

Abstract
Mass spectrometry-based unbiased analysis of the full complement of secretory peptides is expected to facilitate the identification of unknown biologically active peptides. However, tandem MS sequencing of endogenous peptides in their native form has proven difficult because they show size heterogeneity and contain multiple internal basic residues, the characteristics not found in peptide fragments produced by in vitro digestion. Endogenous peptides remain largely unexplored by electron transfer dissociation (ETD), despite its widespread use in bottom-up proteomics. We used ETD, in comparison to collision induced dissociation (CID), to identify endogenous peptides derived from secretory granules of a human endocrine cell line. For mass accuracy, both MS and tandem MS were analyzed on an Orbitrap. CID and ETD, performed in different LC-MS runs, resulted in the identification of 795 and 569 unique peptides (ranging from 1000 to 15000 Da), respectively, with an overlap of 397. Peptides larger than 3000 Da accounted for 54% in CID and 46% in ETD identifications. Although numerically outperformed by CID, ETD provided more extensive fragmentation, leading to the identification of peptides that are not reached by CID. This advantage was demonstrated in identifying a new antimicrobial peptide from neurosecretory protein VGF (non-acronymic), VGF[554–577]-NH2, or in differentiating nearly isobaric peptides (mass difference less than 2 ppm) that arise from alternatively spliced exons of the gastrin-releasing peptide gene. CID and ETD complemented each other to add to our knowledge of the proteolytic processing sites of proteins implicated in the regulated secretory pathway. An advantage of the use of both fragmentation methods was also noted in localization of phosphorylation sites. These findings point to the utility of ETD mass spectrometry in the global study of endogenous peptides, or peptidomics.
Biologically active peptides, commonly known as peptide hormones and antimicrobial peptides, belong to a defined set of endogenous peptides that gain specialized functions not ascribed to original precursor proteins. For a precursor protein to generate such peptides, it must undergo specific cleavages and in some cases needs to be modified at specific sites (1). This limited cleavage, or proteolytic processing, represents an important cellular mechanism by which molecular diversity of proteins is increased at the post-translational level. In the postgenome era, it is being recognized that localization of processing sites in secretory proteins facilitates the identification of biologically active peptides. A standard approach to determining such sites is to use a panel of antibodies directed against different regions of a target protein (2). However, it is practically impossible to prepare antibodies that can thoroughly cover potential processing products arising from the precursor. Alternatively, mass spectrometry-assisted unbiased analysis of endogenous peptides may be a major step toward elucidating proteolytic processing (3).
In neurons and endocrine cells, a majority of biologically active peptides are released via the regulated secretory pathway. They are stored in secretory granules and await secretion until the cells receive an exocytotic stimulus. Owing to their compartmentalization, secretory peptides can be noninvasively recovered in culture supernatant. We have shown that a data set of endogenous peptide sequences that are collected by this procedure is applicable to infer processing sites, as well as to identify bona fide processing products (4). Rather than being digested, every endogenous peptide should be analyzed in its native form to understand how the peptide is generated and subsequently degraded. However, it remains a challenge to identify endogenous peptides because of size heterogeneity (ranging from 3 aa to 100 aa). For example, thyrotropin-releasing hormone is a small 3-aa peptide, human adrenomedullin occurs as a 52-aa peptide, and a 98-aa N-terminal propeptide from the atrial natriuretic peptide precursor is found in the circulation. Unlike digested protein fragments used in bottom-up proteomics, C termini of these endogenous peptides are not restricted to specific residues. Furthermore, proteolytic processing leads to the production of peptides containing multiple internal basic residues, for which collision induced dissociation (CID)1 shows limited performance (5).
A solution to address this issue in endogenous peptide sequencing might be the use of electron transfer dissociation (ETD) tandem mass spectrometry, which has been shown to provide a more complete series of fragment ions and hence a more confident sequence identification, along with the ability to leave labile post-translational modifications intact (6–10). The benefit of ETD in bottom-up proteomics has been increasingly documented, whereas endogenous peptides remain largely unexplored by ETD, despite the expectation that ETD would improve sequencing for larger peptides. In the few studies on endogenous peptides (11, 12), ETD did not cover large peptides exceeding 5000 Da. Because we have used CID to facilitate the discovery of previously unknown biologically active peptides (3, 13, 14), we were interested to see if ETD would be helpful to identify endogenous peptides that have escaped identification by CID. Here we conducted a large-scale identification of endogenous secretory peptides, ranging from 1000 to 15000 Da, using CID and ETD. We describe the merits of using ETD, in connection with CID, in peptidomics studies. The most significant finding is the identification of a previously unknown peptide, VGF[554–577]-NH2, which was sequenced solely by ETD. This peptide was found to have antimicrobial activity.
EXPERIMENTAL PROCEDURES
Sample Preparation
The human islet cell line QGP-1 (∼1 × 107 cells) (15) was stimulated with 10 μm carbachol plus 50 mm potassium chloride in serum-free Hank's balanced solution. Five minutes after stimulation, the medium was harvested to recover released peptides. The supernatant was quickly subjected to solid extraction as previously described (4). The eluate was separated by gel filtration high-performance liquid chromatography (HPLC) to five fractions containing peptides in the approximate Mr range 1000 to 15000.
Liquid Chromatography-Tandem Mass Spectrometry
All data were acquired on an LTQ Orbitrap XL instrument equipped with ETD (Thermo Fisher Scientific, San Jose, CA). A nanoFrontier HPLC system (Hitachi, Japan) was connected to the mass spectrometer for liquid chromatography-tandem mass spectrometry (LC-MS/MS). About 300 to 500 ng of the peptide mixture was loaded onto a trap column (C18, 75 μm × 100 mm) and separated on a MonoSpray C18 tip (GL Sciences) using a 60-min gradient from 10 to 50% acetonitrile in 0.1% formic acid at a flow rate of 200 nl/min. A protonated ion of polycyclodimethylsiloxane with m/z 445.120025 was used for internal calibration throughout. The mass spectrometer alternated between a full FT-MS scan (m/z 400–1500) and subsequent MS/MS scans. Scans were all recorded in the Orbitrap with a resolution of 100,000 at m/z 400. CID and ETD were performed on separate LC-MS runs. Cations were isolated with an isolation window of 5 m/z units and provided on a dynamic exclusion list for 2400 s after selected for at least two MS/MS scans. Singly charged precursors were excluded. Monoisotopic selection was disabled with an exclusion window setting to 1 Da. The four most intense ions were chosen for CID fragmentation. Automatic gain control was used to accumulate sufficient fragment ions (MS/MS target value, 2E5; maximum injection time, 1000 ms). For ETD, the two most intense precursor ions were subjected to MS/MS with an ETD activation time of 90 ms for doubly charged ions. Charge state-dependent activation time was applied. The ETD activation time was optimized for enhancing peptide identification by testing four different periods of time (70, 90, 150, and 200 ms) and also consistent with a previous report conducted on endogenous peptides using an ETD-enabled LTQ-XL (10). Supplemental activation was used for all MS/MS scans (16). ETD spectra were acquired using one microscan per spectrum. Automatic gain control was also used (MS/MS target value, 5E5; fluoranthene, 1E6; maximum fill time, 1500 ms). Samples were not subjected to reductive alkylation. However, an aliquot of the sample was reductive alkylated using dithiothreitol and iodoacetamide to identify peptides derived from chromogranin B (CgB), whose N-terminal region contains two cysteine residues forming a disulfide bond.
Data Analysis and Peptide Identification
Peak picking, deisotoping, and deconvolution of MS/MS spectra were conducted using Mascot Distiller (version 2.2.3) with the default parameters for Orbitrap. Peak lists were searched against NCBI nr human (14,987,464 sequences; 5,132,678,026 residues, as of August 13, 2011) using Mascot (version 2.2.3) with no enzyme specification. Pyroglutamination, C-terminal amidation, N-terminal acetylation, and methionine oxidation were simultaneously allowed as variable modifications. Data were searched with a precursor mass tolerance of 5 ppm and product ion mass tolerance of 50 mmu. Peptides were considered identified with a Mascot expect value of less than 0.05. The false discovery rate for peptide matches above Mascot identity threshold using a decoy database search was 0% in all cases. The highest scored MS/MS spectrum was used to report the score of the unique peptide in supplemental Table S2. The percent fragmentation was calculated as reported (8), with N-terminal proline cleavage also included as a possible fragmentation channel. In Mascot notation, the even-electron c ion corresponds to the b ion plus NH3. Mascot does not try to match radical c-1 ions, which are usually much weaker. The charged radical z+1 ion is the y ion minus NH2. Hence, the most abundant z-type ions observed in ETD are radical z+1 and even-electron z+2. For calculating percent fragmentation, z-type ions were counted only once if both z+1 and z+2 ions were assigned by Mascot for a given N-Cα bond.
Peptide Synthesis
Peptides were synthesized by the solid phase method (Sigma Genosys, Japan) using Fmoc (N-(9-fluorenyl) methoxycarbonyl) strategy, purified by reverse phase HPLC, and verified for correct synthesis by MS and amino acid analysis. Peptide purity was confirmed on separate HPLC systems.
Antibody Preparation and Mass Spectrometric Characterization of Immunoreactivity
Cysteinyl C-terminal 13-residue peptide of human VGF[554–577]-NH2 (CHYHHALPPSRHYP-NH2) was conjugated with maleimide-activated keyhole limpet hemocyanin (Thermo Fisher Scientific). Rabbits were immunized every 3 weeks with the conjugate emulsified with Freund's complete adjuvant. Antiserum (#529–6) was characterized in a radioimmunoassay system (dilution 1/900,000, IC50 = 8 fmol/tube), which completely crossreacted with VGF[554–577]-NH2 but showed no crossreactivity with VGF[554–578] or VGF[554–583]. Furthermore, ten C-terminally amidated peptides (human calcitonin gene-related peptide[23–37]-NH2, pig neuromedin U-8, neurokinin A, vasopressin, human adrenomedullin[22–52]-NH2, human calcitonin, pig peptide histidine isoleucine, ovine corticotropin-releasing factor, proadrenomedullin N-terminal 20-amino acid peptide, calcitonin receptor stimulating peptide-1[24–28]-NH2) showed less than 0.01% cross reactivity up to 100 pmol/tube. Cell culture supernatant was immunorecipitated with the antibody and analyzed on a surface-enhanced laser desorption and ionization mass spectrometer as described (17).
Antibacterial and Antifungal Activity Assay
Each test peptide (up to 10 μm) was assessed with the target microbes Enterococcus hirae (E. hirae), Micrococcus luteus (M. luteus), Staphylococcus aureus (S. aureus) 209P, S. saprophyticus KD, Escherichia coli (E. coli) B, E. coli K12, E. coli kp, and Pichia pastoris (P. pastoris) GS115 using AlamarBlueTM (BioSource International, Camarillo, CA) (14, 18). The optimal growth temperature was 30 °C for M. luteus, S. saprophyticus KD and P. pastoris GS115 and 37 °C for the other microbes. After growth in 3% tryptosoy broth (Eiken Chemical, Tokyo, Japan) for 16 h with shaking at each optimal temperature, cells were washed twice with 10 mm phosphate buffer, pH 7.0, and diluted to 8 × 105 colony-forming units/ml in the same buffer. Twenty-five microliters of bacterial suspension were mixed with an equal volume of sample in the absence or presence of peptides, and incubated for 1 h. After incubation, 200 μl of 3% tryptosoy broth containing 10% alamarBlueTM was added to the reaction mixture and further incubated for the period of time shown in parentheses: E. hirae, S. aureus 209P and E. coli B (4 h), E. coli K12 and E. coli kp (6 h), M. luteus (7 h), S. saprophyticus KD (7.5 h), and P. pastoris GS115 (20 h). Aliquots containing all assay reagents without microbes were used as blank. After incubation, the reactions were monitored by absorbance at 569 and 600 nm. Molar extinction coefficients of OD569 and OD600 in the oxidative condition are 80586 and 117216. Viability (%) was expressed using the following formula: viability (%) = (117216 × OD569 − 80586 × OD600 with peptide/117216 × OD569 − 80586 × OD600 without peptide) × 100. The classical colony formation assay was performed on M. luteus, E. coli K12 and P. pastoris GS115 as described (18). Cathelicidin and β-defensin-2 (Peptide Institute, Osaka, Japan) were used as control.
RESULTS
We studied a complex mixture of peptides secreted by cultured human endocrine cells that received an exocytotic stimulus. The culture supernatant was harvested 5 mins after stimulation and separated by gel filtration HPLC to five fractions containing peptides in the approximate Mr range 1000 to 15000. LC-MS/MS was performed in duplicate for each fraction on an LTQ-Orbitrap equipped with ETD. For comparison, the same sample was independently analyzed by CID in duplicate runs. Both MS and MS/MS scans were recorded with Orbitrap to ensure sequencing of highly charged peptide ions (z>4). The high mass accuracy and resolution enabled by Orbitrap allowed searching the human NCBI nr database with no enzyme specification and acquire significant data, and despite this broad search space the false discovery rate for peptide matches above the identity threshold using a Mascot automatic decoy database search was 0%.
In a total of 20 runs, 2311 and 1733 peptides (including redundant identifications) were identified by CID and ETD, respectively. In Table I, these peptides were sorted according to precursor charge states and fragmentation methods. No peptide was identified using ETD of doubly charged ions although supplemental activation was applied. Aside from this, there was no apparent difference in charge distribution between CID and ETD. More than two-thirds of the peptides were quadruply or more highly charged (69% for CID and 76% for ETD). This charge distribution is skewed, relative to that commonly observed with digested peptides used in bottom-up proteomics, most of which are doubly or triply charged ions. To assess the degree of peptide dissociation, we calculated the percent fragmentation values, defined by Coon et al. (8) as the number of observed fragment ions (b- and y-type ions for CID and c- and z-type ions for ETD), for a peptide sequence, divided by the total theoretically possible number of fragment ions. The percent fragmentation was plotted as a function of m/z values of the identified precursors (Fig. 1, supplemental Table S1). In CID, charge states and precursor m/z values showed little influence on percent fragmentation. Consistent with the previous study (8, 10), ETD percent fragmentation was decreased as precursor ion m/z increases regardless of charge state. Although surpassed by CID in the overall number of identification, ETD provided more extensive fragmentation; nearly 12% of the ETD-identified peptides have percent fragmentation values greater than 75, as compared with 2% of the CID-identified peptides.
Table I. Total peptide identifications sorted by fragmentation method and precursor charge state.
| Precursor charge state | CID | ETD |
|---|---|---|
| +2 | 252 | 0 |
| +3 | 454 | 417 |
| +4 | 451 | 449 |
| +5 | 431 | 300 |
| +6 | 398 | 299 |
| +7 | 220 | 146 |
| +8 | 77 | 60 |
| +9 | 14 | 16 |
| +10 | 6 | 15 |
| +11 | 4 | 23 |
| +12 | 3 | 7 |
| +13 | 1 | 0 |
| +14 | 0 | 1 |
| Total | 2311 | 1733 |
Fig. 1.

Percent fragmentation, defined as the number of observed fragment ions divided by the theoretical number of fragment ions, plotted against precursor m/z. Plotted data are from 2311 CID-identified and 1733 ETD-identified peptides.
After removing redundant identifications caused by either peptide overlaps between two adjacent gel filtration fractions or ionization of the same peptide into different charge states, we concluded that CID and ETD elucidated 795 and 569 unique peptides, respectively, with the overlap being 397 (Fig. 2, supplemental Table S2). Peptides larger than 3000 Da accounted for 54 and 46% in CID and ETD identifications, respectively. Notably, nearly 96% of the peptides arose from secretory precursor proteins. Judged by the number of peptides, our analyte appeared to contain relatively large amounts of VGF and chromogranin B (CgB) (supplemental Table S2). They contain multiple pairs of basic residues, some of which have been described as cleavage sites for processing to biologically active peptides (2, 19).
Fig. 2.
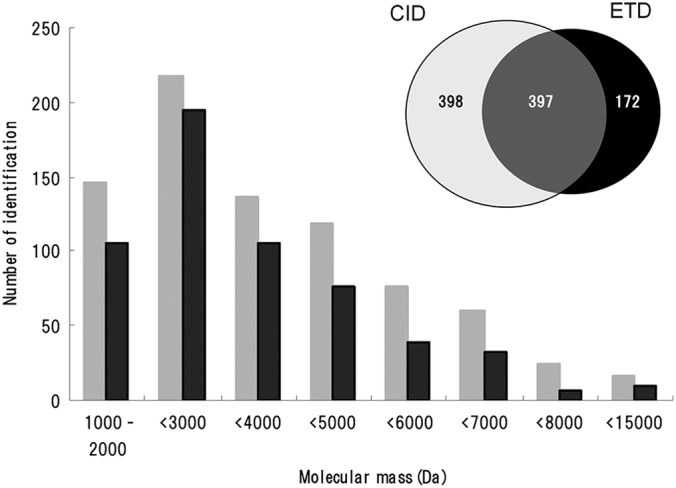
Mass distribution of CID- and ETD-identified peptides. Only unique peptides were counted. (Inset) Venn diagram illustrating the number of unique peptides identified by CID and ETD.
The identified peptides derived from VGF and CgB were aligned to each precursor sequence and illustrated in Fig. 3. They are shown by gray (CID) and black (ETD) lines and stacked to highlight potential processing sites. Multiple lines of the same length indicate redundant identifications. By inspecting the cleavage sites revealed by these lines, we inferred primary processing sites that involve at least one basic residue (dotted lines, Fig. 3). The VGF sequence was almost completely covered by clusters of peptides sharing N termini or C termini, whereas CgB showed less coverage. No peptide was ascribed to the signal sequence. Previously, a limited number of VGF-derived peptides were reported in peptidomics work on human cerebrospinal fluid using CID (20–22). We conducted a more detailed study by analyzing the culture supernatant from a human endocrine cell line and tissue extracts in normal rat brain and gut (3, 4, 23). In earlier work, several CgB-derived peptides have been isolated from extracts of human pheochromocytoma and neuroendocrine tumors (24–26). The specific processing sites that have been proposed by those studies are indicated in Fig. 3. Although differences in species and tissue sources should be taken into account, these sites extensively overlap the potential processing sites revealed by the present study.
Fig. 3.

Identified peptides derived from VGF (GI: 17136078) (A) and CgB/secretogranin-1 (GI: 36439) (B). Gray lines, CID-identified peptides; black lines, ETD-identified peptides. Red boxes, pyroglutamination; yellow boxes, C-terminal amidation. Note that the maps include redundant identifications, excluding oxidized methionine-containing peptides. Processing sites proposed by previous studies in human VGF (20–22) and human CgB (2, 24–26) are indicated by open arrows above the precursor sequence. In (A), arrows indicate processing sites identified in rat brain studies (13, 23). Asterisks indicate processing sites proposed in our previous peptidomics study of a different human endocrine cell line (3). Processing sites inferred from the present study are shown in vertical dotted lines. Magenta bars, consecutive basic residues. Pale magenta bars, single basic residues.
On close inspection some peptides were identified solely by ETD, an example of which is VGF[554–577]-NH2 (Fig. 3, supplemental Table S2). This C-terminally amidated peptide underwent extensive fragmentation at every possible N-Cα bond except the Pro-Ser (positions 572–573), whereas CID showed poor fragmentation (Fig. 4). The lack of ETD cleavage N-terminal to proline is consistent with the previous work on proline-containing synthetic peptides (7). In triplicate analyses, this peptide, detected as quadruply and pentuply charged ions, had a percent fragmentation of 54 to 61% and 56 to 63%, respectively. It might be noteworthy that N-terminally truncated 13- and 14-residue peptides sharing the C-terminal amide structure were solely identified by CID. Antibody specifically recognizing the amide structure was generated to characterize major peptide forms. Major peptides immunoprecipitated with the antibody from the culture supernatant had the molecular masses corresponding to the theoretical masses of VGF[554–577]-NH2 and [565–577]-NH2 (supplemental Fig. S1). Similar results were obtained with the supernatant from the human medullary thyroid carcinoma cell line TT (data not shown). We thus concluded that these two peptides represent major processing products.
Fig. 4.

Deconvoluted CID (A) and ETD (B) MS/MS spectra of pentuply charged VGF[554–577]-NH2.
The three consecutive arginine residues in VGF[554–577]-NH2 prompted us to test antimicrobial activity, along with the related peptides (Table II). VGF[554–583], identified in the present study, was tested because it was C-terminally flanked by the dibasic residues that correspond to the N-terminal processing site of VGF[586–615]/AQEE-30 (Fig. 3a). VGF[554–577]-NH2 showed antimicrobial activity against M. luteus and P. pastoris, comparable to the established antimicrobial peptides β-defensin-2 and cathelicidin, whereas C-terminally extended VGF[554–578] and [554–583] were less potent. Up to the concentrations tested, VGF[565–577]-NH2 was not active against any of the test microbes (Table II and Fig. 5). To examine whether VGF[554–577]-NH2 is bactericidal or just bacteriostatic, we performed a classical colony formation assay. It has been established that the bactericidal peptide concentration revealed by classical colony formation assays and the alamarBlueTM assay shows a good agreement (17). The peptide showed strong antimicrobial activity in the colony assay with IC50 of 1.9 and 1.4 μm against M. luteus and P. pastoris, respectively (data not shown), indicating that it is bactericidal.
Table II. Antimicrobial activity assayed by the alamarBlue method. NI, no inhibition up to 10 μm.
| Peptide | M. luteus | E. coli K12 | P. pastoris GS115 |
|---|---|---|---|
| VGF[554–577]-NH2 | 1.8 | NI | 4.5 |
| VGF[554–578] | 5.6 | NI | NI |
| VGF[554–583] | 5.8 | NI | NI |
| VGF[565–577]-NH2 | NI | NI | NI |
| β-Defensin-2 | 0.7 | 5.6 | 2.6 |
| Cathelicidin | 1.3 | 0.6 | 3.1 |
Fig. 5.

Antimicrobial activity of VGF[554–577]-NH2 and related peptides against P. pastoris GS115. VGF[554–577]-NH2 (closed circle), [554–583] (closed triangle), [565–577]-NH2 (open circle) and [554–578] (open triangle) were assayed using alamarBlue.
It remains a challenge to pinpoint a post-translationally modified site among multiple candidate sites in a sequence, because informative fragment ions for unequivocal localization are not always observed in MS/MS spectra. A total of 13 phosphopeptides were identified using CID and ETD without any phosphopeptide enrichment procedure (Table III). As a consequence, five unique modified sites were identified from six peptides, of which Ser130 of CgB identified in 4684.98-Da and 5067.25-Da peptides and Ser64 of the calcitonin precursor identified in 6297.01-Da peptide have not previously been described. Two phosphorylation sites in CgB, Ser335 of 6109.65-Da and Ser617 of 7049.27-Da peptides, were previously described (27), but represent fortuitous cases where CID, rather than ETD, allowed successful identification. Ser405 of the CgB 3730.65-Da peptide, previously determined as a phosphorylation site through conventional techniques (26), was confidently reconfirmed by ETD (supplemental Fig. S2). We got evidence that the VGF- and secretogranin II-derived peptides are phosphorylated, but failed to localize modified sites.
Table III. Endogenous phosphopeptide identification.
Mascot scores of first to third hits (columns 6 to 8) are accompanied by expectation values shown in parentheses. N-term, N-terminal flanking amino acid (“Signal” indicates that the peptide is flanked by signal sequence); C-term, C-terminal flanking amino acid. Identified phosphorylation sites are marked in bold and underlined. In the last column, the asterisk indicates that either of the two N-terminal serine residues is phosphorylated but not uniquely determined. CT, calcitonin; SgII, secretogranin II.
| Method | Precursor | m/z (obsd.) | z | Mr (calc.) (Da) | 1st Hit | 2nd Hit | 3rd Hit | N-term | Sequence | C-term | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Unequivocal localization | |||||||||||
| CID | CgB | 1175.887 | 6 | 7049.271 | 94 (1.9E-05) | 49 (0.6) | 28 (77) | K | SAEFPDFYDSEEPVSTHQEAENEKDRADQTVLTEDEKKELENLAAMDLELQKIAEKFSQR-NH2 | G | (27) |
| CID | CgB | 1222.937 | 5 | 6109.646 | 60 (3.6E-02) | 9 (4.5E+03) | 6 (8.0E+03) | R | ASEEEPEYGEEIKGYPGVQAPEDLEWERYRGRGSEEYRAPRPQSEESWDEE | D | (27) |
| CID | CgB | 845.549 | 6 | 5067.250 | 82 (2.2E-04) | 45 (1) | 35 (10) | R | LLRDPADASEAHESSSRGEAGAPGEEDIQGPTKADTEKWAEGGGHSRE | R | |
| ETD | CgB | 938.004 | 5 | 4684.981 | 109 (2.9E-07) | 60 (0.023) | 39 (3.1) | R | DPADASEAHESSSRGEAGAPGEEDIQGPTKADTEKWAEGGGHSRE | R | |
| ETD | CgB | 747.137 | 5 | 3730.647 | 136 (6.5E-10) | 12 (1.6E+03) | 4 (8.7E+03) | R | NYPSLELDKMAHGYGEESEEERGLEPGKGRHH | R | (26) |
| ETD | CT | 1260.409 | 5 | 6297.006 | 65 (0.016) | 42 (3.1) | 41 (3.8) | Signal | APFRSALESSPADPATLSEDEARLLLAALVQDYVQMKASELEQEQEREGSSLDSPRS | K | |
| Ambiguous localization | |||||||||||
| ETD | CgB | 845.006 | 5 | 4219.991 | 139 (4.3E-10) | 138 (4.4E-10) | 67 (0.0063) | R | GEDSSEEKHLEEPGETQNAFLNERKQASAIKKEELVA | R | (27) * |
| CID | SgII | 1138.077 | 7 | 7959.485 | 78 (0.00075) | 71 (0.003) | 71 (0.003) | R | ERMDEEQKLYTDDEDDIYKANNIAYEDVVGGEDWNPVEEKIESQTQEEVRDSKENIEKNEQINDEM | K | |
| CID | SgII | 1280.066 | 6 | 7674.341 | 103 (2.5E-06) | 95 (1.5E-05) | 95 (1.5E-05) | R | MDEEQKLYTDDEDDIYKANNIAYEDVVGGEDWNPVEEKIESQTQEEVRDSKENIEKNEQINDEM | K | |
| ETD | SgII | 1030.079 | 5 | 5145.363 | 70 (0.0038) | 70 (0.0038) | 63 (0.018) | R | ALEYIENLRQQAHKEESSPDYNPYQGVSVPLQQKENGDESHLPE | R | |
| CID | VGF | 1117.343 | 6 | 6698.003 | 168 (7.8E-13) | 168 (7.8E-13) | 168 (7.8E-13) | R | SQEETPGHRRKEAEGTEEGGEEEDDEEMDPQTIDSLIELSTKLHLPADDVVSIIEEVEE | K | |
| ETD | VGF | 807.197 | 5 | 4030.954 | 115 (9.2E-08) | 105 (8.1E-07) | 0.5 (2.5E+04) | Signal | APPGRPEAQPPPLSSEHKEPVAGDAVPGPKDGSAPEVRGA | R | |
| ETD | VGF | 937.707 | 4 | 3746.794 | 91 (2.3E-05) | 77 (0.0005) | 6 (7.0E+03) | Signal | APPGRPEAQPPPLSSEHKEPVAGDAVPGPKDGSAPEV | R |
Except for phosphopeptides, the unique peptide sequences listed in supplemental Table S2 excluded any peptide for which the second-ranked sequence also has a Mascot expectation value of 0.05 or lower. However, we encountered one exceptional case; CID of a positively charged ion (+6) with m/z 808.24 resulted in the identification of the amidated peptide from gastrin-releasing peptide (GRP) isoform 1 preproprotein[95–139]-NH2 (calculated monoisotopic mass 4843.377), with a Mascot score of 120 (expt. 3.5E-08). Ranked second was the peptide from GRP isoform 2 preproprotein[95–138] (calculated monoisotopic mass 4843.369), with a score of 96 close to the top (expt. 9.1E-06). The two peptides share N-terminal 27 residues but have alternatively spliced C-terminal halves (Fig. 6). The fragment ions produced by CID were preferentially observed the N-terminal region so that distinguishing these two possibilities was difficult. In ETD, the first rank was the peptide from GRP isoform 1 (score 289, expt. 4.5E-25) and the second was from GRP isoform 2 (score 82, expt. 0.00022). Although both hits still exceed a stringent threshold (expectation < 0.001, rather than a default value of 0.05), we concluded that this peptide arises from the isoform 1 because of the extensive matching to theoretical ions (Fig. 6). It might be worth mentioning that GRP isoform 1 preproprotein[54–139]-NH2 (calculated monoisotopic mass 9514.789), which is N-terminally adjacent to the amidation motif of neuromedin C, was also identified by CID (supplemental Fig. S2).
Fig. 6.

Deconvoluted CID and ETD MS/MS spectra of sextuply charged m/z 808.24. Alternatively spliced exons of the GRP gene produce two nearly isobaric peptides. Residues different from the top peptide are underlined in the lower peptide. Note that the second best matches for both fragmentations are below a Mascot expect value of 0.05, but that ETD showed a wider difference between the best and second best matches because of extensive fragmentations across the sequence.
DISCUSSION
We have shown that mass spectrometric analysis of endogenous secretory peptides provides a basis for identifying biologically active peptides (3, 13, 14). In the present study, our immediate interest is to uncover endogenous peptides that escape identification by CID. Endogenous peptides remain largely unexplored by ETD or electron capture dissociation, whose performance for such peptides has not been thoroughly characterized. ETD was used on a low-end ion trap mass spectrometer to characterize venom peptides extracted from a marine snail (28), resulting in a fraction of small peptides (less than 3000 Da) being sequenced. Shen et al. recently used the same instrumentation as ours to improve endogenous peptide identification, but identified peptides came from doubly to quadruply charged ions in most cases (12). Thus little is known at present about the ability of ETD to dissociate endogenous peptides with higher charge states, despite the occurrence of large endogenous peptides (>5000 Da) in larger amounts than previously thought.
It has been generally accepted that ETD is more effective than CID for sequencing highly charged precursors or larger peptides in bottom-up proteomics (6–9). However, our peptidomic data indicate that CID was as potent as ETD in sequencing precursors having charges greater than three, with their proportions being 69% for CID and 76% for ETD. Furthermore, CID appears to be superior to ETD in total identification; 795 peptides for CID versus 569 peptides for ETD. For peptides larger than 7000 Da, CID uncovered a larger number of peptides than ETD (39 for CID and 15 for ETD). Good et al. reported that a linear decrease in percent fragmentation as a function of increasing precursor m/z was noted for ETD, with few peptides having precursor m/z values exceeding 850 identified (8). We also observed a similar but less remarkable decline; in our study peptides identified with precursors greater than m/z 850 still accounted for 30% (507 of 1733) in the total ETD identification. This difference in ETD performance may be partly explained by the fact that most of our secretopeptidome components have more internal basic residues than in vitro-digested proteins. The current limitation in ETD was particularly noted with precursors having higher m/z values (m/z >1300). Supplemental Fig. S3 shows CID and ETD of two pentuply charged precursors with m/z 1324.6 and 1411.7. They were unequivocally identified by CID as 6618.0-Da VGF-derived peptide and 7053.5-Da N-terminal propeptide of the somatostatin precursor, whereas the corresponding ETD spectra were so dominated by charge reduced species that no significant match was obtained.
We provided the most comprehensive data about VGF and CgB processing. Both fragmentation methods complemented each other to highlight potential cleavage sites more clearly than that provided by either alone. Because they represent major components in neuronal secretory vesicles and several peptides identified here have also been described as neuropeptides (2, 19, 29), our findings will contribute to neuropeptide profiling in neuropeptidomics. The VGF processing sites predicted here are consistent with the antibody-assisted biochemical characterization of major processing products in rat brain (23). For instance, the 7400 Da peptide (also known as TLQP-62 (29)) in the rat study corresponds to human VGF[554–615] (calculated monoisotopic mass 7498.98), and rat 15166 Da and 7047 Da peptides recognized by NERP-2 antibody correspond to human VGF[208–347]-NH2 and VGF[281–347]-NH2 (calculated monoisotopic mass 14832.74 and 7108.77), respectively.
CgB-derived peptides have been isolated from human pituitary and pheochromocytoma tissues without reference to biological activity or immunoreactivity (2, 24–26), including CgB[217–275], [293–323], [326–385], [334–385], [388–437], [440–513](GAWK), [518–537], [575–585](PE-11), [588–597], [600–613](BAM-1745), and [617–676](CCB). All the peptides but CgB[326–385] were identified in the present study. Flanked by either pairs or groups of basic amino acids, they are considered specific processing products. Despite the presence of potential cleavage sites across the sequence, no peptide has been described in the literature for the N-terminal 216-amino acid region. Our data revealed that the precursor undergoes extensive processing across the entire sequence, including previously reported processing sites (Fig. 3B). The reason for the lack of peptide identification for positions 540–572 remains to be elucidated.
Some peptides were solely identified by ETD, including CgB-derived GAWK of 8788.13 Da (24) and VGF[554–577]-NH2. This C-terminally amidated peptide had escaped MS/MS identification because of poor CID fragmentation in our previous peptidomic survey (3). In the present study, ETD was able to provide an almost complete series of ions to ensure confident identification; it has five proline residues, where cleavage N-terminal to proline is missing in ETD (7). Considering this proline effect, the peptide has 18 theoretically cleavable bonds rather than 23. As shown in Fig. 4, 17 of the 18 bonds were cleaved. Of note, related N-terminally truncated peptides were not revealed by ETD, demonstrating the advantage of using different dissociation techniques. The ability of ETD to cleave peptide backbone across the entire sequence was also demonstrated in distinguishing between the peptides derived from two different isoforms of the neuropeptide GRP gene, again reflecting the merits of ETD in peptidomics. The high mass accuracy and resolution attained by Orbitrap enables discrimination between z+1 and z+2 ions, ensuring in some cases that z+2 matches in Mascot are not 13C isotope peaks of z+1 matches.
In rat brain, TLQP-62/rat VGF[556–617] was originally described as a major peptide recognized by antibody against the protein C terminus (29). The N-terminal 556Thr is 554Thr in human VGF. An appreciable number of the peptides starting with 554Thr were revealed by ETD, including VGF[554–577]-NH2, [554–583] and [554–615] (Fig. 3A). Given the sequence identity between human and rat, the cleavage site (PR↓TLQP) may represent a major processing site also in human. These peptides have not been described in peptidomic reports of VGF-derived peptides (3, 20–22). On the other hand, the C-terminal cleavage site has recently been described through MS/MS identification of VGF[565–577]-NH2 (3). Because 574RHYPGR579 agrees with the consensus motif for prohormone convertase (PC) 1/3 or 2 ((K/R)-(X)n -(K/R)↓, where n = 0, 2, 4, or 6 and X is any amino acid other than Cys) (30) and is nested by a canonical amidation signal of 578GR579, generation of the amidated peptides can be explained by the general rule of proteolytic processing in the regulated secretory pathway (30). This is also supported by the fact that the complete set of enzymes involved in C-terminal amidation, PC1/3 or 2, carboxypeptidase E, and peptidylglycine α-amidating monooxygenase were expressed by the cell line used (not shown).
VGF[554–577]-NH2 showed antimicrobial activity against M. luteus and P. pastoris, comparable to major antimicrobial peptides β-defensin-2 and cathelicidin. The N-terminal 11 residues are considered essential for activity, because N-terminally truncated VGF[565–577]-NH2 was inactive. In addition, the decreased activity observed with the two C-terminally extended peptides suggests that the C-terminal amide structure is important as well. It is known that amidated C termini, rather than common carboxyl termini, help to enhance electrostatic interactions between the peptide and a target bacterial membrane (31). This may also be the case with this peptide. The amidation motif is not found in rodent VGF sequences, but shared by primates (supplemental Fig. S1C), suggesting that the peptide is assigned some biological role unique to primates.
We were able to identify phosphopeptides of intermediate size (3500 to 8000 Da). Two phosphorylation sites, not described in the literature, were unequivocally localized. Of note, all but one (6109.65-Da peptide) of the 13 peptides were both N-terminally and C-terminally flanked by previously reported signal sequence cleavage sites or PC1/3 or PC2 consensus sites, strongly suggesting that they are bona fide secretory phosphopeptides. Although not explored, phosphorylated peptides quickly released via exocytosis may have some functional roles in the nervous and endocrine systems. Because larger peptides lead to less confident identification of modification sites than smaller peptides, ETD parameters should be further optimized. We conclude that CID and ETD complement each other to advance our understanding of endogenous peptides, which has received limited attention in the proteomics community.
Supplementary Material
Acknowledgments
We thank Professor Toshifumi Takao (Osaka University, Japan) for his advice on peptide identification by LC-MS/MS. We also thank Junko Kimata, Makoto Takahata, and Morihiko Yoshida (Thermo Fisher Scientific, Japan) for their technical assistance in ETD studies, Itaru Usami and John Cottrell (Matrix Science, United Kingdom) for their help with Mascot, and Masako Matsubara (National Cerebral and Cardiovascular Center) for data analysis.
Footnotes
* This study was supported in part by the Intramural Research Fund of the National Cerebral and Cardiovascular Center, a Health Labor Sciences Research Grant from The Ministry of Health Labor and Welfare, and a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science.
 This article contains supplemental Tables S1 and S2 and Figs. S1 to S3.
This article contains supplemental Tables S1 and S2 and Figs. S1 to S3.
1 The abbreviations used are:
- CID
- collision induced dissociation
- CgB
- chromogranin B
- ETD
- electron transfer dissociation
- GRP
- gastrin-releasing peptide
- PC
- prohormone convertase.
REFERENCES
- 1. Fricker L. D. (2005) Neuropeptide-processing enzymes: applications for drug discovery. AAPS J. 7, E449–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stridsberg M., Eriksson B., Oberg K., Janson E. T. (2005) A panel of 13 region-specific radioimmunoassays for measurements of human chromogranin B. Regul. Pept. 125, 193–199 [DOI] [PubMed] [Google Scholar]
- 3. Sasaki K., Takahashi N., Satoh M., Yamasaki M., Minamino N. (2010) A peptidomics strategy for discovering endogenous bioactive peptides. J. Proteome Res. 9, 5047–5052 [DOI] [PubMed] [Google Scholar]
- 4. Sasaki K., Satomi Y., Takao T., Minamino N. (2009) Snapshot peptidomics of the regulated secretory pathway. Mol. Cell. Proteomics 8, 1638–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tabb D. L., Huang Y., Wysocki V. H., Yates J. R., 3rd (2004) Influence of basic residue content on fragmentation ion peak intensities in low-energy collision-induced dissociation spectra of peptides. Anal. Chem. 76, 1243–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Syka J. E., Coon J. J., Schroeder M. J., Shabanowitz J., Hunt D. F. (2004) Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 101, 9528–9533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mikesh L. M., Ueberheide B., Chi A., Coon J. J., Syka J. E., Shabanowitz J., Hunt D. F. (2006) The utility of ETD mass spectrometry in proteomic analysis. Biochim Biophys Acta. 1764, 1811–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Good D. M., Wirtala M., McAlister G. C., Coon J. J. (2007) Performance characteristic of electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 24, 517–533 [DOI] [PubMed] [Google Scholar]
- 9. Molina H., Horn D. M., Tang N., Mathivanan S., Pandey A. (2007) Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 104, 2199–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Good D. M., Coon J. J. (2009) Mass spectrometric analysis of body fluids for biomarker discovery. Methods Mol. Biol. 566, 277–291 [DOI] [PubMed] [Google Scholar]
- 11. Hui L., Cunningham R., Zhang Z., Cao W., Jia C., Li L. (2011) Discovery and characterization of the Crustacean hyperglycemic hormone precursor related peptides (CPRP) and orcokinin neuropeptides in the sinus glands of the blue crab Callinectes sapidus using multiple tandem mass spectrometry techniques. J. Proteome Res. 10, 4219–4229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shen Y., Tolić N., Purvine S. O., Smith R. D. (2012) Improving collision induced dissociation (CID), high energy collision dissociation (HCD), and electron transfer dissociation (ETD) Fourier transform MS/MS degradome-peptidome identifications using high accuracy mass information. J Proteome Res. 11, 668–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yamaguchi H., Sasaki K., Satomi Y., Shimbara T., Kageyama H., Mondal M. S., Toshinai K., Date Y., González L. J., Shioda S., Takao T., Nakazato M., Minamino N. (2007) Peptidomic identification and biological validation of neuroendocrine regulatory peptide-1 and -2. J. Biol. Chem. 282, 26354–26360 [DOI] [PubMed] [Google Scholar]
- 14. Osaki T., Sasaki K., Minamino N. (2011) Peptidomics-based discovery of an antimicrobial peptide derived from insulin-like growth factor-binding protein 5. J. Proteome Res. 10, 1870–1880 [DOI] [PubMed] [Google Scholar]
- 15. Iguchi H., Hayashi I., Kono A. (1990) A somatostatin-secreting cell line established from a human pancreatic islet cell carcinoma (somatostatinoma): release experiment and immunohistochemical study. Cancer Res. 50, 3691–3693 [PubMed] [Google Scholar]
- 16. Swaney D. L., McAlister G. C., Wirtala M., Schwartz J. C., Syka J. E., Coon J. J. (2007) Supplemental activation method for high-efficiency electron-transfer dissociation of doubly protonated peptide precursors. Anal. Chem. 79, 477–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sasaki K., Sato K., Akiyama Y., Yanagihara K., Oka M., Yamaguchi K. (2002) Peptidomics-based approach reveals the secretion of the 29-residue COOH-terminal fragment of the putative tumor suppressor protein DMBT1 from pancreatic adenocarcinoma cell lines. Cancer Res. 62, 4894–4898 [PubMed] [Google Scholar]
- 18. Osaki T., Omotezako M., Nagayama R., Hirata M., Iwanaga S., Kasahara J., Hattori J., Ito I., Sugiyama H., Kawabata S. (1999) Horseshoe crab hemocyte-derived antimicrobial polypeptides, tachystatins, with sequence similarity to spider neurotoxins. J. Biol. Chem. 274, 26172–26178 [DOI] [PubMed] [Google Scholar]
- 19. Levi A., Ferri G. L., Watson E., Possenti R., Salton S. R. (2004) Processing, distribution, and function of VGF, a neuronal and endocrine peptide precursor. Cell Mol. Neurobiol. 24, 517–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Selle H., Lamerz J., Buerger K., Dessauer A., Hager K., Hampel H., Karl J., Kellmann M., Lannfelt L., Louhija J., Riepe M., Rollinger W., Tumani H., Schrader M., Zucht, H D. (2005) Identification of novel biomarker candidates by differential peptidomics analysis of cerebrospinal fluid in Alzheimer's disease. Comb. Chem. High Throughput Screen. 8, 801–806 [DOI] [PubMed] [Google Scholar]
- 21. Huang J. T., Leweke F. M., Oxley D., Wang L., Harris N., Koethe D., Gerth C. W., Nolden B. M., Gross S., Schreiber D., Reed B., Bahn S. (2006) Disease biomarkers in cerebrospinal fluid of patients with first-onset psychosis. PLoS Med. 3, e428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zougman A., Pilch B., Podtelejnikov A., Kiehntopf M., Schnabel C., Kumar C., Mann M. (2008) Integrated analysis of the cerebrospinal fluid peptidome and proteome. J. Proteome Res. 7, 386–399 [DOI] [PubMed] [Google Scholar]
- 23. Mishiro-Sato E., Sasaki K., Matsuo T., Kageyama H., Yamaguchi H., Date Y., Matsubara M., Ishizu T., Yoshizawa-Kumagaye K., Satomi Y., Takao T., Shioda S., Nakazato M., Minamino N. (2010) Distribution of neuroendocrine regulatory peptide-1 and -2, and proteolytic processing of their precursor VGF protein in the rat. J. Neurochem. 114, 1097–1106 [DOI] [PubMed] [Google Scholar]
- 24. Benjannet S., Leduc R., Lazure C., Seidah N. G., Marcinkiewicz M., Chrétien M. (1985) GAWK, a novel human pituitary polypeptide: isolation, immunocytochemical localization and complete amino acid sequence. Biochem. Biophys. Res. Commun. 126, 602–609 [DOI] [PubMed] [Google Scholar]
- 25. Conlon J.M., Hamberger B., Grimelius L. (1992) Isolation of peptides arising from the specific posttranslational processing of chromogranin A and chromogranin B from human pheochromocytoma tissue. Peptides 13, 639–644 [DOI] [PubMed] [Google Scholar]
- 26. Dahma H., Gourlet P., Vancermeers A., Vandermeers-Piret, M C., Robberecht P. (2001) Evidence that the chromogranin B fragment 368–418 extracted from a pheochromocytoma is phosphorylated. Peptides 22, 1491–1499 [DOI] [PubMed] [Google Scholar]
- 27. Beranova-Giorgianni S., Zhao Y., Desiderio D. M., Giorgianni F. (2006) Phosphoproteomic analysis of the human pituitary. Pituitary 9, 109–120 [DOI] [PubMed] [Google Scholar]
- 28. Ueberheide B. M., Fenyö D., Alewood P. F., Chait B. T. (2009) Rapid sensitive analysis of cysteine rich peptide venom components. Proc. Natl. Acad. Sci. U.S.A. 106, 6910–6915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bartolomucci A., Possenti R., Mahata S. K., Fischer-Colbrie R., Loh Y. P., Salton S. R. (2011) The Extended Granin Family: Structure, Function, and Biomedical Implications. Endocr. Rev. 32, 755–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cameron A., Apletalina E. V., Lindberg I. (2002) The enzymology of PC1 and PC2, in The Enzymes (Dalby R. E., Sigman D. S., eds) pp. 291–328, Academic Press, New York, NY [Google Scholar]
- 31. Dos Santos Cabrera M. P., Arcisio-Miranda M., Broggio Costa S. T., Konno K., Ruggiero J. R., Procopio J., Ruggiero Neto J. (2008) Study of the mechanism of action of anoplin, a helical antimicrobial decapeptide with ion channel-like activity, and the role of the amidated C-terminus. J. Pept. Sci. 14, 661–669 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.