

Abstract
Dried blood spot (DBS) sampling, coupled with multiple reaction monitoring mass spectrometry (MRM-MS), is a well-established approach for quantifying a wide range of small molecule biomarkers and drugs. This sampling procedure is simpler and less-invasive than those required for traditional plasma or serum samples enabling collection by minimally trained personnel. Many analytes are stable in the DBS format without refrigeration, which reduces the cost and logistical challenges of sample collection in remote locations. These advantages make DBS sample collection desirable for advancing personalized medicine through population-wide biomarker screening. Here we expand this technology by demonstrating the first multiplexed method for the quantitation of endogenous proteins in DBS samples. A panel of 60 abundant proteins in human blood was targeted by monitoring proteotypic tryptic peptides and their stable isotope-labeled analogs by MRM. Linear calibration curves were obtained for 40 of the 65 peptide targets demonstrating multiple proteins can be quantitatively extracted from DBS collection cards. The method was also highly reproducible with a coefficient of variation of <15% for all 40 peptides. Overall, this assay quantified 37 proteins spanning a range of more than four orders of magnitude in concentration within a single 25 min LC/MRM-MS analysis. The protein abundances of the 33 proteins quantified in matching DBS and whole blood samples showed an excellent correlation, with a slope of 0.96 and an R2 value of 0.97. Furthermore, the measured concentrations for 80% of the proteins were stable for at least 10 days when stored at −20 °C, 4 °C and 37 °C. This work represents an important first step in evaluating the integration of DBS sampling with highly-multiplexed MRM for quantitation of endogenous proteins.
Dried Blood Spot (DBS)1 samples have many advantages over blood serum or plasma and are the preferred clinical sample for newborn screening for metabolic diseases (1, 2). These samples are collected by pricking a newborn's heel and spotting a drop of blood onto specially designed filter paper collection cards. Samples are then dried under ambient conditions and are usually stored with desiccant at room temperature until analysis. This sampling procedure is simpler and less invasive then intravenous blood draws, which require a trained phlebotomist. Not surprisingly, the majority of adult patients prefer the small lancet used in finger-prick blood sampling methods to the larger needles used in intravenous blood draws (3, 4). Unlike plasma or serum samples, which consume ≥250 μl of blood and must be centrifuged within an hour of collection, DBS samples can be prepared using a volume of only 10 μl, and do not require any specialized equipment at the collection site (5). The simplicity and reduced safety risks associated with DBS sampling enables collection by minimally trained staff or by the patients themselves. In addition, many analytes are stable in the DBS format at room temperature, reducing sample transportation and storage costs, as well as the impact on the environment. Finally, DBS samples are safer to transport and are considered exempt from dangerous goods regulations (6, 7). These advantages make DBS sampling very attractive for advancing personalized medicine and population-based biomarker research (8).
Numerous biomolecular targets covering genomics, metabolomics, and proteomics applications have been quantified in DBS samples using a wide array of analytical techniques (9). The most common clinical application of DBS sampling is screening newborns for metabolomics disorders by targeting small molecule biomarkers. Early screening programs relied on bacterial inhibition assays and later immunoassays, both of which required a different assay for each target of interest (2). However, the time and cost required to perform each assay independently has limited the number of diseases that could be screened nationwide to only a handful. In addition, a single biomarker often lacked the specificity to produce a definitive diagnosis, requiring extensive secondary testing. Hemoglobin is the only protein that is commonly targeted in DBS samples, and primary screening is accomplished by high-performance liquid chromatography (HPLC) or isoelectric focusing (IEF) methods (2). Similar to small-molecule screening methods, these approaches are low-throughput and are not amendable to multiplexing with additional protein targets. In newborn screening programs, these challenges associated with small molecule analysis were overcome with the introduction of multiple reaction monitoring mass spectrometry (MRM-MS) into the clinical laboratories (1, 10). The specificity of MRM enables hundreds of analytes to be monitored during a single experiment to facilitate the development of highly multiplexed assays. The addition of stable isotope-labeled internal standards (SIS) enables the acquisition of highly reproducible results across a variety of instrumentation at different institutions. It is now common for 20–30 small molecule targets including amino acids, fatty acid acylcarnitines, and organic acid acylcarnitines to be analyzed by flow injection MRM-MS, at a cost of $10–20 USD per patient sample (11). Expansion of the screening panel to include additional small-molecule biomarkers on an existing platform may cost less than $1 each. In addition to newborn screening, DBS sampling combined with MS is also gaining acceptance in small-molecule drug development (12, 13). Here the collection of smaller blood volumes allows serial sampling from mice reducing the total number of animals required to generate preclinical toxicology and pharmacokinetic data (5).
Despite the successful use of DBS samples in MS-based experiments for small molecule analysis, there have been few reports of using this technology for protein targets (13). Daniel and coworkers reported a screening method for identifying β-thalassemia using the well-known biomarker HbA2, a hemoglobin variant composed of two alpha- and two delta-globin subunits (14). Proteins were extracted in an aqueous solution, digested with trypsin in 30 min and infused for MRM-MS analysis. Multiple hemoglobin peptides were targeted to measure the abundance of the delta-globin chain, using peptides from the beta-globin chain as an internal standard. This ratio correlated well with the abundance of intact HbA2 as determined by a well-established HPLC method. The shorter acquisition time and the increased specificity of the MS-based method showed promise for improving population-wide screening. Boemer et al. used a similar flow injection MRM-MS strategy to screen newborns for hemoglobin variants associated with sickle cell disease (15). They analyzed more than 2000 DBS samples by targeting tryptic peptides that were unique to four different beta-globin mutations and compared their results with a standard IEF method that measured the intact proteins obtained from corresponding whole blood samples. Their flow injection MRM approach was able to identify the correct phenotype for all targeted variants. Recently, deWilde et al. reported a method for screening newborn DBS samples for ceruloplasmin, a protein linked to Wilson's disease (16). Their method combined SIS peptides with LC/MRM-MS and produced results similar to those from an immunoassay for the analysis of seven patient samples. The monitoring of a therapeutic protein in rat blood was demonstrated with Kehler et al. to evaluate the suitability of this approach for supporting preclinical trials (17). Finally, a multiplexed approach was presented by Sleczka et al. for the simultaneous quantitation of two therapeutic proteins in spiked DBS samples collected from several animal models (18).
In all previous methods, only one to two proteins were targeted in DBS samples and therefore the true multiplexing capabilities of MRM were not realized. MRM-based methods using SIS peptides have already proven proficient at highly multiplexed quantitation of proteins in plasma and serum samples (19–21). Our current work demonstrates the potential for integrating DBS sampling with LC/MRM-MS for highly multiplexed quantitation of endogenous proteins. Many of the 60 proteins that we have targeted have been cleared or approved by FDA, and are already being analyzed one at a time in clinical laboratories. The assay developed in this study includes a highly reproducible method for extracting multiple proteins from DBS samples followed by trypsin digestion and surrogate peptides, along with their SIS analogs, were analyzed by a standard-flow LC/MRM-MS platform that has previously been shown to give accurate, sensitive, and robust analysis of proteotypic peptides in human plasma (21, 22). The quantitative results from whole blood and the corresponding DBS samples were compared, and the integrity of DBS samples stored under various temperatures has been evaluated.
EXPERIMENTAL PROCEDURES
Selection of Proteins and Peptides
The target panel was composed of 60 high-abundance blood proteins, including subunits and fragments of the same protein, as listed in Supplemental Table S1. As noted in our table, 29 of these proteins are already FDA-approved or FDA-cleared analytes in human plasma or serum, and tests for their plasma concentrations are already currently available in clinical laboratories, primarily as immunoassays (23). We have previously described our procedure for selecting surrogate peptides for targeted proteins in MRM-based experiments (24, 25). Briefly, publicly accessible databases (Peptide Atlas and the Global Proteome Machine) are used to select tryptic peptides that are unique for each protein within the human proteome. These peptides are ranked based on the frequency of their observation in MS/MS experiments, lack of easily modified amino acid residues (i.e., methionine, cysteine), suitability for SIS peptide synthesis (moderate hydrophobicity), and ease of MS/MS analysis (doubly- and triply charged precursor ions between m/z 300–1400). We have successfully used many of these same peptide targets to quantify proteins in plasma samples (21, 24). In our DBS experiments, 60 proteins were represented by 65 peptides with two peptides each for alpha-2-HS-glycoprotein, alpha-2-macroglobulin, and complement component C9, and three peptides for transferrin.
Synthesis and Optimization of Peptide Standards
The SIS peptides were synthesized in-house (Prelude, Protein Technologies, Tuscan, AZ) using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry, as described previously (24). All SIS peptides contained a heavy isotope form of an arginine ([13C6] or [13C6, 15N4]) or lysine ([13C6] or [13C6, 15N2]) amino acid residue (Cambridge Isotope Laboratories, Andover, MA) at the C terminus. The crude peptide material was subjected to reversed-phase HPLC separation (Ultimate 3000, Dionex, Idstein, Germany) and fractions containing >80% purity by matrix-assisted laser desorption ionization/time of flight-MS (MALDI-TOF) analysis were combined. The absolute concentration of each purified SIS peptide was then determined by amino acid analysis and capillary zone electrophoresis. To maximize the sensitivity of the assay, the most abundant MRM transitions for each SIS peptide were determined by flow injection analysis on the Agilent 6490 triple quadrupole mass spectrometer with the standard-flow ESI (Jet-Stream) source, as previously described (21). The same optimized MRM parameters were applied to the corresponding natural peptides in the LC/MRM-MS experiments because they exhibit identical electrospray and fragmentation behavior.
DBS Sample Preparation
A pooled sample of whole human blood from five healthy males and five healthy females was purchased from a commercial vendor (Bioreclamation, Westbury, NY). Individual donors were not prescribed fasting and intravenous blood was collected in K2-EDTA coated tubes (367863, Vacutainer Plus, BD Biosciences). The blood was shipped and stored cool (∼4 °C) before use within 1 week. Blood spots of 15 μl volume were made on filter paper (903 Protein Saver Card, Whatman) using an electronic pipette (Proline 100 μl, BioHit). The DBS collection cards were bent so that the back of the card was not in contact with any surface to prevent loss of blood that soaked through the filter paper. All spotting was performed in a biosafety hood (class II, type A, Labconco) and cards were allowed to dry for 4 h. The dried cards were then stored in polyethylene plastic bags with a packet of desiccant (61161–319, sponge humidity indicating, VWR) and a humidity indicator card (VWR International, part 4HIC100). Before removing the DBS samples from the hermetically sealed storage bags, the humidity indicator card was checked to confirm that humidity was less than 10%.
We previously evaluated the effect of 14 different combinations of heat, solvent, chaotropic agents, and surfactants in a quantitative study of their effectiveness in digesting 45 of the 60 blood proteins targeted in this DBS method (26). In this work, we adapted the most successful digestion conditions (1% sodium deoxycholate in an aqueous solution at 37 °C) to also allow for protein extraction from the DBS collection card as follows: Samples were prepared using the entire DBS by excising the 1 cm circle marked on the DBS collection card which was then folded to fit in a 1.5 ml polypropylene vial. Proteins were simultaneously extracted, denatured, and reduced in a 1 ml solution of 25 mm ammonium bicarbonate, 1% sodium deoxycholate, and 5 mm Tris(2-carboxyethyl)phosphine hydrochloride (Thermo Scientific) at 60 °C while vortexing at 1000 rpm (Thermomixer, Eppendorpf) for one hour. An aliquot of the protein extract was then transferred to a new vial and sequentially alkylated with 10 mm iodoacetamide and quenched with 10 mm dithiothreitol for 30 min each at 37 °C. TPCK-treated trypsin (Worthington Biochemical Corporation, Lakewood, NJ) was combined at a concentration of 3.5 μg trypsin per 1 μl of the original blood sample and the digestion was allowed to continue for 16 h at 37°C. Note, DBS samples for the stability experiments were prepared with an alternative trypsin reagent (sequencing grade, Promega, Madison, WI). SIS peptides were spiked in at approximately the same concentrations as the endogenous peptides (as determined from a prior LC/MRM-MS analysis of a nonmatched plasma sample). The sodium deoxycholate was precipitated by the addition of 2% formic acid and pelleted by centrifugation at 4000 × g for 20 min. Samples were desalted by solid-phase extraction (2 mg Oasis HLB μElution plate, Waters, Milford, MA) using the manufacturer's recommended procedure for peptides, and lyophilized overnight (SuperModulyo, Thermo Scientific, Waltham, MA). Before LC/MRM-MS analysis, samples were rehydrated with 0.1% formic acid in water for a 93-fold dilution with respect to the original blood sample.
Samples for calibration curves were generated by serial dilution of whole blood with an isotonic saline solution (0.9% sodium chloride, w/v) before spotting onto DBS collection cards. A total of eight concentration levels spanned a 610-fold range of concentrations, and three different DBS samples were processed in parallel for replicates at each concentration level. As the blood was diluted, the lower viscosity led to an increase in the final spot size on the collection card; however, the entire DBS remained within the 1 cm circle that was removed for analysis. Samples from all concentration levels were spiked with the same amount of concentration-balanced SIS peptides during sample preparation.
LC/MRM-MS Analysis
Peptides were separated using an Agilent 1290 Infinity UHPLC system with a reversed-phase column (Zorbax Eclipse Plus Rapid Resolution HD, 2.1 × 150 mm, 1.8 μm C18 particles, Agilent) heated to 50 °C. Mobile phase A was 0.1% formic acid in water, and mobile phase B was 0.1% formic acid in 90% acetonitrile (LC-MS Chromasolv, Sigma-Aldrich). Partial-loop LC injections of 10 μl corresponded to 107 nL of the original whole blood sample. The flow rate was 0.4 ml/min and the mobile phase gradient was composed of linear steps between the following points (time, %B): 0 min, 3%; 0.1 min, 10%; 20 min, 20%; 25 min, 40%; 26 min, 90%; 29 min, 90%; 30 min, 3%; 35 min, 3%. Peptides were then detected on an Agilent 6490 QQQ mass spectrometer using the following parameters: positive ion mode, 3.5 kV capillary voltage, cone voltage 300 V, drying gas flow rate 15 L/min at 250 °C, nebulizer gas pressure 30 PSI at 250 °C, and Q1 and Q3 set to unit resolution. After screening the transitions for chemical interference (see MRM Transition Screening), the top three interference-free transitions for the natural and SIS forms of the 65 targeted peptides were monitored, giving a total of 390 monitored transitions. The monitoring of each transition was constrained within 1.5 min windows, thus producing a maximum of 60 concurrent transitions. With the total cycle time set to 700 ms, the minimum and maximum dwell times were between 10 ms and 115 ms.
MRM Transition Screening
The five most abundant optimized MRM transitions for each peptide were screened for chemical interference by LC/MRM-MS analysis by monitoring the SIS peptide in buffer, the SIS peptide in digested blood, and the natural peptide in digested blood. The relative intensities for multiple fragments of the same peptide should remain constant for the three conditions listed. Significant changes in the relative intensities indicate the presence of an interfering chemical species. In this study, targeted peptides were considered to have passed this interference screening test if the coefficient of variance (CV) of the relative abundances of multiple transitions per peptide was <25% for at least three transitions per peptide.
Data Analysis
Data was analyzed using MassHunter Quantitative and Qualitative Analysis software (version B.04.00, Agilent). Following MRM transition screening, the most abundant transition was used for quantitation whereas the remaining two transitions served as “qualifiers” to ensure the correct selection of the natural and SIS peptides. All extracted ion chromatograms were manually inspected to ensure correct peak selection and integration by the software. For a concentration level to be included in the calibration curve, the average accuracy of the data points must have been between 80–120% using weighted linear regression (1/x2), with a CV of <20% for the three replicates.
RESULTS AND DISCUSSION
Sample Preparation
DBS sampling is most commonly employed in clinical laboratories to screen newborns for errors in metabolism by targeting small molecule analytes (2). Samples are prepared by spotting a small drop of blood directly onto the DBS collection card and only a portion of the DBS is removed for analysis, using a hole punch. Ideally, the volume of the blood transferred into the assay is defined by the diameter of the punch and therefore does not require precise measurement of the volume of blood that is spotted. There are several variables, however, that may influence how uniformly blood and analytes spread across the filter paper. These include the hematocrit values, blood spot volume, and chromatographic effects. There are conflicting reports in the literature as to whether or not these three variables significantly affect the precision of the analytical result (12, 27). In newborn screening clinics, there is often no correction made for these variables and Lehotay et al. recently estimated that the error associated with the use of DBS samples is ± 30% (28). For applications requiring higher precision, numerous small molecule studies have shown that these variables can be controlled to provide a measurement CV <15% (12). In our initial study of extracting and quantifying multiple proteins from DBS samples, we chose to reduce the number of variables that must be controlled in the sample preparation by spotting precise volumes of blood and removing the entire blood spot for analysis. We also selected the Whatman 903 DBS collection card (formerly the Schleicher and Schuell 903 card) because the properties of this card—including homogenous composition, uniform thickness, purity, and absorbency—have been rigorously characterized through years of use in many nation-wide newborn screening programs (13).
Proteins Observed in DBS Samples
The specificity of an MRM assay results from its ability to isolate a component by the combination of three molecular attributes (LC retention time, Q1 m/z, and Q3 m/z). Because of the complexity of digested blood, even these stringent criteria may not be sufficient to resolve all targeted peptides from other chemical species in the sample matrix. Therefore MRM transitions for each peptide were screened to both confirm the presence of the endogenous peptide in the sample and to identify transitions that contain significant chemical interference. Only natural peptides with at least three interference-free transitions were considered to be “observed” in the sample. Using this criterion, 44 of the 65 targeted peptides, representing 41 proteins, were observed in the DBS sample. A list of the observed peptides along with their interference-free MRM transitions is provided in Supplemental Table S2. Unless otherwise stated, all DBS samples presented in this report were spotted at the same time, with the same original blood sample. Fig. 1 shows extracted ion chromatograms for the 44 observed peptide targets with the natural peptides shown in blue and the SIS peptides shown in red. Although MRM data was acquired for all three interference-free transitions per peptide, only the most abundant transition is shown in the figure for clarity. The inset for Fig. 1 shows the three natural and SIS transitions monitored for a tryptic peptide from alpha-2-HS-glycoprotein, as an example.
Fig. 1.

Extracted ion chromatograms for the 44 observed peptide targets in digested DBS samples with the natural peptides shown in blue and the SIS peptides shown in red. The relative intensity of a natural peptide (LVNEVTEFAK) from albumin, seen at a retention time of 14 min, has been reduced by a factor of 10. The insert shows the 3 transition pairs monitored for a tryptic peptide from alpha-2-HS-glycoprotein.
DBS Protein Quantitation
The overall precision of the assay depends on the reproducibility of both the sample preparation and the method of analysis. When compared with plasma or serum samples, DBS sample preparation contains two additional steps that have the potential to increase the variability. These steps are the spotting of blood onto the DBS collection card and later the extraction of the analytes. Both steps in the procedure must be well-controlled to achieve the precision required for a clinical assay. The precision of this assay was determined by the LC/MRM-MS analysis of eight DBS samples prepared from the same stock of blood. Fig. 2 shows that the measured concentration for all 44 targets had CVs of <15%, with an average CV of 6.4%. These results show that multiple proteins can be extracted from the same DBS sample and analyzed with the high reproducibility required for protein quantitation. In addition, the precision of the LC retention times is directly related to the width of the required MRM transition windows and therefore the total number of transitions that can be monitored in the assay. The average CV for the LC retention times for the 44 peptides was 0.14%. In this experiment, the MRM transition windows could have been reduced by a factor of at least two enabling the expansion of the assay to monitor additional peptides. This excellent retention-time stability is in good agreement with our previous evaluation of standard-flow UHPLC for multiplexed MRM, where the average retention time CV for 85 replicate samples was less than 0.2% (21).
Fig. 2.

The precision of the measured concentration of 44 peptides generated from proteins extracted from replicate DBS samples and analyzed by LC/MRM-MS (n = 8).
The linear dynamic range (LDR) was evaluated by serial dilution of the whole blood before spotting on a DBS collection card, and spiking in a constant amount of concentration-balanced SIS peptides following protein digestion. Three DBS samples for each concentration level were prepared to account for any variation in blood spotting and protein extraction from the DBS collection card, particularly at the lower concentration levels. These samples were analyzed in the order of increasing concentration with one blank injection between different sample concentrations. Fig. 3 shows that the calibration curve for alpha-1-antitrypsin, using the target peptide LSITGTYDLK, was linear across the entire 610-fold range with an R2 value of 0.986. Furthermore, the CV was <15% for the three replicate DBS samples prepared and analyzed for each concentration level. This linear response demonstrates that the protein recovery efficiency and the protein digestion efficiency for the targeted peptide is constant, enabling precise relative protein quantitation. Overall, a linear response was observed for 40 of the 44 observed peptides with an average R2 value of 0.981. The LDR was greater than 10-fold for 27 peptides which represent promising targets for developing a clinical MRM assay. Although many biomarker proteins change by less than 10-fold when comparing disease and healthy populations, it may be necessary to extend the linear dynamic range for some of these targeted proteins by further improvements to the sample preparation procedure.
Fig. 3.
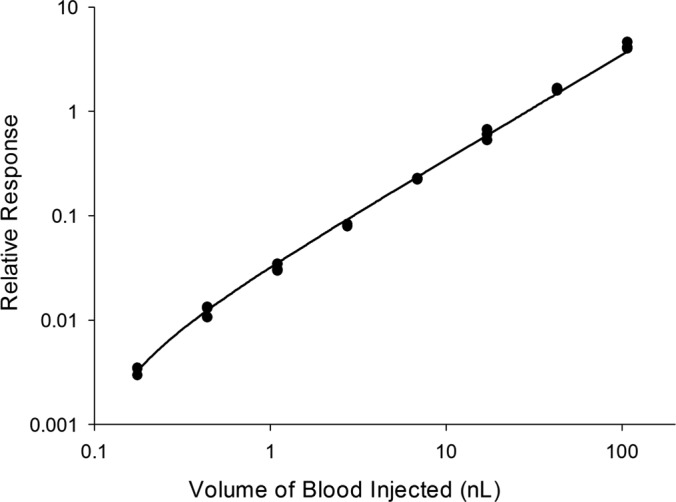
Calibration curve for alpha-1-antitrypsin (target peptide LSITGTYDLK) generated by serial dilution of blood prior to spotting three different DBS samples per concentration level.
Absolute protein concentration in the DBS samples was calculated using the equation derived from each peptide calibration curve, the known SIS peptide concentration, and the processed protein molecule weight (excluding the signaling peptide). Fig. 4 shows the 37 proteins quantified in DBS samples listed in descending order of observed concentration. The overall concentration range of the proteins in this assay spanned more than four orders of magnitude with albumin at 76 mg/ml and apolipoprotein C-I at 5.9 μg/ml. For proteins represented by multiple peptides (alpha-2-HS-glycoprotein and transferrin), the protein concentration reported in Fig. 4 was calculated using the most abundant peptide, which likely corresponds to a portion of the protein that was the most completely digested. Alpha-2-HS-glycoprotein was represented by two peptides, both providing a similar observed protein concentration (APHGPGLIYR 188 μg/ml and HTLNQIDEVK 142 μg/ml). Conversely, the measured concentration of transferrin ranged over 13-fold using three representative peptides (EDPQTFYYAVAVVK 5.1 mg/ml, EGYYGYTGAFR 0.93 mg/ml, and HSTIFENLANK 0.38 mg/ml). It is well-known that the trypsin digestion efficiency for a given sample preparation protocol is not 100% for all proteins (26, 29). As a result, significant discrepancies have been reported for protein concentrations determined using multiple peptides from the same protein (21).
Fig. 4.

Observed concentrations for the 37 proteins quantified in DBS samples listed in descending order of abundance.
We estimated the accuracy of our DBS MRM assay by comparing our measured protein concentration values with established immunoassays. For this comparison, we focused on the 11 FDA approved or cleared protein diagnostic targets with readily available clinical reference ranges from LabCorp that were also quantified by our DBS MRM assay (23, 30). As shown in Fig. 5, four proteins were within the normal reference range (alpha-1-antitrypsin, alpha-2-macroglobulin, ceruloplasmin, and complement C4 beta chain). The protein with the largest deviation was alpha-1-acid glycoprotein 1 which was observed at a concentration 22-fold below the lower reference range. This substantial decrease was expected as glycoproteins are known to be resistant to enzymatic digestion and we previously observed this trend for this particular protein in multiple MRM studies using these digestion conditions (21, 26). Of the six remaining proteins outside the reference ranges, apolipoprotein A-I, complement C1 inactivator, complement C4 gamma chain, and haptoglobin were below the lower reference limit by less than threefold and both Complement C3 and transferrin were observed at an elevated protein concentration within threefold higher than the upper reference limit. Lower protein concentration values may be caused by incomplete digestion and elevated values could be a result of unknown protein isoforms or post-translational modifications. For a further comparison, ceruloplasmin had previously been quantified in DBS samples from seven healthy donors resulting in a range of protein concentrations from 63–180 μg/ml by single-plex LC/MRM-MS and 100–300 μg/ml by immunoassay (16). Although we monitored a different tryptic peptide in our LC/MRM-MS experiments, our calculated ceruloplasmin concentration of 174 μg/ml was similar to the concentrations found in both the previous MRM-based method and the immunoassay. We must remind the reader that a single pooled sample from 10 healthy donors was used to evaluate the analytical metrics of our assay and therefore the comparison of measured protein concentrations to population reference values is only considered to be an estimate.
Fig. 5.

Agreement between protein concentrations measured in DBS samples by MRM (red diamonds) with clinical reference ranges from established immunoassays (black squares) for 11 FDA approved or cleared protein diagnostic targets: 1) alpha-1-acid glycoprotein 1, 2) alpha-1-antitrypsin, 3) alpha-2-macroglobulin, 4) apolipoprotein A-I, 5) ceruloplasmin, 6) Complement C1 inactivator, 7) complement C3, 8) complement C4 beta chain, 9) complement C4 gamma chain, 10) haptoglobin, 11) transferrin.
Ultimately, the value of a clinical assay depends on its ability to distinguish between healthy and diseased patients and this may be accomplished by precise quantitation of the endogenous peptide, relative to the concentration of the internal standard for each analyte. Therefore, the completeness of the enzymatic digestion is not as important as its robustness and reproducibility. We have previously demonstrated that consistent trypsin digestion efficiencies can be achieved across multiple clinical samples in our analysis of 90 cardiovascular disease patient plasma samples by MRM (21). Furthermore, other studies have found that MRM-based quantitation correlated well with immunoassays for several proteins under investigation (31). In this work, we have already demonstrated that this DBS assay meets the first requirement as all quantified peptides were measured with high precision (CVs <15%). Future studies will be required to validate these protein assays on a suitable cohort of patient samples before its clinical value can be directly accessed.
Comparison of DBS and WB Samples
Matching samples of whole blood (WB) were processed in parallel with all of the DBS experiments described above, with the only exception being that the 15 μl of the original blood sample was not spotted on the DBS collection card but rather directly added to the protein extraction buffer. After protein extraction, an aliquot of the WB sample was centrifuged at 600 × g for 10 min and the absence of the pelleted red blood cells indicated that the combination of elevated temperature, denaturing reagents, and vortexing for 1 h was sufficient to lyse these cells before digestion. All MRM transitions were screened for chemical interference using the same procedure applied to DBS samples (see MRM Transition Screening).
Of the 46 peptides observed in the WB sample, 42 were also observed in the DBS samples and the top 3 interference-free MRM transitions for each peptide were exactly the same for both sample formats. This clearly demonstrates that the presence of the filter paper did not add considerable chemical interference to the sample matrix. Alpha-1-acid glycoprotein 1 and fibronectin were exclusive to the DBS sample, whereas apolipoprotein B-100, heparin cofactor II, kiniogen-1, and plasminogen were only observed in the WB sample. An analysis of eight individually prepared WB samples resulted in an average CV of 7.3% for the 46 observed peptide targets, which was similar to the 6.4% observed for the 44 peptide targets in the DBS samples. This confirms that the added steps involved in spotting the sample onto DBS collection cards and later extracting the proteins did not increase the variation in the measurement. Linear calibration curves were obtained for 39 peptides in the whole blood samples, which compares well with the 40 peptides that had a linear response in the DBS samples.
A total of 33 proteins were quantified in both DBS and WB samples and Fig. 6 shows that the measured protein concentration in both samples correlated well with a slope of 0.96 and an R2 value of 0.97. This suggests that highly efficient protein recovery from the DBS collection cards was achieved. Minor differences between the observed protein concentration in DBS and WB samples may have occurred during the drying of the DBS samples. For example, active proteases in the sample may have cleaved our targeted peptides before the samples dried. Although untreated DBS collection cards were used in these experiments, chemically treated DBS cards are available to inactivate endogenous enzymes; however, we have not yet determined their compatibility with MS detection in terms of ionization suppression and chemical interference.
Fig. 6.

Comparison of measured concentration for 33 proteins in matching DBS and WB samples.
Protein Stability in DBS Samples
It is important to determine adequate storage conditions for DBS samples to preserve the integrity of the measured analytes. In our bottom-up approach to quantifying proteins, we are primarily interested in the stability of targeted peptides that serve as surrogates for the proteins and not the intact proteins themselves. We therefore selected peptide targets that lack easily modifiable amino acid residues, such as methionine which may readily undergo oxidation, to improve sample stability. Bowen and coworkers recently monitored the environmental conditions for DBS samples transported without refrigeration on international air cargo shipments (32). Their work demonstrated that samples commonly encounter temperatures between 0 °C and 30 °C and recommended that future method validation cover −20 °C to 40 °C. We evaluated the DBS samples stored at −20 °C, 4 °C and 37 °C for 5 and 10 days, which are two likely time periods required to send samples to a central processing location.
A fresh blood sample was used in this experiment, so a DBS sample was prepared and all MRM transitions were subjected to the same screening for chemical interference as described for the previous DBS sample. Of the 40 peptides quantified in the previous DBS sample, 38 were peptides also observed in these new DBS samples and the most abundant validated MRM transition, used for quantitation, was the same for each peptide. Chemical interference in this new sample most likely prevented the two remaining peptides, corresponding to tryptic fragments from alpha-1-antichymotrypsin and fibronectin, from being observed. An additional 27 DBS samples were spotted at the same time and were evenly divided among the following storage temperatures: −20 °C, 4 °C, and 37 °C. Three replicate DBS samples from each group were processed after 1, 5, and 10 days of storage. Because the DBS samples were spiked with SIS peptides following digestion, any change in the protein recovery or degradation of the target peptide during storage would be clearly observed in the natural/SIS concentration ratios. The “target stability” was calculated by averaging the concentration from the three replicates from day 5 or day 10, normalized to the values obtained on day 1. Peptides were considered stable if their concentration after storage was within 20% of their original measured value.
All 38 peptides were observed in DBS samples after storage for 5 and 10 days at all temperatures investigated. The complete data set is provided in Supplemental Table S4 and is summarized in the histograms in Fig. 7. After 5 days of storage (Fig. 7A), only 3 peptides were observed outside the desired 80–120% range. The concentration of alpha-2-macroglobulin (LLIYAVLPTGDVIGDSAK) increased by 37%, 46%, and 28% after storage at −20 °C, 4 °C, and 37 °C. Likewise, alpha-1-glycoprotein increased by 35% at 37 °C storage and ceruloplasmin increased by 27% at 4 °C storage. An increase in the observed protein concentration could be caused by additional protein denaturation leading to improved tryptic digestion efficiency for the target peptide. Fig. 7B shows that the results obtained after 10 days of storage were similar to those after 5 days. A few peptides are now also observed at concentrations slightly less than 80% of the original concentration. For example, the concentration of complement component C9 decreased by 23 and 21% when stored at 4 °C and 37 °C. The lower concentrations may be caused by reduced protein recovery from the DBS collection card or decomposition of the target peptide.
Fig. 7.

Stability of 38 targeted peptides in digested DBS samples after storage at various temperatures for 5 days (A) and 10 Days (B). The abundance of each peptide is normalized to day 1.
At all temperatures, alpha-2-macroglobulin was observed at elevated levels after 5 days of storage, but was within the targeted 80–120% range after 10 days of storage. The initial signal increase may be because of improved protein denaturation, and therefore digestion efficiency, after short-term storage followed a decrease in signal caused by degradation of the target peptide at longer time intervals. This illustrates the complexity of the multiple degradation pathways that might affect the targeted protein and peptide of interest. Monitoring multiple peptides per protein may help to determine whether the decrease in signal is occurring at the protein or peptide level. In these stability experiments, transferrin was targeted by three peptides and after 10 days of storage at 4 °C, EGYYGYTGAFR was observed to decrease by 23%, whereas HSTIFENLANK increased by 31%. Thus the decrease in EGYYGYTGAFR is peptide-specific and not related to the recovery of transferrin from the DBS collection card. Overall, sample degradation was minimal during storage for 5 and 10 days, and stability was largely independent of storage temperature between −20 °C and 37 °C. We are planning future experiments to monitor the long-term stability of the targeted peptides by re-analyzing the DBS samples after months or possibly years of storage. If necessary, the chemically treated DBS cards mentioned earlier will be evaluated for extending the long-term stability of analytes.
CONCLUSION
The multiplexed quantitation of DBS samples by MS-based methods is an established strategy for population-wide screening for small molecules, and is built on decades of method refinement. Here, we have presented the first evaluation of DBS sampling for highly-multiplexed MRM-based protein quantitation of endogenous proteins. The standard-flow LC/MRM-MS approach provided robust, high-throughput analysis and offers the ability to rapidly expand the target panel. Sample preparation was highly reproducible resulting in high precision measurements of protein concentration that rival those from traditional plasma or serum samples. The majority of the targeted proteins were able to be quantified in DBS samples and exhibited excellent stability over 5–10 days of storage, even at elevated temperatures. These results demonstrate successful MRM-based quantitation of endogenous proteins in DBS samples; however, many steps remain in evaluating the suitability of this technology for clinical applications. The influence of several variables in the blood spotting process (hematocrit, spotting volume, and chromatographic effects) on the precision and accuracy should be determined for each protein in a real-world clinical environment. In addition, larger population sampling and the direct analysis of samples with established methods (e.g., immunoassays) would allow for further assessment of the accuracy of measured protein concentrations. Ultimately, the results of this initial investigation indicate considerable promise for highly-multiplexed, MRM-based protein quantitation in DBS samples and warrant the continued development required for large-scale clinical applications.
Supplementary Material
Acknowledgments
We thank Genome Canada, Genome BC, and the Western Economic Diversification program for funding the Science and Technology Innovation Centre in Proteomics. We are also grateful to Carol E. Parker for helpful discussions during the preparation of this manuscript.
Footnotes
 This article contains supplemental Tables S1 to S4.
This article contains supplemental Tables S1 to S4.
1 The abbreviations used are:
- DBS
- dried blood spot
- IEF
- isoelectric focusing
- HPLC
- high performance liquid chromatography
- SIS
- stable isotope-labeled internal standards
- MRM
- multiple reaction monitoring
- LDR
- linear dynamic range
- WB
- whole blood.
REFERENCES
- 1. Chace D. H., Kalas T. A., Naylor E. W. (2003) Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns. Clin. Chem. 49, 1797–1817 [DOI] [PubMed] [Google Scholar]
- 2. Sahai I., Marsden D. (2009) Newborn Screening. Crit. Rev. Clin. Lab. Sci. 46, 55–82 [DOI] [PubMed] [Google Scholar]
- 3. Merton G., Jones K., Lee M., Johnston A., Holt D. W. (2000) Accuracy of cyclosporin measurements made in capillary blood samples obtained by skin puncture. Therapeutic Drug Monitoring 22, 594–598 [DOI] [PubMed] [Google Scholar]
- 4. Woods K., Douketis J. D., Schnurr T., Kinnon K., Powers P., Crowther M. A. (2004) Patient preferences for capillary vs. venous INR determination in an anticoagulation clinic: a randomized controlled trial. Thrombosis Res. 114, 161–165 [DOI] [PubMed] [Google Scholar]
- 5. Burnett J. E. (2011) Dried blood spot sampling: practical considerations and recommendation for use with preclinical studies. Bioanalysis 3, 1099–1107 [DOI] [PubMed] [Google Scholar]
- 6. (2010) Federal Register. 49 CFR part 173 Shippers General Requirements for Shipments and packaging. Section 173.134(b) http://ecfr.gpoaccess.gov/cgi/t/text/text-idx?c=ecfr&sid=8662f082afd4c81e20b32e6f54361a73&rgn=div8&view=text&node=49:2.1. 1.3.9.4.25.13&idno=49, Access date: June 2012
- 7. (2011–2012) World Health Organization. Guidance on regulations for the transport of infectious substances. Geneva: HO/HSE/IHR/2010.8 [Google Scholar]
- 8. McDade T. W., Williams S., Snodgrass J. J. (2007) What a drop can do: dried blood spots as a minimally invasive method for integrating biomarkers into population-based research. Demography 44, 899–925 [DOI] [PubMed] [Google Scholar]
- 9. Tanna S., Lawson G. (2011) Analytical methods used in conjunction with dried blood spots. Anal. Meth. 3, 1709–1718 [Google Scholar]
- 10. Millington D. S., Kodo N., Norwood D. L., Roe C. R. (1990) Tandem mass spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. Inherited Metabol. Dis. 13, 321–324 [DOI] [PubMed] [Google Scholar]
- 11. Garg U., Dasouki M. (2006) Expanded newborn screening of inherited metabolic disorders by tandem mass spectrometry: clinical and laboratory aspects. Clin. Biochem. 39, 315–332 [DOI] [PubMed] [Google Scholar]
- 12. Li W., Tse F. L. S. (2010) Dried blood spot sampling in combination with LC-MS/MS for quantitative analysis of small molecules. Biomed. Chromatog. 24, 49–65 [DOI] [PubMed] [Google Scholar]
- 13. Keevil B. G. (2011) The analysis of dried blood spot samples using liquid chromatography tandem mass spectrometry. Clin. Biochem. 44, 110–118 [DOI] [PubMed] [Google Scholar]
- 14. Daniel Y. A., Turner C., Haynes R. M., Hunt B. J., Dalto R. N. (2007) Quantification of hemoglobin A2 by tandem mass spectrometry. Clin. Chem. 53, 1448–1454 [DOI] [PubMed] [Google Scholar]
- 15. Boemer F., Ketelslegers O., Minon J. M., Bours V., Schoos R. (2008) Newborn screening for sickle cell disease using tandem mass spectrometry. Clin. Chem. 54, 2036–2041 [DOI] [PubMed] [Google Scholar]
- 16. deWilde A., Sadilkova K., Sadilek M., Vasta V., Hahn S. H. (2008) Tryptic peptide analysis of ceruloplasmin in dried blood spots using liquid chromatography–tandem mass spectrometry: application to newborn screening. Clin. Chem. 54, 1961–1968 [DOI] [PubMed] [Google Scholar]
- 17. Kehler J., Akella N., Citerone D., Szapacs M. (2011) Application of DBS for the quantitative assessment of a protein biologic using on-card digestion LC–MS/MS or immunoassay. Bioanalysis 3, 2283–2290 [DOI] [PubMed] [Google Scholar]
- 18. Sleczka B. G., D'Arienzo C. J., Tymiak A. A., Olah T. V. (2012) Quantitation of therapeutic proteins following direct trypsin digestion of dried blood spot samples and detection by LC/MS-based bioanalytical methods in drug discovery. Bioanalysis, 29–40 [DOI] [PubMed] [Google Scholar]
- 19. Anderson L., Hunter C. L. (2006) Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol. Cell. Proteomics 5, 573–588 [DOI] [PubMed] [Google Scholar]
- 20. Addona T. A., Abbatiello S. E., Schilling B., Skates S. J., Mani D. R., Bunk D. M., Spiegelman C. H., Zimmerman L. J., Ham A. J. L., Keshishian H., Hall S. C., Allen S., Blackman R. K., Borchers C. H., Buck C., Cardasis H. L., Cusack M. P., Dodder N. G., Gibson B. W., Held J. M., Hiltke T., Jackson A., Johansen E. B., Kinsinger C. R., Li J., Mesri M., Neubert T. A., Niles R. K., Pulsipher T. C., Ransohoff D., Rodriguez H., Rudnick P. A., Smith D., Tabb D. L., Tegeler T. J., Variyath A. M., Vega-Montoto L. J., Wahlander A., Waldemarson S., Wang M., Whiteaker J. R., Zhao L., Anderson N. L., Fisher S. J., Liebler D. C., Paulovich A. G., Regnier F. E., Tempst P., Carr S. A. (2009) Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat. Biotechnol. 27, 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Domanski D., Percy A. J., Yang J., Chambers A. G., Hill J. S., Cohen Freue G. V., Borchers C. H. (2012) MRM-based Multiplexed Quantitation of 67 Putative Cardiovascular Disease Biomarkers in Human Plasma. Proteomics 12, 1222–1243 [DOI] [PubMed] [Google Scholar]
- 22. Percy A. J., Chambers A. G., Yang J., Domanski D., Borchers C. H. (2012) Comparison of standard-flow and nano-flow liquid chromatography systems for MRM-based quantitation of putative plasma biomarker proteins. Anal. Bioanal. Chem. 404, 1089–1101 [DOI] [PubMed] [Google Scholar]
- 23. Anderson N. L. (2010) The clinical plasma proteome: a survey of clinical assays for proteins in plasma and serum. Clin. Chem. 56, 177–185 [DOI] [PubMed] [Google Scholar]
- 24. Kuzyk M. A., Smith D., Yang J., Cross T. J., Jackson A. M., Hardie D. B., Anderson N. L., Borchers C. H. (2009) Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics 8, 1860–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kuzyk M. A., Parker C. E., Borchers C. H. (2012, in press) Development of MRM based assays for plasma proteins. In: Backvall H., ed. Meth. Mol. Biol., Humana Press; [DOI] [PubMed] [Google Scholar]
- 26. Proc J. L., Kuzyk M. A., Hardie D. B., Yang J., Smith D. S., Jackson A. M., Parker C. E., Borchers C. H. (2010) A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J. Proteome Res. 9, 5422–5437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mei J. V., Alexander J. R., Adam B. W., Hannon W. H. (2001) Use of filter paper for the collection and analysis of human whole blood specimens. J. Nutrition 131, 1631S–1636S [DOI] [PubMed] [Google Scholar]
- 28. Lehotay D. C., Hall P., Lepage J., Eichhorst J. C., Etter M. L., Greenberg C. R. (2011) LC-MS/MS progress in newborn screening. Clin. Biochem. 44, 21–31 [DOI] [PubMed] [Google Scholar]
- 29. Klammer A. A., MacCoss M. J. (2006) Effects of modified digestion schemes on the identification of proteins from complex mixtures. J. Proteome Res. 5, 695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. LabCorp (2012) LabCorp: Laboratory Corporation of America. https://www.labcorp.com/wps/portal/, Access date: October 2012
- 31. Hoofnagle A. N., Becker J. O., Oda M. N., Cavigiolio G., Mayer P., Vaisar T. (2012) Multiple-reaction monitoring-mass spectrometric assays can accurately measure the relative protein abundance in complex mixtures. Clin. Chem. 58, 777–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bowen C. L., Dopson W., Kemp D. C., Lewis M., Lad R., Overvold C. (2011) Investigations into the environmental conditions experienced during ambient sample transport: impact to dried blood spot sample shipments. Bioanalysis 3, 1625–1633 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.