

Abstract
Snake venom metalloproteases (SVMP) are composed of five domains: signal peptide, propeptide, metalloprotease, disintegrin, and cysteine-rich. Secreted toxins are typically combinatorial variations of the latter three domains. The SVMP-encoding genes of Psammophis mossambicus venom are unique in containing only the signal and propeptide domains. We show that the Psammophis SVMP propeptide evolves rapidly and is subject to a high degree of positive selection. Unlike Psammophis, some species of Echis express both the typical multidomain and the unusual monodomain (propeptide only) SVMP, with the result that a lower level of variation is exerted upon the latter. We showed that most mutations in the multidomain Echis SVMP occurred in the protease domain responsible for proteolytic and hemorrhagic activities. The cysteine-rich and disintegrin-like domains, which are putatively responsible for making the P-III SVMPs more potent than the P-I and P-II forms, accumulate the remaining variation. Thus, the binding sites on the molecule's surface are evolving rapidly whereas the core remains relatively conserved. Bioassays conducted on two post-translationally cleaved novel proline-rich peptides from the P. mossambicus propeptide domain showed them to have been neofunctionalized for specific inhibition of mammalian a7 neuronal nicotinic acetylcholine receptors. We show that the proline rich postsynaptic specific neurotoxic peptides from Azemiops feae are the result of convergent evolution within the precursor region of the C-type natriuretic peptide instead of the SVMP. The results of this study reinforce the value of studying obscure venoms for biodiscovery of novel investigational ligands.
Snake venom metalloproteases (SVMP)1 evolved from ADAM (A disintegrin and metalloprotease) proteins that were recruited into the venom of snakes near the base of the advanced snake (Caenophidia) radiation. They have been identified in the venoms of all lineages of advanced snakes (1–5). The ancestral SVMP (P-III) contains (in downstream order) five domains: signal + propeptide + metalloprotease + disintegrin + cysteine rich. Following the divergence of vipers from the remaining caenophidians, extensive gene duplication, domain loss, and positive selection resulted in generation of the P-I and P-II classes of SVMP within the viperid lineage (6–8). These two derived classes found in viper venoms lack either the cysteine-rich domain (P-II) or the cysteine-rich and disintegrin domains (P-I) (8, 9). The signal peptide and the propeptide domain are typically cleaved off before expression although the latter has been detected in venoms on occasion (9). SVMP are often the dominant venom component in the venom of viperid snakes, but are typically much less significant in the venom of other snake families (10–15). The majority of SVMP principally exhibit hemorrhagic activity, although other functions, such as the activation of prothrombin and Factor X, fibrin(ogen)olysis, apoptosis and the inhibition of platelet aggregation, have also been reported (16). Although SVMP-induced hemorrhage is primarily dependent on the proteolytic activity of the metalloprotease domain, the potency of this activity is increased by the presence of the additional domain structures that are absent from the P-I and P-II class (17). Consequently, P-III SVMP typically exhibits the greatest hemorrhagic activities.
SVMP represent a model system for investigating the evolutionary processes responsible for generating new protein functions. Extensive gene duplication and domain loss has resulted in the generation of a large multilocus gene family that encodes related proteins exhibiting divergent molecular structures (7, 18). Additional diversity has arisen because of accelerated evolution within new SVMP classes following the loss of domains (7, 19). Complicating matters is evidence that some SVMP genes are also capable of selectively expressing specific domains (2, 7, 20); for example, some genes encode” short-coding disintegrins,” which consist solely of a signal peptide and a disintegrin domain (20). There have also been two reports of P-III SVMP that encode only the propeptidedomain of the SVMP gene. Truncated SVMPs that terminate at the end of the prodomain have been transcriptomically identified from the viper genus Echis (7, 11), although the proteins encoded by these genes have yet to be detected proteomically in venom. Notably, Echis venoms contain high levels of SVMP, with representatives of all three SVMP classes present. This prevalence of SVMP in the venom of these snakes is thought to be largely responsible for inducing the severe hemorrhaging observed in envenomated prey (11, 12, 21). SVMP found in the venom of the lamprophiid snake Psammophis mossambicus, on the other hand, consist entirely of selectively expressed propeptide domains, which have evolved via deletion of the metalloprotease, disintegrin, and SVMP domains from the ancestral multidomain P-III SVMP gene (2). Despite this fascinating observation, the evolution and bioactivity of these atypical SVMP remain completely unexplored. Mass spectrometry of P. mossambicus venom revealed an abundance and diversity of peptides with molecular weights consistent to post-translational proteolytically liberated peptides from the propetide precursor region (22).
Here we investigate the evolution of pro-domain SVMP genes in Psammophis and compare the rate of positive selection acting on these genes to propeptide and P-III SVMP genes isolated from Echis venom. We reveal that Psammophis SVMP have accumulated significantly higher numbers of positively selected sites in the propeptide domain than observed in Echis. We also demonstrate that positive selection pressures acting on the truncated structure of Psammophis SVMPs are directly responsible for driving protein neofunctionalization in the form of novel neurotoxic activity. We used molecular phylogenetics to determine if monodomain propeptide expression in Echis shared an evolutionary history with Psammophis, and is thus ancestral to all advanced snakes, or if these were convergent derivations. In addition, it was recently shown that a unique type of proline-rich peptide isolated from the venom of the viperid snake Azemiops feae is neurotoxic like the P. mossambicus Pm1 and Pm2 peptides. The A. feae peptide differs in activity by blocking the neuromuscular nicotinic acetylcholine receptor rather than the neuronal receptor targeted by the peptides in this study (22). Before this study, the molecular evolutionary history of the A. feae peptide remained to be elucidated, as it was known only from the 21-residue post-translationally processed form secreted in crude venom. Thus, we sequenced the full coding region for this neurotoxin from the mRNA of A. feae venom glands to determine whether the proline-rich neurotoxins from Psammophis and Azemiops shared a molecular evolutionary history or if they were convergently-derived.
MATERIALS AND METHODS
Sequence Retrieval and Alignment
Psammophis mossambicus propeptide monodomain, Echis spp propeptide monodomain (E. coloratus and E. pyramidum leakeyi) and E. coloratus multidomain SVMP nucleotide sequences were recovered bioinformatically from the National Center for Biotechnology Information (NCBI: http://www.ncbi.nlm.nih.gov/). The translated nucleotide sequences were aligned using PRANK (24) and then adjusted manually to optimize the alignments. To avoid confusion, previously obtained sequences are given with their UniProt accession numbers whereas ones obtained in this study are given with their Genbank accession numbers.
cDNA Library Construction
Venom glands of an Azemiops feae specimen from Hunan, China were dissected under surgical anesthesia 3 days after stimulation by milking. Total RNA was extracted using the standard TRIzol Plus method (Invitrogen, Carlsbad, CA). Extracts were enriched for mRNA using an RNeasy mRNA mini kit (Qiagen, Valencia, CA). mRNA was reverse transcribed, fragmented and ligated to a unique 10-base multiplex identifier tag prepared using standard protocols and applied to one PicoTitrePlate (PTP) for simultaneous amplification and sequencing on a Roche 454 GS FLX+ Titanium platform (Australian Genome Research Facility). Automated grouping and analysis of sample-specific multiplex identifier reads informatically separated sequences from the other transcriptomes on the plates, which were then post-processed to remove low quality sequences before de novo assembly into contiguous sequences (contigs) using MIRA software (ref.). Assembled contigs were processed using CLC Main Work Bench (CLC-Bio) and the Blast2GO bioinformatic suite (47–50) to provide Gene Ontology, BLAST, and domain and Interpro annotation. The above analyses assisted in rationalization of the large numbers of assembled contigs into phylogenetic “groups” for detailed phylogenetic analyses outlined below.
Test for Recombination
Recombination can mislead phylogenetic and evolutionary selection interpretations (25). Hence, we evaluated the effect of recombination on Psammophis mossambicus and Echis spp. SVMPs by employing single breakpoint recombination implemented in the HyPhy package (26–28). Potential breakpoints were detected using the small sample Akaike information criterion (AIC) and the sequences were compartmentalized before conducting the selection analyses.
Phylogenetic Reconstruction
The best-fit model of nucleotide substitution for each data set was determined using jModeltest (29), according to AIC. Model-averaged parameter estimates of gamma shape parameter (α) and the proportion of invariant sites (pinvar) were used for phylogenetic reconstruction. Phylogenetic relationships were determined using Bayesian and maximum-likelihood approaches. MrBayes version 3.1 (30, 31) was used for Bayesian inference. Tree searches were run using four Markov chains for 10 million generations, sampling every 100th tree. The log likelihood score of each saved tree was plotted against the number of generations to establish the point at which the log-likelihood scores of the analyses reached their asymptote. Twenty-five percent of the total trees sampled were discarded as burn in. The posterior probabilities for clades were established by constructing a majority rule consensus tree for all trees generated after completion of the burn in. The analyses were repeated three times to ensure that the trees generated were not clustered around local optima. An optimal maximum likelihood phylogenetic tree was obtained using PhyML 3.0 (32) and node support was evaluated with 1000 bootstrapping replicates.
Selection Analyses
We employed sophisticated likelihood models of coding-sequence evolution (33, 34) as implemented in CODEML of the PAML (35) package to estimate the selection pressures shaping the E. coloratus multidomain, Echis spp and P. mossambicus monodomain SVMPs. We first employed the lineage-specific one-ratio model that assumes a single ωfor the entire phylogenetic tree. The one-ratio model is very conservative and can only detect positive selection if the ωratio averaged over all the sites along the lineage is significantly greater than one.
The assumption of constant evolutionary selection pressure for the entire phylogenetic tree over millions of years is unrealistic. Thus, lineage-specific models like the one-ratio model fail to identify regions in proteins that might accumulate variation more often than others and hence they can underestimate the strength of selection. We therefore employed site-specific models that estimate positive selection statistically as a nonsynonymous-to-synonymous nucleotide-substitution rate ratio (ω) significantly greater than 1. We compared likelihood values for three pairs of models with different assumed ωdistributions as no a priori expectation exists for the same: M0 (constant ω rates across all sites) versus M3 (allows ω to vary across sites within “n” discrete categories, n ≥ 3); M1a (a model of neutral evolution) in which all sites are assumed to be either under negative (ω <1) or neutral selection (ω = 1) versus M2a (a model of positive selection), which in addition to the site classes mentioned for M1a assumes a third category of sites; sites with ω >1 (positive selection) and, finally, M7 (β) versus M8 (β and ω); models that mirror the evolutionary constraints of M1 and M2 but assumes that ω values are drawn from a β distribution (36). Only if the alternative models (M3, M2a and M8: allow sites with ω >1) show a better fit in the likelihood ratio test relative to their null models (M0, M1a, and M8: do not show allow sites ω >1), are their results considered significant. The likelihood ratio test is estimated as twice the difference in maximum likelihood values between nested models and compared with the χ2 distribution with the appropriate degree of freedom—the difference in the number of parameters between the two models. The Bayes empirical Bayes approach (37) was used to identify amino acids under positive selection by calculating the posterior probabilities that a particular amino acid belongs to a given selection class (neutral, conserved, or highly variable). Sites with greater posterior probability (PP ≥ 95%) of belonging to the “w > 1 class” were inferred to be positively selected.
We employed Single Likelihood Ancestor Counting, Fixed-Effects Likelihood, and Random Effects Likelihood models (38) implemented in HyPhy (39) to provide significant support to the aforementioned analyses and to detect sites evolving under the influence of positive and negative selection. The more advanced Mixed Effects Model Evolution (40) was also used to detect episodic diversifying selection. Mixed Effects Model Evolution employs Fixed-Effects Likelihoodalong the sites and Random-effects likelihood across the branches to detect episodic diversifying selection. For clear depiction of the proportion of sites under selection, an evolutionary fingerprint analysis was carried out using the evolutionary selection distance (ESD) algorithm (41) implemented in Datamonkey.
The direct comparison of omega values computed using the aforementioned methods can be misleading as different proportions of sites may be under selection. Hence, we partitioned the Echis coloratus SVMP domains and computed omega values simultaneously using Mgene (4) and option G test (42) from Codeml to assess the selection pressures on various SVMP domains.
Structural Analyses
To depict the differential selection pressures on various domains of E. coloratus SVMP, we constructed a homology model using the Phyre 2 webserver (43) and mapped the sites under positive selection using Pymol (44). The crystal structure of 2E3X:A was selected as the best-fit template for the target sequence GU012165.1 for homology modeling. The program GETAREA (45) was used to calculate the accessible surface area (ASA) (i.e., solvent exposure) of amino acid side chains. It uses the atom coordinates of the PDB file and indicates if a residue is buried or exposed to the surrounding medium by comparing the ratio between side-chain ASA and the “random coil” values per residue. An amino acid is considered to be buried if it has an ASA less than 20% and exposed if ASA ≥ 50%. The Consurf webserver was used for mapping the evolutionary selection pressures on the three-dimensional homology model of E. coloratus SVMPs (46).
Peptide Synthesis
To explore potential neofunctionalization derivations, we constructed two variations of proline rich peptides encoded by the propeptide domain. Protected Fmoc-amino acid derivatives were purchased from Novabiochem (address) or Auspep (Melbourne, Australia). The following side chain protected amino acids were used: Cys(Trt), His(Trt), Hyp(tBu), Tyr(tBu), Lys(Boc), Trp(Boc), Arg(Pbf), Asn(Trt), Asp(OtBu), Glu(OtBu), Gln(Trt), Ser(tBu), Thr(tBu), Tyr(tBu). All other Fmoc amino acids were unprotected. Peptide-synthesis grade dimethylformamide, dichloromethane, diisopropylethylamine, and trifluoroacetic acid (TFA) were supplied by Auspep. 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, triisopropyl silane, HPLC grade acetonitrile, aceticanhydride and methanol were supplied by Sigma Aldrich. The resin used was Fmoc-Arg(Pbf)-wang resin (0.33 mmol/g) from Peptide International (Louisville, KY). Ethane dithiol was from Merck.
P. mossambicus propeptide SVMPs Pm1 and Pm2 were synthesized on a Protein Technology (Symphony) automated peptide synthesizer using Fmoc-Arg(Pbf)-Wang resin (0.1 mmol). Assembly of the peptides was performed using 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate /diisopropylethylamine in situ activation protocols (Schnolzer et al., 2007) to couple the Fmoc-protected amino acid to the resin (5 equiv. excess, coupling time 20 min). Fmoc deprotection was performed with 30% piperidine/dimethylformamide for 1 min followed by a 2 min repeat. Washes were performed 10 times after each coupling as well as after each deprotection step. After chain assembly and final Fmoc deprotection the peptide resins were washed with methanol and dichloromethane and dried in a stream of nitrogen. Cleavage of peptide from the resin was performed at room temperature in TFA:H2O:triisopropyl silane:Ethane dithiol (87.5:5:5:2.5) for 3 h. Cold diethyl ether (30 ml) was then added to the filtered cleavage mixture and the peptide precipitated. The precipitate was collected by centrifugation and subsequently washed with further cold diethyl ether to remove scavengers. The final product was dissolved in 50% acetonitrile and lyophilized to yield a white solid product. The crude, reduced peptide was examined by reversed-phase HPLC for purity and the correct molecular weight confirmed by electrospray mass spectrometry.
Analytical HPLC runs were performed using a Shimadzu HPLC system LC10A with a dual wavelength UV detector set at 214 nm and 254 nm. A reversed-phase C18 column (Zorbax 300-SB C-18; 4.6 × 50 mm) with a flow rate of 2 ml/min was used. Elution was performed using a 0–80% gradient of Buffer B (0.043% TFA in 90% acetonitrile) in Buffer A (0.05% TFA in water) over 20 min. Crude peptides were purified by semipreparative HPLC on a Shimadzu HPLC system LC8A with a reversed-phase C18 column (Vydac C-18, 25 cm × 10 mm) running at a flow rate of 5 ml/min with a 1%/min gradient of 5–50% Buffer B. The purity of the final product was evaluated by analytical HPLC (Zorbax 300SB C18: 4.6 × 100 mm) with a flow rate of 1 ml/min and a 1.67%/min gradient of Buffer B (5–45%). The final purity of all synthesized peptides was >95%. Electrospray mass spectra were collected inline during analytical HPLC runs on an Applied Biosystems API-150 spectrometer operating in the positive ion mode with an OR of 20, Rng of 220, and Turbospray of 350 degrees. Masses between 300 and 2200 amu were detected (Step 0.2 amu, Dwell 0.3 ms).
Bioactivity Testing
Pharmacological activity of the peptides was assessed using the FLIPRTETRA fluorescence plate reader (Molecular Devices, Sunnyvale, CA) as previously described (51–53). SH-SY5Y human neuroblastoma cells (a kind gift from Victor Diaz, Max Planck Institute for Experimental Medicine, Goettingen, Germany) endogenously expressing human voltage-gated calcium channels (Cav), voltage-gated sodium channels (Nav), and nicotinic acetylcholine receptors (nAChR) were maintained in RPMI medium (Invitrogen, Australia) supplemented with 15% fetal bovine serum and 2 mm l-glutamine and passaged every 3–5 days using 0.25% trypsin/EDTA (Invitrogen).
SH-SY5Y cells were plated at a density of 50,000 cells/well 48 h before fluorescence Ca2+ assays on black-walled clear bottom 384-well plates (Corning) and incubated for 30 min at 37 °C with Calcium-4 NW dye (Molecular Devices) prepared in physiological salt solution (composition in mm: NaCl 140, glucose 11.5, KCl 5.9, MgCl2 1.4, NaH2PO4 1.2, NaHCO3 5, CaCl2 1.8, HEPES 10). Fluorescence responses (excitation 470–495 nm; emission 515–575 nm) were assessed after 5 min pretreatment with peptides. To assess activity at L- and N-type Cav, fluorescence responses after addition of 90 mm KCl/5 mm CaCl2 were assessed in the absence or presence of nifedipine (10 μm), respectively. Activity at Nav was assessed after stimulation with the Nav agonist veratridine. α3β2/4-containing nAChR were activated by addition of nicotine (30 μm), whereas activity at the human α7 nAChR endogenously expressed in SH-SY5Y neuroblastoma cells was assessed in the presence of the α7-selective allosteric modulator PNU120596 (10 μm; Sapphire Bioscience, Waterloo, Australia) after stimulation with the α7 agonist choline (30 μm).
Raw fluorescence readings were converted to response over baseline using the FLIPRTetra software SCREENWORKS 3.1.1.4 (Molecular Devices) and were expressed relative to the maximum increase in fluorescence of control responses.
Male chicks (4–10 days) were killed by CO2 and exsanguination. Both chick biventer cervicis nerve muscle preparations were isolated and mounted on wire tissue holders under 1 g resting tension in 5 ml organ baths containing Krebs solution (NaCl, 118.4 mm; KCl, 4.7 mm; MgSO4, 1.2 mm KH2PO4, 1.2 mm; CaCl2, 2.5 mm; NaHCO3, 25 mm and glucose, 11.1 mm), maintained at 34 °C and bubbled with 95% O2/5% CO2. Indirect twitches were evoked by electrical stimulation of the motor nerve (supramaximal voltage, 0.2 ms, 0.1 Hz) using a Grass S88 stimulator (Grass Instruments, Quincy, MA). d-Tubocurarine (10 μm) was added, and subsequent abolition of twitches confirmed selective stimulation of the motor nerve, after which thorough washing with Krebs solution was applied to re-establish twitches. In the absence of electrical stimulation, contractile responses to acetylcholine (ACh; 1 mm for 30 s), carbachol (CCh; 20 μm for 60 s), and potassium (KCl; 40 mm for 30 s) were obtained before the addition of peptide and at the conclusion of the experiment. The preparation was equilibrated for 30 min before the addition of peptide. Peptides were left in contact with the preparation for a maximum of 3 h to test for slow-developing effects.
Molecular Evolution
Phylogenetic analyses showed that the prepro-only expression in Psammophis and Echis are convergent derivations (Fig. 1). Sequence alignment showed extreme variation in the Psammophis monodomain form whereas the Echis monodomain prepro form was almost identical to the prepro region expressed as part of the multidomain gene (Fig. 2).
Fig. 1.

Snake venom metalloproteinase (SVMP) phylogeny with mid-point rooting.
Fig. 2.

Sequence alignment of Psammophis mossambicus propeptide domain selectively expressed variants (1. A7X4E9, 2. A7X4D4, 3. A7X4B5, 4. A7X4C9, 5. A7X4C4, 6. A7X4C0, 7. A7X443, A7X437, 9. A7X488, 10. A7X473, 11. A7X480, 12. A7X468, 13. A7X452, 14. A7X484, 15. A7X476, 16. A7X457, 17. A7X447, 18. A7X497, 19. A7X4A6, 20. A7X461, 21. A7X493, 22. A7X4B0, 23. A7X465), Echis propeptide domain selectively expressed variants (GR950192 Echis pyramidum leakeyi and GR948204 Echis coloratus) and Echis multidomain SVMP (Q2UXQ6 Echis ocellatus and E9KNB4 Echis carinatus sochureki). Signal peptide is shown in lowercase, metalloprotease domain in straight underline, disintegrin domain in wavy underline.
Selection Analyses
By using the one-ratio model, the simplest of the lineage-specific models that computes a single ω value for all branches in the phylogeny, the global ω was estimated to be 0.94, 0.39, and 0.39 for the Psammophis monodomain, Echis monodomain, and Echis coloratus multidomain SVMP genes, respectively (supplemental Table S2). Because this value is an average over all codons in each lineage, it suggests a rapid accumulation of mutations in the Psammophis SVMP propeptide domain. In contrast, the Echis propeptide domains seem to be under negative selection. The global ω estimates for the Echis SVMP protease, disintegrin-like and cysteine-rich domains were 1.33, 0.92, and 0.97, respectively (supplemental Table S2). This highlights the strong influence of positive selection on the protease domain that is responsible for hemorrhagic activity.
The Bayes empirical Bayes approach implemented in site-model 8 estimated that about 8.5% of sites were under positive selection in the P. mossambicus monodomain propeptide region (ω = 1.23) whereas the Echis monodomain (ω = 0.52) and E. coloratus multidomain (ω = 0.46) SVMPs evolve under negative selection (Fig. 3; Table I). Omega estimates for the protease, disintegrin-like, and cysteine-rich domains of E. coloratus SVMP under this approach were 1.60 (32% of sites), 1.33 (25% of sites), and 1.39 (21% of sites), respectively, highlighting the strong influence of positive selection on the evolution of these domains (Table II and Fig. 4).
Fig. 3.
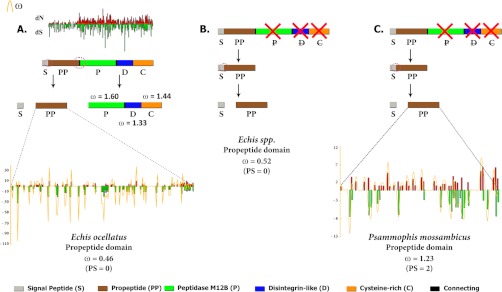
Comparison of evolutionary selection pressures acting on the propeptide regions of (A) Echis coloratus multidomain, (B) Echis spp. monodomain and (C) Psammophis mossambicus monodomain SVMPs. The corresponding site-wise synonymous and non-synonymous mutations are shown in green and red bars, respectively while the omega is shown in gold. Model 8 computed omega values and the number of positively-selected sites (PP ≥ 0.95, Bayes-Empirical Bayes approach) are also indicated. Domains are: C, cysteine-rich; D, disintegrin; p, protease; PP, pro-protein (PP); and S, signal peptide.
Table I. Selection analyses of snake venom metalloproteinase (SVMP) propeptides. ω: mean dN/dS.
| SLACa | FELb | RELc | Integrative analyses | MEMEd | PAMLg | ||
|---|---|---|---|---|---|---|---|
| SLAC + FEL + REL + MEME | M8 | M2a | |||||
| Psammophis | |||||||
| ω > 1e | 0 | 1 | 0 | 2 | 2 | 1 | |
| Monodomain | |||||||
| ω < 1f | 1 | 2 | 4 | 4 | 1 | (0 + 2) | (0 + 1) |
| SVMP | |||||||
| ω = | 0.95 | - | 2.51 | - | 1.23 | 1.24 | |
| Echis | |||||||
| ω > 1e | 0 | 0 | 2 | 2 | 0 | 0 | |
| Monodomain | |||||||
| ω < 1f | 0 | 4 | 1 | 5 | 0 | ||
| SVMP | |||||||
| ω = | 0.32 | - | 0.43 | - | 0.52 | 0.52 | |
| Echis | |||||||
| ω > 1e | 0 | 1 | 2 | 3 | 0 | 0 | |
| Multidomain | |||||||
| ω < 1f | 5 | 12 | 0 | 12 | 1 | ||
| SVMP | |||||||
| ω = | 0.47 | - | 0.82 | - | 0.46 | 0.46 | |
a Single Likelihood Ancestor Counting.
b Fixed-effects likelihood.
c Random-effects likelihood.
d Sites detected as experiencing episodic diversifying selection (0.05 significance) by the Mixed Effects Model Evolution (MEME).
e Number of positively selected sites at 0.05 significance (for SLAC, FEL) or 50 Bayes factor (for REL).
f Number of negatively selected sites at 0.05 significance (for SLAC, FEL) or 50 Bayes factor (for REL).
g Positively selected sites detected using the Bayes Empirical Bayes approach implemented in the site models (M8 and M2a). Sites detected at 0.99 and 0.95 significance are indicated in the parenthesis.
Table II. Selection analyses of Echis coloratus snake venom metalloproteinase (SVMP) domains. ω: mean dN/dS.
| SLACa | FELb | RELc | Integrative | MEMEd | PAMLg | |||
|---|---|---|---|---|---|---|---|---|
| SLAC + FEL + REL + MEME | M8 | M2a | Option Gh | |||||
| Propeptide | ||||||||
| ω > 1e | 0 | 1 | 0 | 2 | 0 | 0 | ||
| ω < 1f | 1 | 2 | 4 | 4 | 1 | 0.38 | ||
| ω = | 0.95 | - | 2.51 | - | 0.46 | 0.46 | ||
| Peptidase M12B | ||||||||
| ω > 1e | 0 | 1 | 8 | 8 | 28 | 15 | ||
| ω < 1f | 2 | 8 | 7 | 10 | 0 | (14 + 14) | (5 + 10) | 1.28 |
| ω = | 0.83 | - | - | - | 1.60 | 1.60 | ||
| Disintegrin | ||||||||
| ω > 1e | 0 | 1 | 3 | 3 | 9 | 6 | ||
| ω < 1f | 2 | 7 | 9 | 10 | 0 | (4 + 5) | (3 + 3) | 0.91 |
| ω = | 0.87 | - | - | - | 1.33 | 1.35 | ||
| Cysteine-rich domain | ||||||||
| ω > 1e | 0 | 2 | 5 | 10 | 15 | 8 | ||
| ω < 1f | 2 | 8 | 16 | 18 | 7 | (6 + 9) | (4 + 4) | 1.04 |
| ω = | 1.04 | - | - | - | 1.44 | 1.43 | ||
| Entire toxin | ||||||||
| ω > 1e | 1 | 8 | 7 | 33 | 34 | 27 | ||
| ω < 1f | 14 | 33 | 15 | 34 | 28 | (14 + 20) | (9 + 18) | - |
| ω = | 1.02 | - | 2.49 | - | 1.15 | 1.16 | ||
a Single Likelihood Ancestor Counting.
b Fixed-effects likelihood.
c Random-effects likelihood.
d Sites detected as experiencing episodic diversifying selection (0.05 significance) by the Mixed Effects Model Evolution (MEME).
e Number of positively selected sites at 0.05 significance (for SLAC, FEL) or 50 Bayes factor (for REL).
f Number of negatively selected sites at 0.05 significance (for SLAC, FEL) or 50 Bayes factor (for REL).
g Positively-selected sites detected using the Bayes Empirical Bayes approach implemented in the site models (M8 and M2a). Sites detected at 0.99 and 0.95 significance are indicated in the parenthesis.
h Mean dN/dS computed using Mgene and option G.
Fig. 4.

Three-dimensional homology model of Echis coloratus SVMP, depicting the locations of positively selected amino acid sites (shown in red) detected by site-model 8 analyses for different domains. The omega values (Codeml: Mgene with option G) and the number of positively selected sites (Model 8, PP ≥ 0.95, Bayes-Empirical Bayes approach) are also indicated for the respective domains. A plot of amino acid positions (x axis) against accessible surface area (ASA) ratio (y axis) indicating the positions of amino-acids (exposed or buried) in the crystal structure of Echis coloratus SVMP is presented. Residues with an ASA ratio of more than 50% (above blue line) are considered to be exposed to the surrounding solvent whereas those with a ratio lesser than 20% (bellow the red line) are considered to be buried. The co-ordinates of positively selected sites in the protease, disintegrin-like and cysteine-rich domains are shown as big green, blue, and orange circles, respectively. Three-dimensional structures of each SVMP domain depicting the locations of positively selected sites (in red) along with the model 8 omega are also presented.
Direct comparison of ω values computed using the aforementioned algorithms could potentially be misleading as these genes may have a different proportion of sites under selection. Hence, for a better comparison of selection pressures acting on different domains of the E. coloratus multidomain SVMP, we employed Mgene with option G analysis of Codeml. This test simultaneously estimated ω of 0.38, 1.28, 0.91, and 1.04 for the propeptide, protease, disintegrin-like and cysteine-rich domains, respectively (Table II). Moreover, the site-specific model 8 analyses using the Bayes empirical Bayes approach identified 28 (∼53% of positively selected sites), 9 (17%) and 15 (∼29%) amino acid sites under positive selection in the protease, disintegrin-like and cysteine-rich domains, respectively, confirming the greater influence of positive selection on the protease domain than any other domain (Table II and Fig. 4). In contrast to the Echis protease domain and the Psammophis monodomain propetide region, the E. coloratus multidomain and the Echis spp monodomain SVMP propeptide regions have evolved under negative selection (ω = 0.38 and ω = 0.52, respectively). We mapped positively selected sites onto the homology model of E ocellatus multidomain SVMP to clearly depict their location in the toxin (Fig. 4). Our analyses show that almost 60% of these positively selected sites are confined to the molecular surface, whereas only 12% are buried in the conserved core of the toxin.
Bioactivity Testing
Assessment of the pharmacological activity of two Psammophis mossambicus proline-rich SVMP propeptide-only domain variants: Pm1 (VYNLHGSVPAPPWQPHARRPRPKNR encoded by uniprot A7X4A6) and Pm2 (VYNLHGSVPAPPWQPHARRPRPKYR encoded by uniprot A7X461) using high-throughput FLIPR assays revealed that these peptides did not exhibit any agonist-like activity at concentrations up to 1 mm, and were inactive at L-type and N-type Cav, Nav and a3b2/4 nAChR (data not shown). However, both peptides caused concentration-dependent inhibition of endogenously expressed human α7 nAChR, with IC50s of 11.99 μm and 11.83 μm (pIC50 4.921 ± 0.058 and 4.927 ± 0.094), respectively (Fig. 5). Psammophis mossambicus SVMP propeptide only domain variants Pm1 and Pm2 had no significant inhibitory effect on twitch height in the chick biventer cervicis nerve-muscle preparation (n = 4, data not shown) at concentrations up to 10 μm over a period of 3 h.
Fig. 5.
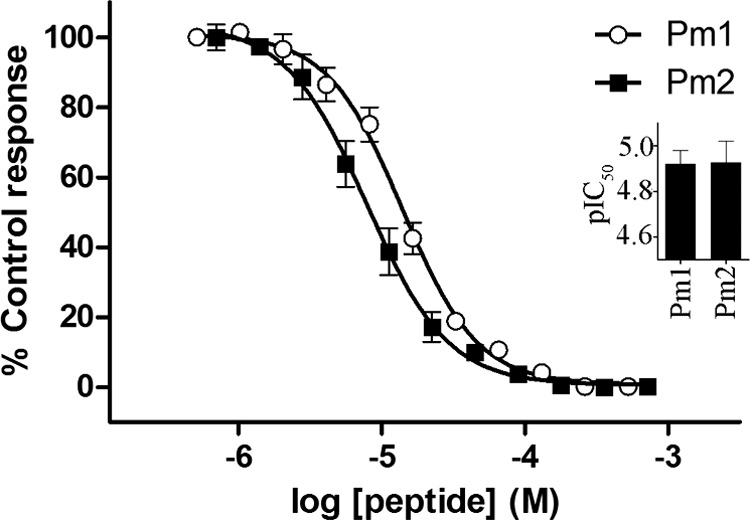
Inhibition of human a7 nAChR by proline-rich SVMP pre-pro only domain variants from Psammophis mossambicus. Pm1 and Pm2 concentration-dependently inhibited a7 nAChR endogenously expressed in SH-SY5Y human neuroblastoma cells in a high-throughput FLIPR Ca2+ imaging assay. Inset, the IC50s for inhibition of a7 nAChR by Pm1 and Pm2 were determined as 11.99 μm (pIC50 4.921 ± 0.058) and 11.83 μm (pIC50 4.927 ± 0.094), respectively. Data are presented as mean ± S.E. with n = 3 replicates from 3 independent assays.
Convergent Derivation of Proline Rich Neurotoxic Peptides
It was revealed that the A. feae proline-rich peptides were not derivations of the SVMP propeptide region but rather the propeptide region of the c-type natriuretic peptide expressed in snake venoms. The A. feae peptide was encoded in tandem repeats, with some forms containing two repeats of the proline-rich neurotoxic domain whereas other transcripts contain three repeats (genbank accession numbers JX467171 and JX467172). Intriguingly, sequence alignment reveals that this region is distinct from the domain that encodes the bradykinin potentiating peptides in the CNP precursor (Fig. 6).
Fig. 6.

Sequence alignment of 1. JX467171 Azemiops feae, JX467172, 2. Azemiops feae, 3.B0VXV8 Sistrurus catenatus edwardsii, 4. Q90Y12 Crotalus durissus terrificus, 5. Q2PE51 Crotalus durissus terrificus, 6. Q27J49 Lachesis muta, 7. P01021 Gloydius blomhoffii, 8. P68515 Bothrops insularis, 9. Q9PW56 Bothrops jararaca, 10. P0C7P5 Protobothrops flavoviridis.
To avoid confusion, previously obtained sequences are given with their UniProt accession numbers whereas ones obtained in this study are given with their Genbank accession numbers (Table S1).
DISCUSSION
Chelation of the Zn2+ ion present in proteases inhibits the proteolytic effect of these enzymes. Although it is unclear which domain of multidomain SVMP is responsible for the hemorrhagic effect of this toxin, chelation of the Zn2+ ion in the protease domain inhibits this activity. It is likely, therefore, that the protease domain of multidomain SVMP is responsible for the hemorrhaging often observed in envenomations by snakes with venoms rich in this toxin type (54, 55). Moreover, it is hypothesized that the presence of the cysteine-rich and disintegrin-like domains results in the increased potency of some forms of SVMP (P-III) in comparison with those that lack them (P-I and P-II). We show that more than half of the positively selected sites detected were confined to the protease domain whereas the remaining variations were shared between the cysteine-rich and disintegrin-like domains, suggesting that these domains play a prominent role in SVMP-induced inflammatory reactions. It has been shown in the past that venom components accumulate mutations in functional regions, on the molecular surface and in the tips of loops of the toxins while preserving the ancient scaffold that provides structural stability (56–59). Most venom novelties are derived from mutations of the protein surface residues, which not only increase the number target sites in the prey for these toxins but could also aid in avoiding the host immune response. We have shown that snake venom metalloproteases exhibit a similar phenomenon with 60% of all positively selected residues being confined to the molecular surface of the toxin whereas only 12% were buried (the remaining 29% could not be assigned into exposed or buried classes (Fig. 4). Moreover, 37 cysteine-residues remain unmodified, which is indicative of the importance of cysteine residues in stabilization of venom proteins (59).
P. mossambicus SVMP consist solely of the propeptide domains, with the additional domains typically found in SVMP having been lost from the ancestral scaffold. Our results demonstrate that the loss of these domains has exposed the propeptide domains to novel evolutionary selection pressures (Table I; Figs. 3 and 4). Importantly, as a logical consequence of the partial loss of weaponry, an increased selection pressure is applied, driving a rapid rate of mutations. Driven by positive selection, this has resulted in neofunctionalization of these venom components. We report here the genesis of a novel postsynaptic neurotoxic activity through inhibition of postsynaptic α7 nicotinic acetylcholine receptors by some of the Psammophis propeptide SVMP forms (uniprot accession numbers A7X4A6 and A7X461). Given the rapid rate of mutations and strong evolutionary selection pressures, the possibility of participation by the other Psammophis SVMP isoforms in envenoming through additional novel mechanisms cannot be ruled out. In contrast to Psammophis, E. coloratus expresses all the SVMP domains specialized for envenoming (protease, cysteine-rich, and disintegrin-like domains - i.e. P-III SVMP). Consequently, the propeptide region, which is post-translationally excised from the final toxin, exhibits no evidence of adaptive evolution (ω = 0.46), suggesting that it lacks a significant role in envenoming (Table I; Figs. 3 and 4). Some species of Echis not only selectively express the propeptide region of the SVMP but also have venom genes encoding highly lethal multidomain SVMP. Hence, they do not exhibit any variation in the propeptide-only toxins, which evolve under the influence of negative selection (ω = 0.52).
It has previously been hypothesized that snake toxins often consist of the smallest functional domain of a large multidomain protein, which acts as a provision for innovations (60). In contrast to this, we show that domains that contribute toward envenoming are still capable of accumulating rapid mutations under the influence of positive selection. Evaluation of selection pressures on different domains of Echis SVMP revealed that most of the mutations were directed toward the protease domain (∼54%; ω = 1.28), highlighting the importance of variation in this region believed to be responsible for inducing hemorrhage (Table II and Fig. 4). We speculate that such mutations may be important for sub-functionalization or increasing the potency of these proteins, or perhaps act as a weapon in the co-evolutionary arms race against prey resistance (61, 62). A major proportion of the remaining variations in Echis SVMP were detected in the cysteine-rich and disintegrin-like domains (∼29%, ω = 1.04 and 17%, ω = 0.91, respectively).
The fact that 60% of all positively selected sites (and associated variations) occur on the molecular surface whereas only 12% correspond to buried residues highlights the importance of changes in the surface chemistry (Fig. 4). Accumulation of surface mutations may facilitate interaction with novel targets and may also aid in avoidance of the host's immune response. The complete conservation of the 37 ancestral cysteine residues highlights the prominent role that disulfide bridges play in stabilizing and structurally scaffolding venom peptides and proteins.
Evidence provided by various analyses [Site-specific model 2a, model 3, model 8, Single Likelihood Ancestor Counting, Fixed-Effects Likelihood, Random-effects likelihood, MEM (Table II and supplemental Table II); Evolutionary fingerprint analyses (supplemental Fig. S1) and Mgene with option G test (Table II) highlights the strong influence of positive Darwinian selection in shaping the various domains of E. coloratus multidomain SVMP responsible for toxicity (protease, disintegrin-like and cysteine-rich). The propeptide domain, which is not known to play a significant role in envenoming, was subject to negative selection. This region is excised from the final protein as part of the post-translational modification and never forms part of the lethal multidomain SVMP toxin. In contrast, P. mossambicus selectively expresses the propeptide region because of the deletion of ancestral domains. Hence, this domain experiences significant selection pressure and evolves under the influence of positive selection (Fig. 3).
We show that positive selection has influenced the Psammophis monodomain SVMP propeptide domain more than its E. corolatus multidomain SVMP counterpart. A few species of Echis that express similar propeptide-only domains also express the regular multidomain SVMPs. Hence, they do not require the same level of variation as the Psammophis protein. Evidently, these Echis monodomain propetide-only SVMP seem to be subject to the same regime of negative selection as the propeptide domain expressed as part of the multidomain gene. Thus, the molecular evolution patterns and expression levels are consistent with a significant role being played by the propeptideonly toxins in the venom of P. mossambicus (extreme diversification and high expression) but not in Echis sp. (little diversification and low expression). This work reveals that the neglected proline-rich peptides are a source of novel ligands that, because of their uniqueness and small size, may prove to be of significant value in drug design and development.
Functional testing of P. mossambicus SVMP propeptide only domain variants Pm1 and Pm2 yielded responses on neuronal nAChRs, specifically on the human α7 neuronal receptor (Fig. 5). Alpha7 neuronal receptors, which are nAChRs comprised solely of α7 subunits, have been documented as being convergently targeted by toxins such as a-conotoxin PnIA (A10L D14K), which expresses a high affinity for the α7 neuronal receptor (63). Eleven subunits of mammalian neuronal nAChRs have been classified to date; eight a subunits (α2–7, α9, α10) and three β subunits (β2–4), with an additional a subunit; a8, present in the chick optic nerve (64). Similarities in receptors are apparent between species as it had been documented that neuronal nAChR subunits in the chicken, in particular α2, α3, and α4 subunits, express a homology of 85% in conserved domains when compared with that of a human or mouse (Nef et al., 1988). Despite the various similarities between chick and mammalian neuronal, and potentially neuromuscular, nAChR subtypes and their respective encoded genes, there are large disparities in the pharmacology of the expressed receptors between the two species (66), which may account for species-specific toxins. One such example is denmotoxin, an avian-specific post-synaptic neurotoxin isolated from Boiga dendrophila, which expresses potent activity at neuromuscular nAChRs in the chick but an effect of lower magnitude in the mouse (67). Dissimilar to neuronal nAChRs, neuromuscular nAChRs consist of five subunits comprising of α and β in addition to γ, δ, and ε subunits with a stoichiometry of (α1)2β1γδ in which the ε subunit replaces the γ subunit in developed forms of the receptor (68). Neuromuscular nAChRs have been documented as being targeted by α-neurotoxins, also referred to as curare-mimetic or post-synaptic neurotoxins (69), an example of which includes acantoxin IVa isolated from Acanthophis sp. seram (70). An absence of any activity of Pm1 and Pm2 at the muscle end plate nAChR in the chick biventer cervicis and activity on human α7 neuronal receptors confirms that both variants affect neuronal opposed to neuromuscular nicotinic acetylcholine receptors.
The two peptides tested in this study are good examples that, because of the extreme selection pressure they are subjected to, venom components often have exquisitely subtle sources of functional variation. Despite varying by only a single amino acid (Y for N at the second to last position) there was a slight but significant difference in potency between the two forms of Psammophis propeptide, with Y-containing Pm2 the more potent of the two. The peptides from Psammophis and Azemiops are a remarkable example of convergent evolution using two different gene types as the starting material (SVMP and CNP respectively). In both cases, there was the de novo evolution of proline-rich peptides in the propeptide domain of a precursor. Addtionally, there was a degree of functional convergence as both target the nicotinic acetylcholine receptor, although each targets a different subtype (neuronal and neuromuscular respectively). Both the variation between the two Psammophis peptides and the convergence of the Psammophis and Azemiops peptides reinforce the wealth of novel peptides to be found in understudied snake venoms.
Supplementary Material
Footnotes
* This work was supported by an Australian Research Council LIEF grant for the FLIPRTetra. NRC was supported by a NERC postdoctoral fellowship (NE/J018678/1). AB was supported by an NHMRC Program Grant. BGF was funded by the Australian Research Council and the University of Queensland. KS was funded by the PhD grant (SFRH/BD/61959/2009) from F.C.T (Fundação para a Ciência e a Tecnologia). IV was financially supported by a NHMRC postdoctoral fellowship (569918). RJL was funded by a NHMRC fellowship (APP1019761).
 This article contains supplemental Fig. S1 and Tables S1 and S2.
This article contains supplemental Fig. S1 and Tables S1 and S2.
1 The abbreviations used are:
- SVMP
- snake venom metalloproteinases
- PP
- posterior probability
- ASA
- accessible surface area
- TFA
- trifluoroacetic acid.
REFERENCES
- 1. Fry B. G., Wüster W. (2004) Assembling an arsenal: origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences. Mol. Biol. Evol. 21, 870–883 [DOI] [PubMed] [Google Scholar]
- 2. Fry B. G., Scheib H., van der Weerd L., Young B., McNaughtan J., Ryan Ramjan S. F., Vidal N., Poelmann R. E., Norman J. A. (2008) Evolution of an arsenal: Structural and functional diversification of the venom system in the advanced snakes (Caenophidia). Mol. Cell. Proteomics 7, 215–246 [DOI] [PubMed] [Google Scholar]
- 3. Weldon C. L., Mackessy S. P. (2012) Alsophinase, a new P-III metalloproteinase with alpha-fibrinogenolytic and hemorrhagic activity from the venom of the rear-fanged Puerto Rican Racer Alsophis portoricensis (Serpentes: Dipsadidae). Biochimie 94, 1189–1198 [DOI] [PubMed] [Google Scholar]
- 4. Peichoto M. E., Leme A. F., Pauletti B. A., Batista I. C., Mackessy S. P., Acosta O., Santoro M. L. (2010) Autolysis at the disintegrin domain of patagonfibrase, a metalloproteinase from Philodryas patagoniensis (Patagonia Green Racer; Dipsadidae) venom. Biochim. Biophys. Acta 1804, 1937–1942 [DOI] [PubMed] [Google Scholar]
- 5. Fry B. G., Scheib H., de L. M. J. d. A. I., Silva D. A., Casewell N. R. (2012) Novel transcripts in the maxillary venom glands of advanced snakes. Toxicon 59, 696–708 [DOI] [PubMed] [Google Scholar]
- 6. Hite L. A., Jia L. G., Bjarnason J. B., Fox J. W. (1994) cDNA sequences for four snake venom metalloproteinases: structure, classification, and their relationship to mammalian reproductive proteins. Arch. Biochem. Biophys. 308, 182–191 [DOI] [PubMed] [Google Scholar]
- 7. Casewell N. R., Wagstaff S. C., Harrison R. A., Renjifo C., Wüster W. (2011) Domain loss facilitates accelerated evolution and neofunctionalization of duplicate snake venom metalloproteinase toxin genes. Mol. Biol. Evol. 28, 2637–2649 [DOI] [PubMed] [Google Scholar]
- 8. Casewell N. R. (2012) On the ancestral recruitment of metalloproteinases into the venom of snakes. Toxicon . 60, 449–454 [DOI] [PubMed] [Google Scholar]
- 9. Fox J. W., Serrano S. M. T. (2008) Insights into and speculations about snake venom metalloproteinase (SVMP) synthesis, folding and disulfide bond formation and their contribution to venom complexity. FEBS J. 275, 3016–3030 [DOI] [PubMed] [Google Scholar]
- 10. Gutierrez J. M., Sanz L., Escolano J., Fernandez J., Lomonte B., Angulo Y., Rucavado A., Warrell D. A., Calvete J. J. (2008) Snake venomics of the Lesser Antillean pit vipers Bothrops caribbaeus and Bothrops lanceolatus: correlation with toxicological activities and immunoreactivity of a heterologous antivenom. J. Proteome Res. 7, 4396–4408 [DOI] [PubMed] [Google Scholar]
- 11. Casewell N. R., Harrison R. A., Wüster W., Wagstaff S. C. (2009) Comparative venom gland transcriptome surveys of the saw-scaled vipers (Viperidae: Echis) reveal substantial intra-family gene diversity and novel venom transcripts. BMC Genomics 10, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wagstaff S. C., Sanz L., Juarez P., Harrison R. A., Calvete J. J. (2009) Combined snake venomics and venom gland transcriptomic analysis of the ocellated carpet viper, Echis ocellatus. J. Proteomics 71, 609–623 [DOI] [PubMed] [Google Scholar]
- 13. Jiang Y., Li Y., Lee W., Xu X., Zhang Y., Zhao R., Wang W. (2011) Venom gland transcriptomes of two elapid snakes (Bungarus multicinctus and Naja atra) and evolution of toxin genes. BMC Genomics 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Petras D., Sanz L., Segura A., Herrera M., Villalta M., Solano D., Vargas M., Leon G., Warrell D. A., Theakston R. D., Harrison R. A., Durfa N., Nasidi A., Gutierrez J. M., Calvete J. J. (2011) Snake venomics of African spitting cobras: toxin composition and assessment of congeneric cross-reactivity of the pan-African EchiTAb-Plus-ICP antivenom by antivenomics and neutralization approaches. J. Proteome Res. 10, 1266–1280 [DOI] [PubMed] [Google Scholar]
- 15. Ching A. T., Paes Leme A. F., Zelanis A., Rocha M. M., Furtado Mde F., Silva D. A., Trugilho M. R., da Rocha S. L., Perales J., Ho P. L., Serrano S. M., Junqueira-de-Azevedo I. L. (2012) Venomics profiling of Thamnodynastes strigatus unveils matrix metalloproteinases and other novel proteins recruited to the toxin arsenal of rear-fanged snakes. J. Proteome Res. 11, 1152–1162 [DOI] [PubMed] [Google Scholar]
- 16. Fox J. W., Serrano S. M. T. (2005) Structural considerations of the snake venom metalloproteinases, key members of the M12 reprolysin family of metalloproteinases. Toxicon 45, 969–985 [DOI] [PubMed] [Google Scholar]
- 17. Fox J. W., Serrano S. M. (2005) Structural considerations of the snake venom metalloproteinases, key members of the M12 reprolysin family of metalloproteinases. Toxicon 45, 969–985 [DOI] [PubMed] [Google Scholar]
- 18. Moura-da-Silva A. M., Theakston R. D., Crampton J. M. (1996) Evolution of disintegrin cysteine-rich and mammalian matrix-degrading metalloproteinases: gene duplication and divergence of a common ancestor rather than convergent evolution. J. Mol. Evolution 43, 263–269 [DOI] [PubMed] [Google Scholar]
- 19. Juárez P., Comas I., González-Candelas F., Calvete J. J. (2008) Evolution of snake venom disintegrins by positive Darwinian selection. Mol. Biol. Evol. 25, 2391–2407 [DOI] [PubMed] [Google Scholar]
- 20. Okuda D., Koike H., Morita T. (2002) A new gene structure of the disintegrin family: a subunit of dimeric disintegrin has a short coding region. Biochemistry 41, 14248–14254 [DOI] [PubMed] [Google Scholar]
- 21. Segura A., Villalta M., Herrera M., Leon G., Harrison R., Durfa N., Nasidi A., Calvete J. J., Theakston R. D., Warrell D. A., Gutierrez J. M. (2010) Preclinical assessment of the efficacy of a new antivenom (EchiTAb-Plus-ICP) for the treatment of viper envenoming in sub-Saharan Africa. Toxicon 55, 369–374 [DOI] [PubMed] [Google Scholar]
- 22. Fry B. G., Wüster W., Ryan Ramjan S. F., Jackson T., Martelli P., Kini R. M. (2003) Analysis of Colubroidea snake venoms by liquid chromatography with mass spectrometry: evolutionary and toxinological implications. Rapid Commun Mass Spectrom 17, 2047–2062 [DOI] [PubMed] [Google Scholar]
- 23. Utkin Y. N., Weise C., Kasheverov I. E., Andreeva T. V., Kryukova E. V., Zhmak M. N., Starkov V. G., Hoang N. A., Bertrand D., Ramerstorfer J., Sieghart W., Thompson A. J., Lummis S. C., Tsetlin V. I. (2012) Azemiopsin from Azemiops feae viper venom, a novel polypeptide ligand of nicotinic acetylcholine receptor. J Biol Chem. 287(32):27079–27086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loytynoja A., Goldman N. (2005) An algorithm for progressive multiple alignment of sequences with insertions. Proc. Natl. Acad. Sci. U. S. A. 102, 10557–10562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Posada D., Crandall K. A. (2002) The effect of recombination on the accuracy of phylogeny estimation. J. Mol. Evol. 54, 396–402 [DOI] [PubMed] [Google Scholar]
- 26. Frost S. D., Liu Y., Pond S. L., Chappey C., Wrin T., Petropoulos C. J., Little S. J., Richman D. D. (2005) Characterization of human immunodeficiency virus type 1 (HIV-1) envelope variation and neutralizing antibody responses during transmission of HIV-1 subtype B. J. Virol. 79, 6523–6527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Delport W., Poon A. F., Frost S. D., Kosakovsky Pond S. L. (2010) Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26, 2455–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kosakovsky Pond S. L., Posada D., Gravenor M. B., Woelk C. H., Frost S. D. (2006) Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 23, 1891–1901 [DOI] [PubMed] [Google Scholar]
- 29. Posada D. (2008) jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256 [DOI] [PubMed] [Google Scholar]
- 30. Huelsenbeck J. P., Ronquist F. (2001) MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 [DOI] [PubMed] [Google Scholar]
- 31. Ronquist F., Huelsenbeck J. P. (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 [DOI] [PubMed] [Google Scholar]
- 32. Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 [DOI] [PubMed] [Google Scholar]
- 33. Goldman N., Yang Z. (1994) A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 11, 725–736 [DOI] [PubMed] [Google Scholar]
- 34. Yang Z. (1998) Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 15, 568–573 [DOI] [PubMed] [Google Scholar]
- 35. Yang Z. (2007) PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 [DOI] [PubMed] [Google Scholar]
- 36. Nielsen R., Yang Z. (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148, 929–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang Z., Wong W. S. W., Nielsen R. (2005) Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118 [DOI] [PubMed] [Google Scholar]
- 38. Kosakovsky Pond S. L., Frost S. D. W. (2005) Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 22, 1208–1222 [DOI] [PubMed] [Google Scholar]
- 39. Kosakovsky Pond S. L., Frost S. D. W., Muse S. V. (2005) HyPhy: Hypothesis testing using phylogenies. Bioinformatics 21, 676–679 [DOI] [PubMed] [Google Scholar]
- 40. Kosakovsky Pond S. L., Murrell B., Fourment M., Frost S. D., Delport W., Scheffler K. (2011) A random effects branch-site model for detecting episodic diversifying selection. Mol. Biol. Evol. 28, 3033–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pond S. L., Scheffler K., Gravenor M. B., Poon A. F., Frost S. D. (2010) Evolutionary fingerprinting of genes. Mol. Biol. Evol. 27, 520–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang Z. (1996) Maximum-Likelihood Models for Combined Analyses of Multiple Sequence Data. J. Mol. Evol. 42, 587–596 [DOI] [PubMed] [Google Scholar]
- 43. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 44. DeLano W. L. (2002) The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA [Google Scholar]
- 45. Fraczkiewicz R., Braun W. (1998) Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Computational Chem. 19, 319–333 [Google Scholar]
- 46. Armon A., Graur D., Ben-Tal N. (2001) ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol. 307, 447–463 [DOI] [PubMed] [Google Scholar]
- 47. Gotz S., Garcia-Gomez J. M., Terol J., Williams T. D., Nagaraj S. H., Nueda M. J., Robles M., Talon M., Dopazo J., Conesa A. (2008) High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Conesa A., Gotz S., Garcia-Gomez J. M., Terol J., Talon M., Robles M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 [DOI] [PubMed] [Google Scholar]
- 49. Conesa A., Gotz S. (2008) Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008, 619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gotz S., Arnold R., Sebastian-Leon P., Martin-Rodriguez S., Tischler P., Jehl M. A., Dopazo J., Rattei T., Conesa A. (2011) B2G-FAR, a species-centered GO annotation repository. Bioinformatics 27, 919–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vetter I., Lewis R. J. (2010) Characterization of endogenous calcium responses in neuronal cell lines. Biochem. Pharmacol. 79, 908–920 [DOI] [PubMed] [Google Scholar]
- 52. Vetter I., Mozar C. A., Durek T., Wingerd J. S., Alewood P. F., Christie M. J., Lewis R. J. (2012) Characterisation of Nav types endogenously expressed in human SH-SY5Y neuroblastoma cells. Biochem. Pharmacol. 83, 1562–1571 [DOI] [PubMed] [Google Scholar]
- 53. Dutertre S., Lewis R. J. (2010) Use of Venom Peptides to Probe Ion Channel Structure and Function. J. Biol. Chem. 285, 13315–13320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bjarnason J. B., Fox J. W. (1994) Hemorrhagic metalloproteinases from snake venoms. Pharmacol. Ther. 62, 325–372 [DOI] [PubMed] [Google Scholar]
- 55. Bjarnason J. B., Tu A. T. (1978) Hemorrhagic toxins from Western diamondback rattlesnake (Crotalus atrox) venom: isolation and characterization of five toxins and the role of zinc in hemorrhagic toxin e. Biochemistry 17, 3395–3404 [DOI] [PubMed] [Google Scholar]
- 56. Sunagar K., Johnson W. E., O'Brien S. J., Vasconcelos V., Antunes A. (2012) Evolution of CRISPs associated with toxicoferan-reptilian venom and mammalian reproduction. Mol. Biol. Evol. 29, 1807–1822 [DOI] [PubMed] [Google Scholar]
- 57. Kini R. M., Chan Y. M. (1999) Accelerated evolution and molecular surface of venom phospholipase A2 enzymes. J. Mol. Evol. 48, 125–132 [DOI] [PubMed] [Google Scholar]
- 58. Fry B. G., Wüster W., Kini R. M., Brusic V., Khan A., Venkataraman D., Rooney A. P. (2003) Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J. Mol. Evol. 57, 110–129 [DOI] [PubMed] [Google Scholar]
- 59. Fry B. G. (2005) From genome to “venome”: Molecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res. 15, 403–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Junqueira-de-Azevedo I. L., Ching A. T., Carvalho E., Faria F., Nishiyama M. Y., Jr., Ho P. L., Diniz M. R. (2006) Lachesis muta (Viperidae) cDNAs reveal diverging pit viper molecules and scaffolds typical of cobra (Elapidae) venoms: implications for snake toxin repertoire evolution. Genetics 173, 877–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Biardi J. E., Coss R. G. (2011) Rock squirrel (Spermophilus variegatus) blood sera affects proteolytic and hemolytic activities of rattlesnake venoms. Toxicon 57, 323–331 [DOI] [PubMed] [Google Scholar]
- 62. Heatwole H., Powell J. (1998) Resistance of eels (Gymnothorax) to the venom of sea kraits (Laticauda colubrina): a test of coevolution. Toxicon 36, 619–625 [DOI] [PubMed] [Google Scholar]
- 63. Celie P. H., Kasheverov I. E., Mordvintsev D. Y., Hogg R. C., van Nierop P., van Elk R., van Rossum-Fikkert S. E., Zhmak M. N., Bertrand D., Tsetlin V., Sixma T. K., Smit A. B. (2005) Crystal structure of nicotinic acetylcholine receptor homolog AChBP in complex with an alpha-conotoxin PnIA variant. Nat. Struct. Mol. Biol. 12, 582–588 [DOI] [PubMed] [Google Scholar]
- 64. Gotti C., Fornasari D., Clementi F. (1997) Human neuronal nicotinic receptors. Prog. Neurobiol. 53, 199–237 [DOI] [PubMed] [Google Scholar]
- 65. Nef P., Oneyser C., Alliod C., Couturier S., Ballivet M. (1988) Genes expressed in the brain define three distinct neuronal nicotinic acetylcholine receptors. EMBO J. 7, 595–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. McGehee D. S., Role L. W. (1995) Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu. Rev. Physiol. 57, 521–546 [DOI] [PubMed] [Google Scholar]
- 67. Pawlak J., Mackessy S. P., Fry B. G., Bhatia M., Mourier G., Fruchart-Gaillard C., Servent D., Ménez R., Stura E., Ménez A., Kini R. M. (2006) Denmotoxin, a three-finger toxin from the colubrid snake Boiga dendrophila (Mangrove Catsnake) with bird-specific activity. J. Biol. Chem. 281, 29030–29041 [DOI] [PubMed] [Google Scholar]
- 68. Boyd R. T. (1997) The molecular biology of neuronal nicotinic acetylcholine receptors. Crit. Rev. Toxicol. 27, 299–318 [DOI] [PubMed] [Google Scholar]
- 69. Kuruppu S., Smith A. I., Isbister G. K., Hodgson W. C. (2008) Neurotoxins from Australo-Papuan elapids: a biochemical and pharmacological perspective. Crit. Rev. Toxicol. 38, 73–86 [DOI] [PubMed] [Google Scholar]
- 70. Wickramaratna J. C., Fry B. G., Loiacono R. E., Aguilar M.-I., Alewood P. F., Hodgson W. C. (2004) Isolation and characterization at cholinergic nicotinic receptors of a neurotoxin from the venom of the Acanthophis sp. Seram death adder. Biochem. Pharmacol. 68, 383–394 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.