

Abstract
Chromatin is a highly dynamic, well-structured nucleoprotein complex of DNA and proteins that controls virtually all DNA transactions. Chromatin dynamicity is regulated at specific loci by the presence of various associated proteins, histones, post-translational modifications, histone variants, and DNA methylation. Until now the characterization of the proteomic component of chromatin domains has been held back by the challenge of enriching distinguishable, homogeneous regions for subsequent mass spectrometry analysis. Here we describe a modified protocol for chromatin immunoprecipitation combined with quantitative proteomics based on stable isotope labeling by amino acids in cell culture to identify known and novel histone modifications, variants, and complexes that specifically associate with silent and active chromatin domains. Our chromatin proteomics strategy revealed unique functional interactions among various chromatin modifiers, suggesting new regulatory pathways, such as a heterochromatin-specific modulation of DNA damage response involving H2A.X and WICH, both enriched in silent domains. Chromatin proteomics expands the arsenal of tools for deciphering how all the distinct protein components act together to enforce a given region-specific chromatin status.
In eukaryotes, genetic information is stored in chromatin, a highly structured nucleoprotein complex that mediates the coordinated regulation of gene expression. The basic unit of chromatin is the nucleosome, which consists of 147 bp of DNA wound around a histone octamer core containing two copies of the histones H2A, H2B, H3, and H4 (1, 2). Changes in chromatin structure and composition that do not involve the nucleotide sequence can translate into transient or heritable adjustments in gene expression (3, 4). Various mechanisms modulate chromatin states; among these, a prominent role is played by covalent histone post-translational modifications (hPTMs),1 for which the repertoires of combinations and positions are extremely varied (5–7). In addition to hPTMs, chromatin is also modulated by the local enrichment of a distinct set of histone variants, binding proteins, including various ATP-dependent remodeling activities, DNA methylation, and differential nucleosome density. Together, these components contribute to the establishment of a specific “chromatin landscape,” enforcing a functional state of the underlying DNA (8).
Chromatin immunoprecipitation (ChIP) and mass spectrometry (MS) are popular and complementary strategies for investigating the epigenetic components of chromatin. ChIP with specific antibodies followed by massively parallel DNA sequencing (ChIP-Seq) allows genome-wide profiling of hPTMs and binders at individual genes and regulatory regions, up to the resolution of individual nucleosomes (9–12). However, ChIP does not provide information about the protein portion of chromatin; such knowledge is instead offered by MS-based proteomics. At the level of individual histones, MS enables one to detect virtually all hPTMs in an unambiguous, unbiased, and global fashion and reveals the interplay among them (13–16). Yet MS analysis of bulk chromatin preparations limits the inspection of PTMs to a global view, with no information about their patterning in distinct functional regions.
“Chromatomics” and “epigenomics” combine these strategies to allow the thorough inspection of both the nucleic acid and the protein component of chromatin, thus yielding a “tridimensional” representation (at the genome, transcriptome, and proteome levels) of the epigenetic phenomena that underlie the establishment and inheritance of specific expression patterns (17, 18). Although extensive ChIP-seq data are already available for virtually all known hPTMs, as well as for a large set of chromatin binders, dissection of the proteomic architecture of chromatin is an ongoing effort (reviewed in Ref. 19).
Affinity-interaction assays using histone-peptide baits in conjunction with proteomics based on stable isotope labeling by amino acids in cell culture (SILAC) were recently developed to pull down and identify proteins specifically interacting with distinct H3 and H4 trimethyl lysines (20, 21). The approach has since been modified into the SILAC nucleosome affinity purification method, whereby in vitro reconstituted nucleosomes are used as bait in affinity purification assays, which are followed by MS in order to obtain a “modification binding profile” of proteins whose interactions are regulated by the cooperative contribution of both DNA methylation and hPTMs (22). Similarly, SILAC-based affinity purification assays also are carried out with recombinant, uniformly modified chromatin templates (23). Finally, the cross-linking assisted and SILAC-based protein identification approach conjugates SILAC with chemical proteomics using photo-cross-linking-based histone peptide probes to detect weak but specific interactions that might be missed with standard pull-down approaches (25).
These analytical strategies represent powerful, established methods to screen for “histone code readers.” However, because of the difficulty of enriching chromatin samples from specific regions in a quantity and purity sufficient for MS analysis, these methods remain limited in terms of the information they provide on histone variants, hPTMs, and their binders in distinct chromatin territories (26, 27).
An elegant proteomics approach that addresses this restraint is mChIP, wherein tandem-affinity-purification-tagged versions of the histone variants Hta2p and Htz1p are used to obtain a qualitative list of co-enriched proteins in S. cerevisiae (28). A global identification of telomere-associated proteins was achieved by Kingston and co-workers through an approach utilizing proteomics of isolated chromatin, wherein cross-linked telomeric regions enriched in repetitive DNA sequences are “captured” and purified with complementary probes immobilized on beads (29).
Recently, a strategy based on the immunoprecipitation of HPLC-purified histone H3 followed by MS was described as a useful tool for both assessing the specificity of antibodies used in ChIP and observing PTM associations within the same molecule (30). Actually, because this method is not an authentic ChIP that immunopurifies intact nucleosomes, it falls short in providing information on hPTM synergisms at the intermolecular level or on functional interactions with histone variants and readers.
Building on these approaches, we describe a global and quantitative proteomic strategy named ChroP, for “chromatin proteomics,” designed to analyze the protein component characterizing distinct chromatin regions, enriched by means of a modified and preparative version of ChIP. Antibodies against trimethylated lysine 9 and lysine 4 on histone H3 (H3K9me3 and H3K4me3) were used to enrich functionally distinct and non-overlapping chromatin regions from HeLa nuclei. High-resolution MS of the fractionated nucleosomes enabled a dissection of the domain-specific composition of histone modifications, variants, and non-histonic proteins, which we refer to as the modificome and interactome.
We observed the expected combinatorial enrichment of silent modifications in H3K9me3 and of active ones in H3K4me3, not only at the intramolecular level within H3, but also across the different core histones, within the same nucleosome. More important, our approach identified a number of histone variants and multi-protein complexes differentially enriched in the two functional territories, suggesting potential novel roles for these proteins and indicating pathways regulating chromatin. A representative case was the variant H2A.X with the WICH chromatin-remodeling complex accumulating in the H3K9me3 regions. We propose a higher local density model for H2A.X in heterochromatin and show that this accumulation correlates with increased phosphorylation of Tyr142, as a consequence of the recruitment of WICH to the same region. As it has been proposed that Tyr142p and Ser139p (γ-H2A.X) function as a molecular switch with regulatory effects on the DNA damage response (31), our data allow speculation that this further level of DNA damage response modulation might be heterochromatin specific.
ChroP delivers comprehensive information on domain-specific chromatin features. Such information will facilitate a deeper understanding of the molecular mechanisms that link the accumulation of a given histone mark, variant, or interactor in a specific region with its functional state.
EXPERIMENTAL PROCEDURES
Cell Culture and SILAC Labeling
HeLa S3 cells were grown in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, catalog number 10270–106), 1% glutamine, 1% Pen/Strep, and 10 mm HEPES pH 7.5. For metabolic labeling, HeLa S3 cells were grown in “heavy” and “light” SILAC media prepared by adding to the SILAC DMEM (M-Medical, catalog number FA30E15086 Pasching, Austria), depleted of lysine and arginine, 10% dialyzed FBS (Invitrogen, catalog number 26400–044), 1% glutamine, 1% Pen/Strep, 10 mm HEPES pH 7.5, and either the light isotope-coded amino acids 12C6 14N2 l-Lysine (Lys 0, Sigma, catalog number L8662) and 12C6 14N4 l-Arginine (Arg0, Sigma, catalog number A6969) or their heavy isotope-counterparts, 13C6 15N2 l-Lysine (Lys 8, Sigma, catalog number 68041) and 13C6 15N4 l-Arginine (Arg10, Sigma, catalog number 608033). Lys and Arg were added at concentrations of 73 mg/l and 42 mg/l, respectively. HeLaS3 were cultivated in SILAC media for nine generations, with careful monitoring of their growth rate, viability, and overall morphology, to ensure that normal physiology was preserved.
NIH 3T3 mouse fibroblasts were grown in DMEM supplemented with 10% calf serum (Lonza, catalog number 14–401F Basel, Switzerland), 1% glutamine, and 1% Pen/Strep.
Native ChIP
The protocol developed was modified from a previously described one (32). Two hundred million HeLa S3 cells were homogenized in lysis buffer (see the supplemental “Experimental Procedures” section for details), and nuclei were separated from the cytoplasm via centrifugation at 3750 rpm (4 °C) for 30 min, laying down cellular lysate on sucrose cushions. Nuclear pellets were washed twice with PBS, re-suspended in digestion buffer (see the supplemental “Experimental Procedures” section for details), and digested with micrococcal nuclease (Roche) at a final concentration of 0.005 U/μl at 37 °C for 60 min. The reaction was stopped by adding 1 mm EDTA and chilling on ice. The soluble fraction of chromatin (S1), comprising small fragments (mono- and di-nucleosomes) (supplemental Fig. S1A, left-hand panel), was collected as the supernatant after the centrifugation of re-suspended nuclei at 10,000 rpm (4 °C) for 10 min. Pellets were re-suspended in dialysis buffer (see the supplemental “Experimental Procedures” section for details) and dialyzed overnight at 4 °C in a dialysis tube (cutoff = 3.5 kDa). The second soluble fraction of chromatin (S2) (supplemental Fig. S1A, left-hand panel), comprising large fragments (tri- to epta-nucleosomes), was the supernatant obtained after centrifugation at 10,000 rpm (4 °C) for 10 min. DNA extracted via Qiaquick columns (Qiagen Hilden, Germany) was run on 1% agarose gel to evaluate fractions of chromatin. The S1 fraction was combined with a small aliquot of S2 fraction (1/100) in order to obtain an input composed of about 95% mono-nucleosomes (supplemental Fig. S1A, middle panel) and diluted in ChIP dilution buffer (50 mm Tris HCl pH 7.6, 50 mm NaCl, and 5 mm EDTA).
Chromatin was immunoprecipitated with 10 μg of the following antibodies: H3K9me3, H3K4me3, and H2A.X. Antibodies were incubated overnight with chromatin; in parallel, 100 μl of G-protein-coupled magnetic beads (Dynabeads, Invitrogen, catalog number 100.04D) were blocked in BSA 0.5% PBS overnight. Blocked beads were washed, added to chromatin, and incubated for 3 h at 4 °C on a rotating wheel. Beads were washed four times (50 mm Tris-HCl pH 7.6, 10 mm EDTA) at increasing salt concentrations (75, 125, and 175 mm NaCl). LDS sample buffer (Invitrogen, catalog number NP0007) supplemented with 50 mm DTT was added to the beads for 5 min at 70 °C to elute the immunoprecipitated proteins from the beads. Proteins were resolved on 4%–12% Bis-Tris acrylamide SDS-PAGE pre-cast gels (Invitrogen, catalog number NP0335BOX) on an Invitrogen system and visualized on the gel using a Colloidal Comassie staining kit (Invitrogen, catalog number LC6025).
Cross-linking ChIP
The protocol developed was a modification of a previously published one (33). Two hundred million SILAC-labeled HeLa S3 cells were harvested, and cell pellets were cross-linked in 0.75% formaldehyde PBS for 20 min at room temperature, with shaking on a rotating wheel, to stabilize protein–DNA and protein–protein interactions. Formaldehyde was quenched by the addition of 125 mm glycine for 5 min. After four washes with cold PBS, cells were suspended in lysis buffer (see the supplemental “Experimental Procedures” section for details) for 10 min at 4 °C. After centrifugation, the nuclear pellets were washed once and then re-suspended in ChIP incubation buffer (see the supplemental “Experimental Procedures” section for details). Chromatin from nuclei was sonicated at 200 W for 15 min (cycles of 30 s “on” and 1 min “off” in a cooled Bioruptor (Diagenode inc. North America, USA)). After sonication, 1% of Triton-100 was added to the sonicated chromatin to pellet debris. Soluble nucleosomes contained in the soluble supernatant after centrifugation at 13,000 rpm (4 °C) for 10 min were immunoprecipitated via the addition of 10 μg of the following antibodies: H3K9me3, H3K4me3, and H2A.X. The immunoprecipitation procedure followed the same steps as the native ChIP (N-ChIP), except for the washes, which were carried out in 20 mm Tris-HCl pH 7.6, 2 mm EDTA, 0.1% SDS, 1% Triton-100, and increasing NaCl concentration (150 and 300 mm). To reverse the cross-linking and elute the immunoprecipitated proteins, SDS-PAGE sample buffer (250 mm Tri-HCl pH 8.8, 0.5 m β-mercaptoethanol, 2% SDS) was added to the beads for 25 min at 95 °C. Proteins were resolved on 4%–12% Bis-Tris acrylamide SDS-PAGE pre-cast gradient gels (Invitrogen) and visualized using a Colloidal Coomassie staining kit (Invitrogen).
Liquid Chromatography and Tandem Mass Spectrometry
Peptide mixtures were separated via nano-flow liquid chromatography (LC) using an Agilent 1100 Series (Agilent Technologies Waldbronn, Germany) coupled to a 7-tesla linear ion trap–Fourier transform–ion cyclotron resonance (LTQ-FT-ICR) Ultra mass spectrometer (ThermoFisher Scientific, Bremen, Germany). The nano-flow LC system was operated in a one-column set-up with a 15-cm analytical column (75 μm inner diameter, 350 μm outer diameter) packed with C18 resin (ReproSil, Pur C18AQ 3 μm, Dr. Maisch, Ammerbuch Germany). Solvent A was 0.1% formic acid and 5% acetonitrile in double-distilled H2O, and solvent B was 95% acetonitrile with 0.1% formic acid. The sample was injected into an aqueous solution at a flow rate of 500 nl/min. Peptides were separated with a gradient of 0%–36% over 120 min followed by gradients of 36%–60% for 10 min and 60%–80% over 5 min at a flow rate of 250 nl/min. For histones, liquid chromatography separation was performed with a gradient of 0%–40% solvent B over 90 min followed by gradients of 40%–60% for 10 min and 60%–80% over 5 min at a flow rate of 250 nl/min. A nanoelectrospray ion source (Proxeon, Odense, Denmark) was used with a spray voltage of 2.4 kV. No sheath, sweep, or auxiliary gasses were used, and the capillary temperature was set at 190 °C. The mass spectrometer was operated in data-dependent mode to automatically switch between MS and MS/MS acquisition. In the mass spectrometer, full scan MS spectra (200–1650 m/z) were acquired with a resolution of 100,000 at 400 m/z, setting an Acquisition Gain Control target of 1,000,000. The five most intense ions were isolated for fragmentation in the linear ion trap using collision-induced dissociation at a target value of 5000. Singly charged precursor ions were excluded. In the MS/MS method, a dynamic exclusion of 60 s was applied, and the total cycle time was ∼2 s. The collision gas pressure was 1.3 millitorr, and the normalized collision energy using wide band activation mode was 35%. The ion selection threshold was 250 counts with an activation q = 0.25. An activation time of 30 ms was applied in MS/MS acquisitions.
Quantitative MS Analysis of hPTMs Co-enriched in Precipitated Chromatin
Raw MS data were converted to mgf files using Raw2MSM software (version 1.10) (34). MS/MS spectra were searched using Mascot Daemon (version 2.2.2, Matrix Science London, UK) against the International Protein Index human database (version 3.68; 87,061 entries). The MS mass tolerance was set at 10 ppm, and the MS/MS mass tolerance was set at 0.5 Da. The search included variables modifications, allowing two missed cleavages: lysine D3-acetylation (+45.0294 Da); lysine monomethylation (calculated as the sum of the masses of D3-acetylation (+45.0294) and monomethylation (+14.016 Da)), dimethylation (+28.031 Da), and trimethylation (42.046 Da); lysine acetylation (+42.010 Da); methionine oxidation (+15.995 Da); and N-terminal protein acetylation (+42.010 Da). Enzyme specificity was set to Arg-C. Low-confidence peptide identifications were filtered from the Mascot results according to the following criteria: peptides with either an ion score lower than 15 or more than five putative PTMs (15) were removed; redundant peptides with the same ID were filtered out by selecting the peptide with the highest Mascot score.
Extracted ion chromatograms were constructed for each precursor based on the m/z value, using a mass tolerance of 10 ppm and a mass precision up to four decimals. Histone PTMs were first quantified by calculating the area under the curve of each peak corresponding to every specific modified peptide (Fig. 1B). Then, their relative abundance was estimated by dividing the area under the curve of each peptide by the sum of the areas corresponding to all observed modified forms of that peptide ((extracted ion chromatogram modified peak/∑ extracted ion chromatogram all peaks) × 100). Modification enrichment was calculated as the ratio between the relative abundance in the ChIPed octamer and the corresponding relative abundance estimated from input. Peptides containing modifications were validated via the manual annotation of MS/MS spectra (see supplemental Fig. S10) using QualBrowser, version 2.0.7 (ThermoFisher Scientific). The theoretical masses of all modified histone peptides analyzed via MS/MS are reported in the supplemental “Experimental Procedures” section.
Fig. 1.
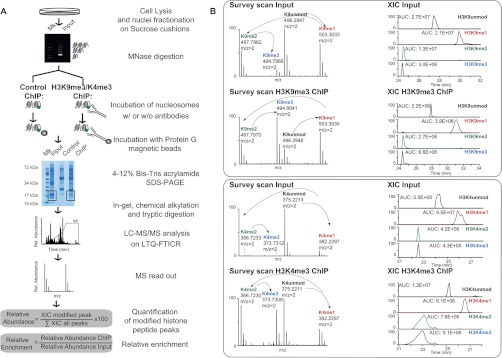
Scheme of N-ChroP strategy combining N-ChIP and MS analysis. A, scheme of the experimental approach. B, zoomed mass spectra and extracted ion chromatograms (XICs) constructed at the corresponding m/z value of the 2+ charge unmodified, mono-, di-, and trimethylated K9 (upper panel) and K4 (lower panel), for both input and ChIP. (See also supplemental Fig. S1.)
Proteomic Analysis of Proteins Co-associated within Precipitated Chromatin Fractions
Protein interactors from the cross-linking ChIPs (X-ChIPs) were identified and quantified using MaxQuant software (version 1.1.1.36) (35). MS/MS spectra were recorded in “centroid” mode, and the six most abundant peaks per 100-Da mass intervals were selected for search. Filtered MS/MS spectra were searched against the International Protein Index human database (version 3.68; 87,061 entries), combined with the standard MaxQuant contaminants database, using the Andromeda search engine (37). MaxQuant analysis included an initial search with a precursor mass tolerance of 20 ppm, and the results were used for subsequent mass recalibration (38). Enzyme specificity was set to trypsin, allowing two missed cleavages and cleavage at the N-terminus of proline. Peptide identification was based on a search with mass deviation of the precursor ion of 6 ppm, and the fragment mass tolerance was set at 0.5 Da. The mass accuracy of the precursor ions was improved by the time-dependent recalibration. MaxQuant was employed to filter identifications at a 1% false discovery rate at three levels, namely, site, peptide, and protein. Carbamidomethylation of cysteine was selected as a fixed modification, and oxidation of methionine and acetylation of the protein N-terminus were included as variable modifications. The modifications corresponding to arginine and lysine labeled with heavy stable isotopes (13C6 15N4 l-Arginine and 13C6 15N2 l-Lysine) were treated as fixed modifications in the Andromeda search. Additional peptides were identified by the “match between run” option in MaxQuant, which matches precursor masses in a 2-min retention time window (after realignment of the runs) based on the accurate mass measurement. Proteins were accepted if they were identified with at least two peptides, one of which was unique, in each experiment. Protein ratios were normalized by standard deviation; complete output tables for the H3K9me3 and H3K4me3 chromatomes are provided as supplemental Tables S1 and S2. Analysis and visualization of the data were performed using the open-source package R with in-house scripts and the Perseus program.2
Quantitative RT-PCR of Immunopurified DNA
DNA from ChIPed material was eluted in TE (10 mm Tris-HCl pH 7.5, 1mM EDTA) containing 2% SDS for 15 min at 65 °C. In X-ChIP, DNA was extensively de-cross-linked via incubation in the same buffer at 65 °C overnight. Subsequently, DNA was purified through Qiaquick columns (Qiagen). 1 μl of purified DNA was used as a substrate for amplification on an Applied Biosystems 7500 Fast Real-time PCR system applying Biosystem Sybr-green. Primers used for ChIP qPCR can be found in the supplemental “Experimental Procedures” section.
ChIP-Seq: Preparation of ChIP DNA Libraries and Sequencing
ChIP DNA was prepared for Solexa 2G sequencing using a standard protocol. For further information about the ChIP-Seq methods and data analysis, see the supplemental “Experimental Procedures” section.
RESULTS
Histone PTM Patterns Co-enriched at Specific Chromatin Regions via N-ChIP Combined with High-resolution MS
Chromatin immunoprecipitation enables profiling of the localization of a protein or a histone modification along the genome via deep sequencing. In a proteomics equivalent of such an experiment, ChIP is followed by the analysis of the immunopurified proteins via MS. We established a preparative version of the classical N-ChIP (namely, native ChIP without crosslinking) to purify chromatin regions enriched in a specific histone modification, in order to characterize by means of MS the hPTM patterns and proteins associated with such regions. We investigated fractions enriched in silent or active chromatin by taking advantage of the well-characterized enrichment of H3K9me3 at both pericentric heterochromatin and repressed euchromatic genes, and of H3K4me3 in active promoters. The protocol is outlined in Fig. 1A. Briefly, chromatin from HeLa S3 cells was digested with micrococcal nuclease; the fraction enriched in mono-nucleosomes (supplemental Fig. S1A, middle panel) was incubated with antibodies recognizing either H3K9me3 or H3K4me3. For immunoprecipitation, we chose two antibodies widely employed by the epigenetic community for ChIP-Seq studies with proved specificity and efficiency (39, 40). Immunopurified chromatin was captured on magnetic beads, and the extracted proteins were separated via SDS-PAGE (supplemental Fig. S1A, right-hand panel). Each core histone from the fractionated nucleosomes was analyzed via LC-MS/MS. PTMs were identified and quantified (Fig. 1B); their basal frequency was estimated in the input (supplemental Fig. S2), and their enrichment in the ChIP was calculated as described under “Experimental Procedures.”
The analysis first focused on histone H3 (9–17 and 3–8) peptides: a specific enrichment of K9me3, as well as of K4me3, associated with the depletion of unmodified H3 and H3 mono-methylated at K9 and K4 demonstrated the efficiency and specificity of the ChIP with the respective antibody (Fig. 2B). The depletion of acetylated H3K9/K14 was also observed in H3K9me3-nucleosomes (Fig. 2B). Western blot (WB) analysis confirmed the MS results, showing a specific enrichment of H3K9me3 and H3K4me3 in the corresponding immunoprecipitates (Fig. 2C). The potential cross-reactivity of the anti-H3K9me3 antibody with other methyl-lysines was excluded via a competition assay in which chromatin was incubated with α-H3K9me3 in the presence of an excess of different soluble peptides bearing the following modifications: K9me2, K4me3, K27me3 in H3, and K20me3 in H4. The unspecific recognition of any other Kme was excluded, except for K9me2, for which mild binding was detected (supplemental Fig. S1B). The comparison between H3K9me3 abundance in flow-through and input demonstrated that about 50% of the chromatin regions of interest were captured in the ChIP, ruling out a potential bias due to the enrichment of only a sub-fraction of chromatin (supplemental Fig. S1C).
Fig. 2.

Estimation of H3K9me3 and H3K4me3 enrichment in the corresponding ChIPs. A, schematic representation of the H3 N-terminal tail, with annotated modification and the peptides produced upon D6-acetic anhydride alkylation followed by in-gel trypsin digestion. Symbols and colors used to annotate different modifications are in the legend. B, relative enrichment of methylations on K9 (co-existing or not with acetylated K14) and on K4 in peptides spanning regions 9–17 and 3–8 of H3 upon N-ChIP using anti-H3K9me3 and anti-H3K4me3 antibodies, respectively. The enrichment is expressed as a log2 ratio of the relative abundance of each methylation in the ChIP sample as compared with the input and represents the averages ± S.E. (standard error of the mean) from three independent experiments. C, WB validation of H3K9me3 and H3K4me3 enrichment in the corresponding ChIPs: aliquots of ChIPed material and of input were loaded on SDS-PAGE and immunoblotted with antibodies H3K9me3 and H3K4me3 (#1 and #2 indicate two replicates). 0.2% and 0.04% of input were loaded for H3K9me3 and H3K4me3, respectively, for a semi-quantitative comparison with immunoprecipitation, using H3 as loading control for normalization. (See also supplemental Figs. S2 and S10.)
Label-free Quantification of hPTMs Enriched in Repressed and Active Chromatin Domains
The hPTMs enriched in silent and active chromatin domains are summarized in the heatmap and histograms in Fig. 3 and supplemental Fig. S3. Focusing on the modifications associated with histone H3, our analysis of intact native nucleosomes validated the selectivity of our antibodies and overall recapitulated the “chromatin canonical states” described by Ernst and Kellis based on a meta-analysis of a large set of data (41). H3K4me1 was found to be depleted in H3K9me3 nucleosomes (supplemental Fig. S3A), as was K4me3 (supplemental Fig. S4A), proving that K4me1, like K4me2 and K4me3, occurs mostly in transcriptionally active chromatin (42). In addition, we observed a depletion of K9 methylation and a parallel increase of K14ac and K9ac/K14ac in H3K4me3-euchromatin (supplemental Figs. S3B and S4B), as previously described (43). H3R26me2, in association with K23Ac, was found to be enriched in H3K4me3 domains (supplemental Fig. S3C); information on arginine methylation was partial, but emerging evidence suggests that H3R26me2 might antagonize Polycomb repression via its proximity to K27 (44, 45). We found H3K18me1 to be enriched in silent domains and depleted in active ones (Fig. 3C, upper left-hand panel, and supplemental Fig. S4C). Indeed, a silencing role for this mark had been already proposed, based on two observations: first, its half-maximal life was reported as significantly lower than those of all other mono-methylations with gene-activating function (46); second, it may antagonize K18ac, an active mark (47).
Fig. 3.
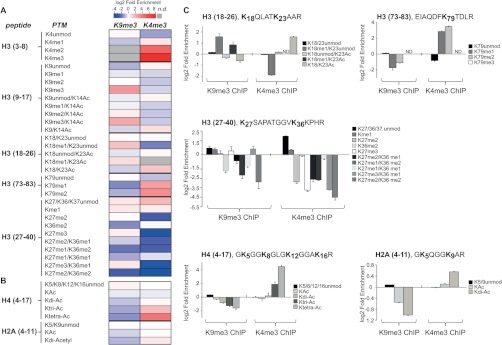
Relative enrichment of modifications in H3K9me3 and H3K4me3 mono-nucleosome. Heatmap of hPTM enrichment for all the modified resides identified on H3 (A), H4, and H2A histones (B). Each row corresponds to a distinct histone modification, and columns correspond to the different antibodies used for the ChIP (n.d., not detected). C, the relative enrichment of each modification is expressed as a log2 ratio between its relative abundance in the immunopurified nucleosomes and that in the input for histone H3, H4, and H2A peptides. Histograms represent the averages ± S.E. (standard error of the mean) from three independent experiments. (See also supplemental Figs. S3, S4, S5, and S10.)
Analysis of methylation profiles on peptide H3 (27–40) identified several distinct modified peptides. Chemical alkylation with D6-acetic anhydride, which labels unmodified and mono-methylated lysines with a deuterated acetyl moiety but does not react with dimethyl, trimethyl, or acetyl lysines, allowed us to distinguish different methylations on K27 and on K36/37. In fact, this chemical reaction results in a delta mass of 45.0294 Da for each D3-acetyl group and causes a different retention time during the chromatography of isobaric modification-bearing peptides, which are thus better separated prior to MS (supplemental Fig. S5). Quantitative analysis showed that H3K27me1 was enriched in heterochromatin (Fig. 3C, middle panel), confirming the findings of prior studies in which constitutive heterochromatin was characterized by the focal enrichment of H3K9me3 and H3K27me1 (48). Moreover, H3K27me3 was slightly enriched in heterochromatin and depleted in euchromatin, as previously described (Fig. 3C, middle panel; supplemental Figs. S4A and S4B) (18). As expected, H3K79me1 and -me2 were depleted in H3K9me3 domains but significantly increased in H3K4me3 domains (Fig. 3C, upper right-hand panel; supplemental Fig. S4B); indeed, H3K79 methylation is proposed to mark active, or at least accessible, genes and to function as a barrier to heterochromatin spreading (49).
Overall, the confirmation of previously described or predicted PTM synergisms confirmed the robustness of the newly established strategy and thus corroborated the discovery of novel associations among histone marks, both intramolecularly and intermolecularly, within the immunopurified nucleosomes. For instance, we observed that dimethylated K36 was depleted in both chromatin domains (Fig. 3C, middle panel; supplemental Figs. S4A and S4B). The unexpected underrepresentation of this mark in H3K4me3 regions can be explained by reasoning that H3K4me3 is typically enriched at proximal promoters, whereas H3K36me2/3 associate with gene elongation and thus are highly overrepresented at the 3′ of genes (50, 51); as such, this evidence further corroborates the high resolution of our analysis in dissecting PTMs clustering at a single nucleosome level.
We then extended the study to the PTMs of the co-purified histone H2A and H4. Acetylations on the H2A N-terminus were reduced in the H3K9me3 domains and enriched in H3K4me3 regions (Fig. 3C, lower right-hand panel); similarly, hyperacetylated H4 was found to be enriched in euchromatin (Fig. 3C, lower left-hand panel; supplemental Figs. S4A and S4B) (52). Methylation at lysine 20 of H4 was overrepresented in H3K9me3 domains but reduced in H3K4me3 domains as determined via WB (supplemental Figs. S4A and S4B); in fact, H4K20me3 follows H3K9 methylation during the initiation of a heterochromatic environment (53).
In conclusion, N-ChroP confirms that “active” modifications (methylated K4, K36, K79; acetylated K9/K14 and K18/K23 of H3; and hyperacetylated H2A and H4) are enriched in H3K4me3 domains, whereas “silent” modifications (methylated K9 and K27 of H3 and K20 of H4) are overrepresented in H3K9me3 territories (18, 42, 54); in addition, the approach shows novel associations among marks not described previously.
Large-scale Study of Chromatin-associated Proteins Employing X-ChIP Combined with High-resolution MS Analysis
To characterize H3K9me3- and H3K4me3-interacting proteins, we modified the cross-linking ChIP protocol to adapt it for SILAC-based quantitative interactomics (20). The protocol is outlined in Fig. 4A: HeLa S3 cells grown in light and heavy media were fixed with formaldehyde, and chromatin was subjected to sonication to generate 300- to 500-bp lengths of DNA fragments (supplemental Fig. S6A, left-hand panel). Both isotope-coded samples (heavy, H; light, L) were incubated with specific antibodies; in one of the two isotope channels, the antibody was saturated with an excess of soluble H3 peptide bearing the modification used as bait (either H3K9me3 or H3K4me3). All nucleosomes exposing those PTMs were competed out, as were all co-associating proteins; unspecific proteins were not selectively competed. Two replicates of the X-ChIP were performed, swapping the H and L channels in which the peptide-competition control was carried out (the so-called forward and reverse formats), in order to increase the discriminating potential of the approach. In the forward experiment, we incubated the specific antibody with an H-labeled chromatin preparation, and the excess of soluble peptide was added to the L-labeled chromatin input; in the reverse format, the two samples were inverted.
Fig. 4.
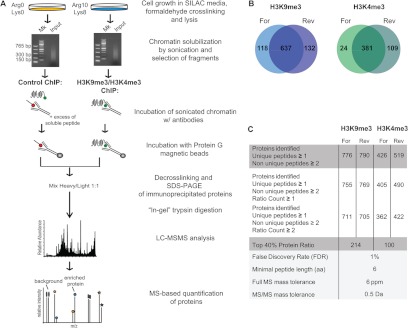
Scheme of the X-ChroP strategy combining X-ChIP and SILAC quantitation. A, scheme of the experimental approach. B, Venn diagrams show the overlap of identified and quantified (ratio count ≥ 1) proteins in two experimental replicates of ChIPs for the modification of interest: H3K9me3 (left) and H3K4me3 (right). C, table summarizing features of identified and quantified proteins for H3K9me3 and H3K4me3 X-ChIPs (For = forward, Rev = reverse). (See also supplemental Fig. S6.)
After incubation, the H and L immunopurified chromatins were pooled, and extracted proteins were separated via SDS-PAGE (supplemental Fig. S6A, right-hand panel). Samples were subjected to trypsin digestion followed by MS analysis. Proteins were identified and quantified using MaxQuant software (35). Using a confidence level of 99% (protein FDR = 1%), 637 and 381 proteins were identified in the H3K9me3 and H3K4me3 X-ChIPs, respectively (Figs. 4B and 4C; supplemental Tables S1 and S2).
The specific enrichment of the modifications used as bait (H3K9me3 and H3K4me3), together with the corresponding depletion of the unmodified and mono-methylated forms, confirmed the efficiency and specificity of the X-ChIP (supplemental Figs. S6B–S6D).
A log2 plot of the H/L ratios of the two sets of identified proteins from forward and reverse experiments allowed the unambiguous identification of binders (Figs. 5A and 5B for H3K9me3 and H3K4me3, respectively). The overall spread distribution of the SILAC ratios observed in the scatter plots can be explained by the fact that completely independent cells and chromatin preparations were used as input for each X-ChIP in the forward and reverse experiments, which thus represent full biological replicates, resulting in increased variability. Yet specific interactors could be selectively distinguished, based on their SILAC H/L ratios (H/L > 1 in the forward experiment and H/L < 1 in the reverse experiment; upper right-hand quadrant of the scatter plot), from the background proteins, which have a constant ratio close to 1. In particular, we considered a protein as specifically enriched when it was present in the top 40% of proteins with H/L ratios > 1, a filter that selected 214 and 100 interactors for H3K9me3 and H3K4me3, respectively (supplemental Figs. S7A and S7B), among which we found several well-known K9me3 and K4me3 binders. For the biological follow-up of novel interactors, however, we chose even higher stringency and focused on proteins present in the top 20% of the ratios.
Fig. 5.

The heterochromatomes and euchromatomes identified via X-ChroP. Proteins are plotted by their SILAC ratios in the first (x-axis) and second (y-axis) SILAC experiments for H3K9me3 (A) and H3K4me3 (B) (For = forward, Rev = reverse). Specific interactors should lie in the upper right-hand quadrant (enlarged), close to the diagonal. Red dotted lines represent the cutoffs, selecting the top 40% and 30% of protein ratios. Already annotated specific interactors are highlighted in color. (See also supplemental Tables S1 and S2 and supplemental Fig. S7.)
X-ChroP Characterizes Novel Players in the H3K9me3 and H3K4me3 Chromatomes
Of the top-40% heterochromatic binders, several had been previously ascribed to this region by independent biochemical experiments and were our positive controls (supplemental Table S1): the three isoforms (α, β, γ) of heterochromatin protein 1 (HP1) (55); CDYL1 (56); SETDB1, the euchromatic HMT of the Suv39 family that trimethylates H3K9 (57, 58); the DNMT1 (59); and its interactor UHRF1 (60). In addition to direct H3K9me3 binders, our approach also enabled the identification of novel and probably indirect interactions among heterochromatic proteins; for instance, we detected the HP1-associated proteins ADNP and POGZ (61, 62) and KDM2A, an H3K36 demethylase (22, 63, 64).
In the H3K4me3 chromatome (supplemental Table S2) we identified Spindlin1 (22) and the subunits of the MLL complexes WDR5, RbPB5, and ASH2 (65, 66). Furthermore, our screening confirmed various proteins also identified by Vermeulen with the SILAC peptide pull-downs, such as BPTF (67), IWS1 (68), PHF2, and various TBP-associated factors of the TFIID complex (20).
These results overall confirmed the robustness of our approach and allowed us to screen for novel interactors with higher confidence. Among the newly identified heterochromatin components (supplemental Table S1), we found HP1-binding protein 3 (HP1-BP74) (69). As the role of HP1-BP74 is still elusive, our result suggests a potential function in the formation and/or maintenance of the compacted chromatin structure through HP1 binding. We also identified different scaffold attachment factors (SAFs), including SAFB1, SAFB2, and SAFA, which bind the AT-rich scaffold/matrix attachment regions of DNA through their scaffold-attachment factor box (70). Other members of the family found in the H3K9me3 interactome are SAFB-like transcriptional modulator (71) and Matrin3, shown to bind to scaffold/matrix attachment regions and localize in heterochromatin regions, similar to SAFA (72, 73). With this as a basis, SAFB proteins could have a role in higher order heterochromatin organization involving interaction with chromatin remodeling complexes to enforce a more compact structure.
Among novel H3K4me3 interactors, we identified the FACT (for “facilitates chromatin transcription”) complex, a heterodimer composed of two subunits of 140 kDa and 80 kDa (74). FACT is essential for Pol II–driven transcription on chromatin templates (74), and it has an intrinsic histone chaperone activity to reassemble histones onto DNA (75). In addition, FACT seems to be associated with actively transcribed Pol II genes in Drosophila (76), and in Arabidopsis FACT co-localizes with actively transcribed genes, whereas it seems excluded by heterochromatin and intragenic regions (77).
When directly comparing the chromatomes of silent and active chromatin, interestingly, we observed also a minor subset of proteins equally, yet specifically, enriched in both fractions (supplemental Fig. S7C)—for instance, SMARCC2 and SMARCA5, subunits of the ATP-dependent chromatin remodeling complex SWI/SNF (78), and HMGB2, a member of the high-mobility-group family (79). In addition, we identified various structural components of chromatin with functions in chromosome condensation and separation, such as SCC112, which binds the cohesin complex and associates with chromatin throughout the cell cycle regulating sister chromatid cohesion during mitosis (80–82). Interestingly, the vast majority of these homogeneously distributed proteins exert more universal structural roles in chromatin homeostasis and dynamics, processes not linked to the transcriptional state. This might be suggestive of similar roles for those proteins in the same class whose function is still uncharacterized.
Accumulation of Histone Variants at Specific Chromatin Domains
A great advantage offered by the ChroP approach with respect to other strategies is the possibility of characterizing histone variants enriched at specific chromatin domains. Histone macro-H2A.1 was among the top 20% interactors of H3K9me3 chromatin, suggesting its prevalent heterochromatic localization (Fig. 6A), in line with evidence of a putative structural role of this variant in organizing constitutive heterochromatin (83, 84).
Fig. 6.
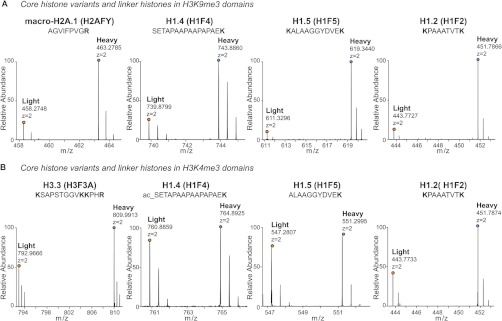
Histone variants enriched in heterochromatin and euchromatin. A, mass spectra of light and heavy SILAC peak pairs from H2AFY, H1.4, H1.5, and H1.2 demonstrating the specific enrichment of these proteins in the K9me3 ChIPed material (heavy) relative to the mock control (light). B, mass spectra of light and heavy SILAC peak pairs from H3.3, H1.4, H1.5, and H1.2 demonstrating the enrichment of these variants in the K4me3 ChIPed material relative to the mock control. (See also supplemental Fig. S8.)
The H3.3 variant was instead overrepresented in the H3K4me3 regions (Fig. 6B, supplemental Fig. S8A). H3.3 is incorporated into chromatin in a replication-independent manner upon chromatin assembly following the active transcription of genes; consequently, it localizes mainly at the promoters of active genes, is decorated by active marks, and contributes to the epigenetic memory maintenance of active regions (85–88). Interestingly, it has been suggested that H3.3 reduces the association of linker histone H1 to chromatin, thus causing a pronounced drop in H1 occupancy at Transcription Start Sites and at more distant cis-regulatory sites of active genes (89). We confirmed this theory, finding that H1.4 and H1.5 were increased in H3K9me3 chromatin but unchanged in H3K4me3 domains (Figs. 6A and 6B, respectively) (90). The SILAC ratio of H1.2 suggested its ubiquitous distribution (Figs. 6A and 6B), in apparent contradiction to previous reports proposing its euchromatic occurrence (91).
The enrichment of H2A.X in heterochromatin (Fig. 7A) was the most unexpected finding, given the prevalent literature focusing only on its Ser139-phosphorylated form (γ-H2A.X), typically described at DNA damage foci (92, 93). Independent validation of the SILAC-based result by means of standard ChIP, followed by WB, confirmed the accumulation of H2A.X in chromatin regions marked by H3K9me3 (Fig. 7B and supplemental Fig. S9A). In addition, the measurement of α-satellite repeats and of two H3K4me3 target genes (94) via qPCR in α-H2A.X ChIPs indicated that this variant enriches for heterochromatic DNA regions but not for active genes (Fig. 7C and supplemental Fig. S9B). Finally, H2A.X profiles in ChIP-Seq strongly correlated with H3K9me3, but not with H3K4me3 (Pearson's r: 0.796 and 0.139, respectively) (Fig. 7D).
Fig. 7.

H2A.X variant is overrepresented in heterochromatin. A, mass spectra of light and heavy SILAC peak pairs from H2A.X in the forward H3K9me3 X-ChIP: an H/L ratio > 1 indicates specific enrichment of this protein. B, Western blot of unmodified H2A.X upon ChIP with α-K9me3 and α-K4me3. Unmodified H3 is the loading control. C, qPCR measurement of α-satellite repeats in H2A.X, H3K9me3, and H3K4me3 domains immunopurified via the N-ChIP strategy relative to the mock control. The euchromatic actively transcribed genes PAICS and HSPD1 were used as negative controls. D, ChIP-Seq profiles of H2A.X, H3K9me3, and H3K4me3 compared with the input across regions of human chromosomes 15 (chr15: 15,396,089–38,421,489) and 19 (chr19: 6,465,305–41,462,000). E, model of higher local density of H2A.X in heterochromatin. F, qPCR measures of α-satellite repeats and genes PAICS and HSPD1 in H2A.X ChIP over total H3. Bars in the graph represent the averages ± S.E. (standard error of the mean) from three replicates. (See also supplemental Fig. S9.)
In order to reconcile this compelling set of results with the general evidence that DNA damage foci marked by γ-H2A.X appear in both silenced and actively transcribed chromatin, we formulated a higher local density model in which H2A.X is present all along the genome, but its accumulation in repressed regions results from a higher frequency of the variant in heterochromatin than in euchromatin, within poly-nucleosomal stretches of the same length (the beads-on-string structure) (Fig. 7E). We observed a specific increase in the level of α-satellite repeats with respect to two active genes when we normalized an H2A.X ChIP to total H3 ChIP, thus confirming the model (Fig. 7F).
Heterochromatic Enrichment of the WICH Complex, Recruited to H2A.X
An in-depth analysis of the H3K9me3 interactome revealed a reproducible overrepresentation (top 20%) of the WSTF-ISWI chromatin remodeling (WICH) complex (Fig. 8A), an observation that we corroborated via immunofluorescence (Fig. 8B). WICH consists of two subunits: ISWI, the ATPase subunit common to several remodeling complexes that mediate nucleosome positioning (95), and WSTF, encoded by the BAZ1B gene. Descriptive immunofluorescence studies had already noted the accumulation of WICH in replicating pericentric heterochromatin (96), but these observations were not followed by studies elucidating the mechanism of its recruitment and function in this region.
Fig. 8.
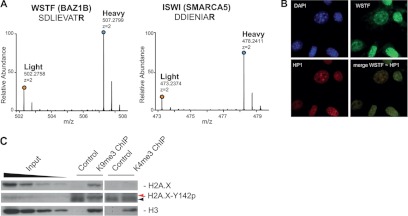
WICH and H2A.X Tyr142p involvement in heterochromatin. A, mass spectra of light and heavy SILAC peak pairs from WSTF and ISWI; H/L ratios > 1 indicate specific enrichment of these proteins. B, WSTF co-localizes with HP1β, a marker of pericentromeric heterochromatin (merge) (1 pixel = 0.172 μm; original magnification = 60×). C, WB of unmodified H2A.X and H2A.X-Tyr142p (red arrow) upon ChIP with α-K9me3 and α-K4me3. Unmodified H3 is the loading control. Black arrow indicates an unspecific band at lower molecular weight, also detected in the control.
The interaction between H2A.X and the WICH complex has been proven recently, together with the phosphorylation of tyrosine 142 of the same variant by WSTF. Tyr142p seems to be present in basal conditions, but its localization at specific chromatin regions/loci has not been explored yet. Based on the dual heterochromatic enrichment of both H2A.X and WICH, we tested whether phosphorylation at Tyr142 was accumulating in H3K9me3 regions. WB analysis (Fig. 8C) corroborated the hypothesis that, at basal conditions, the heterochromatic H2A.X is preferentially phosphorylated in this position, presumably by the accumulating WICH. This result suggests that Tyr142p might have a specific role in heterochromatin.
DISCUSSION
In this work we describe the ChroP approach, a novel analytical strategy for the proteomic investigation of chromatin at the resolution of a few nucleosomes. ChroP uses a modified version of ChIP to isolate native mono-nucleosomes up to cross-linked 500-bp oligonucleosomal stretches derived from distinct chromatin domains. The amount, purity, and quality of the isolated chromatin enable the subsequent mass spectrometric examination of the protein component, providing information on histone modification patterns, variants, and interactors. For the optimization of the method, we focused on two distinct, non-overlapping chromatin compartments: inactive pericentromeric heterochromatin and transcriptionally repressed patches of chromatin in actively transcribed areas, marked by H3K9me3, and active promoters in euchromatin, characterized by H3K4me3. The analysis of the PTM enrichment on histone H3 in the H3K9me3 and H3K4me3 domains confirmed the results recently described using immunoprecipitation of HPLC-purified H3 (30), thus verifying the selectivity of the antibodies used in our study. Overall, the annotation of the H3K9me3 modificome revealed the significant enrichment of known heterochromatic marks, with the corresponding depletion of active modifications, whereas an opposite trend was observed in the H3K4me3 domains, as expected. The agreement of our results with those of prior studies describing PTM patterning within the same histone molecule proves the robustness of the strategy for the following investigation of novel PTMs. More important, ChroP exhibits a unique strength in revealing PTM associations between the different core histones within the same mono-nucleosome, enriched at quasi-purity in N-ChIP. However, a possible drawback might arise from the peptide-centric strategy (bottom-up) undertaken in the MS analysis, wherein the enriched histones are digested with an Arg-C-like cleavage. In fact, such digestion produces relatively short peptides with few co-occurring modifications and thus partially loses information on the long-distance connectivity between the modified sites on the same histone. Therefore, although advantageous because of the high sequence coverage achieved and the resulting confidence in site-specific PTM attribution, the bottom-up strategy offers a partial view of simultaneously occurring long-distance PTMs. In this regard, it would be attractive to expand the MS analysis of the affinity-enriched nucleosomes with middle- and top-down approaches to study longer histone peptides (>20 aa), up to intact proteins (97, 98), so as to distinguish cis/trans and inter/intra-co-occurrence of PTMs on the same histones at specific chromatin regions. In order to make this implemented N-ChroP a more routine approach, specific technical issues must be addressed; for instance, histones need to be separated from IgG and solubilized prior to middle- and top-down MS, and specialized software capable of summarizing the emerging composite modification networks needs to be developed. However, the implementation of N-ChroP with more sophisticated MS analyses holds great potential for finally cracking the combinatorial aspect of the histone code.
The SILAC-based investigation of the corresponding interactomes via X-ChIP confirmed several previously described interactions, thereby validating our method; in addition, we identified numerous novel candidate interactors that had not been experimentally described previously. The remarkable advantage offered by ChroP consists in the fact that homogeneous 300–500-bp nucleosomal stretches in which weak protein–protein interactions are stabilized by formaldehyde cross-linking are purified and MS-analyzed; this enables the dissection of not only modification “readers,” but also more complex architectural structures, resulting from both direct and indirect interactions within intact chromatin domains. For instance, the selected H3K9me3 interactome revealed the enrichment of both KDM2A and HP1, which previously could be functionally associated only by the intersection of distinct independent assays, such as peptide and GFP pull-downs (21). This gain comes at the cost of the simplicity of interpretation of the proteomic readout; thus these new composite hierarchical protein architectures need to be further dissected (99).
The two facets of ChroP, N- and X-ChIP, are highly complementary, with the possibility of arranging in a unique order the different pieces composing chromatin architecture; for instance, the analysis of the H3K9me3 modificome reveals an unexpected depletion of H3K36me2/me3, which is explained by the recruitment of KDM2A found through the interactomics investigation of the same region.
Besides recapitulating known interactors, ChroP indicates putative localization in distinct chromatin compartments for histone variants and binders not previously described. A representative case is offered by the H1 isoforms: the accumulation of individual H1 variants in either active or silenced chromatin suggests their specific contribution to the establishment or maintenance of the functional status of these regions. Intriguingly, when investigating the modification patterns of these variants, we found evidence of at least one novel mono-methylation on lysine 90 of H1.2/H1.4, corresponding to lysine 93 of the H1.5 variant (supplemental Figs. S8B and S8C). This preliminary evidence opens the door to further characterization of PTMs of the H1 variants, at present underinvestigated because of the lack of adequate variant-specific antibodies.
The presence of the WICH complex and H2A.X in the H3K9me3 chromatome is particularly exciting; the “higher local density” of H2A.X in heterochromatin can be further developed including the evidence of WICH heterochromatic enrichment, thus leading to the hypothesis that H2A.X accumulating in heterochromatin is preferentially phosphorylated on Tyr142 by WSTF. WB analysis confirms this assumption and leads to the intriguing possibility that this mark might have a specific role in silent regions. Indeed, there is evidence that H2A.X Tyr142 phosphorylation decreases upon double strand breaks (100), just before the onset of γ-H2A.X, and that the two marks function as a molecular switch to regulate the DNA damage response. Our data suggest that this further level of regulation of the DNA damage response might be heterochromatin specific, with an effect on the genomic stability of this region. Some published evidence is in line with this model: firstly, γ-H2A.X foci are largely excluded from heterochromatin as compared with active euchromatic compartments; secondly, condensed chromatin domains are repaired more slowly than euchromatin (101–104). Finally, the observed correlation between the aberrant expression of Su(var)3–9, with the consequent alteration of the H3K9me3 pattern and HP1 binding and the genomic instability of heterochromatin during mammalian development, is highly suggestive in this context (105–107). Nevertheless, the specific recruitment of WICH to heterochromatin cannot be explained solely by the described binding of WSTF to H2A.X, since the higher density model does not exclude H2A.X from active regions: additional players may be involved. Interestingly, Singh at al. have recently shown that microcephalin (MCPH1) recognizes and binds the di-phosphorylated H2A.X and proposed this protein as mediator in the DNA damage response pathway (108).
Overall, the ChroP approach described here offers the potential to dissect the synergism of hPTMs, variants, and non-histonic interactors at functionally distinct chromatin domains, with a resolution of mono- to oligo-nuclesomes. The approach is relatively easy to set up and implement in the epigenetics groups, given the limited changes made to the conventional N- and X-ChIP protocols used for ChIP-Seq studies. Thus, for relatively straightforward optimization, we predict that ChroP will be amenable to numerous applications in more functional studies—for instance, studies of the dynamic changes of the modificome and intractome of specific chromatin regions upon various perturbations such as global transcriptional activation, differentiation, the depletion of distinct chromatin components, or treatment with epigenetic drugs. Therefore, ChroP emerges as an additional and valuable tool in the arsenal of analytical strategies available for the study of chromatin composition and dynamics in a system-wide fashion.
Supplementary Material
Acknowledgments
We thank S. Ghisletti for help in the optimization of ChIP protocols, M. Bremang for computational support, A. Cuomo for critical discussion and technical help in mass spectrometry optimization, E. Vitale for lab management support, and the Next Generation Sequencing Team at Cogentech for the generation of the ChIP-Seq data. We thank Abcam for providing a set of ChIP-grade antibodies for initial quality control analysis and for testing the best-performing reagents for ChroP. We thank also G. Natoli and T. Straub for critical discussion and G. Natoli, E. Guccione, M. Vermeulen, and M. Bremang for critical reading of the manuscript.
Footnotes
* T.B.'s work is supported by grants from the Giovanni Armenise-Harvard Foundation Career Development Program, the Association of International Cancer Research, the Italian Association for Cancer Research, the Cariplo Foundation, and the Italian Ministry of Health. M.S. is supported by a fellowship from FIRC.
 This article contains supplemental material.
This article contains supplemental material.
2 J. Cox, manuscript in preparation.
1 The abbreviations used are:
- ac
- acetylation
- ChIP
- chromatin immunoprecipitation
- ChIP-Seq
- ChIP-sequencing
- di-Ac
- di-acetylation
- hPTM
- histone post-translational modification
- HP1
- heterochromatin protein 1
- H2A.XTyr142p
- histone H2A.X tyrosine 142 phosphorylation
- K
- lysine
- me1
- mono-methylation
- me2
- di-methylation
- me3
- tri-methylation
- MS
- mass spectrometry
- MS/MS
- tandem mass spectrometry
- N-ChIP
- native chromatin immunoprecipitation
- R
- arginine
- SAF
- scaffold attachment factor
- SDS-PAGE
- sodium dodecyl sulphate polyacrylamide gel electrophoresis
- SILAC
- stable isotope labeling by amino acids in cell culture
- tetra-Ac
- tetra-acetylation
- tri-Ac
- tri-acetylation
- WB
- western blot
- WICH
- WSTF-ISWI chromatin remodeling complex
- X-ChIP
- cross-linking chromatin immunoprecipitation
- γ-H2A.X
- histone H2A.X serine 139 phosphorylation.
REFERENCES
- 1. Kornberg R. D. (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184, 868–871 [DOI] [PubMed] [Google Scholar]
- 2. Luger K., Mader A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 3. Waddington C. H. (2011) The epigenotype. 1942. Int. J. Epidemiol. 41, 10–13 [DOI] [PubMed] [Google Scholar]
- 4. Berger S. L., Kouzarides T., Shiekhattar R., Shilatifard A. (2009) An operational definition of epigenetics. Genes Dev. 23, 781–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spotswood H. T., Turner B. M. (2002) An increasingly complex code. J. Clin. Invest. 110, 577–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jenuwein T., Allis C. D. (2001) Translating the histone code. Science 293, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 7. Kouzarides T. (2007) Chromatin modifications and their function. Cell 128, 693–705 [DOI] [PubMed] [Google Scholar]
- 8. Margueron R., Reinberg D. (2010) Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 11, 285–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pokholok D. K., Harbison C. T., Levine S., Cole M., Hannett N. M., Lee T. I., Bell G. W., Walker K., Rolfe P. A., Herbolsheimer E., Zeitlinger J., Lewitter F., Gifford D. K., Young R. A. (2005) Genome-wide map of nucleosome acetylation and methylation in yeast. Cell 122, 517–527 [DOI] [PubMed] [Google Scholar]
- 10. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 11. Johnson D. S., Mortazavi A., Myers R. M., Wold B. (2007) Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502 [DOI] [PubMed] [Google Scholar]
- 12. Robertson G., Hirst M., Bainbridge M., Bilenky M., Zhao Y., Zeng T., Euskirchen G., Bernier B., Varhol R., Delaney A., Thiessen N., Griffith O. L., He A., Marra M., Snyder M., Jones S. (2007) Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 4, 651–657 [DOI] [PubMed] [Google Scholar]
- 13. Garcia B. A., Shabanowitz J., Hunt D. F. (2007) Characterization of histones and their post-translational modifications by mass spectrometry. Curr. Opin. Chem. Biol. 11, 66–73 [DOI] [PubMed] [Google Scholar]
- 14. Beck H. C. (2010) Mass spectrometry in epigenetic research. Methods Mol. Biol. 593, 263–282 [DOI] [PubMed] [Google Scholar]
- 15. Jung H. R., Pasini D., Helin K., Jensen O. N. (2010) Quantitative mass spectrometry of histones H3.2 and H3.3 in Suz12-deficient mouse embryonic stem cells reveals distinct, dynamic post-translational modifications at Lys-27 and Lys-36. Mol. Cell. Proteomics 9, 838–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taverna S. D., Ueberheide B. M., Liu Y., Tackett A. J., Diaz R. L., Shabanowitz J., Chait B. T., Hunt D. F., Allis C. D. (2007) Long-distance combinatorial linkage between methylation and acetylation on histone H3 N termini. Proc. Natl. Acad. Sci. U.S.A. 104, 2086–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Imhof A., Bonaldi T. (2005) “Chromatomics”: the analysis of the chromatome. Mol. Biosyst. 1, 112–116 [DOI] [PubMed] [Google Scholar]
- 18. Bernstein B. E., Meissner A., Lander E. S. (2007) The mammalian epigenome. Cell 128, 669–681 [DOI] [PubMed] [Google Scholar]
- 19. Eberl H. C., Mann M., Vermeulen M. (2010) Quantitative proteomics for epigenetics. Chembiochem 12, 224–234 [DOI] [PubMed] [Google Scholar]
- 20. Vermeulen M., Mulder K. W., Denissov S., Pijnappel W. W., van Schaik F. M., Varier R. A., Baltissen M. P., Stunnenberg H. G., Mann M., Timmers H. T. (2007) Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell 131, 58–69 [DOI] [PubMed] [Google Scholar]
- 21. Vermeulen M., Eberl H. C., Matarese F., Marks H., Denissov S., Butter F., Lee K. K., Olsen J. V., Hyman A. A., Stunnenberg H. G., Mann M. (2010) Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142, 967–980 [DOI] [PubMed] [Google Scholar]
- 22. Bartke T., Vermeulen M., Xhemalce B., Robson S. C., Mann M., Kouzarides T. (2010) Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 143, 470–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nikolov M., Stutzer A., Mosch K., Krasauskas A., Soeroes S., Stark H., Urlaub H., Fischle W. (2011) Chromatin affinity purification and quantitative mass spectrometry defining the interactome of histone modification patterns. Mol. Cell. Proteomics 10, M110.005371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deleted in proof.
- 25. Li X., Foley E. A., Molloy K. R., Li Y., Chait B. T., Kapoor T. M. (2012) Quantitative chemical proteomics approach to identify post-translational modification-mediated protein-protein interactions. J. Am. Chem. Soc. 134, 1982–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ohta S., Bukowski-Wills J. C., Sanchez-Pulido L., Alves Fde L., Wood L., Chen Z. A., Platani M., Fischer L., Hudson D. F., Ponting C. P., Fukagawa T., Earnshaw W. C., Rappsilber J. (2010) The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell 142, 810–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Torrente M. P., Zee B. M., Young N. L., Baliban R. C., LeRoy G., Floudas C. A., Hake S. B., Garcia B. A. (2011) Proteomic interrogation of human chromatin. PLoS One 6, e24747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lambert J. P., Mitchell L., Rudner A., Baetz K., Figeys D. (2009) A novel proteomics approach for the discovery of chromatin-associated protein networks. Mol. Cell. Proteomics 8, 870–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dejardin J., Kingston R. E. (2009) Purification of proteins associated with specific genomic loci. Cell 136, 175–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peach S. E., Rudomin E. L., Udeshi N. D., Carr S. A., Jaffe J. D. (2012) Quantitative assessment of chromatin immunoprecipitation grade antibodies directed against histone modifications reveals patterns of co-occurring marks on histone protein molecules. Mol. Cell. Proteomics 11, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cook P. J., Ju B. G., Telese F., Wang X., Glass C. K., Rosenfeld M. G. (2009) Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 458, 591–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. O'Neill L. P., Turner B. M. (2003) Immunoprecipitation of native chromatin: NChIP. Methods 31, 76–82 [DOI] [PubMed] [Google Scholar]
- 33. Lee T. I., Johnstone S. E., Young R. A. (2006) Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc. 1, 729–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Olsen J. V., de Godoy L. M., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4, 2010–2021 [DOI] [PubMed] [Google Scholar]
- 35. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 36. Deleted in proof.
- 37. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 38. Cox J., Michalski A., Mann M. (2011) Software lock mass by two-dimensional minimization of peptide mass errors. J. Am. Soc. Mass. Spectrom. 22, 1373–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Austenaa L., Barozzi I., Chronowska A., Termanini A., Ostuni R., Prosperini E., Stewart A. F., Testa G., Natoli G. (2012) The histone methyltransferase Wbp7 controls macrophage function through GPI glycolipid anchor synthesis. Immunity 36, 572–585 [DOI] [PubMed] [Google Scholar]
- 40. Ram O., Goren A., Amit I., Shoresh N., Yosef N., Ernst J., Kellis M., Gymrek M., Issner R., Coyne M., Durham T., Zhang X., Donaghey J., Epstein C. B., Regev A., Bernstein B. E. (2011) Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell 147, 1628–1639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ernst J., Kellis M. (2010) Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 28, 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garcia B. A., Pesavento J. J., Mizzen C. A., Kelleher N. L. (2007) Pervasive combinatorial modification of histone H3 in human cells. Nat. Methods 4, 487–489 [DOI] [PubMed] [Google Scholar]
- 43. Heintzman N. D., Stuart R. K., Hon G., Fu Y., Ching C. W., Hawkins R. D., Barrera L. O., Van Calcar S., Qu C., Ching K. A., Wang W., Weng Z., Green R. D., Crawford G. E., Ren B. (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318 [DOI] [PubMed] [Google Scholar]
- 44. Margueron R., Justin N., Ohno K., Sharpe M. L., Son J., Drury W. J., 3rd, Voigt P., Martin S. R., Taylor W. R., De Marco V., Pirrotta V., Reinberg D., Gamblin S. J. (2009) Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461, 762–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Min J., Zhang Y., Xu R. M. (2003) Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 17, 1823–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zee B. M., Levin R. S., Xu B., LeRoy G., Wingreen N. S., Garcia B. A. (2010) In vivo residue-specific histone methylation dynamics. J. Biol. Chem. 285, 3341–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fischle W., Wang Y., Allis C. D. (2003) Binary switches and modification cassettes in histone biology and beyond. Nature 425, 475–479 [DOI] [PubMed] [Google Scholar]
- 48. Peters A. H., Kubicek S., Mechtler K., O'Sullivan R. J., Derijck A. A., Perez-Burgos L., Kohlmaier A., Opravil S., Tachibana M., Shinkai Y., Martens J. H., Jenuwein T. (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol. Cell 12, 1577–1589 [DOI] [PubMed] [Google Scholar]
- 49. Varga-Weisz P. D., Dalgaard J. Z. (2002) A mark in the core: silence no more! Mol. Cell 9, 1154–1156 [DOI] [PubMed] [Google Scholar]
- 50. Bannister A. J., Schneider R., Myers F. A., Thorne A. W., Crane-Robinson C., Kouzarides T. (2005) Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem. 280, 17732–17736 [DOI] [PubMed] [Google Scholar]
- 51. Li B., Jackson J., Simon M. D., Fleharty B., Gogol M., Seidel C., Workman J. L., Shilatifard A. (2009) Histone H3 lysine 36 dimethylation (H3K36me2) is sufficient to recruit the Rpd3s histone deacetylase complex and to repress spurious transcription. J. Biol. Chem. 284, 7970–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dillon N. (2004) Heterochromatin structure and function. Biol. Cell 96, 631–637 [DOI] [PubMed] [Google Scholar]
- 53. Schotta G., Lachner M., Sarma K., Ebert A., Sengupta R., Reuter G., Reinberg D., Jenuwein T. (2004) A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 18, 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Garcia B. A., Hake S. B., Diaz R. L., Kauer M., Morris S. A., Recht J., Shabanowitz J., Mishra N., Strahl B. D., Allis C. D., Hunt D. F. (2007) Organismal differences in post-translational modifications in histones H3 and H4. J. Biol. Chem. 282, 7641–7655 [DOI] [PubMed] [Google Scholar]
- 55. Bannister A. J., Zegerman P., Partridge J. F., Miska E. A., Thomas J. O., Allshire R. C., Kouzarides T. (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410, 120–124 [DOI] [PubMed] [Google Scholar]
- 56. Franz H., Mosch K., Soeroes S., Urlaub H., Fischle W. (2009) Multimerization and H3K9me3 binding are required for CDYL1b heterochromatin association. J. Biol. Chem. 284, 35049–35059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li H., Rauch T., Chen Z. X., Szabo P. E., Riggs A. D., Pfeifer G. P. (2006) The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells. J. Biol. Chem. 281, 19489–19500 [DOI] [PubMed] [Google Scholar]
- 58. Schultz D. C., Ayyanathan K., Negorev D., Maul G. G., Rauscher F. J., 3rd (2002) SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16, 919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Smallwood A., Esteve P. O., Pradhan S., Carey M. (2007) Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 21, 1169–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rottach A., Frauer C., Pichler G., Bonapace I. M., Spada F., Leonhardt H. (2010) The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res. 38, 1796–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lechner M. S., Schultz D. C., Negorev D., Maul G. G., Rauscher F. J., 3rd (2005) The mammalian heterochromatin protein 1 binds diverse nuclear proteins through a common motif that targets the chromoshadow domain. Biochem. Biophys. Res. Commun. 331, 929–937 [DOI] [PubMed] [Google Scholar]
- 62. Mosch K., Franz H., Soeroes S., Singh P. B., Fischle W. (2011) HP1 recruits activity-dependent neuroprotective protein to H3K9me3 marked pericentromeric heterochromatin for silencing of major satellite repeats. PLoS One 6, e15894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Frescas D., Guardavaccaro D., Kuchay S. M., Kato H., Poleshko A., Basrur V., Elenitoba-Johnson K. S., Katz R. A., Pagano M. (2008) KDM2A represses transcription of centromeric satellite repeats and maintains the heterochromatic state. Cell Cycle 7, 3539–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tsukada Y., Fang J., Erdjument-Bromage H., Warren M. E., Borchers C. H., Tempst P., Zhang Y. (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439, 811–816 [DOI] [PubMed] [Google Scholar]
- 65. Dou Y., Milne T. A., Ruthenburg A. J., Lee S., Lee J. W., Verdine G. L., Allis C. D., Roeder R. G. (2006) Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 13, 713–719 [DOI] [PubMed] [Google Scholar]
- 66. Wysocka J., Swigut T., Milne T. A., Dou Y., Zhang X., Burlingame A. L., Roeder R. G., Brivanlou A. H., Allis C. D. (2005) WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121, 859–872 [DOI] [PubMed] [Google Scholar]
- 67. Li H., Ilin S., Wang W., Duncan E. M., Wysocka J., Allis C. D., Patel D. J. (2006) Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442, 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Krogan N. J., Kim M., Ahn S. H., Zhong G., Kobor M. S., Cagney G., Emili A., Shilatifard A., Buratowski S., Greenblatt J. F. (2002) RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol. Cell. Biol. 22, 6979–6992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hayashihara K., Uchiyama S., Shimamoto S., Kobayashi S., Tomschik M., Wakamatsu H., No D., Sugahara H., Hori N., Noda M., Ohkubo T., Zlatanova J., Matsunaga S., Fukui K. (2010) The middle region of an HP1-binding protein, HP1-BP74, associates with linker DNA at the entry/exit site of nucleosomal DNA. J. Biol. Chem. 285, 6498–6507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Townson S. M., Dobrzycka K. M., Lee A. V., Air M., Deng W., Kang K., Jiang S., Kioka N., Michaelis K., Oesterreich S. (2003) SAFB2, a new scaffold attachment factor homolog and estrogen receptor corepressor. J. Biol. Chem. 278, 20059–20068 [DOI] [PubMed] [Google Scholar]
- 71. Chan C. W., Lee Y. B., Uney J., Flynn A., Tobias J. H., Norman M. (2007) A novel member of the SAF (scaffold attachment factor)-box protein family inhibits gene expression and induces apoptosis. Biochem. J. 407, 355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Malyavantham K. S., Bhattacharya S., Barbeitos M., Mukherjee L., Xu J., Fackelmayer F. O., Berezney R. (2008) Identifying functional neighborhoods within the cell nucleus: proximity analysis of early S-phase replicating chromatin domains to sites of transcription, RNA polymerase II, HP1gamma, matrin 3 and SAF-A. J. Cell. Biochem. 105, 391–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lobov I. B., Tsutsui K., Mitchell A. R., Podgornaya O. I. (2001) Specificity of SAF-A and lamin B binding in vitro correlates with the satellite DNA bending state. J. Cell. Biochem. 83, 218–229 [DOI] [PubMed] [Google Scholar]
- 74. Orphanides G., LeRoy G., Chang C. H., Luse D. S., Reinberg D. (1998) FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92, 105–116 [DOI] [PubMed] [Google Scholar]
- 75. Belotserkovskaya R., Oh S., Bondarenko V. A., Orphanides G., Studitsky V. M., Reinberg D. (2003) FACT facilitates transcription-dependent nucleosome alteration. Science 301, 1090–1093 [DOI] [PubMed] [Google Scholar]
- 76. Saunders A., Werner J., Andrulis E. D., Nakayama T., Hirose S., Reinberg D., Lis J. T. (2003) Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science 301, 1094–1096 [DOI] [PubMed] [Google Scholar]
- 77. Duroux M., Houben A., Ruzicka K., Friml J., Grasser K. D. (2004) The chromatin remodelling complex FACT associates with actively transcribed regions of the Arabidopsis genome. Plant J. 40, 660–671 [DOI] [PubMed] [Google Scholar]
- 78. Phelan M. L., Sif S., Narlikar G. J., Kingston R. E. (1999) Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 3, 247–253 [DOI] [PubMed] [Google Scholar]
- 79. Itou J., Taniguchi N., Oishi I., Kawakami H., Lotz M., Kawakami Y. (2011) HMGB factors are required for posterior digit development through integrating signaling pathway activities. Dev. Dyn. 240, 1151–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hagemann C., Weigelin B., Schommer S., Schulze M., Al-Jomah N., Anacker J., Gerngras S., Kuhnel S., Kessler A. F., Polat B., Ernestus R. I., Patel R., Vince G. H. (2011) The cohesin-interacting protein, precocious dissociation of sisters 5A/sister chromatid cohesion protein 112, is up-regulated in human astrocytic tumors. Int. J. Mol. Med. 27, 39–51 [DOI] [PubMed] [Google Scholar]
- 81. Kumar D., Sakabe I., Patel S., Zhang Y., Ahmad I., Gehan E. A., Whiteside T. L., Kasid U. (2004) SCC-112, a novel cell cycle-regulated molecule, exhibits reduced expression in human renal carcinomas. Gene 328, 187–196 [DOI] [PubMed] [Google Scholar]
- 82. Zheng M. Z., Zheng L. M., Zeng Y. X. (2008) SCC-112 gene is involved in tumor progression and promotes the cell proliferation in G2/M phase. J. Cancer Res. Clin. Oncol. 134, 453–462 [DOI] [PubMed] [Google Scholar]
- 83. Mietton F., Sengupta A. K., Molla A., Picchi G., Barral S., Heliot L., Grange T., Wutz A., Dimitrov S. (2009) Weak but uniform enrichment of the histone variant macroH2A1 along the inactive X chromosome. Mol. Cell. Biol. 29, 150–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ladurner A. G. (2003) Inactivating chromosomes: a macro domain that minimizes transcription. Mol. Cell 12, 1–3 [DOI] [PubMed] [Google Scholar]
- 85. Chow C. M., Georgiou A., Szutorisz H., Maia e Silva A., Pombo A., Barahona I., Dargelos E., Canzonetta C., Dillon N. (2005) Variant histone H3.3 marks promoters of transcriptionally active genes during mammalian cell division. EMBO Rep. 6, 354–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. McKittrick E., Gafken P. R., Ahmad K., Henikoff S. (2004) Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc. Natl. Acad. Sci. U.S.A. 101, 1525–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hake S. B., Garcia B. A., Duncan E. M., Kauer M., Dellaire G., Shabanowitz J., Bazett-Jones D. P., Allis C. D., Hunt D. F. (2006) Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J. Biol. Chem. 281, 559–568 [DOI] [PubMed] [Google Scholar]
- 88. Loyola A., Bonaldi T., Roche D., Imhof A., Almouzni G. (2006) PTMs on H3 variants before chromatin assembly potentiate their final epigenetic state. Mol. Cell 24, 309–316 [DOI] [PubMed] [Google Scholar]
- 89. Braunschweig U., Hogan G. J., Pagie L., van Steensel B. (2009) Histone H1 binding is inhibited by histone variant H3.3. EMBO J. 28, 3635–3645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Oberg C., Izzo A., Schneider R., Wrange O., Belikov S. (2011) Linker histone subtypes differ in their effect on nucleosomal spacing in vivo. J. Mol. Biol. 419, 183–197 [DOI] [PubMed] [Google Scholar]
- 91. Parseghian M. H., Newcomb R. L., Winokur S. T., Hamkalo B. A. (2000) The distribution of somatic H1 subtypes is non-random on active vs. inactive chromatin: distribution in human fetal fibroblasts. Chromosome Res. 8, 405–424 [DOI] [PubMed] [Google Scholar]
- 92. Rogakou E. P., Pilch D. R., Orr A. H., Ivanova V. S., Bonner W. M. (1998) DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868 [DOI] [PubMed] [Google Scholar]
- 93. Fernandez-Capetillo O., Lee A., Nussenzweig M., Nussenzweig A. (2004) H2AX: the histone guardian of the genome. DNA Repair (Amst.) 3, 959–967 [DOI] [PubMed] [Google Scholar]
- 94. Guccione E., Martinato F., Finocchiaro G., Luzi L., Tizzoni L., Dall'Olio V., Zardo G., Nervi C., Bernard L., Amati B. (2006) Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 8, 764–770 [DOI] [PubMed] [Google Scholar]
- 95. Corona D. F., Tamkun J. W. (2004) Multiple roles for ISWI in transcription, chromosome organization and DNA replication. Biochim. Biophys. Acta 1677, 113–119 [DOI] [PubMed] [Google Scholar]
- 96. Bozhenok L., Wade P. A., Varga-Weisz P. (2002) WSTF-ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO J. 21, 2231–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Young N. L., DiMaggio P. A., Plazas-Mayorca M. D., Baliban R. C., Floudas C. A., Garcia B. A. (2009) High throughput characterization of combinatorial histone codes. Mol. Cell. Proteomics 8, 2266–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Boyne M. T., 2nd, Pesavento J. J., Mizzen C. A., Kelleher N. L. (2006) Precise characterization of human histones in the H2A gene family by top down mass spectrometry. J. Proteome Res. 5, 248–253 [DOI] [PubMed] [Google Scholar]
- 99. Poser I., Sarov M., Hutchins J. R., Heriche J. K., Toyoda Y., Pozniakovsky A., Weigl D., Nitzsche A., Hegemann B., Bird A. W., Pelletier L., Kittler R., Hua S., Naumann R., Augsburg M., Sykora M. M., Hofemeister H., Zhang Y., Nasmyth K., White K. P., Dietzel S., Mechtler K., Durbin R., Stewart A. F., Peters J. M., Buchholz F., Hyman A. A. (2008) BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat. Methods 5, 409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Xiao A., Li H., Shechter D., Ahn S. H., Fabrizio L. A., Erdjument-Bromage H., Ishibe-Murakami S., Wang B., Tempst P., Hofmann K., Patel D. J., Elledge S. J., Allis C. D. (2009) WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 457, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kim J. A., Kruhlak M., Dotiwala F., Nussenzweig A., Haber J. E. (2007) Heterochromatin is refractory to gamma-H2AX modification in yeast and mammals. J. Cell Biol. 178, 209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cowell I. G., Sunter N. J., Singh P. B., Austin C. A., Durkacz B. W., Tilby M. J. (2007) gammaH2AX foci form preferentially in euchromatin after ionising-radiation. PLoS One 2, e1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Vasireddy R. S., Karagiannis T. C., El-Osta A. (2010) Gamma-radiation-induced gammaH2AX formation occurs preferentially in actively transcribing euchromatic loci. Cell. Mol. Life Sci. 67, 291–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Goodarzi A. A., Jeggo P., Lobrich M. (2010) The influence of heterochromatin on DNA double strand break repair: getting the strong, silent type to relax. DNA Repair (Amst.) 9, 1273–1282 [DOI] [PubMed] [Google Scholar]
- 105. Peters A. H., O'Carroll D., Scherthan H., Mechtler K., Sauer S., Schofer C., Weipoltshammer K., Pagani M., Lachner M., Kohlmaier A., Opravil S., Doyle M., Sibilia M., Jenuwein T. (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107, 323–337 [DOI] [PubMed] [Google Scholar]
- 106. Ayoub N., Jeyasekharan A. D., Bernal J. A., Venkitaraman A. R. (2008) HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature 453, 682–686 [DOI] [PubMed] [Google Scholar]
- 107. Peng J. C., Karpen G. H. (2009) Heterochromatic genome stability requires regulators of histone H3 K9 methylation. PLoS Genet. 5, e1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Singh N., Basnet H., Wiltshire T. D., Mohammad D. H., Thompson J. R., Heroux A., Botuyan M. V., Yaffe M. B., Couch F. J., Rosenfeld M. G., Mer G. Dual recognition of phosphoserine and phosphotyrosine in histone variant H2A.X by DNA damage response protein MCPH1. Proc. Natl. Acad. Sci. U.S.A. 109, 14381–14386 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.