of m/z 400–1800 with an AGC MS target value of 106 (resolving power of 60,000 at m/z 400). The 10 most intense precursor ions from each MS scan were subjected to collision-induced dissociation (isolation width = 3 Da, normalized collision energy = 35%, activation q = 0.25, activation time = 30 ms) in the ion trap. Each scan included one microscan with a maximum injection time of 200 ms and an AGC MSn target value of 2 × 104.
Data Analysis of MS/MS-derived Data
The MS raw data were loaded in Proteome Discoverer 1.1.0.263 (Thermo Scientific) and run using Mascot 2.3.01 (Matrix Science, London, UK) and Sequest (Thermo Scientific) search algorithms against the B/BL21 theoretical proteome (511693, December 2010) containing 4156 entries (81). A list of common contaminants was included in the database (82). For protein identification, the following search parameters were used: precursor error tolerance = 10 ppm, fragment ion tolerance = 0.8 Da, trypsin full specificity, maximum number of missed cleavages = 3, and cysteine alkylation as a fixed modification. To calculate the protein false discovery rate, a decoy database search (83) was performed simultaneously with strict criteria set to 0.01 and relaxed criteria to 0.05. The resulting dat and msf files were subsequently loaded in Scaffold (version 3.00.07, Proteome Software, Portland, OR) for further processing and validation of the assigned MS/MS spectra. The resultant peptide hits were filtered in Scaffold, taking into account both individual search engine scores and Scaffold calculated probabilities. Peptides were assigned as correct if they matched the following criteria: scaffold peptide probability > 80%, Mascot ion score > 25 (+2) or > 35 (+3, +4), Mascot ion score-identity score > 0, Sequest XCorr > 2.5 (+2) or > 3.5 (+3, +4), and DeltaCn > 0.1. Protein lists were constructed from the respective peptide lists. Technical variation was assessed by calculating coefficients of variation for intra- and inter-experiment analyses (supplemental Table S3A). Coefficients of variation were calculated in Scaffold (quantitative analysis option) for individual proteins across experiments; an average based on individual coefficient of variation values was then calculated per sample. For experiments composed of two technical repeats, Spearman's rank correlation coefficients were used instead (supplemental Table S3A). Coefficients of variation were calculated in the range of 9%–16% for intra-experiments and in the range of 16%–25% for inter-experiments and correlation coefficients in the range of 0.742–0.949. For biological repeats, correlation coefficients were calculated at 0.927 for the untreated and 0.966 for the treated samples (supplemental Table S3A).
Supplementary Material
Acknowledgments
We are grateful to P. Mavroudis for preliminary experiments, W. Studier for insight in the gene annotation of E. coli K-12 and B strains, G. Waldrop for discussions on AccA, I. Tsamardinos for advice on bioinformatics approaches, E. Coudert (Uniprot) for E. coli protein annotation, K. Tsarhopoulos and P. Rigas (Rigas Labs, S.A.) for support with the LTQ-Orbitrap mass spectrometer, S. Ludwigsen (Proteome Software) for advice on Scaffold, and I. Kouklinos for computing infrastructure support.
Footnotes
* The research leading to these results has received funding from the European Commission (EC) through Seventh Framework Programme Agreement No. 229823; Capacities-FP7-REGPOT-2008-1/project “ProFI”; the project “IRAKLITOS II - University of Crete” of the Operational Programme for Education and Lifelong Learning 2007–2013 (E.P.E.D.V.M.) of the NSRF (2007–2013), which is co-funded by the European Union (European Social Fund); the Onassis foundation pre-doctoral program; the Operational Programme “Competitiveness and Entrepreneuriship” (OPCE II - EPAN II) - National Strategic Reference Framework (NSRF 2007–2013), Grant No. 09SYN-13-705; and an Excellence grant (Grant No. 1473) from the General Secretariat of Research (to A.E.).
 This article contains supplemental material.
This article contains supplemental material.
2 Papanastasiou, M., Orfanoudaki, G., Koukaki, M., Kountourakis, N., Tsolis, K., Sardis, M. F., Aivaliotis, M., Karamanou, S., Economou, A., manuscript in preparation.
3 G. L. Waldrop, personal communication.
1 The abbreviations used are:
- CMC
- critical micellar concentration
- DDM
- n-dodecyl β-D-maltoside
- EDTA
- ethylenediaminetetraacetic acid
- IMV
- inverted inner membrane vesicle
- N-PAGE
- native poly-acrylamide gel electrophoresis
- PIM
- peripheral inner membrane.
REFERENCES
- 1. Blattner F., Plunkett G., Bloch C., Perna N., Burland V., Riley M., Collado-Vides J., Glasner J., Rode C., Mayhew G., Gregor J., Davis N., Kirkpatrick H., Goeden M., Rose D., Mau B., Shao Y. (1997) The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1474 [DOI] [PubMed] [Google Scholar]
- 2. Hayashi K., Morooka N., Yamamoto Y., Fujita K., Isono K., Choi S., Ohtsubo E., Baba T., Wanner B. L., Mori H., Horiuchi T. (2006) Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol. Syst. Biol. 2, 2006.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Keseler I. M., Collado-Vides J., Santos-Zavaleta A., Peralta-Gil M., Gama-Castro S., Muniz-Rascado L., Bonavides-Martinez C., Paley S., Krummenacker M., Altman T., Kaipa P., Spaulding A., Pacheco J., Latendresse M., Fulcher C., Sarker M., Shearer A. G., Mackie A., Paulsen I., Gunsalus R. P., Karp P. D. (2011) EcoCyc: a comprehensive database of Escherichia coli biology. Nucleic Acids Res. 39, D583–D590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sayers E. W., Barrett T., Benson D. A., Bolton E., Bryant S. H., Canese K., Chetvernin V., Church D. M., Dicuccio M., Federhen S., Feolo M., Fingerman I. M., Geer L. Y., Helmberg W., Kapustin Y., Krasnov S., Landsman D., Lipman D. J., Lu Z., Madden T. L., Madej T., Maglott D. R., Marchler-Bauer A., Miller V., Karsch-Mizrachi I., Ostell J., Panchenko A., Phan L., Pruitt K. D., Schuler G. D., Sequeira E., Sherry S. T., Shumway M., Sirotkin K., Slotta D., Souvorov A., Starchenko G., Tatusova T. A., Wagner L., Wang Y., Wilbur W. J., Yaschenko E., Ye J. (2012) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 40, D13–D25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Consortium T. U. (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 40, D71–D75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singer S. J., Nicolson G. L. (1972) The fluid mosaic model of the structure of cell membranes. Science 175, 720–731 [DOI] [PubMed] [Google Scholar]
- 7. Dowhan W., Bogdanov M., Mileykovskaya E. (2008) Chapter 1: functional roles of lipids in membranes. In Biochemistry of Lipids, Lipoproteins and Membranes (Dennis E. V., Jean E. V., eds) 5th Ed., pp. 1-I, Elsevier, San Diego [Google Scholar]
- 8. Adelman M. R., Sabatini D. D., Blobel G. (1973) Ribosome-membrane interaction. J. Cell Biol. 56, 206–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fujiki Y., Hubbard A. L., Fowler S., Lazarow P. B. (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ohlendieck K. (2003) Extraction of membrane proteins. In Protein Purification Protocols (Cutler P., ed) 2nd Ed., pp. 283–290, Humana Press, Totowa, NJ [Google Scholar]
- 11. Kreibich G., Sabatini D. D. (1974) Selective release of content from microsomal vesicles without membrane disassembly. J. Cell Biol. 61, 789–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Speers A. E., Wu C. C. (2007) Proteomics of integral membrane proteins: theory and application. Chem. Rev. 107, 3687–3714 [DOI] [PubMed] [Google Scholar]
- 13. Han M. J., Lee S. Y. (2006) The Escherichia coli proteome: past, present, and future prospects. Microbiol. Mol. Biol. R. 70, 362–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bernsel A., Daley D. (2009) Exploring the inner membrane proteome of Escherichia coli: which proteins are eluding detection and why? Trends Microbiol. 17, 444–449 [DOI] [PubMed] [Google Scholar]
- 15. Spelbrink R. E. J., Kolkman A., Slijper M., Killian J. A., de Kruijff B. (2005) Detection and identification of stable oligomeric protein complexes in Escherichia coli inner membranes. J. Biol. Chem. 280, 28742–28748 [DOI] [PubMed] [Google Scholar]
- 16. Stenberg F., Chovanec P., Maslen S. L., Robinson C. V., Ilag L. L., von Heijne G., Daley D. O. (2005) Protein complexes of the Escherichia coli cell envelope. J. Biol. Chem. 280, 34409–34419 [DOI] [PubMed] [Google Scholar]
- 17. Huang C. Z., Lin X. M., Wu L. N., Zhang D. F., Liu D., Wang S. Y., Peng X. X. (2006) Systematic identification of the subproteome of Escherichia coli cell envelope reveals the interaction network of membrane proteins and membrane-associated peripheral proteins. J. Proteome Res. 5, 3268–3276 [DOI] [PubMed] [Google Scholar]
- 18. Lasserre J. P., Beyne E., Pyndiah S., Lapaillerie D., Claverol S., Bonneu M. (2006) A complexomic study of Escherichia coli using two-dimensional blue native/SDS polyacrylamide gel electrophoresis. Electrophoresis 27, 3306–3321 [DOI] [PubMed] [Google Scholar]
- 19. Pan J. Y., Li H., Ma Y., Chen P., Zhao P., Wang S. Y., Peng X. X. (2010) Complexome of Escherichia coli envelope proteins under normal physiological conditions. J. Proteome Res. 9, 3730–3740 [DOI] [PubMed] [Google Scholar]
- 20. Li H., Pan J. Y., Liu X. J., Gao J. X., Wu H. K., Wang C., Peng X. X. (2012) Alterations of protein complexes and pathways in genetic information flow and response to stimulus contribute to Escherichia coli resistance to balofloxacin. Mol. Biosyst. 8, 2303–2311 [DOI] [PubMed] [Google Scholar]
- 21. Maddalo G., Stenberg-Bruzell F., Götzke H., Toddo S., Björkholm P., Eriksson H., Chovanec P., Genevaux P., Lehtiö J., Ilag L. L., Daley D. O. (2011) Systematic analysis of native membrane protein complexes in Escherichia coli. J. Proteome Res. 10, 1848–1859 [DOI] [PubMed] [Google Scholar]
- 22. Jeong H., Barbe V., Lee C. H., Vallenet D., Yu D. S., Choi S. H., Couloux A., Lee S. W., Yoon S. H., Cattolico L., Hur C. G., Park H. S., Segurens B., Kim S. C., Oh T. K., Lenski R. E., Studier F. W., Daegelen P., Kim J. F. (2009) Genome sequences of Escherichia coli B strains REL606 and BL21(DE3). J. Mol. Biol. 394, 644–652 [DOI] [PubMed] [Google Scholar]
- 23. Horler R. S. P., Butcher A., Papangelopoulos N., Ashton P. D., Thomas G. H. (2009) EchoLOCATION: an in silico analysis of the subcellular locations of Escherichia coli proteins and comparison with experimentally derived locations. Bioinformatics 25, 163–166 [DOI] [PubMed] [Google Scholar]
- 24. Futai M. (1974) Orientation of membrane vesicles from Escherichia coli prepared by different procedures. J. Membrane Biol. 15, 15–28 [DOI] [PubMed] [Google Scholar]
- 25. Steck T. L. (1974) The organization of proteins in the human red blood cell membrane. J. Cell Biol. 62, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pieper R., Huang S. T., Clark D. J., Robinson J. M., Alami H., Parmar P. P., Suh M. J., Kuntumalla S., Bunai C. L., Perry R. D., Fleischmann R. D., Peterson S. N. (2009) Integral and peripheral association of proteins and protein complexes with Yersinia pestis inner and outer membranes. Proteome Sci. 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Silva J. C., Gorenstein M. V., Li G. Z., Vissers J. P. C., Geromanos S. J. (2006) Absolute quantification of proteins by LCMSE: a virtue of parallel MS Acquisition. Mol. Cell. Proteomics 5, 144–156 [DOI] [PubMed] [Google Scholar]
- 28. Butland G., Peregrin-Alvarez J. M., Li J., Yang W., Yang X., Canadien V., Starostine A., Richards D., Beattie B., Krogan N., Davey M., Parkinson J., Greenblatt J., Emili A. (2005) Interaction network containing conserved and essential protein complexes in Escherichia coli. Nature 433, 531–537 [DOI] [PubMed] [Google Scholar]
- 29. Arifuzzaman M., Maeda M., Itoh A., Nishikata K., Takita C., Saito R., Ara T., Nakahigashi K., Huang H. C., Hirai A., Tsuzuki K., Nakamura S., Altaf-Ul-Amin M., Oshima T., Baba T., Yamamoto N., Kawamura T., Ioka-Nakamichi T., Kitagawa M., Tomita M., Kanaya S., Wada C., Mori H. (2006) Large-scale identification of protein-protein interaction of Escherichia coli K-12. Genome Res. 16, 686–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Regonesi M. E., Del Favero M., Basilico F., Briani F., Benazzi L., Tortora P., Mauri P., Deho G. (2006) Analysis of the Escherichia coli RNA degradosome composition by a proteomic approach. Biochimie (Paris) 88, 151–161 [DOI] [PubMed] [Google Scholar]
- 31. Wu T., McCandlish A. C., Gronenberg L. S., Chng S. S., Silhavy T. J., Kahne D. (2006) Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 103, 11754–11759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kerrien S., Aranda B., Breuza L., Bridge A., Broackes-Carter F., Chen C., Duesbury M., Dumousseau M., Feuermann M., Hinz U., Jandrasits C., Jimenez R. C., Khadake J., Mahadevan U., Masson P., Pedruzzi I., Pfeiffenberger E., Porras P., Raghunath A., Roechert B., Orchard S., Hermjakob H. (2012) The IntAct molecular interaction database in 2012. Nucleic Acids Res. 40, D841–D846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Masuda T., Saito N., Tomita M., Ishihama Y. (2009) Unbiased quantitation of Escherichia coli membrane proteome using phase transfer surfactants. Mol. Cell. Proteomics 8, 2770–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ishihama Y., Schmidt T., Rappsilber J., Mann M., Hartl F. U., Kerner M., Frishman D. (2008) Protein abundance profiling of the Escherichia coli cytosol. BMC Genomics 9, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Taniguchi Y., Choi P. J., Li G. W., Chen H., Babu M., Hearn J., Emili A., Xie X. S. (2010) Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science 329, 533–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Szeto T. H., Rowland S. L., Rothfield L. I., King G. F. (2002) Membrane localization of MinD is mediated by a C-terminal motif that is conserved across eubacteria, archaea, and chloroplasts. Proc. Natl. Acad. Sci. U.S.A. 99, 15693–15698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shih Y. L., Le T., Rothfield L. (2003) Division site selection in Escherichia coli involves dynamic redistribution of Min proteins within coiled structures that extend between the two cell poles. Proc. Natl. Acad. Sci. U.S.A. 100, 7865–7870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou H., Lutkenhaus J. (2003) Membrane binding by MinD involves insertion of hydrophobic residues within the C-Terminal amphipathic helix into the bilayer. J. Bacteriol. 185, 4326–4335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walz A. C., Demel R. A., de Kruijff B., Mutzel R. (2002) Aerobic sn-glycerol-3-phosphate dehydrogenase from Escherichia coli binds to the cytoplasmic membrane through an amphipathic alpha-helix. Biochem. J. 365, 471–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang G., Keifer P. A., Peterkofsky A. (2003) Solution structure of the N-terminal amphitropic domain of Escherichia coli glucose-specific enzyme IIA in membrane-mimetic micelles. Protein Sci. 12, 1087–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang G., Peterkofsky A., Clore G. M. (2000) A novel membrane anchor function for the N-terminal amphipathic sequence of the signal-transducing protein IIAglucose of the Escherichia coli phosphotransferase system. J. Biol. Chem. 275, 39811–39814 [DOI] [PubMed] [Google Scholar]
- 42. Kall L., Krogh A., Sonnhammer E. L. (2007) Advantages of combined transmembrane topology and signal peptide prediction—the Phobius web server. Nucleic Acids Res. 35, W429–W432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang R., Ou H. Y., Zhang C. T. (2004) DEG: a database of essential genes. Nucleic Acids Res. 32, D271–D272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johnson M., Zaretskaya I., Raytselis Y., Merezhuk Y., McGinnis S., Madden T. L. (2008) NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. De Leeuw E., Graham B., Phillips G. J., Ten Hagen-Jongman C. M., Oudega B., Luirink J. (1999) Molecular characterization of Escherichia coli FtsE and FtsX. Mol. Microbiol. 31, 983–993 [DOI] [PubMed] [Google Scholar]
- 46. Lutkenhaus J., Addinall S. G. (1997) Bacterial cell division and the Z ring. Annu. Rev. Biochem. 66, 93–116 [DOI] [PubMed] [Google Scholar]
- 47. Meile J., Wu L., Ehrlich S., Errington J., Noirot P. (2006) Systematic localisation of proteins fused to the green fluorescent protein in Bacillus subtilis: identification of new proteins at the DNA replication factory. Proteomics 6, 2135–2146 [DOI] [PubMed] [Google Scholar]
- 48. Choi-Rhee E., Cronan J. E. (2003) The biotin carboxylase-biotin carboxyl carrier protein complex of Escherichia coli acetyl-CoA carboxylase. J. Biol. Chem. 278, 30806–30812 [DOI] [PubMed] [Google Scholar]
- 49. Perez-Bercoff A., McLysaght A., Conant G. C. (2011) Patterns of indirect protein interactions suggest a spatial organization to metabolism. Mol. Biosyst. 7, 3056–3064 [DOI] [PubMed] [Google Scholar]
- 50. Huthmacher C., Gille C., Holzhutter H.-G. (2008) A computational analysis of protein interactions in metabolic networks reveals novel enzyme pairs potentially involved in metabolic channeling. J. Theor. Biol. 252, 456–464 [DOI] [PubMed] [Google Scholar]
- 51. Klein C., Garcia-Rizo C., Bisle B., Scheffer B., Zischka H., Pfeiffer F., Siedler F., Oesterhelt D. (2005) The membrane proteome of Halobacterium salinarum. Proteomics 5, 180–197 [DOI] [PubMed] [Google Scholar]
- 52. Alexandersson E., Saalbach G., Larsson C., Kjellbom P. (2004) Arabidopsis plasma membrane proteomics identifies components of transport, signal transduction and membrane trafficking. Plant Cell Physiol. 45, 1543–1556 [DOI] [PubMed] [Google Scholar]
- 53. Aivaliotis M., Haase W., Karas M., Tsiotis G. (2006) Proteomic analysis of chlorosome-depleted membranes of the green sulfur bacterium Chlorobium tepidum. Proteomics 6, 217–232 [DOI] [PubMed] [Google Scholar]
- 54. Aivaliotis M., Karas M., Tsiotis G. (2007) An alternative strategy for the membrane proteome analysis of the green sulfur bacterium Chlorobium tepidum using blue native PAGE and 2-D PAGE on purified membranes. J. Proteome Res. 6, 1048–1058 [DOI] [PubMed] [Google Scholar]
- 55. Sinz A. (2010) Investigation of protein–protein interactions in living cells by chemical crosslinking and mass spectrometry. Anal. Bioanal. Chem. 397, 3433–3440 [DOI] [PubMed] [Google Scholar]
- 56. Bruce J. E. (2012) In vivo protein complex topologies: sights through a cross-linking lens. Proteomics 12, 1565–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rappsilber J. (2011) The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J. Struct. Biol. 173, 530–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Raffaelli N., Lorenzi T., Mariani P. L., Emanuelli M., Amici A., Ruggieri S., Magni G. (1999) The Escherichia coli NadR regulator is endowed with nicotinamide mononucleotide adenylyltransferase activity. J. Bacteriol. 181, 5509–5511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ostrovsky de Spicer P., Maloy S. (1993) PutA protein, a membrane-associated flavin dehydrogenase, acts as a redox-dependent transcriptional regulator. Proc. Natl. Acad. Sci. U.S.A. 90, 4295–4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lopian L., Nussbaum-Shochat A., O'Day-Kerstein K., Wright A., Amster-Choder O. (2003) The BglF sensor recruits the BglG transcription regulator to the membrane and releases it on stimulation. Proc. Natl. Acad. Sci. U.S.A. 100, 7099–7104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lopez-Campistrous A., Semchuk P., Burke L., Palmer-Stone T., Brokx S. J., Broderick G., Bottorff D., Bolch S., Weiner J. H., Ellison M. J. (2005) Localization, annotation, and comparison of the Escherichia coli K-12 proteome under two states of growth. Mol. Cell. Proteomics 4, 1205–1209 [DOI] [PubMed] [Google Scholar]
- 62. Khemici V., Poljak L., Luisi B. F., Carpousis A. J. (2008) The RNase E of Escherichia coli is a membrane-binding protein. Mol. Microbiol. 70, 799–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kobayashi R., Suzuki T., Yoshida M. (2007) Escherichia coli phage-shock protein A (PspA) binds to membrane phospholipids and repairs proton leakage of the damaged membranes. Mol. Microbiol. 66, 100–109 [DOI] [PubMed] [Google Scholar]
- 64. Adams H., Teertstra W., Demmers J., Boesten R., Tommassen J. (2003) Interactions between phage-shock proteins in Escherichia coli. J. Bacteriol. 185, 1174–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jeffery C. J. (1999) Moonlighting proteins. Trends Biochem. Sci. 24, 8–11 [DOI] [PubMed] [Google Scholar]
- 66. Li Y., Stewart N. K., Berger A. J., Vos S., Schoeffler A. J., Berger J. M., Chait B. T., Oakley M. G. (2010) Escherichia coli condensin MukB stimulates topoisomerase IV activity by a direct physical interaction. Proc. Natl. Acad. Sci. U.S.A. 107, 18832–18837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Petrushenko Z. M., Lai C. H., Rybenkov V. V. (2006) Antagonistic interactions of kleisins and DNA with bacterial Condensin MukB. J. Biol. Chem. 281, 34208–34217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. de Boer P. A. (2010) Advances in understanding E. coli cell fission. Curr. Opin. Microbiol. 13, 730–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hayden J. D., Ades S. E. (2008) The extracytoplasmic stress factor, sigma(E), is required to maintain cell envelope integrity in Escherichia coli. PLoS One 3, e1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bordes P., Lavatine L., Phok K., Barriot R., Boulanger A., Castanie-Cornet M. P., Dejean G., Lauber E., Becker A., Arlat M., Gutierrez C. (2011) Insights into the extracytoplasmic stress response of Xanthomonas campestris pv. campestris: role and regulation of sigma(E)-dependent activity. J. Bacteriol. 193, 246–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. van den Ent F., Johnson C. M., Persons L., de Boer P., Lowe J. (2010) Bacterial actin MreB assembles in complex with cell shape protein RodZ. EMBO J. 29, 1081–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bendezu F. O., Hale C. A., Bernhardt T. G., de Boer P. A. (2009) RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E. coli. EMBO J. 28, 193–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Salje J., van den Ent F., de Boer P., Lowe J. (2011) Direct membrane binding by bacterial actin MreB. Mol. Cell 43, 478–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kruse T., Blagoev B., Lobner-Olesen A., Wachi M., Sasaki K., Iwai N., Mann M., Gerdes K. (2006) Actin homolog MreB and RNA polymerase interact and are both required for chromosome segregation in Escherichia coli. Gene Dev. 20, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ingerson-Mahar M., Briegel A., Werner J. N., Jensen G. J., Gitai Z. (2010) The metabolic enzyme CTP synthase forms cytoskeletal filaments. Nat. Cell Biol. 12, 739–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schryvers A., Lohmeier E., Weiner J. H. (1978) Chemical and functional properties of the native and reconstituted forms of the membrane-bound, aerobic glycerol-3-phosphate dehydrogenase of Escherichia coli. J. Biol. Chem. 253, 783–788 [PubMed] [Google Scholar]
- 77. Cozzarelli N. R., Koch J. P., Hayashi S., Lin E. C. (1965) Growth stasis by accumulated L-alpha-glycerophosphate in Escherichia coli. J. Bacteriol. 90, 1325–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gitai Z., Dye N., Shapiro L. (2004) An actin-like gene can determine cell polarity in bacteria. Proc. Natl. Acad. Sci. U.S.A. 101, 8643–8648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Soufo H., Reimold C., Linne U., Knust T., Gescher J., Graumann P. L. (2010) Bacterial translation elongation factor EF-Tu interacts and colocalizes with actin-like MreB protein. Proc. Natl. Acad. Sci. U.S.A. 107, 3163–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Candiano G., Bruschi M., Musante L., Santucci L., Ghiggeri G. M., Carnemolla B., Orecchia P., Zardi L., Righetti P. G. (2004) Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 25, 1327–1333 [DOI] [PubMed] [Google Scholar]
- 81. UniProt (2010) The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 38, D142–D148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rappsilber J., Ryder U., Lamond A. I., Mann M. (2002) Large-scale proteomic analysis of the human spliceosome. Genome Res. 12, 1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Elias J. E., Haas W., Faherty B. K., Gygi S. P. (2005) Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat. Meth. 2, 667–675 [DOI] [PubMed] [Google Scholar]
- 84. Keseler I. M., Bonavides-Martínez C., Collado-Vides J., Gama-Castro S., Gunsalus R. P., Johnson D. A., Krummenacker M., Nolan L. M., Paley S., Paulsen I. T., Peralta-Gil M., Santos-Zavaleta A., Shearer A. G., Karp P. D. (2009) EcoCyc: a comprehensive view of Escherichia coli biology. Nucleic Acids Res. 37, D464–D470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rudd K. E. (2000) EcoGene: a genome sequence database for Escherichia coli K-12. Nucleic Acids Res. 28, 60–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotech. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 87. Riley M., Abe T., Arnaud M. B., Berlyn M. K. B., Blattner F. R., Chaudhuri R. R., Glasner J. D., Horiuchi T., Keseler I. M., Kosuge T., Mori H., Perna N. T., Plunkett G., Rudd K. E., Serres M. H., Thomas G. H., Thomson N. R., Wishart D., Wanner B. L. (2005) Escherichia coli K-12: a cooperatively developed annotation snapshot—2005. Nucleic Acids Res. 34, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Aranda B., Achuthan P., Alam-Faruque Y., Armean I., Bridge A., Derow C., Feuermann M., Ghanbarian A. T., Kerrien S., Khadake J., Kerssemakers J., Leroy C., Menden M., Michaut M., Montecchi-Palazzi L., Neuhauser S. N., Orchard S., Perreau V., Roechert B., van Eijk K., Hermjakob H. (2010) The IntAct molecular interaction database in 2010. Nucleic Acids Res. 38, D525–D531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chatzi K., Sardis M. F., Karamanou S., Economou A. (2013) Breaking on through to the other side: protein export through the bacterial Sec system. Biochem. J. 449, 25–37 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.