

Abstract
Hypoxia-inducible factors (HIFs) are heterodimeric transcription factors that play a key role in cellular adaptation to hypoxia. HIF proteins are composed of an α subunit regulated by oxygen pressure (essentially HIF1α or HIF2α) and a constitutively expressed β subunit. These proteins are often overexpressed in cancer cells, and HIF overexpression frequently correlates with poor prognosis, making HIF proteins promising therapeutic targets. HIF proteins are involved in melanoma initiation and progression; however, the specific function of HIF2 in melanoma has not yet been studied comprehensively. Identifying protein complexes is a valuable way to uncover protein function, and affinity purification coupled with mass spectrometry and label-free quantification is a reliable method for this approach. We therefore applied quantitative interaction proteomics to identify exhaustively the nuclear complexes containing HIF2α in a human melanoma cell line, 501mel. We report, for the first time, a high-throughput analysis of the interactome of an HIF subunit. Seventy proteins were identified that interact with HIF2α, including some well-known HIF partners and some new interactors. The new HIF2α partners microphthalmia-associated transcription factor, SOX10, and AP2α, which are master actors of melanoma development, were confirmed via co-immunoprecipitation experiments. Their ability to bind to HIF1α was also tested: microphthalmia-associated transcription factor and SOX10 were confirmed as HIF1α partners, but the transcription factor AP2α was not. AP2α expression correlates with low invasive capacities. Interestingly, we demonstrated that when HIF2α was overexpressed, only cells expressing large amounts of AP2α exhibited decreased invasive capacities in hypoxia relative to normoxia. The simultaneous presence of both transcription factors therefore reduces cells' invasive properties. Knowledge of the HIF2α interactome is thus a useful resource for investigating the general mechanisms of HIF function and regulation, and here we reveal unexpected, distinct roles for the HIF1 and HIF2 isoforms in melanoma progression.
Hypoxia-inducible transcription factors (HIFs)1 are central mediators of cellular adaptation to hypoxia that control the expression of genes involved in anaerobic metabolism, intracellular pH, angiogenesis, and cell growth and survival (1). HIF proteins are heterodimers consisting of an oxygen-regulated α subunit and a constitutively expressed β subunit (also referred to as the arylhydrocarbon receptor nuclear translocator, or ARNT). Three isoforms have been described for each subunit; most HIF function, however, has been attributed to HIF1α, HIF2α, and HIF1β (2). HIF1α is detected in almost all tissues, whereas HIF2α (also known as EPAS1, for endothelial PAS protein 1) is restricted to certain tissues. HIF2 is expressed notably in hypoxic endothelium but also in distinct cell populations in the brain, liver, kidney, lung, and intestine (3). HIFα proteins are regulated post-translationally as a function of the partial pressure of oxygen. In normoxic conditions, the protein is rapidly hydroxylated by prolyl hydroxylases and then recognized by the von Hippel-Lindau protein, a component of an E3-ubiquitin ligase complex that targets the α subunit for degradation by the proteasome. When the partial pressure of oxygen is low, prolyl hydroxylase activity is inhibited, allowing α subunit accumulation (4).
In the past decade, HIF protein expression has been examined extensively in tumor cells. Many immunohistochemical studies have clearly demonstrated high expression of HIF1α and/or HIF2α in many types of human primary tumors (5, 6). Moreover, numerous recent studies have revealed a significant correlation between HIFα subunit expression in human tumors and a poor prognosis (1). Other studies, however, have demonstrated distinct effects of the two HIFα subunits in some tumor cell types. For instance, in renal cell carcinoma models, HIF1α expression has been shown to decrease tumor size in a nude mouse xenograft model, whereas HIF2α expression has the opposite effect (7). Therefore, despite similarities in their structure and regulation, the two HIFα subunits display comparable but not identical behaviors in physiological and physiopathological conditions (8, 9).
Malignant melanoma is a cancer arising from pigment-producing cells called melanocytes, present predominantly in the skin. It is a very aggressive form of cancer that accounts for almost 80% of the casualties associated with skin cancer, and its incidence is currently growing faster than that of any other cancer worldwide, especially among young adults. Melanoma is treatable by surgical resection when caught in the early stages, yet it still causes tens of thousands of deaths each year worldwide. Although genetic and epigenetic alterations contribute to tumorigenesis (10), various stimuli coming from the tumor microenvironment appear to be required for the development of a malignant phenotype (11). Among the different elements of the melanoma microenvironment, oxygen availability is of major importance. Both HIF1 and HIF2 are up-regulated in melanoma cells (12, 13). Although the importance of HIF1 in melanocyte survival and proliferation in hypoxia (14) and in melanoma carcinogenesis (15, 16), progression (17), and invasion (18, 19) has been studied extensively, the function of HIF2α in melanoma still remains unclear.
Protein–protein interactions play a key role in modulating protein functions. Thus, the identification of a protein's interactome might be a relevant way to uncover the role of proteins within the cell. Mass-spectrometry-based proteomics is the method of choice for addressing this issue in a systematic and relatively unbiased manner. Several strategies may be applied, including affinity purification with one or several tags, with or without prior cross-linking, combined or not with quantitative spectrometry (20–22).
Here, we report an extensive study of HIF2α protein partners in the 501mel melanoma cell line using affinity purification and quantitative mass spectrometry. With this approach, we have described, for the first time, the global nuclear interactome of one HIF subunit. Among the HIF2α partners that we found quantitatively enriched in hypoxic melanoma cells, certain proteins relevant to melanoma biology were confirmed as HIF2α partners, leading to a better knowledge of melanoma biology.
EXPERIMENTAL PROCEDURES
Cell Culture
Melanoma cell lines (provided by Dr. Lionel Larue, Institut Curie, Orsay, France) were grown in RPMI 1640 (Invitrogen) containing 10% fetal calf serum (FCS) (Invitrogen), 50 μg/ml penicillin-streptomycin mix (Invitrogen), and 500 μg/ml hygromycin in order to select stably transfected cell lines. All cell lines were used within two months after the resuscitation of frozen aliquots and were authenticated on the basis of viability, recovery, growth, and morphology.
HIFα expression was induced by incubating cells for 12 h in a hypoxic chamber (0.1% O2) or by adding 125 μm CoCl2 (Sigma), a hypoxia-mimicking reagent, to the cell culture medium for 12 h. For longer hypoxic assays, mild hypoxia was used (1% O2 in a hypoxic chamber or 25 μm CoCl2). Cell number, viability, and proliferation rate were evaluated either simply by counting or by quantifying metabolically active cells (MTT assay). Briefly, 10,000 cells were plated in 0.1 ml of medium in 96-well plates. At the indicated times, 100 μl of 1 mg/ml MTT (Sigma) solution in PBS was added to each well for 2 h. After removal of the medium, 100 μl of DMSO was added to induce cell lysis and dissolve formazan crystals. The absorbance at 570 nm was determined using a microplate spectrophotometer (μQuant, Bio-Tek, Winooski, VT).
Plasmid Construction and Establishment of Stable Cell Lines
The full-length coding region of human HIF2α or HIF1α (gifts from Dr. Richard Bruick of UT Southwestern Medical Center, Dallas, TX, and from Dr. Amato Giaccia of Stanford University, Stanford, CA, respectively) was cloned in-frame with a linker encoding an HA tag in a pCMV-hygro 2xFLAG expression vector (supplemental Figs. S1 and S6). The resulting vectors were transfected into 501mel cells, and stable transfectants were obtained after four weeks of hygromycin selection. A control cell line (Mock) was produced after transfection of a 501mel cell line with the empty pCMV-hygro 2xFLAG vector. For each construct, single clones were obtained by limiting dilution and were genotyped via PCR.
Immunoaffinity Purification for Mass Spectrometry Analysis
Nuclear extracts from stably transfected 501mel cells were prepared as described elsewhere (23), with minor modifications: before hypertonic treatment, nuclei were incubated for 10 min in buffer B (20 mm Tris-HCl pH 7.5, 5 mm MgCl2, 0.1 mm EDTA, 0.5 μm CaCl2, complete protease inhibitor mixture (Roche Applied Science), and 0.5 mm Pefabloc in the presence of PSC protector solution (Roche Applied Science)). After centrifugation, nuclear proteins were extracted by incubating the pellet in the same buffer containing 400 mm NaCl and 10% glycerol for 30 min at 4 °C. The protein solution was concentrated via ultrafiltration (Vivaspin10, Vivascience Sartorius, Stonehouse, UK) at 4 °C. Nuclear extracts were loaded onto a 4-ml 10%–50% glycerol gradient in buffer B containing 400 mm NaCl and subsequently were centrifuged at 200,000g for 4 h at 4 °C. The glycerol gradients were then divided into 16 fractions of 0.25 ml and tested for HIF2α via immunoblotting. Fractions 2–8, which contained HIF2α protein, were pooled. The salt concentration was adjusted to 200 mm NaCl, and HIF2α protein complexes were purified via immunoprecipitation using anti-FlagM2 antibody-conjugated agarose beads (Sigma-Aldrich, France). Immunopurified complexes were washed twice with IP0.2 buffer (20 mm Tris-HCl pH 7.5, 200 mm NaCl, 0.5 mm MgCl2, 0.5% Nonidet P-40, 10 mm β-mercaptoethanol, and protease inhibitor mixture) and twice in IP0.4 (IP0.2 containing 400 mm NaCl) and eluted with a solution of 500 μg/ml 3xFlag peptide in IP0.4 buffer (Sigma-Aldrich, France). As a negative control, the same purification was undertaken starting from the Mock cell line. Samples from HIF2α and Mock cell lines were separated by means of SDS-PAGE and submitted for proteomic analysis.
One-dimensional SDS-PAGE Fractionation, In-gel Digestion, and Nano–liquid Chromatography–electrospray Ionization LTQ-Orbitrap MS/MS Analysis
Before SDS-PAGE separation, proteins were reduced with 25 mm DTT in 25 mm Tris-HCl pH 6.8, 5% glycerol, and 1% SDS for 30 min at 56 °C. Then, proteins were alkylated by the addition of 90 mm iodoacetamide for 30 min at room temperature in the dark. After protein separation via Nu-PAGE on a precast gradient 4%–12% acrylamide gel followed by Coomassie Blue staining, gel lanes were cut into 10 slices and incubated 3-fold on a shaker for 15 min at 37 °C in 100 mm ammonium bicarbonate in 50% acetonitrile (ACN). The gel slices were then vacuum dried and rehydrated with 40 μl modified sequencing-grade trypsin solution (10 ng/μl; Promega, Madison, WI) in 50 mm ammonium bicarbonate for in-gel tryptic digestion. After absorption of the trypsin solution, 100 μl of 50 mm ammonium bicarbonate was added. Trypsin digestion was performed overnight at 37 °C. The resulting peptides were extracted from the gel slices through three incubations (15 min at 37 °C) with shaking: a first incubation in 50 mm ammonium bicarbonate, and two subsequent incubations in 10% formic acid and ACN (1:1). The three successive extractions were pooled with the initial digestion supernatant, vacuum dried in a SpeedVac, and re-suspended in 14 μl of 2% ACN and 0.05% TFA.
Peptide mixtures were analyzed via nano-LC-MS/MS using an Ultimate3000 system (Dionex, Amsterdam, The Netherlands) coupled to an LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) operating in positive mode. Five microliters of each sample were loaded onto a C18 precolumn (300 μm inner diameter × 5 mm; Dionex) at 20 μl/min in 2% ACN and 0.05% TFA. After 5 min of desalting, the precolumn was switched online with the analytical C18 column (75 μm inner diameter × 15 cm; PepMap C18, Dionex) equilibrated in 100% solvent A (5% ACN, 0.2% formic acid) and 0% solvent B (80% ACN, 0.2% formic acid). Peptides were eluted by using a 0%–50% gradient of solvent B for 60 min at a flow rate of 300 nl/min. The LTQ-Orbitrap XL was operated in data-dependent acquisition mode with Xcalibur software (version 2.0 SR2, Thermo Fisher Scientific). Survey scan MS spectra were acquired in the Orbitrap in the 300–2000 m/z range with the resolution set to a value of 60,000 at m/z 400. Up to five of the most intense multiply charged ions (2+ and 3+) per survey scan were selected for collision-induced dissociation fragmentation, and the resulting fragments were analyzed in the linear trap (LTQ). Dynamic exclusion was used within 60 s to prevent repetitive selection of the same peptide.
Database Search and Data Analysis
Mascot Daemon software (version 2.3.2, Matrix Science, London, UK) was used to perform database searches in batch mode with all of the raw files acquired for each sample. To extract peak lists automatically from Xcalibur raw files, the Extract msn.exe macro provided with Xcalibur was used through the Mascot Daemon interface. The following parameters were set for the creation of the peak lists: parent ions in the mass range of 300–4500, no grouping of MS/MS scans, and no threshold. A peak list was created for each analyzed fraction (i.e. gel slice), and Mascot searches were performed for each fraction. Data were searched against all entries in the International Protein Index human protein database (v3.86, released September 7, 2011; 91,522 sequences). Cysteine carbamidomethylation was set as a fixed modification; methionine oxidation and protein N-terminal acetylation were set as variable modifications for all Mascot searches. Proteins were digested by trypsin, and two missed trypsin cleavage sites were allowed. The mass tolerances in MS and MS/MS were set at 10 ppm and 0.6 Da, respectively, and the instrument setting was specified as “ESI-Trap.” To calculate the false discovery rate (FDR), the search was performed using the “decoy” option in Mascot. Mascot results were parsed and quantified with the in-house-developed software Mascot File Parsing and Quantification (MFPaQ), version 4.0.0 (24). Peptide identifications extracted from Mascot result files were validated at a final peptide FDR of 1 (at least one peptide with a score of 40, or at least two peptides with a score of 20). The FDR was calculated at the protein level (FDR = number of validated decoy hits/number of validated target hits × 100). If the same set of peptides belongs to several proteins, the first protein listed is the one that contains the most peptides. Unambiguous protein identification is provided by a proteotypic peptide (i.e. a peptide with a unique amino acid sequence among all proteins). Known contaminants excluded were keratins and trypsin.
Label-free Quantification
The quantification of proteins was performed using the label-free module implemented in the MFPaQ v4.0.0 software (25). Briefly, for each sample, the software used the validated identification results and extract ion chromatograms of the identified peptide ions in the corresponding raw nano-LC/MS files, based on their experimentally measured retention time and monoisotopic m/z values. In order to perform protein relative quantification in different samples, a protein abundance index (PAI) was calculated and defined as the average of the extract ion chromatogram area values from the three most intense peptides identified for the protein in Mock (501mel cell line stably transfected with an empty vector) and H2 (501mel cell line stably transfected with an HIF2α vector) samples. The integration of quantitative data across the fractions was performed by summing the PAI values for fractions adjacent to the fraction with the best PAI (the same three consecutive fractions for all the samples to be compared). Four biological replicates were analyzed in order to minimize potential experimental deviations. Proteins were subsequently screened for the following: (1) known nuclear localization, (2) proteins detected in at least two independent experiments with FDRs lower than 1%, and (3) proteins enriched more than 4-fold in H2 relative to Mock assays (see supplemental Fig. S3 and Table I). The retained proteins were further analyzed for gene ontology and protein interaction network based on literature investigations and Ingenuity Pathway Analysis.
Table I. HIF2α protein partners identified via mass spectrometry.
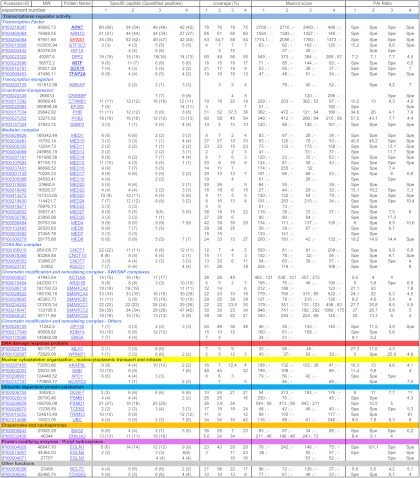
The proteins listed have been identified and quantified in at least two of four independent experiments with (i) an FDR < 1 and (ii) a ratio of PAI assay/PAI control > 4. Accession IDs, molecular weights (MW), protein names, the number of Specific peptides identified (the number of quantified peptides is indicated in parentheses), sequence coverage, Mascot score, and PAI ratios are indicated in the table. Score column: two score values are provided, separated by a semicolon. The left-hand value corresponds to the protein score obtained in the H2 assay. The right-hand value corresponds to the protein score obtained in the Mock assay. A dash (-) indicates that protein peptides have not been sequenced or detected. Ratio column: ratios are the PAI of the protein in the assay divided by the PAI of the protein in the control. The PAI is the average of the extract ion chromatogram area values for the three most intense peptides identified for a protein. Spe: proteins were not detected in the Mock protein extract and were thus Specific for H2 cells. Proteins have been categorized according to their biological function. The bait (HIF2α = EPAS1) is in bold red. Proteins whose interaction with HIF2α has been further validated in the current study are in bold black (TFAP2A = AP2α). Annotated MS/MS Spectra of single peptide-based protein identifications are provided in supplemental Fig. S9.
Co-immunoprecipitation for Western Blot Analysis
Cell extracts were prepared via the lysis of 10 million cells in 0.5 ml RIPA buffer (50 mm Tris-HCl pH 8.0, 0.1% SDS, 0.01% sodium deoxycholate, 150 mm NaCl, 1% Triton X-100, and complete protease inhibitor mixture) for 15 min on ice and clarified via centrifugation. Protein extracts were diluted 4-fold with IP buffer (50 mm Tris-HCl pH 8.0, 0.01% sodium deoxycholate, 150 mm NaCl, 1% Triton X-100, and complete protease inhibitor mixture). Flag-immunoprecipitations were performed as described elsewhere (26), except that the beads were washed in WIP buffer (50 mm Tris-HCl pH 8.0, 0.01% sodium deoxycholate, 350 mm NaCl, 1% Triton X-100, and complete protease inhibitor mixture).
Alternatively, 5 μg of antibody (supplemental Table S3) was incubated with the protein extract for 3 h at 4 °C with rotation prior to the addition of 40 μl of protein G Plus agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). The mixture was incubated for 1 h at 4 °C with rotation to capture immune complexes, and then the beads were pelleted via brief centrifugation and washed three times with WIP buffer. Bound proteins were eluted by means of boiling with 50 μl of SDS-PAGE loading buffer. Protein content was analyzed via Western blot (antibodies are listed in supplemental Table S3).
Fluorescence Microscopy of the Actin Cytoskeleton
Cells were grown on coverslips for at least 16 h and were subsequently washed, fixed for 15 min in 3.7% paraformaldehyde, permeabilized, blocked, and incubated for 2 h with rhodamine-conjugated phalloidin (Molecular Bioprobes, Eugene, Oregon, USA) in phosphate-buffered saline (PBS) containing 1% BSA (PBS-BSA). Subsequently, the coverslips were washed with PBS-BSA, air dried, and embedded in Dako Cytomation Fluorescent Mounting Medium. Fluorescence was detected using a Leica microscope (Leitz DMRB; 100× objective). Images were acquired as sets of color images and prepared using Photoshop software.
Cell Migration and Invasion Assays
Equal numbers of cells (1 × 105) in serum-free RPMI were added to the upper compartments of the chamber (ThinCerts®, 12 wells, 8-μm pores (Greiner Bio-One, Frickenhausen, Germany)) and kept in an incubator at 37 °C for 20 h. As a chemoattractant, the lower compartment contained RPMI supplemented with 10% FCS. At the end of the incubation time, cells from the upper surface of the filter were wiped off with a cotton swab. The lower surface of the filter was stained with Toluidine Blue O (1% w/v, Sigma) in 1% sodium tetraborate. Cells that had migrated to the bottom of the chamber were quantified via optical density measurement (570 nm) after solubilization in lysis buffer (6.25 mm Tris-HCl pH 6.8, 10% glycerol, 2% SDS, 5% β-mercaptoethanol). At least three independent sets of experiments were carried out, each in duplicate. For Matrigel assays, the filter separating the two compartments was pre-coated with 50 μl of 0.125 μg/μl Matrigel (BD Biosciences).
siRNA-mediated Knockdown of HIFα Subunit
Validated siRNA duplexes targeting human HIFα subunits (ON-TARGETplus SMARTpool J-004018–07 for HIF1α and ON-TARGETplus SMARTpool J-004814–06 for HIF2α) and control non-targeting siRNA (ON-TARGETplus non-targeting Pool d-001810–10) were synthesized by Dharmacon (Dharmacon, Inc., Lafayette, CO). 501mel cells were seeded in six-well plates at 1.0 × 106 cells per well and were treated 24 h after seeding with 25 nm HIF1α siRNA, 25 nm HIF2α siRNA, or 25 nm control non-targeting siRNA using the DharmaFECT 1® transfection reagent (Dharmacon) according to the manufacturer's instructions. CoCl2 (125 μm final concentration) was added 36 h after siRNA transfection for an additional 12 h. Transfection efficiency was verified using fluorescent siRNA (siGLO green d-001630–01-02, Dharmacon), and the inhibition of HIFα expression was confirmed by means of both RT-qPCR and Western blot (supplemental Fig. S7).
Statistical Analyses
The values reported in the tables and figures are means ± S.E. of the mean. The percentages of melanoma cells in the migration and invasion assays were compared between Mock-, HIF1α-, and HIF2α-transfected cells using Student's t test.
RESULTS
Identification of HIF2α-interacting Proteins
501mel cells express the HIF2α subunit when cultured under hypoxic conditions or in the presence of the hypoxia-mimicking agent cobalt chloride (supplemental Fig. S1D). However, to facilitate HIF2α-interacting protein purification, we chose to stably transfect this human melanoma cell line with a plasmid construction that allowed the expression of a Flag/HA fusion protein. This epitope-tagging strategy allows a generic purification procedure that results in cleaner samples and a higher yield than traditional immunoprecipitation experiments (22). Transient interactions between transcription factors and most of their partners make the identification of this type of interactome harder (22). To address this problem, we chose to perform a one-step Flag-affinity-based protein purification protocol rather than tandem affinity purification, and we combined this with quantitative MS analysis. To discriminate between genuine partners and contaminants, label-free quantitative MS analysis was performed on immunoprecipitates of Flag-HIF2α prepared from stably transfected cells and from a control cell line that does not express the tagged bait. The label-free MS method allows the identification of contaminants, which are present in similar amounts in both control and Flag-HIF2α-expressing cells, and their differentiation from true interactors, which are significantly enriched in Flag- HIF2α expressing cells.
The expression, correct functionality, and nuclear location of exogenous tagged HIF2α protein were verified via Western blot analysis, reporter gene assay, and immunofluorescence microscopy (supplemental Fig. S1). One clone (named H2) exhibiting a high total HIF2α expression (about 10-fold higher than in the Mock cell line, which was stably transfected with the empty vector) was selected. Exogenous tagged HIF2α protein overexpression partially bypasses the proteasomal degradation capacity of the cell; therefore, the recombinant protein was also detected in normoxic conditions. As expected, higher amounts of the recombinant HIF2α protein were produced under hypoxic conditions (supplemental Fig. S1D). Accordingly, as normoxic and hypoxic HIF proteins present different functional characteristics, we performed co-immunoprecipitation experiments under hypoxia.
The strategy followed in order to identify nuclear proteins associated with HIF2α is presented in Fig. 1A. Nuclear extracts were prepared from H2 cells cultured in hypoxia-mimicking conditions, and soluble nuclear protein complexes were separated using a glycerol gradient to reduce sample complexity. The HIF2α content of the different fractions of the glycerol gradient was analyzed via Western blotting using an anti-HIF2α antibody (Fig. 1B). HIF2α-positive fractions were pooled (fractions 2 to 8). Immunoprecipitation of HIF2α with its protein partners was then performed using an anti-Flag monoclonal antibody, and the proteins were subsequently eluted by Flag peptide competition. Nuclear extracts from the Mock cell line were treated similarly and used as a negative control. The Flag-tagged protein was not expressed in this cell line. HIF2α was identified in the H2-immunopurification (IP) but not in the Mock-IP control lane (Fig. 1C). Aliquots of the IP fractions were loaded for SDS-PAGE and silver stained. A protein band that might correspond to HIF2α and specifically enriched protein bands corresponding to HIF2 partners were observed in the H2-IP lane (Fig. 1D). The low background in the Mock-IP samples and the presence of specific bands in the H2-IP samples indicate the specificity of the IP protocol. The remaining IP fractions were subjected to analytical SDS-PAGE. The gel was cut into slices and subsequently treated with trypsin. Peptides were extracted and analyzed via nano-LC-MS/MS on an LTQ-Orbitrap-XL. The identification of peptides was performed using the Mascot Daemon software for searching databases, combined with a label-free protein quantification analysis with in-house-developed MFPaQ software (24). Prior to quantification, a list of proteins exhibiting an FDR lower than 1% was automatically validated. Protein quantification was then performed using the listed proteins. Label-free quantification is based on a comparison of the PAI of each protein in two different conditions. The PAI (average of area under the curve) is calculated from the three most intense peptides of each protein. To minimize biological and analytical variations, four independent experiments were performed, leading to the identification of, on average, 624 proteins (643, 597, 663, and 592 proteins, respectively, for the first, second, third, and fourth experiments). The reproducibility among these four experiments was evaluated using a four-way Venn diagram that compared the proteins identified in each biological replicate (supplemental Fig. S2A). Of these 624 proteins, 57% were common among the four experiments. Multiple-step filtering was performed to retain high-confidence interacting partners. First, proteins lacking nuclear gene term ontology were excluded. Then, proteins had to be detected in at least two independent experiments with FDRs lower than 1%. Additionally, proteins had to display a greater than 4-fold enrichment between H2 and Mock assays (supplemental Fig. S3 and Table I). This quality-control filtering validated 70 proteins as specific direct or indirect HIF2α-interacting proteins (HIF2α = EPAS1; Table I). Of the selected proteins, 71% were encountered in all experiments (supplemental Fig. S2B), with more than 87% being identified in at least three of them.
Fig. 1.

Purification of HIF2α-interacting proteins. A, schematic presentation of the strategy used to identify the HIF2α nuclear interactome in 501mel cells. B, nuclear extracts from 501mel cells expressing Flag/HA-tagged HIF2α (H2) or from Mock cells, cultured with CoCl2, were fractionated on a glycerol gradient. Fractions were analyzed via Western blotting with an anti-HIF2α antibody. I/20, input/20. C, immunoblot analysis of nuclear extract (NE), unbound (U), wash (W), and Flag immunoaffinity-purified fractions (IP) of Mock and H2 cell lines using an antibody against HIF2α. D, purified HIF2α complexes (IP) were separated on a 4%–12% gradient protein gel and revealed by means of silver staining. The band that might correspond to HIF2α protein is indicated by an arrowhead. Nuclear extracts (NE) were used as loading controls.
The identified HIF2α protein partners were grouped according to their biological function based on literature investigations and function analysis using Ingenuity Pathway Analysis from Ingenuity. Proteins belonged to diverse biological categories and were known to participate in various cellular functions and activities, including transcription regulation (n = 51), nuclear cytoskeleton organization, mitosis and nucleocytoplasmic transport (n = 4), ubiquitin-dependent protein catabolism (n = 6), and protein modification (n = 3) (Fig. 2A). Thirteen proteins found in the present study had been previously reported to interact directly or indirectly with HIF2α, HIF1α, or HIF1β (ARNT1) (supplemental Table S1). Moreover, peptides belonging to several previously characterized partners of HIF subunits were also detected in our conditions but did not fulfill all the screening criteria. For example, peptides belonging to HDAC1–3, Hsp90 (HSP90A), and Cullin B (CUL2), well-known interacting partners of HIFα subunits, were detected in only one of four experiments or were insufficiently enriched relative to the control (<4-fold enriched) (supplemental Table S1). These proteins therefore did not meet our stringent criteria and could not be firmly assigned as HIF2α partners. A functional network analysis was performed to test for possible complex formation between several HIF2α partners (Fig. 2C). Among the 70 nuclear protein partners of HIF2α identified in the present study, 60 were already known to interact directly with at least one other protein on the list, and 182 direct interactions were revealed in this protein interaction network. Among these interactions, some well-known complexes such as mediator or SMARC complexes were detected (Fig. 2C). Indeed, 20 proteins belonging to the multiprotein mediator complex (27) were identified as HIF2α partners (Table I; Figs. 2B and 2C). Almost all of the subunits thought to be part of the mediator complex were identified, which is remarkable, as these proteins are usually found in small amounts in cells (28). MED21 was the only missing subunit; this might be explained by its relatively small size (144 residues). Also, the two subunits recovered with the lowest scores, MED18 and MED20, are not part of the mediator core complex and therefore are more labile (29) (supplemental Fig. S4A). Similarly, subunits involved in SWI/SNF (SMARC) complex formation (30), and thus in chromatin remodeling, have also been shown to be HIF2α protein partners. One of these SMARC complexes has been entirely identified in our study: BRM/BRG1 complex (BRM = SMARCA2, BRG1 = SMARCA4; Figs. 2B and 2C, supplemental Fig. S4B). CBP (CREBBP) and P300 (EP300) were also identified as HIF2α interacting partners. These proteins, which exhibit histone acetyltransferase activity, are major partners of both HIFα subunits and interact with their C-terminal transactivation domains (31). CBP and P300 displayed more than 60% sequence identity. In the four independent proteomic experiments carried out in the current study, only 8 and 11 peptides among the 15 and 20 identified peptides for CBP and P300, respectively, strictly discriminated both proteins (Table I and supplemental Fig. S5). CBP/P300, mediator, and the SMARC complex are ubiquitous, and some of these proteins have already been identified as HIF1 and/or HIF2 partners (supplemental Table S1). Indeed, these interactions might not be specific to melanoma cells. Nevertheless, the presence of these partners not only confirms the quality and biological relevance of the identified interactome, but also suggests that they might play a role in melanoma progression, as discussed below. Interestingly, among all the identified HIF2α partners, several proteins were already known to play key roles in melanoma development, such as microphthalmia-associated transcription factor (MITF), SOX10, and AP2α (TFAP2A) (32–34). Because of the novelty of these results, we decided to focus our study on these interactions.
Fig. 2.
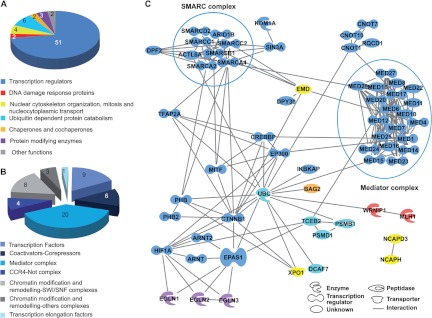
Classification of identified HIF2α-interacting proteins. A, pie chart representing the distribution in functional groups of the 70 proteins identified as HIF2α nuclear partners in the human 501mel melanoma cell line. B, pie chart depicting classification into subcategories of the 51 identified transcription regulator proteins. C, network analysis of HIF2α interacting proteins. Only proteins that are known to have direct interactions with each other are indicated in the diagram. The color code is common for A and C. EPAS1 = HIF2α, ARNT = HIF1β, TFAP2A = AP2α, CREBBP = CBP, EP300 = P300, SMARCA2 = BRM, and SMARCA4 = BRG1. Analysis was performed using Ingenuity Pathway Analysis software. Two main complexes (SMARC and mediator) are boxed.
Validation of HIF2α Interaction with MITF, SOX10, and AP2α
Binding of MITF, SOX10, and AP2α to HIF2α was confirmed by means of either co-immunoprecipitation using both tagged and endogenous proteins or reciprocal co-immunoprecipitation using endogenous proteins, followed by Western blot analysis. Cell lysates from Mock and H2 cells cultured under hypoxia-mimicking conditions were prepared and immunoprecipitated with an anti-FLAG antibody targeting exogenous HIF2α protein. Immunoprecipitated complexes were then separated via SDS-PAGE and analyzed via Western blot. Antibodies raised against HIF1β (as a positive control), actin (as a negative control), MITF, SOX10, and AP2α were used. As expected, all proteins, except actin, were detected in the immunoprecipitate of H2-nuclear extract (Fig. 3A). No signal was observed in the Mock-nuclear extract immunoprecipitate (Fig. 3A). Note that no significant difference in the levels of expression of these proteins was observed in any of the 501mel clones used in the present study, except for HIF2α (see “Input” lanes in Fig. 3A). The interaction between HIF2α and its partners was further confirmed in hypoxic 501mel cells using an anti-HIF2α antibody targeting endogenous HIF2α (Fig. 3B). Again, all proteins were detected in the immunoprecipitate of 501mel protein extracts, clearly demonstrating that endogenous HIF2α was also present in protein complexes containing HIF1β, MITF, SOX10, or AP2α. Reciprocal co-immunoprecipitations were carried out in untransfected 501mel cells to confirm the interactions between HIF2α and its newly identified partners. Lysates from hypoxic 501mel cells were immunoprecipitated with anti-SOX10, anti-MITF, or anti-AP2α antibodies. Immunoprecipitated complexes were separated via SDS-PAGE and analyzed via Western blot (Fig. 3C). HIF2α was detected in the three different immunoprecipitates, confirming the interaction between HIF2α and these three proteins.
Fig. 3.
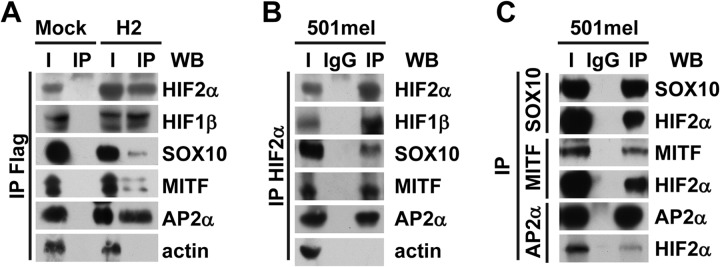
Western blot validation of mass spectrometry data. A, Flag/HA-tagged proteins from cell lysates of either Mock (left-hand panel) or H2 (right-hand panel) clones cultured with CoCl2 were immunoprecipitated with anti-Flag beads. Tagged HIF2α or co-immunoprecipitated partners were detected via immunoblotting with anti-HIF2α, anti-HIF1β, anti-SOX10, anti-MITF, anti-AP2α, or anti-actin antibodies. I, input (cell lysate); IP, immunoprecipitated fraction. B, endogenous HIF2α protein was immunoprecipitated from lysate of untransfected 501mel cells cultured under hypoxic conditions using anti-HIF2α antibodies (IP, immunopurified) or a control isotype-matched antibody (IgG) followed by immunoblotting with the indicated antibodies. I, input (cell lysate). C, lysate from untransfected 501mel cells cultured in hypoxia was immunoprecipitated with anti-SOX10, anti-MITF, anti-AP2α antibodies (IP, immunoprecipitated), or a control isotype-matched antibody (IgG). I, input (cell lysate). Immunoprecipitates were analyzed via Western blotting with the indicated antibodies. For all panels, a representative blot is shown from at least three independent experiments.
Interaction of Newly Identified HIF2α Partners with HIF1α
To see whether HIF1α could also interact with HIF2α partners in melanoma cells, similar co-immunoprecipitation experiments were performed with the HIF1α protein as bait. The strategy to assess the presence of proteins in HIF1α-containing protein complexes was similar to that used to validate interactions between HIF2α and its partners. First, stable 501mel cell lines overexpressing tagged HIF1α were established. The expression, functionality, and subcellular localization of the tagged protein were verified (supplemental Figs. S6A, S6B). A clone, named H1, was selected that expressed about 2-fold more HIF1α than the Mock cell line (supplemental Fig. S6C). Immunoprecipitation of tagged HIF1α protein was performed on lysates of both Mock and H1 clones cultured under hypoxia-mimicking conditions (Fig. 4A). As expected, HIF1β co-immunoprecipitated with HIF1α, whereas actin did not. SOX10 and MITF both co-immunoprecipitated, although less than when HIF2α was used as bait. This might be explained by a lower expression of the HIF1α tagged protein in the H1 clone than of HIF2α in the H2 clone. Interestingly, we did not detect AP2α as an HIF1α partner. Co-immunoprecipitations and reciprocal co-immunoprecipitations were also performed with protein extracts from untransfected 501mel cells cultured under hypoxia (Figs. 4B, 4C), from which we conclude that in 501mel cells, SOX10 and MITF interact with both HIFα subunits, whereas AP2α is specifically associated with HIF2α protein complexes.
Fig. 4.
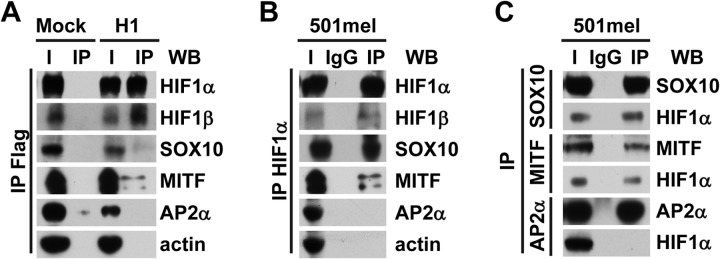
Do the HIF2α partners identified via MS/MS also interact with HIF1α? A, Flag/HA-tagged HIF1α proteins from lysates of either Mock (left-hand panel) or H1 (right-hand panel) cells cultured with CoCl2 were immunoprecipitated with anti-Flag beads. Detection of tagged HIF1α or co-immunoprecipitated partners was performed via immunoblotting with anti-HIF1α, anti-HIF1β, anti-SOX10, anti-MITF, anti-AP2α, or anti-actin antibodies. I, input (cell lysate); IP, immunoprecipitated fraction. B, endogenous HIF1α protein was immunoprecipitated from 501mel cells cultured under hypoxia using anti-HIF1α antibodies (IP) or a control isotype-matched antibody (IgG) followed by immunoblotting with the indicated antibodies. C, lysate from untransfected 501mel cells cultured in hypoxic conditions was immunoprecipitated with anti-SOX10, anti-MITF, anti-AP2α antibodies (IP), or a control isotype-matched antibody (IgG). Immunoprecipitates were analyzed via Western blotting using the indicated antibodies. I, input (cell lysate). In all panels, a representative blot is shown from at least three independent experiments.
Interactions between HIFα Subunits and AP2α, MITF, and SOX10 Proteins Are Observed in Other Melanoma Cell Lines
To verify that the interactions between HIF1α or HIF2α and their partners also occur in other melanoma cell lines, co-immunoprecipitations were performed in a panel of melanoma cell lines expressing both HIF1 and HIF2 proteins. The G1, SK-MEL-28, and Lu1205 cell lines were cultured under hypoxia-mimicking conditions. Cell lysates were prepared and co-immunoprecipitation was carried out using either HIF1α- or HIF2α-specific antibodies. As observed in the 501mel cell line, MITF and SOX10 co-immunoprecipitated with both subunits, whereas AP2α interacted only with HIF2α (Fig. 5). Note that neither MITF nor AP2α was detected in the Lu1205 cell line.
Fig. 5.
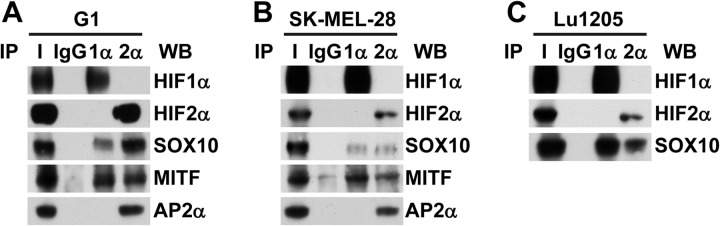
Interactions between HIFα subunits and AP2α, MITF, or SOX10 are observed in other melanoma cell lines. G1 (A), SK-MEL-28 (B), or Lu1205 (C) melanoma cell lines were treated with CoCl2. Lysates from the three different cell lines were immunoprecipitated with the indicated antibodies (1α for anti-HIF1α, 2α for anti-HIF2α, and IgG for a control isotype-matched antibody). HIF1α, HIF2α, or co-immunoprecipitated partners were detected via immunoblotting with anti-HIF1α, anti-HIF2α, anti-SOX10, anti-MITF, and anti-AP2α antibodies. I, input (cell lysate).
HIF1α and HIF2α Overexpressing Cell Lines Exhibit Different Invasive Potentials
AP2α transcription factor expression is known to decrease cell growth, migration, and invasion (34). In 501mel cells, AP2α interacts with HIF2α but not with HIF1α. To determine whether the presence of HIF proteins modifies AP2α functions, we first analyzed the effect of HIF2α or HIF1α overexpression on the proliferation, invasion, and migration of 501mel cells. The growth rates of untransfected 501mel, Mock-, HIF1α-, and HIF2α-transfected cells were compared under hypoxia-mimicking conditions. Numbers of viable cells were evaluated daily using the MTT assay. No statistical significance was observed between the growth rate curves of the different cell lines (Fig. 6A); therefore, in our conditions, HIFα subunit overexpression did not affect cell proliferation. Migrating cells are characterized by being more spread on the substratum and by remodeling of the actin cytoskeleton with the formation of stress fibers (35). Rhodamin-phalloidin staining was used to assess actin cytoskeleton organization in the cell lines overexpressing HIFα subunits (Fig. 6B). Interestingly, whereas the H2 cells (overexpressing HIF2α) displayed a more dendritic phenotype (a hallmark of differentiated melanocytes) and only few stress fibers, the H1 cells (overexpressing HIF1α) were more spread and had an extensive network of stress fibers.
Fig. 6.

HIF2α- and HIF1α-transfected 501mel melanoma cell lines exhibit distinct invasive properties. A, growth rates of the untransfected 501mel cell line or stably transfected clones were compared under hypoxia-mimicking conditions. The number of viable cells was evaluated daily using the MTT assay. B, 501mel, Mock, H2, or H1 cell lines were seeded on glass coverslips, incubated at 37 °C, and allowed to grow for 16 h in the presence of CoCl2. Actin was labeled with rhodamine-phalloidin and visualized by means of fluorescence microscopy. C, equal numbers of human 501mel and 501mel-transfected melanoma cells cultured under hypoxia-mimicking conditions were subjected to cell migration assays. Total and migratory cells were evaluated. The results were expressed as the ratio of the number of migratory cells of the indicated cell lines to the number of migratory cells of the Mock cell line (left-hand panel). Invasion assays were performed using Matrigel invasion chambers (right panel). D, wild-type 501mel melanoma cells were transfected with the indicated siRNA (siC for non-targeting siRNA and siH1 or siH2 for, respectively, HIF1α or HIF2α targeting siRNA). Thirty-six hours after transfection, cells were depleted in FCS and treated with CoCl2 for 12 additional hours. Cell invasion assays were performed as above. The results were expressed as the ratio of the number of invasive cells of the indicated cell lines to the number of invasive cells of the control siRNA-transfected cell line. Error bars represent S.E. (n = 4 for migration assay, n = 3 for invasion assay). *p value < 0.01; **p value < 0.001.
Untransfected or stably transfected 501mel cells were subjected to cell migration assays under hypoxia-mimicking conditions. Interestingly, cell migration was significantly increased in HIF1α-transfected cells (1.39-fold, p value = 0.0008) and reduced (0.81-fold, p value = 0.003) in HIF2α-transfected cells relative to Mock-transfected or untransfected 501mel cells (Fig. 6C, left-hand panel). In order to metastasize, melanoma cells must invade the extracellular matrix. The invasion of 501mel cells under hypoxia-mimicking conditions into a Matrigel matrix was therefore assayed. Again, HIF1α-transfected cells showed a significantly higher invasion capacity than untransfected 501mel or Mock cells (1.4-fold, p value = 0.0006). In comparison, HIF2α-transfected cells were less able to invade a Matrigel matrix (0.68-fold, p value = 0.0005) (Fig. 6C, right-hand panel).
In HIF1α and HIF2α stably transfected cell lines, both HIFα isoforms are expressed. To decipher the role of each HIFα isoform in the invasive capacity of the 501mel cell line, siRNA-mediated knock-down experiments were carried out. siRNA targeting HIF1α or HIF2α or an irrelevant siRNA was transfected into 501mel cells. Cells were placed under hypoxia-mimicking conditions 36 h after transfection, and invasion assays were performed. The knock-down efficiency was assayed using RT-qPCR and Western blotting (supplemental Figs. S7A, S7B). For both HIF1α and HIF2α, more than 95% of the protein was depleted. As expected, whereas HIF1α knock-down moderately reduced the invasive capacity of cells relative to the control (1.26-fold, p value = 0.006), HIF2α knock-down strongly enhanced invasion (2.14-fold, p value = 0.002) (Fig. 6D).
In conclusion, HIF1 enhanced the invasive capacity of 501mel cells, whereas HIF2 severely decreased it, possibly through its interaction with AP2α.
Melanoma Cells Co-expressing AP2α and HIF2α Exhibit Decreased Invasive Capacities
Six different human melanoma cell lines were used in an attempt to correlate the expression of both AP2α and HIF2α proteins with the cells' invasive capacities. Cells were cultured under normoxia or hypoxia-mimicking conditions. Western blot analysis of AP2α expression discriminated two groups of melanoma cell lines: G1, 501mel, Daju, and SK-MEL-28 clearly expressed AP2α, and Lyse and Lu1205 had no or little detectable AP2α protein. Hypoxia did not change AP2α expression levels in any cell line considered (Fig. 7A). All melanoma cell lines tested expressed HIF1α under hypoxia, whereas only a subset expressed HIF2α. G1 and 501mel exhibited large amounts of HIF2α under hypoxic conditions, whereas both SK-MEL-28 and Lu1205 cell lines expressed small amounts, and Lyse and Daju cell lines had no detectable HIF2α (Fig. 7A). In order to evaluate the possible role of HIFα subunit expression on the invasive properties of melanoma cell lines in relation to AP2α status, Matrigel invasion assays were performed with cells cultured for 24 h either under normoxia (no expression of HIFα subunits) or under hypoxia. For each cell line, the percentage of invasive cells under hypoxia was divided by the percentage of invading cells measured under normoxia. Melanoma cells expressing high levels of HIF2α under hypoxia (G1 and 501mel) exhibited a decrease in their invasive capacities under low oxygen pressure relative to normoxic conditions. Conversely, melanoma cells expressing only HIF1α under hypoxia (Lyse and Daju) showed a greater capacity to invade Matrigel under low oxygen pressure. Cells expressing low amounts of HIF2α under hypoxia (SK-MEL-28 and Lu1205) showed an intermediate modification of their invasive abilities (Fig. 7B). Contrary to an earlier report (34), we found no correlation between AP2α expression and invasive capacity in either normoxic or hypoxic conditions (supplemental Fig. S8). We therefore postulated that simultaneous expressions of both AP2α and HIF2α were required in order to alter cell invasion potential. To test this hypothesis, cell lines naturally expressing high or low amounts of AP2α (Daju or Lu1205 cell lines, respectively) were transiently transfected with an HIF2α expression vector or an empty vector (Mock). The transfected cells strongly expressed HIF2α even under normoxia, as confirmed by Western blot analysis. In these conditions, the amount of HIF2α detected in the transfected cells was comparable to the physiological level found in the G1 cell line cultured under hypoxia (Fig. 7C; see inset). Matrigel invasion assays were performed under normoxia 36 h after transfection. HIF2α overexpression in the AP2α-negative cell line Lu1205 did not change its invasive ability. In contrast, the HIF2α-transfected Daju cell line exhibited a significantly altered invasion capacity (0.51-fold, p value = 0.001), suggesting that the simultaneous high expression of AP2α and HIF2α weakens the invasive capacities of melanoma cells (Fig. 7C).
Fig. 7.
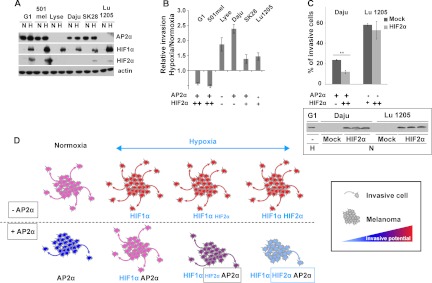
Melanoma cell lines expressing both AP2α and HIF2α exhibit poor invasive properties. A, Western-blot analysis of AP2α, HIF1α, and HIF2α expression in six different human melanoma cell lines. Actin was used as a loading control. N, normoxic condition; H, hypoxia-mimicking condition. The blot presented is representative of three distinct experiments. SK28, SK-MEL-28. B, evaluation of the invasion capacity of six human melanoma cell lines. Data are calculated as the ratio of the number of cells invading Matrigel under hypoxia-mimicking conditions divided by the number of cells invading the gel under normoxic conditions. Data are the means of three independent experiments. C, analysis of the invasion potential of Daju and Lu1205 melanoma cells transiently transfected with either a plasmid expressing HIF2α or the empty vector (Mock). Data are presented as the relative invasion of HIF2α transfected cells normalized to Mock transfected cells. Experiments were performed in triplicate. Error bars represent S.E. (n = 3). **p value < 0.001. The inset presents a Western blot analysis of HIF2α expression in wild-type unstranfected (−) G1 cells cultured under hypoxic conditions (H) and Daju and Lu1205 cells transfected either with an empty vector (Mock) or with a plasmid-expressing HIF2α and cultured under normoxic conditions (N). The three lanes for each condition correspond to protein extracts from three independent transfections. D, schematic illustration of AP2α, HIF1α, and HIF2α contributions to invasive capacities in melanoma cells growing under normoxic and hypoxic conditions.
As summarized in the model proposed in Fig. 7D, our compelling results demonstrate that a high HIF2α level under hypoxia leads to a decrease in the invasive capacities only of cells expressing high levels of AP2α. In other cases, hypoxia increases the invasive properties of melanoma cell lines as already described (19).
DISCUSSION
The oxygen distribution in skin is highly heterogeneous. Oxygen tension at the dermal–epithelial junction where melanocytes reside is about 5%, whereas it is about 0.5% around hair follicles (36). Oxygen availability has important physiological consequences, including the mediation of cellular transformation, especially during the melanocyte-melanoma transition (14). Cellular adaptation to hypoxia is facilitated by the expression of HIFα subunits, mainly HIF1α and HIF2α. HIF proteins are transcription factors that, under hypoxia, enable the activation of target genes involved in metabolism regulation, stress adaptation, growth, migration and invasion, drug resistance, and apoptotic cell death (37).
Protein function is frequently regulated by assembly into complexes. This aspect is particularly important for transcription factors, which interact with co-activators or repressors and with the transcription machinery to regulate gene expression. Indeed, transcription factor functions are profoundly remodeled by protein–protein interactions. Although several partners of HIF1 or HIF2 have already been described (supplemental Table S1), no global interactome analysis of HIF proteins is currently available. Furthermore, none of the well-characterized protein partners of these subunits have been identified in melanoma cells. In the present study, we performed a full screening of the HIF2α nuclear interactome in 501mel cells. The majority of the HIF2α protein partners identified are involved in the initiation of gene transcription. Mediator and SMARC complexes were found associated with the HIF2α subunit. Both protein complexes have already been shown to regulate melanoma development. The deletion of MED1, a component of the mediator complex, is known to increase melanoma aggressiveness (38). In the same way, when underexpressed in a cell line exhibiting a BRAF mutation (V600E) as in 501mel cells, SMARCB1, a component of the SMARC complex, prevents senescence and promotes melanomagenesis (39, 40).
The current study also shows for the first time that HIF2α interacts with the products of two well-characterized melanoma master genes, MITF and SOX10. These direct or indirect interactions were also observed with the HIF1α subunit. SOX10 plays a key role in the development of the melanocyte lineage, mediated in part by transcriptional control of MITF expression (41). SOX10 is expressed in most, if not all, primary and metastatic melanoma cells. Moreover, this transcription factor regulates the expression of nestin, a marker of melanocytic stem cells that correlates with poor prognosis (33, 42). MITF also has a key role regarding melanocytes and melanoma in that it regulates cell cycle progression, survival, and differentiation (19, 43, 44). Under hypoxia, MITF stimulates HIF1α expression in melanocytes, resulting in enhanced cell survival and improved resistance to UVB mutations leading to the malignant transformation of melanocytes to melanoma (45). In melanoma, HIF1α also indirectly represses MITF expression (19, 46). In the 501mel cell line used in the current study, MITF down-regulation was not observed under hypoxia, even though this cell line carries a BRAF V600E mutation known to stimulate HIF1α expression (47).
As reported above, the capacity of 501mel cells to invade an extracellular matrix is modified when the expression of the HIFα subunits is modulated. MITF is also known to regulate the aggressiveness of melanoma cell lines. Cells expressing small amounts of MITF are more invasive, and cells expressing large amounts of MITF are more differentiated (48, 49). However, several data argue against a key role of the regulation of MITF expression and function by HIF isoforms in explaining the observed effect on cell invasion. We found that neither HIF overexpression nor HIF down-regulation using specific siRNA lead to a modification of MITF expression (supplemental Fig. S7). Concerning its function, we found that the expression of a panel of MITF target genes was not globally modified by HIF1α or HIF2α overexpression or down-regulation (data not shown). Therefore, the biological significance of the interaction between HIF1α or HIF2α and MITF, and also SOX10, deserves further investigation. In contrast to MITF and SOX10, the transcription factor AP2α was identified in the current study as a partner of HIF2α, but not of HIF1α, in the 501mel melanoma cell line (Figs. 3 and 4). We further demonstrated that the co-expression of AP2α and HIF2α has functional consequences for the invasive properties of tumor cells (Figs. 6 and 7). In many tumor types, including melanoma, AP2α acts as a tumor suppressor gene by activating p21Waf1/Cip1 expression and thereby inducing cell cycle arrest (50). Quantitative analysis of a large retrospective cohort of patients suffering from melanoma reveals that a high cytoplasmic-to-nuclear ratio of AP2α correlates with a poor prognosis. In addition, a loss of AP2α expression has been associated with malignant transformation and melanoma progression (34, 51). Also, a recent in vitro study demonstrated that AP2α transcription factor overexpression could reduce cell invasion (52). Consistent with this observation, the very aggressive melanoma A375SM exhibits greatly reduced invasive capacities, tumor growth rates, and ability to metastasize when transfected with a plasmid allowing AP2α overexpression (53). Conversely, the overexpression of a dominant-negative mutant of AP2α in a non-metastatic melanoma cell line, SB2, increases the aggressiveness of the cell line (53). In 501mel cells, AP2α interacts with HIF2α but not with HIF1α (Figs. 3 and 4). HIF2α overexpression in 501mel cells notably reduced cell invasion, whereas HIF1α overexpression significantly enhanced this property (Fig. 6). Similar defects in invasion potential have been observed in other human melanoma cell lines expressing HIF2α under hypoxia. Strikingly, HIF2α overexpression in a cell line expressing very small amounts of AP2α had no effect on cell invasion, whereas HIF2α overexpression in AP2α-expressing cells was associated with a decrease in invasion potential (Fig. 7). Thus, we propose that the interaction between these two transcription factors in hypoxic conditions might modify the target genes they interact with and, consequently, reduce cell invasion properties.
In summary, our data strongly suggest that the simultaneous presence of both AP2α and HIF2α in melanoma cells might be a useful prognostic factor for patients. It will be very interesting to test this hypothesis in immunohistochemistry studies on large collections of melanoma tumors. Drugs targeting HIF proteins are currently under clinical assessment in patients. Our work underlines that, although important, the function of the HIFα subunits might be modulated by partners that confer opposing properties. Therefore, the functions of HIF1α and HIF2α must be investigated specifically in each human tumor type.
Supplementary Material
Acknowledgments
We acknowledge Dr. Irwin Davidson for his suggestions of MITF target genes and Dr. Lionel Larue for providing melanoma cell lines and for very useful discussions about this project. We also thank Pamela Houston for critical reading of the manuscript and Françoise Viala and David Bouyssié for, respectively, iconographic and bioinformatics assistance.
Footnotes
* Studies performed in our laboratories were supported by the University of Toulouse (AO CS 2008 to L.N.) and the Centre National de la Recherche Scientifique (CNRS), the Advanced Technology Institute in Life Sciences (ALMA project to M.E.), the Association contre le Cancer (ARC 2008-1102 to C.M.), and the Radioprotection Committee of EDF (C.M.). O.S. and B.M. were supported by grants from the Agence Nationale de la Recherche (ANR Plates-formes technologiques du vivant), Fondation pour la Recherche Médicale (Programme Grands Equipements), Région Midi-Pyrénées, and Infrastructures en Biologie Santé et Agronomie (IBISA).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- ACN
- acetonitrile
- FDR
- false discovery rate
- HIF
- hypoxia-inducible transcription factor
- IP
- immunopurification
- MITF
- microphthalmia-associated transcription factor
- PAI
- protein abundance index.
REFERENCES
- 1. Semenza G. L. (2011) Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 365, 537–547 [DOI] [PubMed] [Google Scholar]
- 2. Gordan J. D., Simon M. C. (2007) Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr. Opin. Genet. Dev. 17, 71–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wiesener M. S., Jurgensen J. S., Rosenberger C., Scholze C. K., Horstrup J. H., Warnecke C., Mandriota S., Bechmann I., Frei U. A., Pugh C. W., Ratcliffe P. J., Bachmann S., Maxwell P. H., Eckardt K. U. (2003) Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 17, 271–273 [DOI] [PubMed] [Google Scholar]
- 4. Maxwell P. H., Wiesener M. S., Chang G. W., Clifford S. C., Vaux E. C., Cockman M. E., Wykoff C. C., Pugh C. W., Maher E. R., Ratcliffe P. J. (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275 [DOI] [PubMed] [Google Scholar]
- 5. Semenza G. L. (2003) Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732 [DOI] [PubMed] [Google Scholar]
- 6. Semenza G. L. (2012) Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 33, 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raval R. R., Lau K. W., Tran M. G., Sowter H. M., Mandriota S. J., Li J. L., Pugh C. W., Maxwell P. H., Harris A. L., Ratcliffe P. J. (2005) Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol. Cell. Biol. 25, 5675–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ratcliffe P. J. (2007) HIF-1 and HIF-2: working alone or together in hypoxia? J. Clin. Invest. 117, 862–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Loboda A., Jozkowicz A., Dulak J. (2010) HIF-1 and HIF-2 transcription factors—similar but not identical. Mol. Cells 29, 435–442 [DOI] [PubMed] [Google Scholar]
- 10. Bennett D. C. (2008) How to make a melanoma: what do we know of the primary clonal events? Pigment Cell Melanoma Res. 21, 27–38 [DOI] [PubMed] [Google Scholar]
- 11. Postovit L. M., Seftor E. A., Seftor R. E., Hendrix M. J. (2006) Influence of the microenvironment on melanoma cell fate determination and phenotype. Cancer Res. 66, 7833–7836 [DOI] [PubMed] [Google Scholar]
- 12. Giatromanolaki A., Sivridis E., Kouskoukis C., Gatter K. C., Harris A. L., Koukourakis M. I. (2003) Hypoxia-inducible factors 1alpha and 2alpha are related to vascular endothelial growth factor expression and a poorer prognosis in nodular malignant melanomas of the skin. Melanoma Res. 13, 493–501 [DOI] [PubMed] [Google Scholar]
- 13. Valencak J., Kittler H., Schmid K., Schreiber M., Raderer M., Gonzalez-Inchaurraga M., Birner P., Pehamberger H. (2009) Prognostic relevance of hypoxia inducible factor-1alpha expression in patients with melanoma. Clin. Exp. Dermatol. 34, e962–e964 [DOI] [PubMed] [Google Scholar]
- 14. Bedogni B., Powell M. B. (2009) Hypoxia, melanocytes and melanoma—survival and tumor development in the permissive microenvironment of the skin. Pigment Cell Melanoma Res. 22, 166–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bedogni B., Welford S. M., Cassarino D. S., Nickoloff B. J., Giaccia A. J., Powell M. B. (2005) The hypoxic microenvironment of the skin contributes to Akt-mediated melanocyte transformation. Cancer Cell 8, 443–454 [DOI] [PubMed] [Google Scholar]
- 16. Monsel G., Ortonne N., Bagot M., Bensussan A., Dumaz N. (2010) c-Kit mutants require hypoxia-inducible factor 1alpha to transform melanocytes. Oncogene 29, 227–236 [DOI] [PubMed] [Google Scholar]
- 17. Sun B., Zhang D., Zhang S., Zhang W., Guo H., Zhao X. (2007) Hypoxia influences vasculogenic mimicry channel formation and tumor invasion-related protein expression in melanoma. Cancer Lett. 249, 188–197 [DOI] [PubMed] [Google Scholar]
- 18. Spinella F., Rosano L., Di Castro V., Decandia S., Nicotra M. R., Natali P. G., Bagnato A. (2007) Endothelin-1 and endothelin-3 promote invasive behavior via hypoxia-inducible factor-1alpha in human melanoma cells. Cancer Res. 67, 1725–1734 [DOI] [PubMed] [Google Scholar]
- 19. Cheli Y., Giuliano S., Botton T., Rocchi S., Hofman V., Hofman P., Bahadoran P., Bertolotto C., Ballotti R. (2011) Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene 30, 2307–2318 [DOI] [PubMed] [Google Scholar]
- 20. Bousquet-Dubouch M. P., Baudelet E., Guerin F., Matondo M., Uttenweiler-Joseph S., Burlet-Schiltz O., Monsarrat B. (2009) Affinity purification strategy to capture human endogenous proteasome complexes diversity and to identify proteasome-interacting proteins. Mol. Cell. Proteomics 8, 1150–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cravatt B. F., Simon G. M., Yates J. R., 3rd (2007) The biological impact of mass-spectrometry-based proteomics. Nature 450, 991–1000 [DOI] [PubMed] [Google Scholar]
- 22. Gavin A. C., Maeda K., Kuhner S. (2011) Recent advances in charting protein-protein interaction: mass spectrometry-based approaches. Curr. Opin. Biotechnol. 22, 42–49 [DOI] [PubMed] [Google Scholar]
- 23. Nakatani Y., Ogryzko V. (2003) Immunoaffinity purification of mammalian protein complexes. Methods Enzymol. 370, 430–444 [DOI] [PubMed] [Google Scholar]
- 24. Bouyssie D., Gonzalez de Peredo A., Mouton E., Albigot R., Roussel L., Ortega N., Cayrol C., Burlet-Schiltz O., Girard J. P., Monsarrat B. (2007) Mascot file parsing and quantification (MFPaQ), a new software to parse, validate, and quantify proteomics data generated by ICAT and SILAC mass spectrometric analyses: application to the proteomics study of membrane proteins from primary human endothelial cells. Mol. Cell. Proteomics 6, 1621–1637 [DOI] [PubMed] [Google Scholar]
- 25. Gautier V., Mouton-Barbosa E., Bouyssie D., Delcourt N., Beau M., Girard J. P., Cayrol C., Burlet-Schiltz O., Monsarrat B., Gonzalez de Peredo A. (2012) Label-free quantification and shotgun analysis of complex proteomes by one-dimensional SDS-PAGE/NanoLC-MS: evaluation for the large scale analysis of inflammatory human endothelial cells. Mol. Cell. Proteomics 11, 527–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mazars R., Gonzalez-de-Peredo A., Cayrol C., Lavigne A. C., Vogel J. L., Ortega N., Lacroix C., Gautier V., Huet G., Ray A., Monsarrat B., Kristie T. M., Girard J. P. (2010) The THAP-zinc finger protein THAP1 associates with coactivator HCF-1 and O-GlcNAc transferase: a link between DYT6 and DYT3 dystonias. J. Biol. Chem. 285, 13364–13371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Conaway R. C., Conaway J. W. (2011) Function and regulation of the Mediator complex. Curr. Opin. Genet. Dev. 21, 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Flanagan P. M., Kelleher R. J., 3rd, Sayre M. H., Tschochner H., Kornberg R. D. (1991) A mediator required for activation of RNA polymerase II transcription in vitro. Nature 350, 436–438 [DOI] [PubMed] [Google Scholar]
- 29. Tsai C. J., Nussinov R. (2011) Gene-specific transcription activation via long-range allosteric shape-shifting. Biochem. J. 439, 15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hargreaves D. C., Crabtree G. R. (2011) ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Rourke J. F., Tian Y. M., Ratcliffe P. J., Pugh C. W. (1999) Oxygen-regulated and transactivating domains in endothelial PAS protein 1: comparison with hypoxia-inducible factor-1alpha. J. Biol. Chem. 274, 2060–2071 [DOI] [PubMed] [Google Scholar]
- 32. Palmieri G., Capone M., Ascierto M. L., Gentilcore G., Stroncek D. F., Casula M., Sini M. C., Palla M., Mozzillo N., Ascierto P. A. (2009) Main roads to melanoma. J. Transl. Med. 7, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harris M. L., Baxter L. L., Loftus S. K., Pavan W. J. (2010) Sox proteins in melanocyte development and melanoma. Pigment Cell Melanoma Res. 23, 496–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Melnikova V. O., Bar-Eli M. (2008) Transcriptional control of the melanoma malignant phenotype. Cancer Biol. Ther. 7, 997–1003 [DOI] [PubMed] [Google Scholar]
- 35. Kaverina I., Krylyshkina O., Small J. V. (2002) Regulation of substrate adhesion dynamics during cell motility. Int. J. Biochem. Cell Biol. 34, 746–761 [DOI] [PubMed] [Google Scholar]
- 36. Evans S. M., Schrlau A. E., Chalian A. A., Zhang P., Koch C. J. (2006) Oxygen levels in normal and previously irradiated human skin as assessed by EF5 binding. J. Invest. Dermatol. 126, 2596–2606 [DOI] [PubMed] [Google Scholar]
- 37. Pouyssegur J., Dayan F., Mazure N. M. (2006) Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441, 437–443 [DOI] [PubMed] [Google Scholar]
- 38. Ndong Jde L., Jean D., Rousselet N., Frade R. (2009) Down-regulation of the expression of RB18A/MED1, a cofactor of transcription, triggers strong tumorigenic phenotype of human melanoma cells. Int. J. Cancer 124, 2597–2606 [DOI] [PubMed] [Google Scholar]
- 39. Wajapeyee N., Serra R. W., Zhu X., Mahalingam M., Green M. R. (2008) Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 132, 363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson J. P. (1999) Cell adhesion molecules in the development and progression of malignant melanoma. Cancer Metastasis Rev. 18, 345–357 [DOI] [PubMed] [Google Scholar]
- 41. Murisier F., Guichard S., Beermann F. (2007) The tyrosinase enhancer is activated by Sox10 and Mitf in mouse melanocytes. Pigment Cell Res. 20, 173–184 [DOI] [PubMed] [Google Scholar]
- 42. Tanabe K., Amoh Y., Kanoh M., Takasu H., Sakai N., Sato Y., Katsuoka K. (2010) Prognostic significance of the hair follicle stem cell marker nestin in patients with malignant melanoma. Eur. J. Dermatol. 20, 283–288 [DOI] [PubMed] [Google Scholar]
- 43. Vachtenheim J., Borovansky J. (2010) “Transcription physiology” of pigment formation in melanocytes: central role of MITF. Exp. Dermatol. 19, 617–627 [DOI] [PubMed] [Google Scholar]
- 44. Pinner S., Jordan P., Sharrock K., Bazley L., Collinson L., Marais R., Bonvin E., Goding C., Sahai E. (2009) Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 69, 7969–7977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Busca R., Berra E., Gaggioli C., Khaled M., Bille K., Marchetti B., Thyss R., Fitsialos G., Larribere L., Bertolotto C., Virolle T., Barbry P., Pouyssegur J., Ponzio G., Ballotti R. (2005) Hypoxia-inducible factor 1{alpha} is a new target of microphthalmia-associated transcription factor (MITF) in melanoma cells. J. Cell Biol. 170, 49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Feige E., Yokoyama S., Levy C., Khaled M., Igras V., Lin R. J., Lee S., Widlund H. R., Granter S. R., Kung A. L., Fisher D. E. (2011) Hypoxia-induced transcriptional repression of the melanoma-associated oncogene MITF. Proc. Natl. Acad. Sci. U.S.A. 108, E924–E933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kumar S. M., Yu H., Edwards R., Chen L., Kazianis S., Brafford P., Acs G., Herlyn M., Xu X. (2007) Mutant V600E BRAF increases hypoxia inducible factor-1alpha expression in melanoma. Cancer Res. 67, 3177–3184 [DOI] [PubMed] [Google Scholar]
- 48. Carreira S., Goodall J., Denat L., Rodriguez M., Nuciforo P., Hoek K. S., Testori A., Larue L., Goding C. R. (2006) Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 20, 3426–3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lekmine F., Chang C. K., Sethakorn N., Das Gupta T. K., Salti G. I. (2007) Role of microphthalmia transcription factor (Mitf) in melanoma differentiation. Biochem. Biophys. Res. Commun. 354, 830–835 [DOI] [PubMed] [Google Scholar]
- 50. Zeng Y. X., Somasundaram K., el-Deiry W. S. (1997) AP2 inhibits cancer cell growth and activates p21WAF1/CIP1 expression. Nat. Genet. 15, 78–82 [DOI] [PubMed] [Google Scholar]
- 51. Berger A. J., Davis D. W., Tellez C., Prieto V. G., Gershenwald J. E., Johnson M. M., Rimm D. L., Bar-Eli M. (2005) Automated quantitative analysis of activator protein-2alpha subcellular expression in melanoma tissue microarrays correlates with survival prediction. Cancer Res. 65, 11185–11192 [DOI] [PubMed] [Google Scholar]
- 52. Braeuer R. R., Zigler M., Villares G. J., Dobroff A. S., Bar-Eli M. (2011) Transcriptional control of melanoma metastasis: the importance of the tumor microenvironment. Semin. Cancer Biol. 21, 83–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huang S., Jean D., Luca M., Tainsky M. A., Bar-Eli M. (1998) Loss of AP-2 results in downregulation of c-KIT and enhancement of melanoma tumorigenicity and metastasis. EMBO J. 17, 4358–4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.